

Abstract
Glutamate racemases (GR) are members of the family of bacterial enzymes known as cofactor-independent racemases and epimerases and catalyze the stereoinversion of glutamate. D-amino acids are universally important for the proper construction of viable bacterial cell walls, and thus have been repeatedly validated as attractive targets for novel antimicrobial drug design. Significant aspects of the mechanism of this challenging stereoinversion remain unknown. The current study employs a combination of MD and QM/MM computational approaches to show that the GR from H. pylori must proceed via a pre-activation step, which is dependent on the enzyme’s flexibility. This mechanism is starkly different than previously proposed mechanisms. These findings have immediate pharmaceutical relevance, as the H. pylori GR enzyme is a very attractive allosteric drug target. The results presented in this study offer a distinctly novel understanding of how AstraZeneca’s lead series of inhibitors cripple the H. pylori GR’s native motions, via prevention of this critical chemical pre-activation step. Our experimental studies, using SPR, fluorescence and NMR WaterLOGSY show that H. pylori GR is not inhibited by the uncompetitive mechanism originally put forward by Lundqvist et al. The current study supports a deep connection between native enzyme motions and chemical reactivity, which has strong relevance to the field of allosteric drug discovery.
Keywords: Glutamate racemase, carbon acids, MD, QM/MM, allostery, allosteric inhibition, SPR, enzyme flexibility
Graphical Abstract

Glutamate racemases (GR) are members of the family of bacterial enzymes known as cofactor-independent racemases and epimerases. Using a combination of computational and experimental approaches we show that the GR from H. pylori must proceed via a pre-activation step and that H. pylori GR is not inhibited by the uncompetitive mechanism originally put forward. The current study supports a deep connection between native enzyme motions and chemical reactivity, which has strong relevance to the field of allosteric drug discovery.
Introduction
There has been great interest in the source of the catalytic power of cofactor independent racemases and epimerases, which represent a variety of highly attractive antimicrobial drug targets. Glutamate racemase (GR) is an enzyme ubiquitous to all bacteria and is responsible for the production of d-glutamate (d-Glu), which is an essential component of the peptidoglycan layer of bacterial cell walls. GR performs a highly exotic “two-base” racemization reaction; in which it catalyzes the stereoinversion of glutamate without the assistance of any cofactor. Two cysteine residues flank the Cα of the glutamate substrates bound in the active site of GR. The current consensus of over 25 years of research on this enzyme is that a minimal mechanism entails abstraction of the Cα-proton by a thiolate of Cys74 in the d-to-l direction with concomitant protonation of Cα by the Cys185 thiol in the other direction.[1] This double proton transfer racemization is fully reversible, with the two catalytic Cys switching ionization states in the l-to-d direction, and has been the topic of much research;[2] analogous mechanisms are seen in the proline racemase and diaminopimelate (DAP) epimerases as well.[3] However, this simplistic picture is not consistent with a more detailed atomistic framework, since it entails the acidification of a Cα-proton bond that has a pKa value projected to be ~ 29, and a catalytic base (a Cys thiolate) that has a pKa dramatically lower (normally ~ 9).[4] Several previous studies have noted the importance of GR’s flexibility to achieving catalytic turnover. [4a, 5]
If one examines similar enzyme catalyzed reactions, which are exceedingly more well characterized than any of the cofactor independent racemases, such as mandelate racemase (MR), muconate lactonizing enzyme (MLE), and enolase,[6] we always see the very important role of Cα acidification by either metal chelation or close placement of a general acid to the α-carboxylate in these systems. Fascinatingly, this approach to Cα-proton acidification has not previously been proposed for any of the cofactor independent racemases, though (as described below) this is very feasible and reasonable. In the enolase family of enzymes, acidification is mostly achieved through coordination of α-carboxylate to a divalent metal (e.g., Mg2+), while in PLP-dependent enzymes this is achieved via the electron withdrawing potential of the aldimine intermediate formed with the substrate. Additionally, there is some evidence of low barrier hydrogen bonding (strong hydrogen bonding) in the form of hydrogen bond donation to the substrate carboxylate oxygen. [2e, 7]
A variety of past computational studies on GR have employed molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) geometry optimizations on five different GR enzymes from the species M. tuberculosis, B. subtilis, B. anthracis, A. pyrophilus, and H. pylori. Puig et al., used relatively short simulation times (100s of ps) with semi empirical basis sets; [8] Malapati et al., employed simulation times in the tens of nanoseconds, and also, used semi empirical basis sets; [9] Spies et al., employed ~ 2 ns of simulation time, and used a 6–31G** basis set, but with a relatively small QM region. [4a]
In the current study we expand on these previous efforts, employing a larger DFT (B3LYP/6–31G*) basis set, and enhanced MD sampling and snapshot clustering on the GR from H. pylori. We find a mandatory, pre-activation step that shows mechanistic features in common with more well understood systems, such as MR, MLE, and enolase. Our current computational study provides the longest atomistic MD simulations (150 ns) to date, the highest level of QM/MM theory, and a larger QM region than the previous ab initio study. The QM/MM geometry optimizations reveal a pre-activation step that acidifies the Cα-proton via a protonation of the α-carboxylate oxygen from the catalytic acid/base Cys185.
Since this obligatory pre-activation step (protonation of the α-carboxylate) occurs through a relatively large conformational change (a near 180° reorientation of the S-Cβ dihedral bond of the catalytic Cys185) which occurs in the 10s of nanosecond time scale, it is unlikely that this would have been captured in the previous MD/QM/MM computational studies.
The immediate pharmaceutical importance of these findings is that they provide a distinct alternative hypothesis about how H. pylori GR (a drug target for gastric cancer) is allosterically inhibited by small molecules that bind in a cryptic pocket, located on the opposite face to the entrance to the active site. Indeed, these findings also provide a new rationale for why GR needs to be so flexible, and by extension how the cofactor independent racemase class of enzymes may acidify Cα-proton bonds without the usual metal and/or electron sink capabilities. Flexibility is key to reactivity for this enzyme, for a number of reasons, including these initial acid/base reactions, as well as the desolvation of the active site upon substrate binding. We experimentally tested the hypothesis that an allosteric drug named “compound A” (Figure 1a), identified by AstraZeneca in an HTS campaign, is not working by the mechanism proposed by Lundqvist et al.,[5] Briefly, the mechanistic hypothesis for inhibition put forth by Lundqvist et al., was focused on a physical step of hinge motion (between two different monomer domains of H. pylori GR), which was thought to control substrate access to the active site. It was suggested that Compound A (the allosteric inhibitor) only binds to a form of the enzyme that is already bound to substrate (an uncompetitive mechanism), essentially stabilizing and trapping the product (i.e., a physical step in the catalytic cycle). The current study describes the elucidation of a radically different mechanism for allosteric inhibition, which works by dampening the flexibility of H. pylori GR in such a way that the enzyme is unable to acidify Cα-proton in this critical pre-activation step. Taken together, the computational and experimental studies (employing surface plasmon resonance (SPR), fluorescence spectroscopy, and NMR WaterLOGSY) presented herein, disprove the uncompetitive inhibition model of compound A-inhibition of GR, and suggests that the allosteric inhibitor restricts the protonation of the substrate carboxylate, crippling the enzyme’s ability to stereoinvert the d-glutamate substrate. These findings change the way we think about the mechanism of action of remote allosteric inhibition of enzymes.
Figure 1.
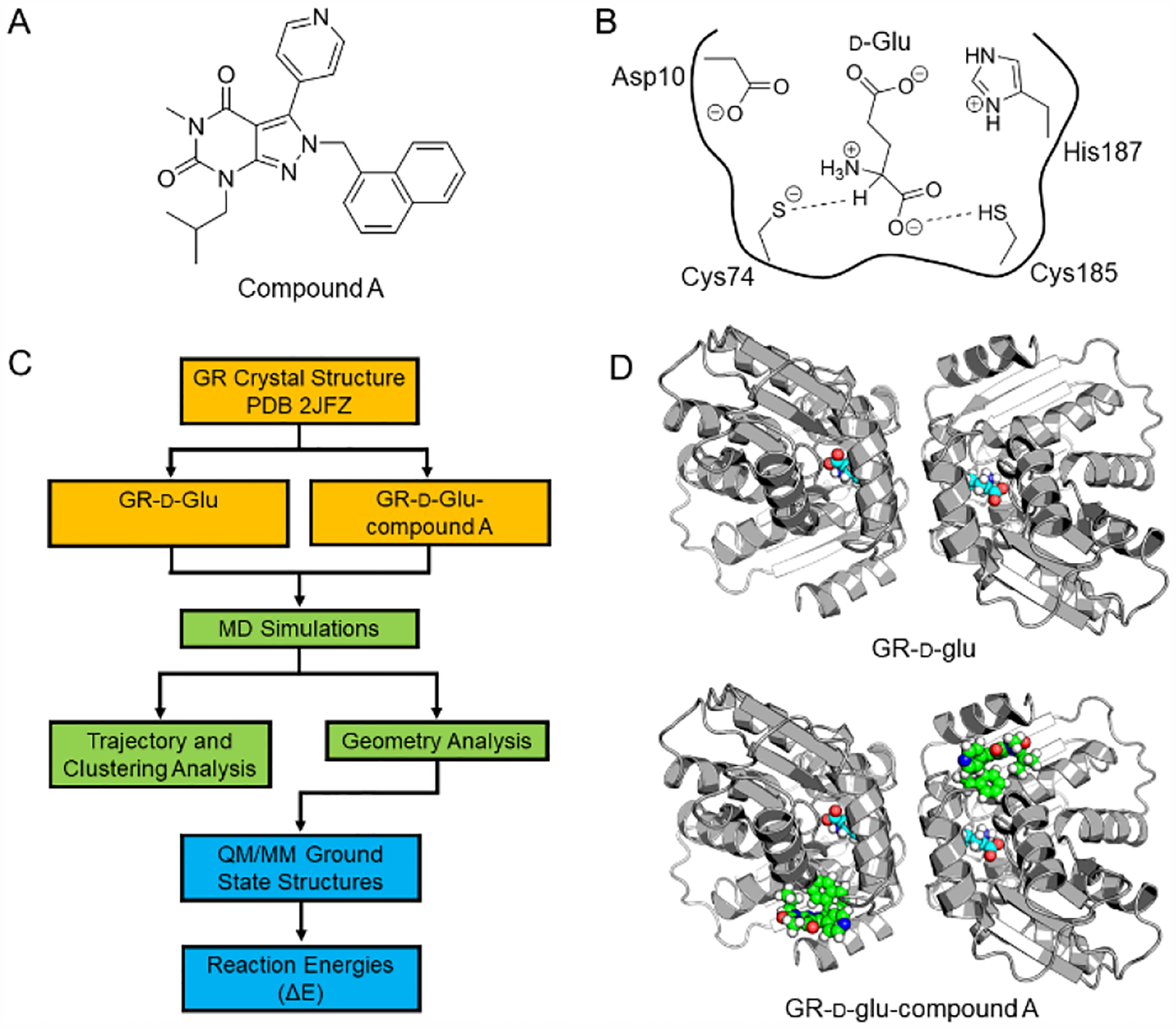
a) The pyrazolopyrimidinedione GR inhibitor (compound A) discovered by AstraZeneca. [5] Compound A binds to a cryptic allosteric pocket remote from the active site. b) The active site of GR with the d-glutamate substrate. The dashed lines represent the two proton transfer steps discussed in this manuscript. Before the Cα proton removal by the Cys74 catalytic base occurs, Cys185 acts as a general acid, protonating the α-carboxylate of the glutamate to dramatically lower the Cα pKa. The four residues shown, along with the d-glutamate, are the atoms modeled in the QM region of our QM/MM computational studies. c) The multi-layered computational workflow implemented in this study. The crystal structure (PDB 2JFZ) of GR-d-glu-compound A was used as the starting structure for all calculations. For the GR-d-glu complex, compound A was removed before MD equilibration. The YAMBER3 force field was employed in the YASARA Structure package (YASARA Biosciences), [10] while the QM/MM calculations were performed with QSite/Jaguar (Schrödinger LLC).[11] See the full details in the text and the SI. d) The dimeric structure of GR, in which the two active sites face one another (in the upper right panel, one can see the two glutamate substrates rendered in space filling mode). The lower panel depicts compound A (green space filling mode) bound in the allosteric pocket for each monomer.
Results and Discussion
Computational Findings.
A multi-layered computational approach was utilized to study the Cα-proton transfer between d-glutamate and the thiolate of Cys74 (Figure 1b). All atom classical MD simulations, structural clustering of MD snapshots and data mining to identify near attack conformations, followed by QM/MM (DFT/B3LYP/6–31G*) geometry optimizations were employed on the GR system. This layered approach provided both the ability to obtain greater sampling than previous studies, while applying a high level basis set to promising geometries for the QM region (Figure 1c). This layered approach is necessary in GR, due to its notable flexibility and the need to provide a relatively rigorous analysis of transferring protons in a very polar, yet mostly desolvated active site.
MD simulations of GR-d-Glu.
MD simulations were performed on the GR dimer with d-glutamate using the YAMBER3 knowledge-based force field implemented in YASARA Biosciences [10b, 10c] v.17.1.28 (Figure 1d). The Cys74 was represented as a thiolate in the MD simulations to capture the ground state for the abstraction of the cα proton in the QM/MM geometry optimizations. The simulation ran in triplicate for a total of 150 ns, and snapshots were collected every 100 ps. The ensemble achieved a global root-mean-square deviation (RMSD) convergence in each replicate (Figure S1). To study the proton transfer between Cα(d-Glu) and S(Cys74), geometries of Cys74 and d-glutamate were analyzed to identify structures for QM/MM geometry optimizations; the key geometries tabulated were the distance between S(Cys74)---Hα(d-Glu) and S(Cys74)---Cα(d-Glu) as well as the transfer angle between S(Cys74), Hα(d-Glu), and Cα(d-Glu). The likelihood of the Cα(d-Glu) and S(Cys74) proton transfer to occur was highest when the distance between proton donor and acceptor was ~ 3 Å. While the transfer angle also plays a role, there was more variability between the snapshots selected for QM/MM geometry optimizations. The optimal transfer angle has been shown to be between 120° and 140° for other GR-d-glu ground state snapshots,17 however, for H. pylori GR the minimum angle was found to be smaller, 107 ± 5°. These geometries are highlighted in the 2-D histogram plot of the distances and angles between d-Glu/Cys74 (Figure 2a).
Figure 2.
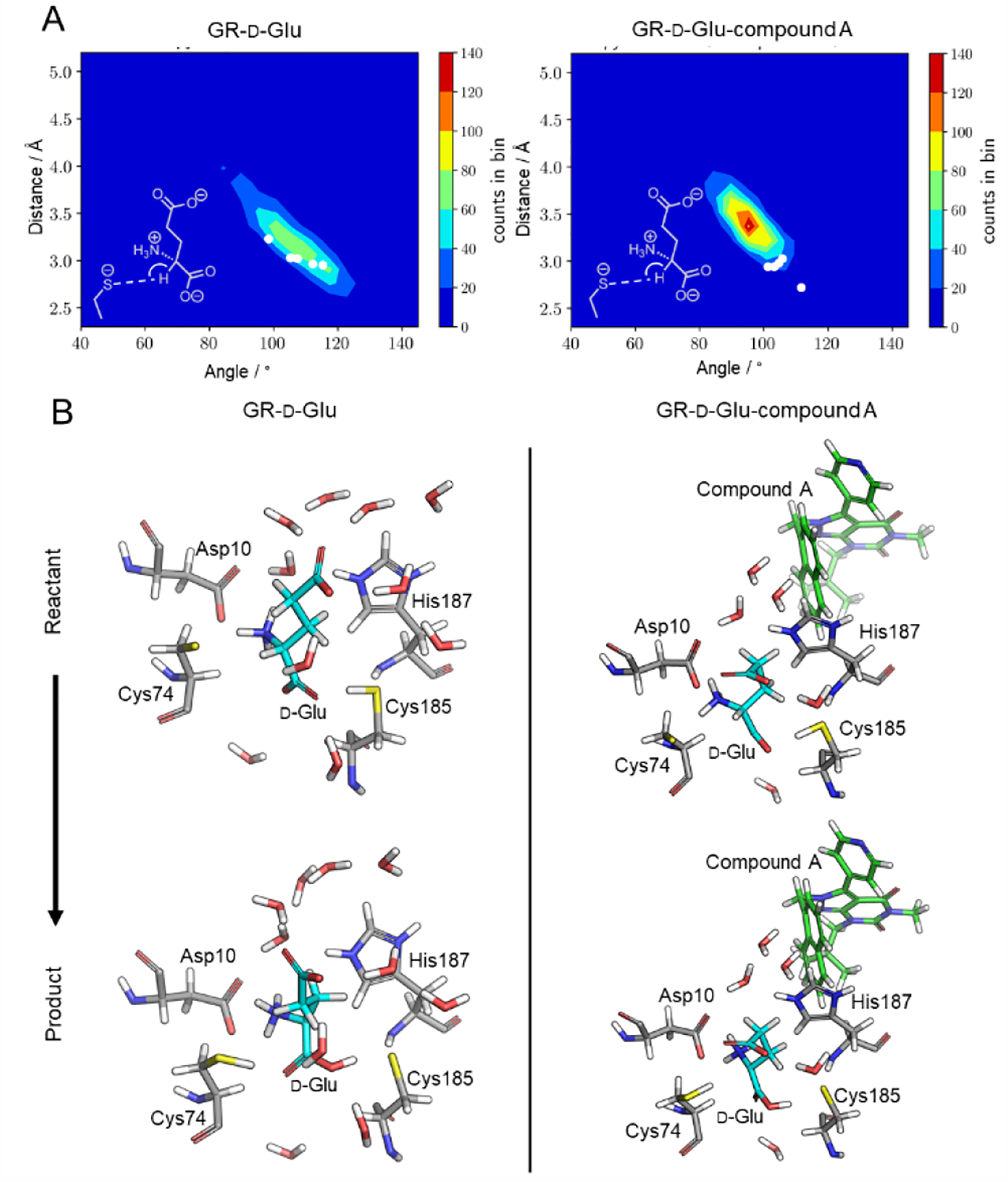
a) 3-D contour plots of the proton transfer distances and angles between Cys74 and d-glutamate in GR-d-glu (left) and GR-d-glu-compound A (right). The plotted distance is between S(Cys74)---Hα(d-Glu) (the dashed line in the Lewis structure inset) and the plotted angle is the transfer angle between S(Cys74)-Hα-Cα (the arch in the Lewis structure inset). The white dots represent the snapshots from the MD simulations selected for QM/MM geometry optimizations. Reasonable proton transfer geometry for QM/MM optimization is usually < 3.0 Å between donor and acceptor atoms, and > 105°. b) Representative QM/MM geometry optimized ground state reactant (top) and product (bottom) structures for GR-d-glu (left) and GR-d-glu-compound A (right). QM/MM geometry optimizations were performed in QSite (Schrödinger LLC) [11b] at the DFT/B3LYP/6–31G* level, full details in the text and the SI. The active site is shown using a stick representation. Compound A is represented by green sticks and water molecules are represented by the white and red sticks, both molecules were not included in the QM region. Structures are shown for the QM/MM geometry optimization for one snapshot while all other snapshots are shown in the SI.
Ab initio QM/MM geometry optimizations of GR-d-Glu.
MD snapshots were selected to study the proton transfer between Cα(d-Glu) and S(Cys74) using QM/MM geometry optimizations, performed in QSite (Schrödinger LLC) [11b] on five structures from the GR-d-Glu ensemble. These five structures were selected based on the distance between S(Cys74)---Hα(d-Glu) and Hα(d-Glu)---Cα(d-Glu) as well as the transfer angle between S(Cys74), Hα(d-Glu), and Cα(d-Glu) (Table S1), and are represented by white dots in Figure 2a. These starting structures from MD snapshots where then used as starting points for QM/MM geometry optimizations of ground state reactants and ground state products. The reactant structures contain the proton bonded to the Cα of d-glutamate while the product structures have the proton abstracted and bound to S(Cys74). For all optimizations, the QM region (Figure 1b) consisted of d-glutamate and the side chains of residues Asp10, Cys74, Cys185, and His187. Vibrational frequency calculations at the B3LYP/6–31G* level were performed on all ground state structures to ensure that a ground state was indeed captured (Table S2).
Ground state reactant and product structures were obtained for all structures. Representative ground state reactant and product structures collected from the QM/MM geometry optimization for GR-d-Glu are shown in Figure 2b, while the other reactant and product structures obtained are shown in Figure S2. A major finding was the degree to which the orientation of the proton on S(Cys185) significantly impacted the convergence for a ground state product structure to be captured. To acquire a ground state product structure the proton on S(Cys185) must be donated to the α-carboxylate of d-glutamate during the overall reaction.
This proton transfer from S(Cys185) can be classified as a type of pre-activation step, which has been identified in other systems such as MR, MLE, and enolase. The pKa value of the Cα-proton in d-glutamate has been projected to be ~ 29. [4] Using the pKa utility of Jaguar (Schrödinger LLC), [11a] we calculated the pKa of the Cα-proton of free d-glutamate in aqueous solution with the α-carboxylate being protonated and deprotonated, respectively. As shown in Figure 3a, the pKa of the Cα-proton in d-glutamate decreases from 22.8 to 14.4 when the α-carboxylate is protonated. Lowering the pKa of the Cα-proton allows for an enormous reduction in the unfavourability of proton transfer between Cα(d-Glu) and S(Cys74). As in the case of related enzymes, such as MR, MLE and enolase, these are integral parts of the carbon acidification. The significant contribution of this key pre-activation step may provide a rationale for the extreme flexibility of GR, as described below.
Figure 3.

a) The pKa of the Cα-proton was calculated using Jaguar (Schrödinger LLC). [11a] This ab initio calculation employed the DFT/B3LYP/6–31G* method. The pKa of the Cα-proton was calculated for d-glutamate with the α-carboxylate not protonated (left) and protonated (right), highlighted in yellow. When the α-carboxylate is protonated, the pKa of the Cα-proton is significantly lower, which radically changes the difficulty of the Cα proton abstraction by the thiolate of Cys74. b) The RMSF (root mean square fluctuation about the average atomic position for each heavy atom, calculated using crystallographic B-factors) values for the crystal structure 2JFX (red; GR-D-glu) and 2JFZ (black; GR-D-glu-compound A), middle panel. The RMSF values fluctuate more in the uninhibited system, red line. Additionally, RMSF values calculated from MD simulations for GR-d-glu (red) and GR-d-glu-compound A (black) follow similar trends as the crystal structures (middle panel).
With the α-carboxylate of d-glutamate protonated, the Cα-proton is able to be transferred to S(Cys74) (i.e., the overall reaction includes two proton transfers) and the ΔE values were calculated for this reaction. Ground state product complexes could not be obtained in the absence of the initial proton transfer to the α-carboxylate oxygen. Four out of five of the GR-d-Glu structures resulted in favorable ΔE values for the transfer of Cα-proton to Cys74, Table S3.
The chemically productive snapshots critically depended on a number of geometries, which exist in only a small subset of the MD ensemble. The high degree of flexibility of GR, exhibited both in MD simulations and in terms of the experimental literature, suggest that small molecule allosteric inhibitors may be acting by restricting these key native motions. The H. pylori GR enzyme itself has been the target of a drug discovery campaign by AstraZeneca, which identified an allosteric inhibitor (compound A) that binds to a cryptic allosteric pocket that is remote from the active site.[5] Interestingly, the crystal structures of GR-d-glu and GR-d-glu-compound A are nearly identical with an ~ 0.8 Å RMSD based on the CA/CB distances (see SI for full details). Comparing the root-mean-square fluctuation (RMSF) (Figure 3b) values of the two crystal structures (PDBs 2JFX and 2JFZ), calculated from their B-factor values, shows there are distinct differences when the inhibitor is present. This was not addressed in the original publication,[5] and lends further support for the idea that dynamics may be an important and unexamined aspect of the allosteric inhibition of compound A. The findings discussed below show that these differences of motions are found to not only impair the chemistry of the active site but the pre-activation step as well, which provides a completely fresh perspective on small molecule allosteric control of this class of enzymes.
MD simulations of GR- d-glu-compound A.
The global dynamics of the enzyme were found to be highly restricted in the GR- d-glu-compound A complex, relative to the GR- d-glu complex. The radius of gyration for the dimer demonstrated that when compound A is present, there is overall less movement and flexibility throughout the entire enzyme (Figure 4a). The GR- d-glu-compound A ensemble has an average radius of gyration of 24.8 ± 0.1 Å compared to 25.3 ± 0.2 Å for the GR-d-Glu ensemble.
Figure 4.

a) Radius of gyration (Rg) determined using the MDAnalysis toolkit. [12] The Rg is much more consistent for the GR-d-glu-compound A structure (black) compared to the GR-d-glu structure (red). b) A heat map of coupled motion between the catalytic base, Cys74, and every other residue in the GR-d-glu (upper panel) and GR-d-glu-compound A (lower panel) mapped onto the time averaged structures from MD simulations, respectively. Blue indicates a positive correlation, while red indicates negative correlation. The heat maps were generated from dynamic cross correlation matrices (DCCM), and the details of this are described in the Computational Methods section. When compound A is absent, there is more correlation between Cys74 and the surrounding residues. c) Clustering results from the MD simulation, determined from UCSF Chimera, [13] shows that there is more similarity between the GR-d-glu-compound A structures (black) compared to the GR-d-glu structures (red).
The dynamic relationship between all residues was observed through the dynamic cross-correlation matrix (DCCM). This matrix displays how each residue’s motion correlates with every other residue during the MD simulation. This is usually represented as a full matrix, capturing all the individual residue’s correlated motions (Figure S3). However, in order to show how the presence of compound A affects the global motions coupled to the catalytic base (Cys74), we graphically present the DCCM values with respect to Cys74 for the GR-d-glu and GR-d-glu-compound A complexes, respectively, Figure 4b. The DCCM values have been normalized, anti-correlation values are represented by shades of red and positive correlation values are represented by shades of blue. Cys74 is the darkest blue since Cys74 is perfectly correlated with itself. It is clear from a visual inspection that there is a much stronger correlation between Cys74 and other residues from the GR-d-glu ensemble compared to the GR-d-Glu-compound A ensemble. The superimposition of these correlated motion values onto the structure itself is very illuminating. Indeed, it suggests that the native enzyme contains strong coupling with very distant residues, extending out in a 12 Å sphere in the native GR. The dynamics of the allosterically inhibited system are starkly different, and show very dampened positive correlated motions, in which only a few neighboring residues’ motions are coupled to the catalytic Cys74. Additionally, only in the allosterically inhibited system does one observe a negatively coupled motion.
The restriction of enzyme dynamics in the GR-d-Glu-compound A structures led to less conformational freedom between all snapshots. Using the method of Pettersen et al, [13] the snapshots from the GR-d-Glu and GR-d-Glu-compound A MD simulations were clustered based on the active site residues: Asp10, Cys74, Cys185, and His187, our clustering parameters are detailed in the SI. The MD clustering analysis resulted in 24 different clusters for the GR-d-Glu ensemble but only 16 clusters for the GR-d-Glu-compound A ensemble (Figure 4c). The top three clusters from the GR-d-Glu system represented 47% of all the snapshots while the top three clusters from the GR-d-Glu-compound A simulation represented 78% of the overall snapshots.
A major finding from the native QM/MM geometry optimizations was the importance of the orientation of the proton on the Cys185 thiol. This proton must transfer to the α-carboxylate of d-glutamate for the Cα-proton transfer to occur between Cα(d-Glu) and S(Cys74). In the GR-d-Glu compound A system, the proton on the Cys185 thiol frequently rotates between conformers facing towards and away from the α-carboxylate of d-glutamate, respectively. Once again, when selecting which structures to use for QM/MM to study the proton transfer between Cα(d-Glu) and S(Cys74), the same key geometries between Cys74 and d-glutamate were tabulated and are shown in a 2-D histogram (Figure 2a, right panel). The key geometries tabulated were the distance between S(Cys74)---Cα(d-Glu) and S(Cys74)---Hα(d-Glu) as well as the transfer angle between S(Cys74), Hα(d-Glu), and Cα(d-Glu). Additionally, a critical geometry was the orientation of the proton on the Cys185 thiol and only snapshots where the proton was pointing towards the α-carboxylate of d-glutamate were selected for further analysis, since only these systems lead to Cα-proton transfer.
Ab initio QM/MM geometry optimizations of GR-d-Glu-compound A.
To determine how compound A affects Cα-proton abstraction, the proton transfer between Cα(d-Glu) and S(Cys74) was once again studied using QM/MM geometry optimizations. Five structures from the GR-d-Glu-compound A ensemble were selected based on the distances between S(Cys74)---Hα(d-Glu) and S(Cys74)---Cα(d-Glu) as well as the transfer angle between S(Cys74), Hα(d-Glu), and Cα(d-Glu) (Table S1), and are represented by white dots in Figure 2a (right panel). The selected MD snapshots were used as starting points for QM/MM geometry optimizations to determine reactant and product ground states. A representative illustration of the reactant and product ground state structures is shown in Figure 2b, while snapshots 2–5 are shown in Figure S4 for the reactant and product structures, respectively.
From the native QM/MM geometry optimizations, it was revealed that the orientation of the thiol proton on S(Cys185) significantly impacted the convergence to the ground state product structure. Surprisingly, in the GR-d-Glu-compound A complex this key thiol was very dynamic, and often was located pointing in a non-productive orientation (i.e., unable to protonate the α-carboxylate oxygen of glutamate). The thiol of Cys185 in the GR-d-Glu complex, on the other hand, was mostly poised to donate its proton to the α-carboxylate oxygen of glutamate. Additionally, when the S(Cys185) proton is pointing towards the α-carboxylate of d-glutamate the H---O distances are longer in the snapshots with compound A present (Figure 5a).
Figure 5.

a) A key finding is that the thiol of Cys185 must be angled towards the α-carboxylate of d-glutamate to achieve Cα proton abstraction. QM/MM geometry optimization of GR-d-Glu leads to a double proton transfer, in which the d-glutamate α-carboxylate is protonated and the Cα proton is transferred to the thiolate of Cys74. The distance between S(Cys185) and the α-carboxylate oxygen atom of d-glutamate was tabulated and converted into a frequency histogram to yield the distributions of distance values for GR-d-Glu (red) and GR-d-Glu-compound A (black). It is starkly apparent that the distance between S(Cys185) and the α-carboxylate oxygen atom is much smaller in the absence of compound A. b) Reaction coordinates for the proton transfer between the Ca of d-glutamate to S(Cys74) performed using QSite (DFT/B3LYP/6–31G*)/OPLS and described fully in the text and SI. The Cα(d-Glu) proton is driven in 0.2 Å increments from d-Glu to S(Cys74), and a single point calculation was performed. The most favorable starting structures were selected from the QM/MM optimizations for Cα proton transfer (see Table S2). The reaction profiles show that there is a 12.02 kcal/mol smaller barrier in the GR-d-glu complex (red) than in the GR-d-glu-compound A complex (black).
To further compare the geometries of the two ensemble’s active sites, proton transfer geometries were tabulated for the ground state reactant and product structures. For the ground state reactant structures, the geometries tabulated were the distance between S(Cys74) and Hα(d-Glu), the distance between Hα(d-Glu) and Cα(d-Glu), and the angle between S(Cys74), Hα(d-Glu), and Cα(d-Glu), Table S4. For the ground state product structures, the geometries tabulated were the distance between S(Cys74) and H(Cys74), the distance between Hα(d-Glu) and Cα(d-Glu), and the distance between H(Cys185) and O(d-Glu), Table S5. There was no statistical difference between these average distances in the product structures. For example, the average distance between S(Cys74) and H(Cys74) was 1.41 ± 0.03 Å for the GR-d-Glu complex compared to 1.39 ± 0.03 Å in the GR-d-Glu-compound A complex.
Although the proton geometries in the reactant and product structures were similar, the presence of compound A affected the change in QM/MM potential energy (ΔE) for this transformation. As previously stated, four of the five tested Cα-proton abstractions in the GR-d-Glu system had negative (favorable) ΔE values. On the other hand, four out of five of GR-d-Glu-compound A structures resulted in a positive (unfavorable) ΔE, Table S3.
We were unable to obtain a concerted transition state structure involving both protons in flight. Nevertheless, we were able to use reaction coordinate driving of the α-carboxylic acid (i.e., activated) product, in order to obtain the transition structure for the Cα proton transfer (Figure 5b). In this coordinate driving scheme, the Cα(d-Glu) proton shifted in 0.2 Å increments from d-Glu to the S(Cys74) thiolate. The energy difference between the reactant structure and the top of the reaction coordinate barrier for the most favorable snapshot was calculated for the GR-d-Glu and GR-d-Glu-compound A systems. The ΔΔG‡ between these two systems was determined to be 12.02 kcal/mol, with GR-d-Glu having the smaller barrier.
The potential energy difference and the ΔΔG‡ difference between the GR-d-Glu and GR-d-Glu-compound A systems could be due to the placement of the protonated His187 residue (also known as Hip187) in the active site. In the GR-d-Glu and GR-d-Glu-compound A systems, the Hip187 residue is oriented differently. In the GR-d-Glu system, Hip187 is aligned parallel to the plane of Cys185, compared to being oriented perpendicular to the plane of Cys185 as in the GR-d-Glu-compound A system. When Hip187 is in plane with Cys185 this results in a closer distance between one of the nitrogen atoms from Hip187 and S(Cys185), Table S6. On the other hand, when Hip187 is perpendicular to Cys185 the two nitrogen atoms are at a farther distance away from the S(Cys185). A closer S(Cys185)/N(Hip187) is ideal to complete the catalytic reaction and is hindered in the presence of compound A. [2g, 4a]
This pre-activation step observed in H. pylori GR is likely also be a major contributor to the catalytic power in proline racemases as well. A crystal structure of proline racemase (PDB 1W61) contains pyrrole-2-carboxylic acid (P2C) in the active site; this is a flat molecule, thought to be a transition state mimic, which is in a conformation much closer to Cα deprotonation chemistry than the many ground state crystal structures of GR. [14] It can be seen from this transition state structural mimic that the distance between Cys300 and the α-carboxylate from the P2C carboxylate oxygen is 3.67 Å (Figure S5), suggesting that the actual chiral substrate should yield a very close distance between these two heteroatoms. Indeed, when we built the d-proline into PDB 1W61, the distance between the two S(Cys) atoms and the α-carboxylate oxygen is ~ 3.1 Å. This strengthens the possibility that the entire cofactor independent family of racemases are employing such a pre-activation step.
Experimental Findings.
Based on our MD simulations and QM/MM Cα-proton transfer mechanistic studies, we chose to re-investigate the proposed mechanism of action of the allosteric inhibitor, compound A. The nature of the inhibition we see in MD/QM/MM studies is not consistent with the uncompetitive model originally proposed by Lundqvist et al, [5] in which product release limits the catalytic cycle. Thus, to clarify the role of this allosteric inhibitor, we employ SPR, intrinsic fluorescence quenching, and NMR WaterLOGSY studies of binding compound A to H. pylori GR. Our studies reveal with unprecedented detail that the inhibition of compound A does not follow an uncompetitive model. These results are remarkable as they raise serious questions about the mechanism of action of a drug candidate that was extensively employed through Phase 1. [15] Three different experimental techniques show that d-glutamate does not need to be present for compound A to successfully bind to H. pylori GR.
SPR.
SPR experiments were performed to study ligand binding to H. pylori GR. A combination of HisCapture [16] and amine coupling was used to fix the enzyme to the surface of the SPR biosensor. A fixed concentration of ligand was injected to determine the binding affinity of compound A, d-glutamate, and l-glutamate (Figure 6a). Using a 2:1 Langmuir binding model, consistent with substrate inhibition observed by Lundqvist et al., the Kd was determined to be 24 ± 1 μm and 830 ± 20 μm for the two d-glutamate sites and 1300 ± 300 μm and 1600 ± 400 μm for the two l-glutamate sites and using a 1:1 Langmuir binding model, a Kd of 52 ± 2 μm for compound A was obtained (Table S7). The Kd value for l-glutamate is significantly larger than d-glutamate; this large difference is also true of the KM values. The previously obtained KM values by Lundqvist et al. were determined to be 63 ± 0.4 μm and 740 ± 50 μm, for d-glutamate and l-glutamate respectively. [5]
Figure 6.

a) Representative binding of d-glutamate (left), l-glutamate (middle), and compound A (right) to GR as measured by SPR. Data was fit with a 2:1 or 1:1 binding model shown in red with raw data shown in black. Using a 2:1 The Kd was determined to be 24 ± 1 μm and 830 ± 20 μm for the two d-glutamate sites and 1300 ± 300 μm and 1600 ± 400 μm for the two l-glutamate sites and using a 1:1 Langmuir binding model, a Kd of 52 ± 2 μm for compound A was obtained. Error values represent standard deviation (SD). b) Binding curve of compound A to GR using intrinsic tryptophan fluorescence ran in triplicates; compound A was titrated into the reaction buffer containing d-glutamate (black line) and repeated without d-glutamate in the buffer (blue line). The Kd values obtained for GR-compound A were 0.63 ± 0.08 μm and 6.00 ± 0.59 μm in the presence and absence of d-glutamate, respectively. Error values represent SD. c) 1D 1H NMR spectrum of compound A superposed on the WaterLOGSY spectrum (GR-compound A), which establishes that compound A does, indeed bind to apo-GR. All three lines of evidence presented in this figure serve to refute the uncompetitive inhibition model of compound A.
Intrinsic Fluorescence Quenching.
Intrinsic fluorescence quenching experiments monitored the binding of compound A to H. pylori GR with and without d-glutamate present in the reaction buffer. The addition of compound A to H. pylori GR led to a decrease in the intrinsic tryptophan fluorescence (Figure 6b). Compound A bound to H. pylori GR with and without d-glutamate present in the reaction buffer. The Kd values obtained for GR-compound A were 0.63 ± 0.08 μm and 6.00 ± 0.59 μm in the presence and absence of d-glutamate, respectively. A Kd of 1.4–6.3 μm has been previously obtained for GR-d-glu-compound A through multiple biochemical assays.[5] Using a different methodology, our Kd was slightly outside the published range, however, the Kd values are significantly different in the presence and absence of d-glutamate.
WaterLOGSY.
Lastly, NMR WaterLOGSY experiments also confirmed that compound A binds to H. pylori GR in the absence of substrate. The WaterLOGSY spectrum (Figure 6c) for compound A with H. pylori GR contained positive peaks, which indicate that compound A binds to H. pylori GR without d-glutamate present. All three experimental techniques concluded that compound A binds well to H. pylori GR in the absence or presence of d-glutamate, which invalidates the proposed uncompetitive model of inhibition. The presence of compound A alters the hinge motion of the enzyme. Crippling these coupled motions restricts the enzyme’s ability to stereoinvert the d-glutamate substrate.
Conclusion
Our current understanding of how small molecule allosteric inhibitors work on the atomistic level is poor. Unfortunately, enzyme cocrystal structures alone simply do not provide a clear picture of what the mechanism of action is for an allosteric drug. This is despite the fact that some of the most successful drugs in recent times (e.g., Gleevec and Nevirapine) have proceeded by nebulous allosteric mechanisms. [17] This problem is ubiquitous in flexible drug targets, and certainly is manifested in the family of cofactor independent racemases and epimerases. In this paper, we carried out the longest MD simulations and the highest level of theory of QM/MM geometry optimizations to date in studying the Cα-proton transfer in GR.
Our calculations at the DFT/B3LYP/6–31G* level show that H. pylori GR takes advantage of the very large pKa difference between the α-carboxylic acid form of glutamate and the α-carboxylate form, to facilitate a proton transfer from Cα to the enzyme’s catalytic thiolate, Cys74. This pre-activation step of GR is similar to a number of cofactor dependent enzymes that catalyze Cα proton transfers (e.g., mandelate racemase, MLE, and enolase). Additionally, there is strong structural evidence that such a mechanism may be occurring in proline racemase as well.
Our MD simulations clearly show that this acid-catalyzed pre-activation step is tied to enzyme dynamics. An extended network of coupled motions is nested around the critical Cys185 residue, which may or may not be in position to protonate the α-carboxylate. Importantly, simulations of GR bound to compound A reveal that Cys185 is mostly pointing ~ 180° away from the α-carboxylate oxygen, in a non-productive position (i.e., the pre-activation step is conformationally hindered). Even when oriented in the correct direction for proton transfer, the GR-d-Glu-compound A ensemble was found to have significantly increased proton donor and acceptor distances. The MD simulations revealed with unprecedented detail that compound A dampens the dynamic motions of the enzyme, which impacts the flexibility of the catalytic residues. QM/MM geometry optimizations reveal that the proton transfer is more energetically favorable when compound A is absent.
These computational findings raised significant concerns with the proposed mechanism of action of small molecule allosteric inhibitors outlined by Lundqvist et al.,[5] which focused on a highly exotic uncompetitive inhibition model, in which product release was crippled by the filling of the allosteric pocket by compound A. The uncompetitive model is not consistent with our proposed model of acid catalyzed pre-activation, in that the former depends on a slow physical step, while the latter invokes inhibition of a necessary chemical step (albeit one that depends on differences in flexibility between the H. pylori GR and H. pylori GR-compound A systems) for stereoinversion. An analysis of the experimental RMSF values calculated using crystallographic B-factors from the structures of the GR-d-Glu and GR-d-Glu-compound A crystal structures, together with a similar comparison of the RMSF values from our MD analyses on these systems, shows a clear pattern; the GR-d-Glu complex is much more flexible than the GR-d-Glu-compound A complex (Figure 3b, bottom panel). While the crystallographic data provides indirect support for our computational observations, they still leave the possibility that H. pylori GR is potentially inhibited by the physical step proposed by Lundqvist et al.[5] Our experimental investigation of compound A binding to H. pylori GR, via SPR and NMR WaterLOGSY, and intrinsic fluorescence quenching were all in agreement, and invalidate the originally proposed uncompetitive inhibition model of compound A on H. pylori GR. Taken together, this study provides both a novel and exciting atomistic basis for remote allosteric control by a small molecule drug lead. Additionally, this study should serve as a cautionary tale regarding the hasty assignment of inhibitory mechanisms of small molecule effectors in the absence of direct biophysical data on complex formation.
Computational & Experimental Section
Computational Methods.
The full details of the computational methods are provided in the Supporting Information (SI). The layered computational approach employed comprised of MD simulations and QM/MM geometry optimizations. MD simulations were performed in triplicate for a total of 150 ns using the YASARA Structure package (YASARA Biosciences) [10b, 10c] version 17.1.28 on GR-d-Glu and GR-d-Glu-compound A. The YAMBER3 [10a] force field was employed at a temperature of 298 K and a pH of 7.4. All MD snapshots were analyzed using MDAnalysis and UCSF Chimera. [12–13] MD structures were next ported into QSite (Schrödinger LLC) [11b] to further analyze the protein dynamics and investigate the proton transfer between the Cα-proton of d-glutamate and the S of Cys74. These structures were selected based on proton donor and acceptor distances and the transfer angle between d-glutamate and Cys74 (Cα-H-SCys74), further detailed below. Ab initio QM/MM geometry optimizations were performed using the frozen orbital method of Phillip et al [18] at the DFT/B3LYP/6–31G* level to capture ground state reactants and product structures for the GR-d-Glu and GR-d-Glu-compound A snapshots.
Experimental Methods. Expression and Purification of H. pylori GR.
A 5 mL starter culture of Luria Broth (LB) medium with 50 μg/mL ampicillin, 34 μg/mL chloramphenicol, and 25 μg/mL kanamycin was prepared from the stock E. coli BL21-DE3 cells containing pET-15b plasmid with the gene of choice and grown overnight at 37°C with rotation. The 5 mL starter culture was back-diluted into 750 mL of LB medium with antibiotics. Cells were grown at 37°C with shaking until the OD600 reached 0.8–1.0. Protein expression was induced upon addition of a final concentration of 0.1 mm IPTG. Following induction, cells were grown for an additional 16–18 hr at 20 °C with shaking. Cells were harvested by centrifugation at 5,000 × g at 4°C for 15 min. Supernatant was discarded and cell pellets were resuspended in buffer A (100 mm Tris, 100 mm NaCl, 10 mm imidazole, 1mm TCEP, pH 8.0). An emulsiflex microfluidizer was used to lyse the cells. Insoluble materials were pelleted by centrifugation at 30,000 × g for 75 min at 4°C to remove insoluble material and passed through a 0.22 μm filter. His-tagged proteins were loaded onto a 1 mL HiTrap IMAC HP (GE Healthcare) cobalt resin column equilibrated with buffer A. Pooled fractions were then exchanged into protein storage buffer (50 mm Tris, 100 mm NaCl, 0.2 mm DTT, pH 8.0) and concentrated utilizing a 10,000 MWCO Amicon centrifugal filter device. Protein stocks were stored at a final concentration of 5–7 mg/mL with 20% glycerol at −20°C.
Surface Plasmon Resonance.
Experiments were carried out using a SensiQ Pioneer SPR. H. pylori GR was immobilized to the biosensor using a HisCapture couple method detailed in the SI. Serial two-fold dilutions of d-glutamate, l-glutamate, and compound A were prepared in running buffer and injected in the test and reference biosensor chip channels at 50 μL/min, followed by a 100 sec dissociation phase at 25°C. Serial dilutions of each compound were injected in triplicate along with several injections of running buffer in random order. Data was analyzed using the program Qdat (2.6.3.0) and fit to a model of simple binding of 1:1 Langmuir binding model for compound A or a 2:1 Langmuir binding model for d-glutamate and l-glutamate to fit the microscopic rate of association and dissociation, ka and kd, respectively. (Table S7).
Intrinsic Fluorescence.
Compound A binding to H. pylori GR was characterized by measuring intrinsic tryptophan fluorescence using a Cary Eclipse Fluorescence Spectrophotometer. H. pylori GR at a 1 μm concentration in buffer (50 mM Tris, 1 mM TCEP at pH 8.0) was excited at 280 nm (5 nm slit width) and emission was monitored at 350 nm (10 nm slit width). Compound A was titrated into the H. pylori GR reaction buffer and mixed as fluorescence was monitored. Relative fluorescence compared to the initial fluorescence of 1 μm H. pylori GR was recorded and compared to the compound A concentration. The experiment was performed with and without 100 mm d-glutamate in the reaction buffer (50 mm Tris, 1 mm TCEP at pH 8.0). The data was fit using a one-site binding model with GraphPad v. 7.00.
WaterLOGSY NMR.
The WaterLOGSY spectra of H. pylori GR was acquired using a water NOE mixing time of 1 sec and a T2 relaxation filter of 50 ms just before data acquisition to suppress the broad signals derived from protein.[19] The purity and structure of compound A was assessed from 1H NMR spectra collected on a Varian Unity Inova 600 MHz NMR spectrometer at a 10 mm concentration diluted into DMSO-d6. The protein buffer used in the experiments contained 25 mm Tris-d11, 50 mm KCl, and 1 mm TCEP at a pH of 8.0. All NMR data was acquired on a Bruker Avance II 800 MHz NMR spectrometer equipped with a sensitive cryoprobe and recorded at 25°C. The 1H chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS). NMR spectra were processed using NMR Pipe and analyzed using NMR View. [20]
Supplementary Material
Acknowledgements
This work was supported by NIH grants awarded to M.A.S. (R01-GM09737). This research was supported in part through computational resources provided by the University of Iowa, Iowa City, IA.
Footnotes
Supporting information for this article can be found under: https://doi.org/10.1002/cmdc.201900642.
References
- [1].a) Tanner ME, Gallo KA, Knowles JR, Biochemistry 1993, 32, 3998–4006; [DOI] [PubMed] [Google Scholar]; b) Gallo KA, Tanner ME, Knowles JR, Biochemistry 1993, 32, 3991–3997; [DOI] [PubMed] [Google Scholar]; c) Gallo KA, Knowles JR, Biochemistry 1993, 32, 3981–3990; [DOI] [PubMed] [Google Scholar]; d) Whalen KL, Pankow KL, Blanke SR, Spies MA, Acs Medicinal Chemistry Letters 2010, 1, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Spies MA, Woodward JJ, Watnik MR, Toney MD, J Am Chem Soc 2004, 126, 7464–7475; [DOI] [PubMed] [Google Scholar]; b) Spies MA, Toney MD, Biochemistry 2003, 42, 5099–5107; [DOI] [PubMed] [Google Scholar]; c) Major DT, Gao JL, J Am Chem Soc 2006, 128, 16345–16357; [DOI] [PubMed] [Google Scholar]; d) Major DT, Nam K, Gao JL, J Am Chem Soc 2006, 128, 8114–8115; [DOI] [PubMed] [Google Scholar]; e) Gerlt JA, Babbitt PC, Annual Review of Biochemistry 2001, 70, 209–246; [DOI] [PubMed] [Google Scholar]; f) Liu HY, Zhang YK, Yang WT, J Am Chem Soc 2000, 122, 6560–6570; [Google Scholar]; g) Mixcoha E, Garcia-Viloca M, Lluch JM, Gonzalez-Lafont A, J Phys Chem B 2012, 116, 12406–12414; [DOI] [PubMed] [Google Scholar]; h) Mobitz H, Bruice TC, Biochemistry 2004, 43, 9685–9694; [DOI] [PubMed] [Google Scholar]; i) Malabanan MM, Amyes TL, Richard JP, Curr Opin Struc Biol 2010, 20, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Pillai B, Moorthie VA, van Belkum MJ, Marcus SL, Cherney MM, Diaper CM, Vederas JC, James MNG, J Mol Biol 2009, 385, 580–594; [DOI] [PubMed] [Google Scholar]; b) Pillai B, Cherney M, Diaper CM, Sutherland A, Blanchard JS, Vederas JC, James MNG, Biochem Bioph Res Co 2007, 363, 547–553; [DOI] [PubMed] [Google Scholar]; c) Richard JP, Amyes TL, Goryanova B, Zhai X, Curr Opin Chem Biol 2014, 21, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Spies MA, Reese JG, Dodd D, Pankow KL, Blanke SR, Baudry J, J Am Chem Soc 2009, 131, 5274–5284; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Williams G, Maziarz EP, Amyes TL, Wood TD, Richard JP, Biochemistry 2003, 42, 8354–8361. [DOI] [PubMed] [Google Scholar]
- [5].Lundqvist T, Fisher SL, Kern G, Folmer RHA, Xue YF, Newton DT, Keating TA, Alm RA, de Jonge BLM, Nature 2007, 447, 817–822. [DOI] [PubMed] [Google Scholar]
- [6].a) Ruzheinikov SN, Taal MA, Sedelnikova SE, Baker PJ, Rice DW, Structure 2005, 13, 1707–1713; [DOI] [PubMed] [Google Scholar]; b) Hegeman GD, Rosenber.Ey, G. L. Kenyon, Biochemistry 1970, 9, 4029–4036; [DOI] [PubMed] [Google Scholar]; c) Landro JA, Gerlt JA, Kozarich JW, Koo CW, Shah VJ, Kenyon GL, Neidhart DJ, Biochemistry 1994, 33, 635–643; [DOI] [PubMed] [Google Scholar]; d) Mitra B, Kallarakal AT, Kozarich JW, Gerlt JA, Clifton JG, Petsko GA, Kenyon GL, Biochemistry 1995, 34, 2777–2787; [DOI] [PubMed] [Google Scholar]; e) Larsen TM, Wedekind JE, Rayment I, Reed GH, Biochemistry 1996, 35, 4349–4358. [DOI] [PubMed] [Google Scholar]
- [7].Cleland WW, Frey PA, Gerlt JA, Journal of Biological Chemistry 1998, 273, 25529–25532. [DOI] [PubMed] [Google Scholar]
- [8].Puig E, Garcia-Viloca M, Gonzalez-Lafont A, Lluch JM, J. Phys Chem A 2006, 110, 717–725. [DOI] [PubMed] [Google Scholar]
- [9].Malapati P, Krishna VS, Nallangi R, Srilakshmi RR, Sriram D, Eur J Med Chem 2018, 36, 996–1007. [DOI] [PubMed] [Google Scholar]
- [10].a) Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G, Proteins 2004, 57, 678–683; [DOI] [PubMed] [Google Scholar]; b) Krieger E, Vriend G, J Comput Chem 2015, 36, 996–1007; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Krieger E, Vriend G, Bioinformatics 2014, 30, 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a; Schrödinger Release 2016–4: Jaguar, Schrödinger, LLC, New York, NY, 2016; [Google Scholar]; b. Schrödinger Release 2016–4: QSite, Schrödinger, LLC, New York, NY, 2016. [Google Scholar]
- [12].Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O, J Comput Chem 2011, 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J Comput Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [14].Buschiazzo A, Goytia M, Schaeffer F, Degrave W, Shepard W, Gregoire C, Chamond N, Cosson A, Berneman A, Coatnoan N, Alzari PM, Minoprio P, Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 1705–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fisher SL, Microbial Biotechnology 2008, 1, 345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Affinity Capture Surface for Histidine-Tagged Recombinant Proteins. https://www.sensiqtech.com/uploads/file/brochures/1421-DM-8083-RF.
- [17].a) Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J, Science 2000, 289, 1938–1942; [DOI] [PubMed] [Google Scholar]; b) Spence RA, Kati WM, Anderson KS, Johnson KA, Science 1995, 267, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Philipp DM, Friesner RA, Journal of Computational Chemistry 1999, 20, 1468–1494; [Google Scholar]; b) Murphy RB, Philipp DM, Friesner RA, Journal of Computational Chemistry 2000, 21, 1442–1457. [Google Scholar]
- [19].a) Dalvit C, Pevarello P, Tato M, Veronesi M, Vulpetti A, Sundstrom M, J Biomol Nmr 2000, 18, 65–68; [DOI] [PubMed] [Google Scholar]; b) Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B, J Biomol Nmr 2001, 21, 349–359. [DOI] [PubMed] [Google Scholar]
- [20].a) Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, J Biomol Nmr 1995, 6, 277–293; [DOI] [PubMed] [Google Scholar]; b) Johnson BA, Blevins RA, J Biomol Nmr 1994, 4, 603–614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.