

Abstract
Background
Bone marrow is a common site of metastasis for a number of tumor types, including breast, prostate, and lung cancer, but the mechanisms controlling tumor dormancy in bone are poorly understood. In breast cancer, while advances in drug development, screening practices, and surgical techniques have dramatically improved survival rates in recent decades, metastatic recurrence in the bone remains common and can develop years or decades after elimination of the primary tumor.
Recent Findings
It is now understood that tumor cells disseminate to distant metastatic sites at early stages of tumor progression, leaving cancer survivors at a high risk of recurrence. This review will discuss mechanisms of bone lesion development and current theories of how dormant cancer cells behave in bone, as well as a number of processes suspected to be involved in the maintenance of and exit from dormancy in the bone microenvironment.
Conclusions
The bone is a complex microenvironment with a multitude of cell types and processes. Many of these factors, including angiogenesis, immune surveillance, and hypoxia, are thought to regulate tumor cell entry and exit from dormancy in different bone marrow niches.
Keywords: angiogenesis, bone marrow, dormancy, hypoxia, immune surveillance, metastasis
1. INTRODUCTION
Tumor cells frequently home to the bone marrow, where they encounter a unique microenvironment that contains a milieu of growth factors and cell types and is orders of magnitude more rigid than the primary tumor. Bone metastases are often observed in patients with breast, prostate, and lung cancer, but have been reported for a number of other tumor types1, 2, 3, 4 and will likely increase as treatments that target the primary tumor continue to improve. Upon dissemination into the bone marrow, tumor cells come in contact with and compete for the perivascular and endosteal niches, and may either grow and colonize the marrow or enter a dormant state. In breast cancer patients, and in patients with estrogen receptor positive (ER+) disease in particular, this period of dormancy can last anywhere from months to decades.5
Upon dissemination into the bone marrow, tumor cells frequently secrete parathyroid hormone related protein (PTHrP), which signals through the parathyroid hormone (PTH) type 1 receptor (PTH1R) on the surface of osteoblast lineage cells to promote their activity and differentiation. PTHrP:PTH1R signaling results in increased expression of receptor activator of nuclear factor kappa‐B ligand (RANKL) on osteoblasts, which binds to its receptor RANK on pre‐osteoclasts to promote osteoclastogenesis.6 Tumor cells may also secrete interleukins (e.g. IL‐6 and IL‐8) to stimulate osteoclastogenesis at the tumor‐bone interface. This increase in the number and activity of osteoclasts results in localized bone resorption and the release of growth factors from the bone matrix, such as transforming growth factor β (TGF‐β), insulin‐like growth factor I and II (IGF‐I/II), platelet derived growth factor (PDGF), and bone morphogenetic proteins (BMPs). These growth factors normally function to couple osteoclast and osteoblast activity, but in the context of the tumor‐bone microenvironment can also stimulate tumor cell proliferation to further enhance the secretion of osteolytic factors and promote tumor cell growth7 (Figure 1). These interactions were coined the “vicious cycle” by Greg Mundy6 and this theory has been demonstrated by multiple groups over the last few decades.8, 9, 10, 11, 12 While many of these interactions are well‐defined, the steps prior to the induction of osteolysis, and specifically, how tumor cells enter and exit dormancy and initiate osteolysis in the bone marrow, are less clear but actively being explored.
Figure 1.
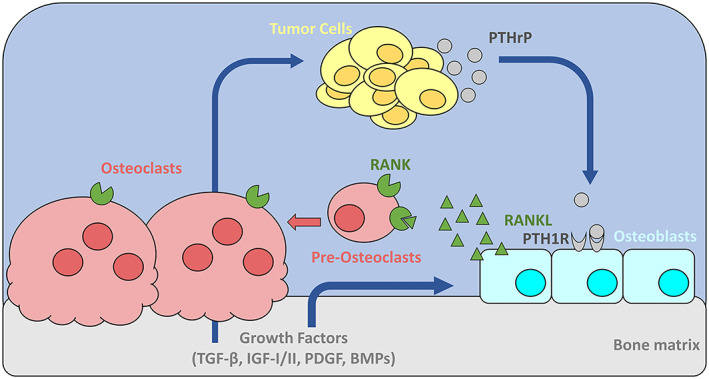
Mechanisms of tumor‐induced bone destruction. Tumor cells secrete parathyroid hormone related protein (PTHrP), which signals through parathyroid hormone receptor type 1 (PTH1R) on the surface of osteoblasts to promote their activity and differentiation. Increased osteoblast activity results in increased secretion of the receptor activator of nuclear factor kappa‐B ligand (RANKL), which binds to RANK on pre‐osteoclasts to promote osteoclastogenesis and on mature osteoclasts to stimulate their activity. Increased number and activity of osteoclasts results in more bone resorption and release of growth factors from the bone matrix, including transforming growth factor β (TGF‐β), insulin‐like growth factor I and II (IGF‐I/II), platelet derived growth factor (PDGF), and bone morphogenetic proteins (BMPs). These growth factors normally function to couple osteoclast and osteoblast activity, but can also stimulate further tumor cell proliferation and PTHrP production by the tumor cells to feed this cycle of bone destruction
Prior to the process of colonization, bone‐disseminated tumor cells may enter a prolonged state of dormancy, where the cells survive in the bone but do not grow into clinically detectable metastases.13 The mechanisms controlling dormancy are still poorly understood, but many groups are working to identify factors that regulate dormancy in various tissues.14 A clearer understanding of these mechanisms is fundamental to tumor biology and our understanding of metastatic colonization of distant sites, and could also lead to new therapeutic strategies to prevent the development of bone metastases and the establishment of the vicious cycle.
The bone is a complex organ responsible for hematopoiesis, protection of vital organs, and facilitation of movement. When discussing the interaction of tumor cells with other bone‐resident cell types, it is important to appreciate the different niches that are present in the bone marrow: the perivascular niche, hematopoietic niche, and the endosteal niche (Figure 2).5 The perivascular niche of stable microvessels can promote tumor dormancy, while neovascular tips support rapid tumor cell proliferation.15 The hematopoietic niche, which is replete with hematopoietic stem cell (HSC) maintenance factors produced by osteoblasts and endothelial cells, is thought to contribute to the quiescence of tumor cells as well as the resident stem cells.16, 17, 18 Endothelial cells in the bone marrow support the hematopoietic niche by expressing factors such as E‐selectin, fibroblast growth factor 2 (FGF2), delta‐like 1 (DLL1), insulin‐like growth factor‐binding protein 2 (IGFBP2), angiopoietin 1 (ANGPT1), desert hedgehog (DHH) and epidermal growth factor (EGF).19 The endosteal niche is home to osteoblasts and osteoclasts that are responsible for bone remodeling and healthy skeletal homeostasis.20 Importantly, these niches may overlap considerably in the bone and therefore simultaneously influence tumor cell dormancy and proliferation status in bone, as observed by the multiple supportive roles for endothelial cells and potential overlap between the hematopoietic and endosteal niche. While osteoblasts secrete RANKL, to drive osteoclastogenesis and the bone remodeling cycle, and osteoprotegerin (OPG), to maintain the balance of osteoclast and osteoblast activity, the role of osteocytes, or terminally differentiated osteoblasts, in signaling to the various osteogenic cells to maintain bone homeostasis is now appreciated.20 While there is considerably less known about the role for osteocytes in tumor dormancy in bone, it is well recognized that osteoblasts and osteoclasts contribute to tumor‐induced bone destruction and can also produce factors to maintain tumor dormancy.
Figure 2.
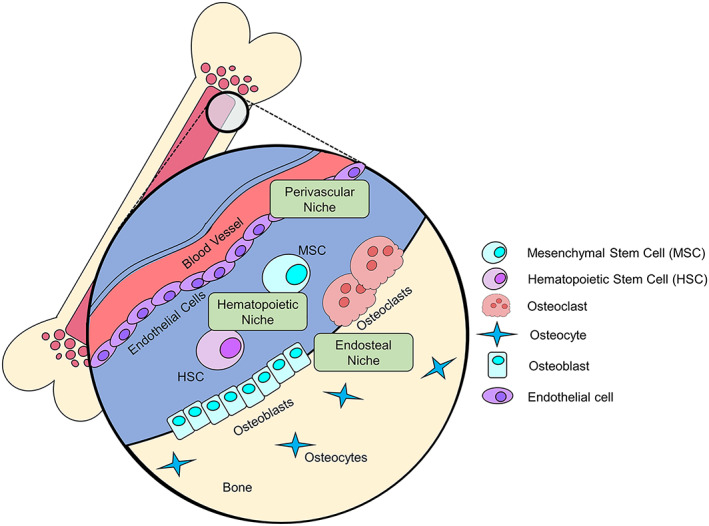
The bone is composed of three distinct niches that can exert pro‐ or anti‐dormancy effects. The perivascular niche contains blood vessel lining endothelial cells that can secrete factors to push cell into, or out of, dormancy, and signaling between endothelial cells and hypoxic tumor cells can drive tumor vascularization and escape from dormancy. Hematopoietic stem cells (HSC) and mesenchymal stem cells (MSC) that reside in the hematopoietic niche secrete signaling factors or produce extracellular vesicles that can influence a cell's dormancy status. The endosteal niche contains osteoclasts and osteoblasts that play a role in the vicious cycle, but can also secrete factors to promote dormancy. Since these niches are in close proximity to one another, tumor cells will often seed in the bone marrow and experience the effects of multiple niches
1.1. Clinical implications of bone metastasis
Despite improvements in early detection and the development of tumor‐targeted therapies, breast cancer remains the most common cancer diagnosis for women and accounts for the second most cancer‐related deaths in the United States.21 Many of these deaths are due to metastatic disease, with one of the most common sites of metastasis being the bone. Up to 70% of patients that succumb to metastatic breast cancer are found to have bone metastases upon autopsy, and many other cancer types, including prostate cancer, renal cell carcinoma, and lung cancer are also known to commonly metastasize to bone.22
Metastatic lesions in the bone can be classified as osteolytic, where bone is resorbed at the site of tumor colonization, or osteoblastic, where excessive bone formation follows the lytic phase and results in poorly organized bone. Breast cancer, for example, most commonly forms osteolytic lesions, while prostate cancer commonly induces the formation of osteoblastic lesions.23 It has been proposed that the formation of osteoblastic lesions in prostate cancer is preceded by a resorptive phase, given the success of anti‐resorptive therapies (e.g. bisphosphonates) in reducing bone pain in prostate cancer patients.24 Mixed lesions (both osteolytic and osteoblastic) have also been observed in patients with bone metastatic prostate cancer upon autopsy.25 It is important to note, however, that even in osteoblastic lesions, where more bone is formed, the quality of the bone is compromised and these patients are still more prone to fracture.26 The bone is thus weakened at the site of tumor growth, leaving patients at an increased risk of skeletal‐related events (SREs), including pathologic fracture and spinal cord compression, which require surgery or radiation.27
Anti‐resorptive therapies such as bisphosphonates, which inhibit osteoclast function, and the anti‐RANKL antibody denosumab, which blocks osteoclastogenesis, have been used clinically in patients who are at increased risk of fracture in order to prevent further bone resorption.28 Early clinical trials demonstrated that bisphosphonates effectively reduced skeletal complications of breast cancer bone metastasis, but did not improve disease progression or overall survival.29, 30 A later clinical study, the AZURE trial, confirmed that bisphosphonates had no effect on disease‐free survival,31 but further sub‐group analysis revealed that post‐menopausal women specifically had a significant increase in disease‐free survival and reduction in bone metastases,32 findings that were later confirmed in another study.33 Teriparatide and abaloparatide are PTH/PTHrP analogues that can be used to stimulate bone formation and are the only FDA‐approved anabolic therapies available34; however, anabolics are not commonly used in cancer patients with bone‐metastatic disease due to the risk of exacerbating tumor‐induced bone disease and since both therapies have been shown to increase the risk of osteosarcoma in rats,35 although this has not been observed in patients. In castration‐resistant prostate cancer patients with symptomatic bone metastases, intravenous injections of radium‐223 in addition to the best standard of care (BSoC) regimen available at each center (for example, local external beam radiation therapy, corticosteroids, or antiandrogens, but excluding chemotherapy, hemibody external radiation, or other systemic radionuclides) have been found to be well tolerated by patients36 and prolong overall survival compared to placebo plus BSoC and prolong the time to first symptomatic skeletal event.37 However, none of the therapies used against established bone metastases are curative. Thus, the current therapeutic options serve to slow or prevent the progression of osteolysis and the occurrence of SREs38 in patients with skeletal metastases.
1.2. Models of tumor dormancy
The mechanisms that control dormancy and reactivation are not well defined, but two models have been proposed to describe the behavior of dormant tumor cells: single‐cell/cellular dormancy and micrometastatic dormancy. The cellular dormancy model proposes that each disseminated tumor cell (DTC) enters a non‐proliferative, quiescent state and is arrested in G0‐G1, as indicated by negative staining for Ki‐67 and/or PCNA.39, 40 In the micrometastatic dormancy model, the size of the micrometastatic lesion as a whole does not grow, since the cells are proposed to either cycle at a very slow rate, or to undergo apoptosis and proliferation at similar rates, resulting in static tumor burden until the tumor cells are reactivated by exogenous processes, such as angiogenesis.41 These models are not mutually exclusive, since quiescent cells may exist in a micrometastasis, and both models likely contribute to the observed phenotype of cell survival and resistance to conventional chemotherapeutic agents. It is also possible that a cell could escape quiescence and then enter a state of micrometastatic dormancy before growing out into an overt metastasis, making the two models sequential steps in the process of dormancy escape. It is worth noting that it can be technically challenging to detect single, dormant tumor cells in patient samples and in mouse models of tumor dormancy, making it difficult to prove whether all dormant tumor cells are in fact arrested in G0‐G1.
While there is not a clear definition of tumor cell dormancy, the main features that define a dormant cell are resistance to chemotherapy42, 43 and a lack of proliferation (i.e. non‐ or slow‐cycling). These two processes are linked, since increased chemoresistance is at least in part due to a lack of proliferation. Human colon adenocarcinoma (DLD‐1) cells that have high expression of the quiescence marker p27 were found to have increased resistance to vinblastine, doxorubicin, cisplatin, and 5‐fluorouracil.43 p27 is a cyclin‐dependent kinase inhibitor involved in halting cell cycle in the G0‐G1 transition,44 making it a commonly used marker of quiescent cells. Systemic therapies have been found to be less effective against tumors that have poor vascularization and high extracellular matrix (ECM) density due to insufficient permeation of drugs into the tumors. Increased lysyl oxidase (LOX) expression in fibrosarcomas that grew from subcutaneously injected MT6 cells was found to contribute to this dense, highly crosslinked ECM that resulted in resistance to therapy.45 Since LOX expression is known to drive pre‐metastatic niche formation and alter ECM composition,46 these results suggest that disseminated cells or micrometastases in these niches may experience lower drug doses due to a similar mechanism, resulting in their persistence following therapy.
The definition of dormancy and what factors cause dormancy in the bone remain ambiguous and contested partly due to the limited models that are available to study the complex interactions that occur in the bone marrow.47 In vivo models are most commonly employed to study dormancy in bone, but live imaging of tumor cells in bone remains challenging, limiting the ability to track dormant cells over time. Recently, several groups have developed ossicle models that overcome some of these limitations.48, 49, 50 While in vivo models more fully recapitulate all of the microenvironmental factors that lead to tumor cell dormancy, in vitro models are often easier to manipulate and analyze. Furthermore, more sophisticated in vitro techniques are being developed that more accurately model the bone. For example, 3D bone mimicking scaffolds have been synthesized based on human bone structure from microCT scans and these scaffolds can be seeded with tumor cells and bone‐resident cells, such as mesenchymal stem cells, to study their interactions; the scaffolds recapitulate the rigidity and structural nuances of trabecular bone and thus have advantages over 2D co‐culture models.51 These scaffold cultures can be used for many applications and can even be sectioned for histological analysis.
While new models such as these are in development, the most common models to study tumor dormancy in bone are intratibial and intracardiac injections into various mouse strains.52, 53, 54, 55, 56 These injection types are more commonly used to investigate tumor dormancy in bone than orthotopic injections into the primary tumor site, since intratibial and intracardiac innoculations allow the delivery of sufficient numbers of cells to the bone marrow, making the detection of the tumor cells in the bone more feasible.57 Intracardiac inoculation, or the injection of tumor cells into the left ventricle of the heart of a mouse, will not model invasion or intravasation, but will recapitulate the later stages of systemic dissemination including extravasation and colonization of the bone marrow.47 Intratibial injections allow the isolated investigation of the colonization capabilities of the cells. Intrailiac inoculation of tumor cells has also been used to deliver human MCF7 cells to the bone marrow with greater frequency than intracardiac injection,58 but the procedure is more technically complicated and invasive than intracardiac injection, and has therefore been slower in its adoption. While it is not used to model dormancy in the bone, the CAM model, where tumor cells are implanted onto chicken egg chorioallantoic membranes, is frequently used to study dormancy and metastasis, since it provides an easy to work with in vivo environment.59, 60, 61, 62, 63 PC3 cells are also commonly used to study prostate cancer dormancy in bone, and MDA‐MB‐231 cells (human ER‐) or 4T1 cells (murine ER‐) are commonly used to study breast cancer dormancy in bone since all of these cell lines readily colonize and are easily detectable in bone; however, these aggressive cell lines rapidly form osteolytic lesions and do not model the prolonged dormancy that is observed in many human patients. MCF7 cells, a human ER+ breast cancer cell line, on the other hand are non‐proliferative in bone and induce minimal osteolysis,12, 15, 52, 58 making it a more clinically relevant model; however, MCF7 cells do not proliferate in vivo without estrogen supplementation, which causes abnormal bone density.64 More sensitive detection methods for human and murine breast cancer cell lines that lie dormant in vivo (MCF7 +/− exogenous 17β‐estradiol [E2], D2.0R +/− E2, SUM159) are in development, making it more feasible to move away from the use of aggressive cell lines when modeling dormancy.57
Metastasis has classically been considered to occur in the late stages of tumor growth, but dissemination of tumor cells to distant metastatic sites is now understood to be an early event. DTCs in the bone are detectable as early as 4–9 weeks of age in HER2/neu and PyMT transgenic mice, at which point only atypical ductal hyperplasia (ADH) or ductal carcinoma in situ (DCIS) was detectable in the mammary gland.65 Interestingly, while mice with larger primary tumors did have higher numbers of DTCs, the number of DTCs detected did not increase at the same rate as the growth of the primary tumor. Although the primary tumor area grew exponentially, the number of DTCs only grew 4–7 fold, indicating that tumor cell dissemination may not rely upon primary tumor progression. Another study found that 57% of cytokeratin‐positive cells isolated from the bone marrow of breast cancer patients without clinically evident metastases did not display chromosomal aberrations, whereas the matched primary tumors of these patients regularly harbored multiple chromosomal alterations.66 This suggests that these cells disseminated to the bone marrow before extensive chromosomal instability occurred at the primary site, supporting the argument that dissemination is an early step in cancer progression. This is consistent with the clinical difficulty in preventing metastasis, since tumor cells are likely to have disseminated by the time the cancer is diagnosed. It is clear that treatments that target metastatic lesions are necessary, and targeting dormant cells to prevent reactivation or make them more chemo‐sensitive are attractive approaches.
Chronic dormancy induction is one of the proposed methods to therapeutically target dormant tumor cells.67 This is an attractive approach, since suppressing reactivation may prevent relapses later in life. However, patients would be placed on a chronic therapy regimen, requiring strict adherence and long‐term expense. Furthermore, potential side effects from chronic stimulation of dormancy promoting pathways or inhibition of proliferation stimulating factors would have to be managed. An alternative method is to clear dormant tumor cells from the patient's system by mobilizing the cells out of the marrow and making them more sensitive to chemotherapy.67 One advantage to this approach is that this type of treatment could be combined with systemic neoadjuvant or adjuvant therapies and has the potential to create a cancer‐free state for the patient. When identifying factors to target in this manner it will be crucial to find targets that will increase the cell's susceptibility to therapy without stimulating their proliferative capacity, since this may lead to reactivation and the potential for further metastases to develop.
Disrupting cell polarization by expressing a form of β4 integrin that lacks the hemidesmosome targeting domain was found to interfere with tissue polarity and NFκB activation and made S‐1 or T4–2 breast epithelial cells sensitive to apoptosis without inducing proliferation in 2D monolayer or 3D spheroid cultures.68 Similar findings have been reported for other integrin molecules or cancer types: α4β1 or α5β1 integrins on myeloma cells binding to fibronectin,69 α5β1 integrin on breast cancer cells binding to fibronectin,70 and small cell lung cancer cells binding to ECM (particularly through β1 integrin)71, 72 have all been shown to increase drug resistance, emphasizing the potential clinical utility of cancer cell mobilization. There is still debate surrounding whether it is better to keep cells dormant or mobilize and eliminate them, and no therapeutic strategies have been developed, based on either strategy, that have been translated to the clinic or tested in clinical trials. It is clear that a deeper understanding of the signals that push cells into, or out of, dormancy will enable us to better target these cells therapeutically, as will improved methods to detect dormant, disseminated tumor cells in patients.
2. FACTORS CONTROLLING DORMANCY OR REACTIVATION
2.1. Immune surveillance
The model of immune surveillance and immune editing of tumors proposes that tumor cells are cleared by the immune system through an initial elimination phase, which is followed by an equilibrium phase in which tumor growth is inhibited by a balance of tumor cell elimination and proliferation.73 Given what we know about the model of micrometastatic dormancy, it is possible that immune surveillance may promote tumor dormancy during the equilibrium phase, or that the summation of pro‐tumor and pro‐clearance pathways that are active in the tumor microenvironment may promote micrometastatic dormancy (Figure 3A). If the proliferation rate of the micrometastasis surpasses the rate of elimination by immune mechanisms, or if the immunosuppressive signals in the microenvironment are amplified, the tumor would enter the escape phase of the immune surveillance process, similar to the process of dormancy escape. These immune modulatory signals, including cytokines and chemokines, can be produced by the tumor cells or by the microenvironment.
Figure 3.
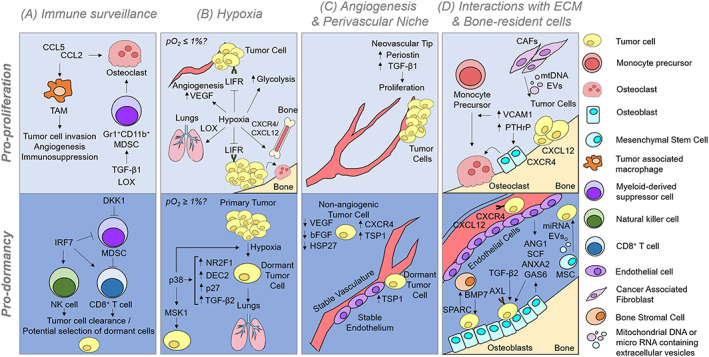
Tumor cells enter or exit dormancy depending on the micro‐environmental signals they encounter. A, Immune surveillance mechanisms can stimulate angiogenesis, immunosuppression, and osteoclastogenesis to stimulate tumor growth, but immune cells can also clear tumor cells and potentially select for non‐immunogenic or, potentially, dormant clones. B, Hypoxia promotes dissemination of tumor cells to distant sites and their ability to colonize the bone marrow. Hypoxia has also been shown to stimulate the expression of several dormancy‐associated genes. We speculate that severe hypoxia (pO2 ≤ 0.5%) may stimulate tumor cell exit from dormancy while moderate hypoxia (pO2 < 4% and ≥ 0.5%) may favor dormancy. C, According to the angiogenic dormancy model, micrometastases that cannot induce angiogenesis will remain dormant. It is now clear that the spatial relationship of tumor cells in relation to endothelial derived factors can control the proliferation or dormancy of disseminated tumor cells, in that cells located along neovascular tips are stimulated to proliferate, while cells residing along the stable vasculature will remain dormant. D, the bone is an organ packed with many cell types. Tumor cells in bone can induce the vicious cycle of bone destruction due to PTHrP expression, as described in more detail in Figure 1. Additionally, if tumor cells have upregulated VCAM1 expression, they can stimulate osteoclastogenesis by recruiting monocyte precursors that can differentiate into osteoclasts. Endothelial cells and osteoblasts in particular secrete many factors that are critical for bone homeostasis and hematopoietic stem cell quiescence and maintenance, which can be coopted by some tumor cells. Thus, once tumor cells home to the bone marrow, they are exposed to many signals that promote their survival and quiescence. CCL2 = C‐C motif chemokine ligand 2 (monocyte chemotactic protein 1); CCL5 = C‐C motif chemokine ligand 5; TAM = tumor associated macrophage; MDSC = monocyte‐derived suppressor cell; TGF‐B1/2 = transforming growth factor Beta 1/2; LOX = lysyl oxidase; DKK1 = dikkopf‐1; IRF7 = interferon regulatory factor 7; NK cell = natural killer cell; LIFR = leukemia inhibitory factor receptor; VEGF = vascular endothelial growth factor; NR2F1 = nuclear receptor subfamily 2 group F member 1; DEC2 = differentially expressed in chondrocytes 2; MSK1 = mitogen‐ and stress‐activated protein kinase 1; TSP1 = thrombospondin 1; HSP27 = heat shock protein 27; bFGF = basic fibroblast growth factor; VCAM1 = vascular cell adhesion molecule 1; PTHrP = parathyroid hormone related protein; mtDNA = mitochondrial DNA; EV = extracellular vesicle; CXCR4 = C‐X‐C motif chemokine receptor 4; CXCL12 = C‐X‐C motif chemokine ligand 12 (stromal cell‐derived factor 1); miRNA = micro RNA; ANG1 = angiopoietin 1; SCF = stem cell factor; ANXA2 = Annexin II; GAS6 = growth arrest‐specific 6; BMP7 = bone morphogenetic protein 7; SPARC = secreted protein rich in cysteine
Tumor cells frequently upregulate the expression of certain cytokines to modulate the activity of immune cells and other cells in the microenvironment to make it more amenable to proliferation and lesion growth.74, 75 For example, CCL2 and CCL5 produced by breast cancer cells in the primary tumor site are correlated with poor prognosis and high tumor grade in patients.76, 77, 78 CCL2 and CCL5 produced by T47D breast cancer cells recruit metastasis promoting myeloid cells in vitro and their expression is correlated to an increased number of tumor associated macrophages (TAMs) in primary breast tumors from patients.74, 79, 80, 81, 82 CCL2 and CCL5 act as chemoattractants for monocytes,83, 84 which are precursors of macrophages. From human breast tumor samples it is known that these TAMs can release vascular endothelial growth factor (VEGF), which is correlated with increased microvessel density and significantly worse disease‐free survival.85 Interestingly, bisphosphonates have been demonstrated in multiple clinical studies of breast cancer to have anti‐tumor effects31, 86, 87, 88 and intravital imaging has recently demonstrated that TAMs engulf bisphosphonates in 4T1 mammary fat pad tumors, possibly leading to the anti‐tumor effects that have previously been observed.89 Although the functional role for bisphosphonate uptake by TAMs in 4T1 tumors still needs to be elucidated, these data suggest that TAM engulfment of bisphosphonates may either mitigate the pro‐tumor effects of TAMs or confer anti‐tumor activity. TAMs also promote breast cancer cell invasion, and thus likely metastasis, as demonstrated by live imaging of PyMT mammary tumors in mice.90, 91 CCL2 expression by breast cancer cells promotes bone metastatic lesion growth, through enhanced osteoclastogenesis.92 Furthermore, breast cancer cells can increase the expression of CCL2 by osteoblasts, which further stimulates osteoclastogenesis.93, 94 While this has not been directly linked to tumor dormancy studies, it is understood that tumor cells that reside in the osteoblast niche may re‐activate with bone turnover13 suggesting that CCL2 may promote tumor cell exit from dormancy.
Interferon signaling has been shown to regulate bone metastasis through its effects on the CD8+ T cell population and NK cells. Interferon regulatory factor 7 (IRF7) reduced bone metastases in a 4T1 mouse model and breast cancer patient samples, suggesting that patients with bone metastases might benefit from interferon‐targeting therapies.95 The authors of this study speculate that IRF7‐induced interferon production might prevent the accumulation of myeloid‐derived suppressor cells (MDSCs), thus releasing the inhibition on NK cells and CD8+ T cells, allowing them to attack and clear breast tumor cells in the bone. It has also been demonstrated that CD11+Gr‐1+ myeloid cells are expanded in MDA‐MB‐231 breast cancer bone metastases by TGF‐β signaling independent of the T cell population and can differentiate into osteoclasts in the bone marrow.96 Blocking dikkopf‐1 (DKK1), which is elevated in the bone marrow in mice with extraskeletal tumors that form following subcutaneous injection of Lewis lung carcinoma cells, reduces tumor growth by restoring T cell recruitment to the primary tumor site.97 Lysyl oxidase (LOX) expression, which is associated with poor overall survival in multiple tumor types,98, 99, 100, 101 can promote the formation of the pre‐metastatic niche for MDA‐MB‐231 cells through recruitment of CD11b+ myeloid cells and c‐Kit+ myeloid progenitor cells to the lungs.46 Since many of these studies were carried out with aggressive tumor models of breast cancer it is difficult to determine how immune recruitment to the pre‐metastatic niche may impact dormant disseminated tumor cells, but given that many of these cells originate in the bone marrow and that altering the recruitment of multiple immune populations influences bone colonization by tumor cells, further studies into the effects of immune editing on tumor dormancy and entry/exit into dormancy are warranted.
2.2. Hypoxia
Hypoxia, or low oxygen tensions, triggers numerous gene expression changes through stabilization of the hypoxia‐inducible factor (HIF) transcription factors.102, 103 HIF1 binds to hypoxia response elements (HREs) to drive the expression of many target genes, including VEGF, which stimulates angiogenesis by binding to VEGF receptor (VEGFR) on the surface of endothelial cells and causing them to migrate and assemble into new blood vessels.104, 105 Increased microvessel density improves the delivery of oxygen and nutrients to tumor cells, which in turn promotes tumor cell proliferation. Hypoxia also triggers a HIF‐mediated increase in glycolysis by stimulating the expression of glucose transporters and glycolytic enzymes.106, 107, 108, 109 This enables tumor cells to survive in nutrient poor microenvironments.
Hypoxia alters the expression of genes involved in dormancy maintenance (Figure 3B), such as the leukemia inhibitory factor receptor (LIFR), which acts as a breast cancer tumor suppressor in MCF7 human breast cancer cells110 and breast cancer pulmonary metastasis suppressor in SUM159 human breast cancer cells,111 and confers a dormant phenotype in bone‐disseminated MCF7 breast cancer cells.52 Hypoxia negatively regulates LIFR expression in MCF7 and SUM159 breast cancer cells in vitro and is negatively correlated with LIFR mRNA levels in patient samples,52 suggesting that hypoxia may be a mechanism by which LIFR is down‐regulated in breast cancer. In the context of the bone marrow, where oxygen levels fluctuate between approximately <1–6% partial oxygen,112, 113, 114, 115 this suggests that the microenvironment may promote tumor cell exit from dormancy. It is important to note that the negative effect of hypoxia on LIFR expression in breast cancer cells was only observed in extremely low oxygen levels (<0.5% pO2), suggesting that the bone marrow microenvironment in general would not be sufficient to down‐regulate LIFR; this would presumably only happen in regions of extreme hypoxia, such as has been observed in the sinusoidal region.116 Furthermore, as tumors grow in the bone marrow, they will rapidly become hypoxic, further perpetuating the loss of LIFR. The concept that hypoxia may also promote some bone‐disseminated tumor cells to exit dormancy is supported by data indicating that loss of HIF signaling in osteoblast lineage cells (osterix‐positive cells) reduces metastasis and growth of PyMT mouse mammary carcinoma cells in the bone in vivo, while activation of HIF signaling through osteoblast‐specific deletion of the HIF negative regulator VHL increases tumor burden in bone.117
Hypoxia has also been reported to induce dormancy in disseminated tumor cells (DTCs). In this study, the dormancy markers NR2F1 (Nuclear Receptor Subfamily 2 Group F Member 1) and DEC2 were found to be co‐expressed with hypoxia markers such as HIF1α and GLUT1 in MDA‐MB231 cells injected into the mammary fat pad. Additionally, non‐cycling cells in the CAM‐implanted HEp3 head and neck squamous cell carcinoma cells had more intense pimonidozole staining, which is a marker of hypoxia, indicating that quiescent cells were more commonly localized to hypoxic regions.118 Similar effects were seen in models where CAM‐implanted tumors were treated with desferrioxamine (DFOM, a hypoxia mimicking drug that causes the accumulation of HIF1α). DFOM treated tumors had significantly more quiescent cells and DTCs from the DFOM treated tumors were also resistant to cisplatin. Thus, hypoxic signaling can induce quiescence and chemotherapy resistance, which are two hallmarks of dormancy. NR2F1 expression was previously shown to be elevated in dormant HEp3 cells62 and high NR2F1 has been shown to correlate with longer disease‐free survival in prostate cancer patients who received hormone ablation therapy,119 suggesting a pro‐dormancy role for NR2F1. DEC2 (Differentially Expressed In Chondrocytes 2, also known as SHARP1) causes the degradation of HIF1α and HIF1β proteins and thus inhibits migration, invasion, and metastasis of MDA‐MB‐231 cells in vitro and in vivo and low DEC2 expression in the tumors of triple‐negative breast cancer patients is associated with worse metastasis‐free survival.120
p38 is a MAP kinase that has been shown to be activated downstream of TGF‐β2 signaling. Interestingly, it is the ratio of ERK to p38 signaling that appears to influence dormancy, where a high ERK/p38 ratio (i.e. high ERK and low p38 signaling) drives tumor cells out of dormancy, while a low ERK/p38 ratio (i.e. low ERK and high p38 signaling) was characteristic of dormant tumor cells.63 p38 activation is proposed to drive NR2F1 expression62 and can lead to the induction of HIF1α,121 suggesting that p38 signaling may orchestrate the co‐expression of these genes. Mitogen‐ And Stress‐Activated Protein Kinase 1 (MSK1), a downstream target of p38 MAPK, has also been found to drive dormancy in estrogen receptor positive latent breast cancer cells.53 MSK1 was identified as a dormancy regulator from a whole‐genome shRNA screen in dormant bone metastatic (DBM) T47D human breast cancer cells that were isolated following intracardiac inoculation. Lentiviral knockdown or CRISPR‐Cas9 deletion of MSK1 in DBM T47D cells as well as ZR75 cells, a poorly metastatic ER+ human breast cancer cell line, increased bone homing as indicated by bioluminescence imaging (BLI) following intracardiac and mammary fat pad injection, respectively. Furthermore, high MSK1 expression was associated with increased bone metastasis free survival in ER+ breast cancer patients, supporting the conclusion that MSK1 expression reduces tumor homing to the bone marrow. Interestingly, the mechanism of MSK1 on tumor dormancy was found to be independent of tumor cell quiescence, survival in hypoxia, adhesion, migration, or invasion, but rather induced epigenetic modifications of luminal differentiation genes. Thus loss of MSK1 prevents tumor cell differentiation and the maintenance of a dormant population in the bone marrow.
Lysyl oxidase (LOX) is a hypoxia induced pro‐invasion and pro‐metastasis factor that plays a role in the formation of the pre‐metastatic niche (PMN) in distant organs to make these sites hospitable for colonization. LOX secreted from primary breast tumors localizes to the lungs where it can trigger remodeling of the ECM and the formation of the PMN46, 122 along with bone marrow‐derived cells (BMDCs), which have been found to be critical in PMN establishment and promoting distant metastasis.123 LOX knockdown in MDA‐MB‐231 cells reduced the rate of dissemination to lung following mammary fat pad injections in mice, and reduced CD11b + myeloid cells and c‐Kit+ myeloid progenitor cell migration to the lungs,46 suggesting that LOX is required for tumor dissemination to the lungs through recruitment of these progenitor cells to the PMN. In vitro invasion assays also showed that the presence of LOX in the ECM increases BMDC invasiveness.46 LOX increased collagen IV crosslinks in matrigel cultures, which increased the adhesion of CD11b + cells and stimulated MMP‐2 production in vitro, possibly resulting in a microenvironment that is more permissive to further invasion by BMDCs and the establishment of a PMN and eventual metastatic outgrowth. This model has so far been proposed in pulmonary metastasis; whether LOX plays a distinct role in tumor cell dissemination to bone remains to be determined. Since BMDCs that normally establish the PMN are already resident in bone, it is difficult to tease out the effects of the PMN in the bone marrow. The formation of a PMN provides a microenvironment that is more hospitable to tumor cells and thus will ultimately promote metastasis and tumor cell growth; however, it has been speculated that tumor cells that disseminate while the PMN is still being established may be kept dormant until the microenvironment is sufficiently altered to support their growth.124 However, if the proposed model is operative in bone, the myeloid cells that are abundant in the PMN may lead to increased osteoclastogenesis and fuel tumor growth.46, 96 Thus, while it is possible that the PMN may support temporary tumor dormancy, bone‐disseminated tumor cells in the PMN are likely to reactivate and colonize the distant site.
Together, these data suggest that hypoxia may have different regulatory roles in different phases of tumor progression and in different metastatic sites. For instance, differences in oxygen tensions may regulate the dormancy phenotype, where oxygen levels >1% promote tumor dormancy, while oxygen levels <1% promote tumor cell exit from dormancy, and this may be further dependent upon the niche in which bone‐disseminated tumor cells are currently residing. It is also possible that hypoxia may induce dormancy in the primary tumor site, and promote exit from dormancy at sites of bone metastasis. Both of these mechanisms are consistent with poor overall survival in breast cancer patients who have increased HIF1α staining in the primary tumor.102, 125, 126, 127 In the bone marrow, aged bones have less vasculature128 and may therefore experience lower oxygen tensions, and some studies suggest that age is a risk factor for bone metastatic disease in breast cancer.129 Thus, extreme hypoxic compartments in the bone may stimulate bone‐disseminated tumor cells to exit dormancy, also reducing patient survival. Furthermore, it is known that hypoxia in PyMT mouse mammary carcinoma tumors promotes dissemination to the lung.130 Whether hypoxia in the primary tumor also stimulates dissemination of breast cancer cells to the bone marrow remains unknown; however, in vivo data from multiple groups indicates that activation of HIF1α signaling in MDA‐MB‐231 breast cancer cells promotes bone colonization,131 and inhibition of HIF1α signaling reduces bone colonization.11, 131, 132 What is well appreciated is that in all cases hypoxic effects on tumor cells at any stage of the disease is detrimental to patient outcome.
2.3. Angiogenesis and the perivascular niche
Proliferating tumor cells possess increased oxygen and nutrient demands and stimulate angiogenesis to meet these needs. Insufficient angiogenesis has been recognized as a dormancy‐inducing mechanism, particularly in the model of micrometastatic dormancy.133 In the “angiogenic dormancy” model, the inability of a micrometastasis to induce angiogenesis limits its ability to grow past approximately 1 mm in diameter. This may be due to either a lack of intra‐tumoral microvessels or from an active repulsion of existing vessels in the microenvironment, due to increased expression of anti‐angiogenic factors such as thrombospondin 1 (TSP1 or THBS1) and low expression of angiogenic proteins such as VEGF and basic fibroblast growth factor (bFGF).134, 135 As tumor cells proliferate and tumor mass increases, low oxygen supplies induce hypoxia and VEGF production by the tumor cells.136 This stimulates angiogenesis, increases oxygen supply, and promotes dormancy escape; however, if the tumor cells reside in a microenvironment rich in anti‐angiogenic factors, the cells may not be able to induce angiogenesis and escape dormancy (Figure 3C). Non‐angiogenic tumors in a MDA‐MB‐436 breast cancer xenograft model were found to have lower expression of heat shock protein 27 (HSP27), which was associated with reduced endothelial proliferation and decreased secretion of VEGF and bFGF. Conversely, HSP27 overexpression in MDA‐MB‐436 cells that were originally non‐angiogenic stimulated proliferation in vivo.137 A set of 19 microRNAs that govern the phenotypic switch of dormant MDA‐MB‐436 breast cancer cells, T98G glioblastoma cells, KHOS‐24OS osteosarcoma cells, and SW872 liposarcoma cells to fast‐growing angiogenic tumors has also been identified by testing for the expression of 378 microRNAs in dormant or angiogenic/fast‐growing tumors of each cell line with high‐throughput real time qRT‐PCR based miR expression analysis.138 Interestingly, SW872 liposarcoma tumor cells that had an angiogenic phenotype in vivo, meaning they have the capacity to promote angiogenesis and tumor progression, can transition into a non‐angiogenic phenotype when injected into a different mouse, and these dormant non‐angiogenic cells were able to then spontaneously switch back to an angiogenic phenotype, suggesting that angiogenic dormancy may be a fluid state.139 “Revertant clones” that were non‐angiogenic but had been derived from an angiogenic clone were found to have decreased VEGF expression but increased TSP1 expression, supporting the fact that they are not able to induce angiogenesis.
TSP1 is an angiogenesis inhibitor140 and matricellular protein known to interact with dozens of factors, leading to a wide variety of effects on cellular activity by aggregating, sequestering, or activating many proteins, and modifying the extracellular matrix.141 In the context of tumor dormancy, in addition to limiting angiogenesis, TSP1 acts as an endothelial cell‐derived tumor suppressor in the perivascular niche in both the lungs and the bone, by suppressing the proliferation of tumor cells that are adjacent to stable microvessels.15 Using bone marrow vasculature mimicking in vitro cultures, MDA‐MB‐231 or T4–2 breast cancer cells that settled adjacent to TSP1‐rich, stable microvessels did not proliferate for the duration of the 17 day culture and remained Ki‐67 negative, unlike breast cancer cells that settled next to actively growing vascular tips. TSP1 has a known role in metastasis suppression in breast cancer, but these actions have been exclusively attributed to its angiogenesis‐inhibiting function,142 since it is known to stabilize microvascular endothelium by inhibiting endothelial cell motility and growth.143 Since tumors can only grow to 1–2 mm3 without additional blood supply,144 micrometastases that are beginning to grow into active lesions would likely have increased angiogenesis and high neovascular tip density. Periostin (POSTN) and transforming growth factor beta (TGF‐β1), which both enhance tumor cell proliferation, are enriched in the neovascular tip.15 The role for TSP1 in this process was demonstrated in bone marrow‐like vasculature, and although the role for POSTN and TGF‐β1 were not specifically tested in the bone marrow, given the extensive body of literature demonstrating that TGF‐β1 promotes tumor cell growth in the bone145, 146, 147, 148 these data are consistent with the pro‐tumorigenic role for TGF‐β1 in the bone marrow. Taken together, these findings suggest active angiogenesis awakens cells from dormancy, while DTCs that settle adjacent to stable microvessels are maintained in a more quiescent state.
2.4. Interactions with the extracellular matrix and bone‐resident cells
In 1889 Stephen Paget put forth the “seed and soil” hypothesis; if the soil is fertile and conducive to growth, the seed will grow; however, if the soil that the seed lands in is hostile or incompatible with the needs of the seed, the seed will not germinate.149 Thus, a cancer cell that disseminates to an incompatible microenvironment in a distant site may become dormant150; however, the microenvironment may change over time, making the soil more fertile for the disseminated tumor cell.
Several factors have been identified that exert their quiescence‐inducing effects by modulating cell interactions with the extracellular matrix (ECM) (Figure 3D). Reduced urokinase plasminogen activator receptor (uPAR) expression was found to induce G0‐G1 arrest in HEp3 cells grown on a CAM, as measured by FACS analysis of DNA content. This growth arrest was found to be due to the lack of uPAR interacting with integrin α5β1. 151 Integrin α5β1 and uPAR interact to recruit focal adhesion kinase (FAK) and epidermal growth factor receptor (EGFR) to the cell surface. FAK and EGFR signal through the Ras‐extracellular signal‐regulated kinase (ERK) pathway in a fibronectin‐dependent manner, which drives cell proliferation. Thus, decreased uPAR expression leads to decreased integrin α5β1 mediated adhesion to the ECM and reduced ERK activation, inducing a dormant state.152 Although it is not clear whether uPAR plays a role in tumor dormancy in the bone marrow, PC3 prostate cancer cells that were previously grown as subcutaneous tumors in vivo and were able to spontaneously disseminate to the bone marrow expressed elevated levels of uPAR protein by cytokine array in comparison to parental PC3 cells only grown in 2D culture,153 and uPAR was detected at a higher frequency in bone‐disseminated prostate cancer cells harvested from patients compared to tumor cells found in circulation and was associated with a poor Gleason score.154
Integrins can also play a role in the cross talk of cancer cells and bone resident cell types. MDA‐MB‐231 breast cancer cells that have aberrant vascular cell adhesion molecule 1 (VCAM1) expression can promote monocytic osteoclast progenitor cell recruitment by interacting with integrin α4β1 on their surface, leading to increased osteolysis and the progression of tumor‐induced bone disease in vivo.155 It is not clear whether VCAM1 expression on breast cancer cells promotes their exit from dormancy, since these experiments utilized a breast cancer cell line that readily colonizes the bone, but this interaction demonstrates how tumor cells directly communicate with bone‐resident cells to promote their growth and survival.
The expression of CXCR4 on the surface of tumor cells is recognized as a key mechanism for tumor cell homing to the bone marrow. CXCR4 is overexpressed in many cancer types including hematological malignancies, breast cancer, colorectal cancer, esophageal cancer, renal cancer, gynecologic cancer, pancreatic cancer, and liver cancer and is correlated with poor progression free survival.156 CXCR4 binding to its ligand CXCL12 (SDF‐1) has also been shown to regulate tumor cell migration; DU4475 and MDA‐MB‐231 breast cancer cell migration was found to increase in a dose‐dependent manner when treated with CXCL12.157 Thus, tumor cells that overexpress CXCR4 have an increased ability to colonize niches in the bone marrow that are rich in CXCL12.158, 159, 160 CXCL12 is expressed by bone marrow stromal cells and osteoblasts,161 which recruits these tumor cells to the endosteal niche. CXCL12 has also been shown to stimulate osteoclast precursor cell migration.162, 163 PC3 cells, which aggressively colonize the bone marrow when inoculated in vivo, have been shown to have higher endogenous levels of CXCR4 compared to less aggressive LNCaP cells, and CXCR4 is further stimulated in PC3 cells following exogenous treatment with CXCL12.162 This CXCR4 upregulation allowed the cells to adhere to human umbilical vein endothelial cell monolayer in culture and enhanced transendothelium migration, suggesting that CXCR4‐ECM interactions in CXCL12‐rich niches facilitate the entrance of prostate cancer cells into the bone marrow. Abundant CXCR4 expression on prostate cancer cells also mediates interactions with α5 and β3 integrins,161 which may make CXCR4 another attractive therapeutic target to mobilize cells and increase their chemosensitivity. Comparison of “angiogenic” and “revertant” SW872 liposarcoma tumors found that revertant clones that remained dormant and did not grow into large tumors when implanted subcutaneously had much higher expression of CXCR4 than the angiogenic clones,139 supporting a potential pro‐dormancy role for CXCR4 that may be dependent upon the vasculature. In contrast, cells isolated from breast cancer patient‐derived xenografts (either in the orthotopic tumor or from lung metastases) that exhibited undetectable CXCR4 expression were more frequently non‐proliferative.164 Thus, it is clear that CXCR4‐CXCL12 signaling promotes tumor homing to the bone marrow, but its role in dormancy is not well defined.
Bone morphogenetic protein 7 (BMP7) produced by bone stromal cells has been shown to induce dormancy in an apparently reversible manner.55 Treatment of prostate cancer cell lines LNCaP, C4, C4–2, C4–2B, PC3, DU145, and ALVA with conditioned media (CM) from HS5 bone stromal cells induced p21, p27 and p38 MAPK, which are markers of quiescence. Treating PC3 mm cells, which are a highly metastatic variant of the human PC3 prostate cancer cell line,165 with HS5 CM also induced senescence‐associated β‐galactosidase protein expression as well as N‐MYC downstream‐regulated gene 1 (NDRG1), which is a metastasis suppressor, at the mRNA and protein level. Of the most abundant known bone‐derived factors, including FGFs and TGF‐β, only BMP7 activated all of these pro‐dormancy factors in vitro. Treatment with BMP7 reduced the incidence of PC3 mm bone metastases following intracardiac inoculation in vivo, and knockdown of BMP7 in intratibial‐inoculated PC3 mm cancer stem cells resulted in increased tumor burden by BLI in vivo. It remains unclear whether BMP7 causes the PC3 cells to enter a non‐proliferative or low‐cycling state in vivo, but these data indicate that BMP7 inhibits bone colonization by prostate cancer cells.
Interestingly, BMP7 expression from bone stromal cells can be induced by Secreted Protein Rich in Cysteine (SPARC) secreted by dormant prostate cancer cells.56 Microarray analysis revealed that SPARC was significantly up‐regulated in indolent PC3 mm CSCs compared to aggressive PC3 mm CSCs. When SPARC expression was knocked down in indolent cells using shRNA and inoculated into mice by intratibial or intracardiac injection, tumor burden was increased by BLI and the incidence of bone metastases was significantly increased. Injection of recombinant SPARC also reduced the incidence of bone metastasis by aggressive tumor cells, which in vitro studies suggest is likely to influence tumor cell proliferation through modification of the bone marrow stromal cells. In vitro, the SPARC promoter was found to be more heavily methylated in aggressive compared to indolent cells. SPARC induced BMP7 expression, and this was reversibly modified through treatment with DNA de‐methylating agents. In support of these findings, SPARC and BMPR2 were significantly down‐regulated in the primary tumor samples of patients who developed bone metastases compared to those with no bone metastases, suggesting that these molecules may be important in the formation of bone metastases and subsequent colonization. These data further support the previous finding that BMP7 prevents bone colonization in vivo.55
Cancer associated fibroblasts (CAFs), while not specific to tumors growing in the bone, are an important cell type that can alter the behavior of tumor cells. Previous work has demonstrated that fibroblasts promote the growth of RWGT2 human lung cancer cells and MDA‐MB‐231 human breast cancer cells in the bone in vivo, likely due to their elevated secretion of Wnt ligands in vitro, which stimulates subsequent GLI2 and PTHrP production by bone‐disseminated tumor cells166; however, the effect of Wnts secreted by CAFs has not been directly evaluated in the context of tumor dormancy in bone. Extracellular vesicles (EVs) secreted by CAFs and containing mitochondrial DNA (mtDNA) have recently been demonstrated to transfer mtDNA to estrogen receptor positive breast cancer cells, and promote their exit from metabolic dormancy induced by the hormone therapy fulvestrant.167 While several of the CAF patient samples from this study were derived from a patient bone metastasis, the effect of CAF EVs on tumor exit from dormancy in the bone marrow has not been directly evaluated. The specific cargo packaged within EVs is believed to play an important role in controlling how EVs alter disseminated tumor cells. Exosomes derived from mesenchymal stem cells have been shown to push a subset of MDA‐MB‐231 and T47D human breast cancer cells into the S phase and another subset into quiescence. The exosomes that promote quiescence were found to be mediated by miRNA‐222 in vitro and anti‐miR‐222/223 reduced the presence of i.p. inoculated breast cancer cells disseminating to the femur, as indicated by an absence of GFP staining and reduction in Ki67‐positive cells.168 Other microRNAs have also been shown to play a role in promoting dormancy; BCLC9 human hepatocellular carcinoma cells engineered to express miR‐122 exhibited reduced proliferation in vitro, formed smaller tumors when injected subcutaneously, and exhibited a low ERK/p38 activation ratio, suggesting the cells were in a dormant state.168, 169 Furthermore, it has been demonstrated that miR‐135 and miR‐203 reduce spontaneous dissemination of MDA‐MB‐231 cells to the bone marrow,170 and microRNAs are currently being investigated as a means to target bone‐disseminated tumor cells.171
As metastatic cells disseminate to the bone marrow and settle in the HSC niche, it has been proposed that they may coopt the quiescence inducing signals produced by osteoblasts and perivascular cells, such as angiopoietin‐1 (ANG1),16 stem cell factor (SCF),17 annexin II (ANXA2) and growth arrest‐specific 6 (GAS6).18 Some HSCs express TIE2, a receptor tyrosine kinase that binds ANG1 expressed by osteoblasts. This interaction induces quiescence in the HSCs and protects against apoptosis.16 Thus, if tumor cells express TIE2, they may be able to take advantage of this quiescence‐inducing interaction. SCF is expressed by perivascular cells throughout the bone marrow, and SCF deletion in either endothelial cells or perivascular stromal cells lead to a depletion of HSCs, indicating that HSCs reside in a perivascular niche where there are likely many other factors that contribute to their maintenance and quiescence.17 ANXA2 in particular has been shown to promote prostate cancer cell homing to HSC niches by binding to the annexin II receptor (ANXA2R). Once in the bone marrow, the high ANXA2 levels stimulate the expression of AXL, a receptor tyrosine kinase, in the prostate cancer cells. Osteoblast‐derived GAS6 signaling through its receptor AXL on the prostate cancer cells was found to promote a semi‐dormant phenotype, leading to decreased proliferation and chemo‐sensitivity.18 GAS6/AXL signaling appears to drive TGF‐β receptor expression (TGFBR2 and TGFBR3), and knockdown of GAS6 or AXL resulted in dampened p27 expression in prostate cancer cells following TGF‐β2 stimulation in vitro, indicating that GAS6/AXL signaling may control the dormant phenotype by altering the cell's response to TGF‐β2,172 which has been demonstrated to drive dormancy.63 In addition to AXL, GAS6 can signal through two other receptors, TYRO3 and MER. Interestingly, when TYRO3 expression is higher than AXL expression, an expression pattern dominant in primary tumors or bone metastatic lesions, prostate cancer cells are proliferative. However, when AXL expression is predominant, an expression state common in bone disseminated cells, they remain dormant.173 Thus, GAS6 may have opposing effects on tumor cell growth depending on disease state and receptor expression patterns.
While there are multiple molecules that stimulate the growth and survival of tumor cells in the bone through its interactions with bone resident cells, one of the most well‐studied is parathyroid hormone‐related protein (PTHrP). PTHrP was originally discovered as the factor that causes humoral hypercalcemia of malignancy174 and is well established to promote tumor‐induced bone destruction through its stimulation of RANKL on osteoblast lineage cells to promote osteoclast‐mediated bone resorption.8, 9, 10, 11, 12 More recently, it has been suggested that PTHrP may also play a role in the awakening of tumor cells in the bone marrow.52, 54 Overexpression of PTHrP, which normally binds to the parathyroid hormone receptor type 1 (PTHR1) to induce cAMP signaling, induces osteolytic bone destruction in cells that otherwise lie dormant in the bone marrow12 and down‐regulates a number of pro‐dormancy genes52 through a cAMP‐independent mechanism.54 Interestingly, PTHrP is regulated by the bone matrix itself, in that tumor cells that come in contact with the rigid bone surface up‐regulate PTHrP; however, this is specific for tumor cells that are already aggressive in nature.175 In breast cancer cells that do not readily induce osteolysis (e.g. MCF7), the rigid bone microenvironment is not sufficient to induce PTHrP expression, but significantly and dose‐dependently increases PTHrP levels in aggressive bone metastatic cells (e.g. MDA‐MB‐231).175 This is consistent with the concept that not all tumor cells that disseminate to the bone marrow switch on PTHrP and immediately induce osteolysis; rather, those cells that are already primed are fueled by the rigidity of the bone matrix they encounter.
2.5. The cancer stem cell hypothesis
Dormant tumor cells are able to give rise to clinically significant metastases, and it is therefore likely that they also possess self‐renewal capacities. These characteristics are notably similar to those of a cancer stem cell (CSC). Similar to pluripotent stem cells, CSCs are long‐lived, generally quiescent, and have the ability to self‐renew and differentiate.176 CSCs are thought to play a role in tumor initiation, progression and metastasis, since sorted CSCs from human patient derived xenografts (PDX) are able to form tumors in immunocompromised mice.177 CSCs have been identified in a number of tumor types including melanoma, breast, colon, prostate, and pancreatic cancers,177, 178, 179, 180, 181 and several groups have found a strong correlation between the number and existence of slow‐cycling cancer cells, frequency of CSCs and their ability to form tumors.182 DTCs have been proposed to adopt a CSC phenotype due to their shared characteristics with pluripotent stem cells, such as self‐renewal and differentiation, quiescence, and chemotherapeutic resistance.152 It is therefore theorized that the DTCs that survive and grow into clinically detectable metastases after escaping dormancy may be CSCs. These properties may confer the necessary characteristics for the cell to survive in circulation and grow from a single DTC into a metastasis in the secondary site. While the connection between dormancy and CSCs has not been extensively investigated in the context of bone disseminated cells, this recently proposed paradigm warrants discussion.
CSCs have the capacity for self‐renewal, giving rise to uncontrolled growth of differentiated cell populations that may lead to primary and metastatic tumor growth. One of the key defining properties of CSCs is their ability to “seed” the growth of a tumor, or their tumor initiating capacity (TIC). This feature of CSCs has been demonstrated in the context of breast cancer, where cells from human patient derived xenografts (PDXs) were sorted for human breast CSCs (CD44hi/CD24lo cells). Breast CSCs were able to form palpable tumors in immunocompromised mice with fewer than 100 cells.177 Several regulatory signaling pathways, such as Notch,183 Hedgehog,184 transforming growth factor‐beta (TGF‐β),185 estrogen receptor/progesterone receptor (ER/PR),186 epidermal growth factor (EGF) /EGF receptor (EGFR/HER2),187 and leukemia inhibitory factor (LIF)188 have been reported to promote self‐renewal in mammary stem cells and CSCs. It is therefore possible that the signaling pathways that promote a stem‐like state could also promote a chronic state of dormancy in DTCs.
CSCs and DTCs can reside in a quiescent, non‐proliferative state. Clonal tracking and membrane dyes have been used to identify slow‐cycling or dormant pools of colon cancer and breast cancer cells from patient biopsies.182, 189 In the context of breast cancer, human mammary gland cells that were identified as slow‐cycling, through the retention of membrane dye PKH26, were used to develop an expression profile that identified human normal mammary stem cells (hNMSCs). PKH‐positive and ‐negative hNMSCs were immunophenotypically characterized for expression of markers indicating epithelial or myoepithelial characteristics as well as terminal differentiation. PKH‐positive cells expressed markers of both epithelial and myoepithelial cells but did not express terminal differentiation markers. This expression signature was applied to human breast cancer samples and demonstrated the ability to identify cells with the capacity for tumor initiation, which was found to be predictive of biological and molecular features of CSCs.182 For colon cancer, clonal tracking using lentiviral transduction and serial transplantation were used to track tumor initiating cells from primary human colon cancer samples. These studies found that serially transplanted tumors from xenografts contributed to tumor initiation in both the primary site and metastatic sites after subsequent transplantations.189 These studies established a positive correlation between slow‐cycling/dormant tumor cell populations and CSCs,182, 189 highlighting the shared functional and molecular characteristics.
The connection between DTCs and CSCs is also observed in their ability to resist cancer therapy. DTCs are often spared by current treatment modalities due to their low proliferative phenotype, and similar effects are seen in CSCs across tumor types. Dormant HEp3 head and neck squamous cell carcinoma cells, defined by their active p38 signaling, low uPAR expression, and reduced growth in vivo, proved to be highly resistant to etoposide and doxorubicin in vitro when compared to tumorigenic controls.190 In multiple myeloma (MM), chemotherapeutic agents can induce a slow cycling/dormant phenotype. RPMI 8226 MM cells treated with either bortezomib or a proteasome inhibitor (MG‐132) strongly induced cell death in a time dependent manner in vitro. However, the absolute number of MM cells after 24‐hour treatment with either chemotherapeutic agent remained constant for several days post‐treatment, due to the G0‐G1 arrest of surviving cells.191 Quiescence is a hallmark of DTCs, especially in the context of chemo/radio‐therapy and is an important mechanism for underlying CSC resistance to therapy.192 In acute myeloid leukemia (AML), leukemic cells isolated from peripheral blood and normal bone marrow samples taken from patients were able to engraft in the bone marrow of NOD/SCID mice. However, quiescent leukemic stem cells (LSCs), identified as slow‐cycling cells in G0, had significantly better engraftment into the bone marrow of mice in comparison to cells in G1, and S/G2 + M,193 suggesting that dormant cells may possess a survival advantage in the bone marrow. In chronic myeloid leukemia (CML), resistance to second generation tyrosine kinase inhibitors against BCR‐ABL has been linked to the failure of the chemotherapeutic regimen to deplete quiescent fractions of LSCs. Primary CML samples acquired from patients were treated with tyrosine kinase inhibitors dasatinib and nilotinib, resulting in an increase in the less proliferative CD34+ CD38− CFSEMax (carboxyflurorescein succinimidly ester) cell population, which have been identified as stem/progenitor cells when cultured in vitro..194, 195 Breast CSCs from patients with HER2 amplification were found to accumulate after cytotoxic chemotherapy had eliminated the bulk of tumor cells. These data are further corroborated by patient biopsies before and after neoadjuvant chemotherapy and HER2 targeted therapies that showed an increase in the percentage of CD44Hi/CD24Lo breast CSCs and their ability to form mammospheres post‐treatment, with similar results observed in colorectal cancer patients.196, 197 It is important to note that other mechanisms for the therapeutic resistance of DTCs may be attributable to mechanisms that are intrinsic to CSCs, such as impairment of apoptosis pathways,198 reduced drug accumulation,199, 200 and regulation of DNA repair pathways.201, 202, 203
What is lacking in the field is clear evidence for whether bone‐disseminated dormant tumor cells are in fact cancer stem cells. This is largely due to a lack of studies that examine the CSC population in bone‐disseminated populations; however, there are a few studies that have done so. It has been found that the majority of bone‐disseminated cancer cells from breast cancer patients exhibited CD44hi/CD24lo CSC markers.204 In contrast, it has been observed that when human MCF7 breast cancer cells exit dormancy in vivo through knockdown of LIFR, which is part of the receptor complex for the pro‐stemness factor LIF, the CD44Hi/CD24Lo population does not decrease, as would be hypothesized if there were overlap between DTCs and CSCs; however, the MCF7 cells harbor an extremely low CSC population, making it difficult to detect a decrease.52 Interestingly, PC3 prostate cancer cells that readily colonize the bone in vivo express the CD44 and CD133 CSC markers at an equal rate as cells that do not colonize the bone.205 This study also found that PC3 cells that were mitotically quiescent in vitro formed more bone metastases following intracardiac inoculation than cells that were proliferative in vitro. This suggests that the ability of prostate cancer cells to induce osteolytic bone destruction is not due to an enrichment of the CSC phenotype.
It remains unclear whether bone‐disseminated tumor cells are in fact CSCs or have adopted their quiescent properties as a survival mechanism to persist long term in foreign sites. Given the overlap of CSC and DTC properties, robust models that can identify dormant populations and their potential overlap with the CSC population could shed light on the mechanisms that drive tumor cell entry and exit from dormancy. Their shared characteristics support the hypothesis that DTCs adopt CSC features in distant sites, which may ultimately contribute to latent disease and delayed cancer recurrence.
3. REFLECTIONS AND REMAINING QUESTIONS
There are many unanswered questions surrounding the fundamental features of bone‐disseminated tumor cells and their entry and exit from dormancy. Are bone‐disseminated cells predisposed to enter dormancy, or it purely dependent on environmental cues that trigger dormancy upon arrival in the bone marrow? Factors such as TSP1 suggest that the perivascular niche encountered by disseminated tumor cells may induce dormancy,15 but a cell autonomous model of dormancy may still be possible. How do dormancy‐maintaining signals and reactivating signals determine the fate of a disseminated cell? Factors that promote proliferation are often co‐expressed with dormancy‐promoting factors, but it is unclear exactly how these two processes overlap. For example, hypoxia stimulates tumor‐induced osteolysis by downregulating LIFR52 and tumor cell proliferation by promoting angiogenesis,136 while inducing the expression of pro‐dormancy factors such as NR2F1, DEC2, p27, and TGF‐β2.118 Is there a critical mass for the proliferation signals that outweigh the expression of dormancy‐promoting factors, and is this dependent upon microenvironmental cues? In other words, does a dormant tumor cell require multiple “hits” to exit dormancy? Furthermore, is tumor dormancy fluid? Can cells fluctuate between cellular and micrometastatic dormancy? If the loss of pro‐dormancy factors is driving the reactivation, what is causing this downregulation? In the example of the loss of LIFR expression, extremely hypoxic regions in the bone marrow are proposed to suppress its expression and drive cells out of dormancy,52 but these regulatory mechanisms need to be characterized for many other factors. The niches that tumor cells home to in the bone marrow also require a greater degree of characterization and investigation at the spatial, temporal, and molecular level.
4. CONCLUSIONS
Dormancy maintaining factors include both microenvironmental signals and intrinsic expression of factors on tumor cells that enable them to receive pro‐dormancy signals. While the list of dormancy regulating factors is growing, many of the targets are not therapeutically actionable, or translational aspects have not yet been explored. Further studies are needed to identify new targets and facilitate potential new treatments. Additionally, the identification of pro‐proliferative signals that drive tumor cells out of dormancy into a reactivated state is needed in order to better understand what triggers this phenotypic switch.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, V.M., R.W.J.; Writing ‐ Original Draft, V.M., T.O.; Writing ‐ Review & Editing, R.W.J.; Supervision, R.W.J.; Funding Acquisition, R.W.J.
ACKNOWLEDGEMENTS
The authors wish to thank Miranda Sowder for critical reading of the manuscript. Rachelle Johnson is supported in part by NIH R00CA194198 (RWJ) and DoD CDMRP award W81XWH‐18‐1‐0029 (RWJ). Vera Mayhew is supported by the Cellular, Biochemical and Molecular Sciences Training Program 5T32GM008554‐22 (JGP). Tolu Omokehinde is supported by HHMI Gilliam Fellowship GT10833.
Mayhew V, Omokehinde T, Johnson RW. Tumor dormancy in bone. Cancer Reports. 2020;3:e1156. 10.1002/cnr2.1156
REFERENCES
- 1. Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7(4):208‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20 Pt 2):6243s‐6249s. [DOI] [PubMed] [Google Scholar]
- 3. Riihimaki M, Hemminki A, Fallah M, et al. Metastatic sites and survival in lung cancer. Lung Cancer. 2014;86(1):78‐84. [DOI] [PubMed] [Google Scholar]
- 4. Katakami N, Kunikane H, Takeda K, et al. Prospective study on the incidence of bone metastasis (BM) and skeletal‐related events (SREs) in patients (pts) with stage IIIB and IV lung cancer‐CSP‐HOR 13. J Thorac Oncol. 2014;9(2):231‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gomis RR, Gawrzak S. Tumor cell dormancy. Mol Oncol. 2016;11:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mundy GR. Mechanisms of bone metastasis. Cancer. 1997;80(8 Suppl):1546‐1556. [DOI] [PubMed] [Google Scholar]
- 7. Sims NA, Martin TJ. Coupling signals between the osteoclast and osteoblast: how are messages transmitted between these temporary visitors to the bone surface? Front Endocrinol (Lausanne). 2015;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin JJ, Selander K, Chirgwin JM, et al. TGF‐beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103(2):197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sterling JA, Oyajobi BO, Grubbs B, et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone‐related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006;66(15):7548‐7553. [DOI] [PubMed] [Google Scholar]
- 10. Johnson RW, Nguyen MP, Padalecki SS, et al. TGF‐beta promotion of Gli2‐induced expression of parathyroid hormone‐related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling. Cancer Res. 2011;71(3):822‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunn LK, Mohammad KS, Fournier PGJ, et al. Hypoxia and TGF‐beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4(9):e6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas RJ, Guise TA, Yin JJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140(10):4451‐4458. [DOI] [PubMed] [Google Scholar]
- 13. Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer. 2016;16(6):373‐386. [DOI] [PubMed] [Google Scholar]
- 14. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol. 2017;11(1):40‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15(7):807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin‐1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149‐161. [DOI] [PubMed] [Google Scholar]
- 17. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shiozawa Y, Pedersen EA, Patel LR, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12(2):116‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20(8):833‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen H, Senda T, Kubo KY. The osteocyte plays multiple roles in bone remodeling and mineral homeostasis. Med Mol Morphol. 2015;48(2):61‐68. [DOI] [PubMed] [Google Scholar]
- 21. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 22. Johnson RW, Schipani E, Giaccia AJ. HIF targets in bone remodeling and metastatic disease. Pharmacol Ther. 2015;150:169‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David Roodman G, Silbermann R. Mechanisms of osteolytic and osteoblastic skeletal lesions. Bonekey Rep. 2015;4:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bienz M, Saad F. Management of bone metastases in prostate cancer: a review. Curr Opin Support Palliat Care. 2015;9(3):261‐267. [DOI] [PubMed] [Google Scholar]
- 25. Morrissey C, Roudier MP, Dowell A, et al. Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration‐resistant prostate cancer: results from the University of Washington Rapid Autopsy Series. J Bone Miner Res. 2013;28(2):333‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL, Ott SM. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180(3):1154‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Major PP, Cook RJ, Lipton A, Smith MR, Terpos E, Coleman RE. Natural history of malignant bone disease in breast cancer and the use of cumulative mean functions to measure skeletal morbidity. BMC Cancer. 2009;9(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48(4):677‐692. [DOI] [PubMed] [Google Scholar]
- 29. Lipton A, Theriault RL, Hortobagyi GN, et al. Pamidronate prevents skeletal complications and is effective palliative treatment in women with breast carcinoma and osteolytic bone metastases: long term follow‐up of two randomized, placebo‐controlled trials. Cancer. 2000;88(5):1082‐1090. [DOI] [PubMed] [Google Scholar]
- 30. Hillner BE. The role of bisphosphonates in metastatic breast cancer. Semin Radiat Oncol. 2000;10(3):250‐253. [DOI] [PubMed] [Google Scholar]
- 31. Coleman RE, Marshall H, Cameron D, et al. Breast‐cancer adjuvant therapy with zoledronic acid. N Engl J Med. 2011;365(15):1396‐1405. [DOI] [PubMed] [Google Scholar]
- 32. Coleman R, Cameron D, Dodwell D, et al. Adjuvant zoledronic acid in patients with early breast cancer: final efficacy analysis of the AZURE (BIG 01/04) randomised open‐label phase 3 trial. Lancet Oncol. 2014;15(9):997‐1006. [DOI] [PubMed] [Google Scholar]
- 33. Early Breast Cancer Trialists' Collaborative, G . Adjuvant bisphosphonate treatment in early breast cancer: meta‐analyses of individual patient data from randomised trials. Lancet. 2015;386(10001):1353‐1361. [DOI] [PubMed] [Google Scholar]
- 34. Leder BZ. Parathyroid hormone and parathyroid hormone‐related protein analogs in osteoporosis therapy. Curr Osteoporos Rep. 2017;15(2):110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andrews EB, Gilsenan AW, Midkiff K, et al. The US postmarketing surveillance study of adult osteosarcoma and teriparatide: study design and findings from the first 7 years. J Bone Miner Res. 2012;27(12):2429‐2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parker CC, Coleman RE, Sartor O, et al. Three‐year safety of Radium‐223 dichloride in patients with castration‐resistant prostate Cancer and symptomatic bone metastases from phase 3 randomized Alpharadin in symptomatic prostate Cancer trial. Eur Urol. 2017;73(3):427‐435. [DOI] [PubMed] [Google Scholar]
- 37. Sartor O, Coleman R, Nilsson S, et al. Effect of radium‐223 dichloride on symptomatic skeletal events in patients with castration‐resistant prostate cancer and bone metastases: results from a phase 3, double‐blind, randomised trial. Lancet Oncol. 2014;15(7):738‐746. [DOI] [PubMed] [Google Scholar]
- 38. Martin M, Bell R, Bourgeois H, et al. Bone‐related complications and quality of life in advanced breast Cancer: results from a randomized phase III trial of Denosumab versus Zoledronic acid. Clin Cancer Res. 2012;18(17):4841‐4849. [DOI] [PubMed] [Google Scholar]
- 39. Naumov GN, MacDonald I, Weinmeister PM, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002;62(7):2162‐2168. [PubMed] [Google Scholar]
- 40. Ghiso JAA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147(1):89‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Banys M, Hartkopf AD, Krawczyk N, et al. Dormancy in breast cancer. Breast Cancer (Dove Med Press). 2012;4:183‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li S, Kennedy M, Payne S, et al. Model of tumor dormancy/recurrence after short‐term chemotherapy. PLoS One. 2014;9(5):e98021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mellor HR, Ferguson DJ, Callaghan R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br J Cancer. 2005;93(3):302‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eblen ST, Fautsch MP, Anders RA, Leof EB. Conditional binding to and cell cycle‐regulated inhibition of cyclin‐dependent kinase complexes by p27Kip1. Cell Growth Differ. 1995;6(8):915‐925. [PubMed] [Google Scholar]
- 45. Rohrig F, Vorlová S, Hoffmann H, et al. VEGF‐ablation therapy reduces drug delivery and therapeutic response in ECM‐dense tumors. Oncogene. 2017;36(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Erler JT, Bennewith KL, Cox TR, et al. Hypoxia‐induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell JP, Merkel AR, Masood‐Campbell SK, Elefteriou F, Sterling JA. Models of bone metastasis. J Vis Exp. 2012;67:e4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reinisch A, Hernandez DC, Schallmoser K, Majeti R. Generation and use of a humanized bone‐marrow‐ossicle niche for hematopoietic xenotransplantation into mice. Nat Protoc. 2017;12(10):2169‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reinisch A, Thomas D, Corces MR, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. 2016;22(7):812‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dondossola E, Alexander S, Holzapfel BM, et al. Intravital microscopy of osteolytic progression and therapy response of cancer lesions in the bone. Sci Transl Med. 2018;10(452):eaao5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vanderburgh JP, Guelcher SA, Sterling JA. 3D bone models to study the complex physical and cellular interactions between tumor and the bone microenvironment. J Cell Biochem. 2018;119(7):5053‐5059. [DOI] [PubMed] [Google Scholar]
- 52. Johnson RW, Finger EC, Olcina MM, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. 2016;18(10):1078‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gawrzak S, Rinaldi L, Gregorio S, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat Cell Biol. 2018;20(2):211‐221. [DOI] [PubMed] [Google Scholar]
- 54. Johnson RW, Sun Y, Ho PWM, et al. Parathyroid hormone‐related protein negatively regulates tumor cell dormancy genes in a PTHR1/cyclic AMP‐independent manner. Front Endocrinol (Lausanne). 2018;9:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kobayashi A, Okuda H, Xing F, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem‐like cells in bone. J Exp Med. 2011;208(13):2641‐2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sharma S, Xing F, Liu Y, et al. Secreted protein acidic and rich in cysteine (SPARC) mediates metastatic dormancy of prostate Cancer in bone. J Biol Chem. 2016;291(37):19351‐19363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sowder ME, Johnson RW. Enrichment and detection of bone disseminated tumor cells in models of low tumor burden. Sci Rep. 2018;8(1):14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang H, Yu C, Gao X, et al. The osteogenic niche promotes early‐stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nowak‐Sliwinska P, Segura T, Iruela‐Arispe ML. The chicken chorioallantoic membrane model in biology, medicine and bioengineering. Angiogenesis. 2014;17(4):779‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li M, Pathak RR, Lopez‐Rivera E, Friedman SL, Aguirre‐Ghiso JA, Sikora AG. The in Ovo Chick Chorioallantoic membrane (CAM) assay as an efficient xenograft model of hepatocellular carcinoma. J Vis Exp. 2015;(104):52411. 10.3791/52411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bragado P, Estrada Y, Sosa MS, et al. Analysis of marker‐defined HNSCC subpopulations reveals a dynamic regulation of tumor initiating properties. PLoS One. 2012;7(1):e29974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sosa MS, Parikh F, Maia AG, et al. NR2F1 controls tumour cell dormancy via SOX9‐ and RARbeta‐driven quiescence programmes. Nat Commun. 2015;6(1):6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bragado P, Estrada Y, Parikh F, et al. TGF‐beta2 dictates disseminated tumour cell fate in target organs through TGF‐beta‐RIII and p38alpha/beta signalling. Nat Cell Biol. 2013;15(11):1351‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gustafsson KL, Farman H, Henning P, et al. The role of membrane ERalpha signaling in bone and other major estrogen responsive tissues. Sci Rep. 2016;6(1):29473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58‐68. [DOI] [PubMed] [Google Scholar]
- 66. Schmidt‐Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S a. 2003;100(13):7737‐7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. 2015;15(4):238‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weaver VM, Lelièvre S, Lakins JN, et al. beta4 integrin‐dependent formation of polarized three‐dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2(3):205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM‐DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658‐1667. [PMC free article] [PubMed] [Google Scholar]
- 70. Korah R, Boots M, Wieder R. Integrin alpha5beta1 promotes survival of growth‐arrested breast cancer cells: an in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004;64(13):4514‐4522. [DOI] [PubMed] [Google Scholar]
- 71. Fridman R, Giaccone G, Kanemoto T, Martin GR, Gazdar AF, Mulshine JL. Reconstituted basement membrane (matrigel) and laminin can enhance the tumorigenicity and the drug resistance of small cell lung cancer cell lines. Proc Natl Acad Sci U S a. 1990;87(17):6698‐6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sethi T, Rintoul RC, Moore SM, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5(6):662‐668. [DOI] [PubMed] [Google Scholar]
- 73. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137‐148. [DOI] [PubMed] [Google Scholar]
- 74. Soria G, Ben‐Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267(2):271‐285. [DOI] [PubMed] [Google Scholar]
- 75. Coniglio SJ. Role of tumor‐derived chemokines in osteolytic bone metastasis. Front Endocrinol (Lausanne). 2018;9:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ueno T, Toi M, Saji H, et al. Significance of macrophage chemoattractant protein‐1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6(8):3282‐3289. [PubMed] [Google Scholar]
- 77. Chavey C, Bibeau F, Gourgou‐Bourgade S, et al. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9(1):R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Steiner JL, Murphy EA. Importance of chemokine (CC‐motif) ligand 2 in breast cancer. Int J Biol Markers. 2012;27(3):e179‐e185. [DOI] [PubMed] [Google Scholar]
- 79. Saji H, Koike M, Yamori T, et al. Significant correlation of monocyte chemoattractant protein‐1 expression with neovascularization and progression of breast carcinoma. Cancer. 2001;92(5):1085‐1091. [DOI] [PubMed] [Google Scholar]
- 80. Azenshtein E, Luboshits G, Shina S, et al. The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res. 2002;62(4):1093‐1102. [PubMed] [Google Scholar]
- 81. Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003;63(23):8360‐8365. [PubMed] [Google Scholar]
- 82. Adler EP, Lemken CA, Katchen NS, Kurt RA. A dual role for tumor‐derived chemokine RANTES (CCL5). Immunol Lett. 2003;90(2–3):187‐194. [DOI] [PubMed] [Google Scholar]
- 83. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res. 2009;29(6):313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347(6294):669‐671. [DOI] [PubMed] [Google Scholar]
- 85. Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage infiltration and its prognostic implications in breast cancer: the relationship with VEGF expression and microvessel density. Oncol Rep. 2005;14(2):425‐431. [PubMed] [Google Scholar]
- 86. Coleman R, Gnant M, Morgan G, Clezardin P. Effects of bone‐targeted agents on cancer progression and mortality. J Natl Cancer Inst. 2012;104(14):1059‐1067. [DOI] [PubMed] [Google Scholar]
- 87. Gnant M, Mlineritsch B, Schippinger W, et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N Engl J Med. 2009;360(7):679‐691. [DOI] [PubMed] [Google Scholar]
- 88. Coleman R, de Boer R, Eidtmann H, et al. Zoledronic acid (zoledronate) for postmenopausal women with early breast cancer receiving adjuvant letrozole (ZO‐FAST study): final 60‐month results. Ann Oncol. 2013;24(2):398‐405. [DOI] [PubMed] [Google Scholar]
- 89. Junankar S, Shay G, Jurczyluk J, et al. Real‐time intravital imaging establishes tumor‐associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. 2015;5(1):35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wyckoff JB, Wang Y, Lin EY, et al. Direct visualization of macrophage‐assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649‐2656. [DOI] [PubMed] [Google Scholar]
- 91. Williams CB, Yeh ES, Soloff AC. Tumor‐associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Khan UA, Hashimi SM, Bakr MM, Forwood MR, Morrison NA. CCL2 and CCR2 are essential for the formation of osteoclasts and foreign body Giant cells. J Cell Biochem. 2016;117(2):382‐389. [DOI] [PubMed] [Google Scholar]
- 93. Bussard KM, Venzon DJ, Mastro AM. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J Cell Biochem. 2010;111(5):1138‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kinder M, Chislock E, Bussard KM, Shuman L, Mastro AM. Metastatic breast cancer induces an osteoblast inflammatory response. Exp Cell Res. 2008;314(1):173‐183. [DOI] [PubMed] [Google Scholar]
- 95. Bidwell BN, Slaney CY, Withana NP, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012;18(8):1224‐1231. [DOI] [PubMed] [Google Scholar]
- 96. Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA. Myeloid‐derived suppressor cells expand during breast cancer progression and promote tumor‐induced bone destruction. Oncoimmunology. 2012;1(9):1484‐1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. D'Amico L, Mahajan S, Capietto AH, et al. Dickkopf‐related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. 2016;213(5):827‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia‐induced metastasis. Nature. 2006;440(7088):1222‐1226. [DOI] [PubMed] [Google Scholar]
- 99. Sakai M, Kato H, Sano A, et al. Expression of lysyl oxidase is correlated with lymph node metastasis and poor prognosis in esophageal squamous cell carcinoma. Ann Surg Oncol. 2009;16(9):2494‐2501. [DOI] [PubMed] [Google Scholar]
- 100. Erler JT, Giaccia AJ. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006;66(21):10238‐10241. [DOI] [PubMed] [Google Scholar]
- 101. Le QT, Harris J, Magliocco AM, et al. Validation of lysyl oxidase as a prognostic marker for metastasis and survival in head and neck squamous cell carcinoma: radiation therapy oncology group trial 90‐03. J Clin Oncol. 2009;27(26):4281‐4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S a. 1995;92(12):5510‐5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia‐initiated angiogenesis. Nature. 1992;359(6398):843‐845. [DOI] [PubMed] [Google Scholar]
- 105. Tsuzuki Y, Fukumura D, Oosthuyse B, Koike C, Carmeliet P, Jain RK. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia‐inducible factor‐1alpha‐‐> hypoxia response element‐‐> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000;60(22):6248‐6252. [PubMed] [Google Scholar]
- 106. Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase a. Interaction between hypoxia‐inducible factor 1 and cAMP response elements. J Biol Chem. 1995;270(36):21021‐21027. [DOI] [PubMed] [Google Scholar]
- 107. Gleadle JM, Ratcliffe PJ. Induction of hypoxia‐inducible factor‐1, erythropoietin, vascular endothelial growth factor, and glucose transporter‐1 by hypoxia: evidence against a regulatory role for Src kinase. Blood. 1997;89(2):503‐509. [PubMed] [Google Scholar]
- 108. Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen‐regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase a genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci U S a. 1994;91(14):6496‐6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter‐1 via distinct cis‐acting sequences. J Biol Chem. 1995;270(49):29083‐29089. [DOI] [PubMed] [Google Scholar]
- 110. Iorns E, Ward TM, Dean S, et al. Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res Treat. 2012;135(1):79‐91. [DOI] [PubMed] [Google Scholar]
- 111. Chen D, Sun Y, Wei Y, et al. LIFR is a breast cancer metastasis suppressor upstream of the hippo‐YAP pathway and a prognostic marker. Nat Med. 2012;18(10):1511‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Johnson RW, Sowder ME, Giaccia AJ. Hypoxia and bone metastatic disease. Curr Osteoporos Rep. 2017;15(4):231‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I Krogh's model Biophys J. 2001;81(2):675‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99(1):394. [DOI] [PubMed] [Google Scholar]
- 116. Branemark P. Experimental Investigation of Microcirculation in Bone Marrow. Angiology. 1961;12(7):293‐305. [Google Scholar]
- 117. Devignes CS, Aslan Y, Brenot A, et al. HIF signaling in osteoblast‐lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc Natl Acad Sci U S a. 2018;115(5):E992‐E1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fluegen G, Avivar‐Valderas A, Wang Y, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19(2):120‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Thompson VC, Day TK, Bianco‐Miotto T, et al. A gene signature identified using a mouse model of androgen receptor‐dependent prostate cancer predicts biochemical relapse in human disease. Int J Cancer. 2012;131(3):662‐672. [DOI] [PubMed] [Google Scholar]
- 120. Montagner M, Enzo E, Forcato M, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia‐inducible factors. Nature. 2012;487(7407):380‐384. [DOI] [PubMed] [Google Scholar]
- 121. Shemirani B, Crowe DL. Hypoxic induction of HIF‐1alpha and VEGF expression in head and neck squamous cell carcinoma lines is mediated by stress activated protein kinases. Oral Oncol. 2002;38(3):251‐257. [DOI] [PubMed] [Google Scholar]
- 122. Wong CC, Zhang H, Gilkes DM, et al. Inhibitors of hypoxia‐inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med (Berl). 2012;90(7):803‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature. 2005;438(7069):820‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu Y, Cao X. Characteristics and significance of the pre‐metastatic niche. Cancer Cell. 2016;30(5):668‐681. [DOI] [PubMed] [Google Scholar]
- 125. Dales JP, Garcia S, Meunier‐Carpentier S, et al. Overexpression of hypoxia‐inducible factor HIF‐1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int J Cancer. 2005;116(5):734‐739. [DOI] [PubMed] [Google Scholar]
- 126. Xiang L, Liu ZH, Huan Q, et al. Hypoxia‐inducible factor‐2a is associated with ABCG2 expression, histology‐grade and Ki67 expression in breast invasive ductal carcinoma. Diagn Pathol. 2012;7(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Giatromanolaki A, Koukourakis MI, Simopoulos C, et al. C‐erbB‐2 related aggressiveness in breast cancer is hypoxia inducible factor‐1alpha dependent. Clin Cancer Res. 2004;10(23):7972‐7977. [DOI] [PubMed] [Google Scholar]
- 128. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Coleman RE, Smith P, Rubens RD. Clinical course and prognostic factors following bone recurrence from breast cancer. Br J Cancer. 1998;77(2):336‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia‐inducible factor‐1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67(2):563‐572. [DOI] [PubMed] [Google Scholar]
- 131. Hiraga T, Kizaka‐Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia‐inducible factor‐1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67(9):4157‐4163. [DOI] [PubMed] [Google Scholar]
- 132. Lu X, Yan CH, Yuan M, Wei Y, Hu G, Kang Y. In vivo dynamics and distinct functions of hypoxia in primary tumor growth and organotropic metastasis of breast cancer. Cancer Res. 2010;70(10):3905‐3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Almog N, Henke V, Flores L, et al. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. 2006;20(7):947‐949. [DOI] [PubMed] [Google Scholar]
- 134. Naumov GN, Folkman J, Straume O, Akslen LA. Tumor‐vascular interactions and tumor dormancy. APMIS. 2008;116(7–8):569‐585. [DOI] [PubMed] [Google Scholar]
- 135. Almog N, Ma L, Raychowdhury R, et al. Transcriptional switch of dormant tumors to fast‐growing angiogenic phenotype. Cancer Res. 2009;69(3):836‐844. [DOI] [PubMed] [Google Scholar]
- 136. Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26(2):281‐290. [DOI] [PubMed] [Google Scholar]
- 137. Straume O, Shimamura T, Lampa MJG, et al. Suppression of heat shock protein 27 induces long‐term dormancy in human breast cancer. Proc Natl Acad Sci U S a. 2012;109(22):8699‐8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Almog N, Ma L, Schwager C, et al. Consensus micro RNAs governing the switch of dormant tumors to the fast‐growing angiogenic phenotype. PLoS One. 2012;7(8):e44001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Rogers MS, Novak K, Zurakowski D, et al. Spontaneous reversion of the angiogenic phenotype to a nonangiogenic and dormant state in human tumors. Mol Cancer Res. 2014;12(5):754‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Good DJ, Polverini PJ, Rastinejad F, et al. A tumor suppressor‐dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S a. 1990;87(17):6624‐6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin‐1 interactome. Matrix Biol. 2014;37:83‐91. [DOI] [PubMed] [Google Scholar]
- 142. Weinstat‐Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994;54(24):6504‐6511. [PubMed] [Google Scholar]
- 143. Roberts DD. Regulation of tumor growth and metastasis by thrombospondin‐1. FASEB J. 1996;10(10):1183‐1191. [PubMed] [Google Scholar]
- 144. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007;26(3–4):489‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor‐beta stimulates parathyroid hormone‐related protein and osteolytic metastases via Smad and mitogen‐activated protein kinase signaling pathways. J Biol Chem. 2002;277(27):24571‐24578. [DOI] [PubMed] [Google Scholar]
- 146. Javelaud D, Mohammad KS, McKenna CR, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67(5):2317‐2324. [DOI] [PubMed] [Google Scholar]
- 147. Mohammad KS, Javelaud D, Fournier PGJ, et al. TGF‐beta‐RI kinase inhibitor SD‐208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71(1):175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Biswas S, Nyman JS, Alvarez JA, et al. Anti‐transforming growth factor beta antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS One. 2011;6(11):e27090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Paget S. The distribution of secondary growths in cancer of the breast. The Lancet. 1889;133(3421):571‐573. [PubMed] [Google Scholar]
- 150. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46(7):1181‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147(1):89‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Aguirre‐Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7(11):834‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Shi J, Wang L, Zou C, et al. Tumor microenvironment promotes prostate cancer cell dissemination via the Akt/mTOR pathway. Oncotarget. 2018;9(10):9206‐9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Thomas C, Wiesner C, Melchior SW, et al. Urokinase‐plasminogen‐activator receptor expression in disseminated tumour cells in the bone marrow and peripheral blood of patients with clinically localized prostate cancer. BJU Int. 2009;104(1):29‐34. [DOI] [PubMed] [Google Scholar]
- 155. Lu X, Mu E, Wei Y, et al. VCAM‐1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1‐positive osteoclast progenitors. Cancer Cell. 2011;20(6):701‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over‐expression and survival in cancer: a system review and meta‐analysis. Oncotarget. 2015;6(7):5022‐5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Fernandis AZ, Prasad A, Band H, Klösel R, Ganju RK. Regulation of CXCR4‐mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. 2004;23(1):157‐167. [DOI] [PubMed] [Google Scholar]
- 158. Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50‐56. [DOI] [PubMed] [Google Scholar]
- 159. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540‐550. [DOI] [PubMed] [Google Scholar]
- 160. Singh S, Singh UP, Grizzle WE, Lillard JW. CXCL12‐CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84(12):1666‐1676. [DOI] [PubMed] [Google Scholar]
- 161. Engl T, Relja B, Marian D, et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006;8(4):290‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kukreja P, Abdel‐Mageed AB, Mondal D, Liu K, Agrawal KC. Up‐regulation of CXCR4 expression in PC‐3 cells by stromal‐derived factor‐1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway‐dependent NF‐kappaB activation. Cancer Res. 2005;65(21):9891‐9898. [DOI] [PubMed] [Google Scholar]
- 163. Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF‐1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006;25(4):573‐587. [DOI] [PubMed] [Google Scholar]
- 164. Nobutani K, Shimono Y, Mizutani K, et al. Downregulation of CXCR4 in metastasized breast Cancer cells and implication in their dormancy. PLoS One. 2015;10(6):e0130032. 10.1371/journal.pone.0130032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Delworth M, Nishioka K, Pettaway C, et al. Systemic administration of 4‐amidinoindanon‐1‐(2′‐amidino)‐hydrazone, a new inhibitor of s‐adenosylmethionine decarboxylase, produces cytostasis of human prostate‐cancer in athymic nude‐mice. Int J Oncol. 1995;6(2):293‐299. [DOI] [PubMed] [Google Scholar]
- 166. Johnson RW, Merkel AR, Page JM, Ruppender NS, Guelcher SA, Sterling JA. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin Exp Metastasis. 2014;31(8):945‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sansone P, Savini C, Kurelac I, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy‐resistant breast cancer. Proc Natl Acad Sci U S a. 2017;114(43):E9066‐E9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bliss SA, Sinha G, Sandiford OA, et al. Mesenchymal stem cell‐derived exosomes stimulate cycling quiescence and early breast Cancer dormancy in bone marrow. Cancer Res. 2016;76(19):5832‐5844. [DOI] [PubMed] [Google Scholar]
- 169. Boix L, López‐Oliva JM, Rhodes AC, Bruix J. Restoring miR122 in human stem‐like hepatocarcinoma cells, prompts tumor dormancy through Smad‐independent TGF‐beta pathway. Oncotarget. 2016;7(44):71309‐71329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Taipaleenmaki H, Browne G, Akech J, et al. Targeting of Runx2 by miR‐135 and miR‐203 impairs progression of breast Cancer and metastatic bone disease. Cancer Res. 2015;75(7):1433‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Haider MT, Taipaleenmaki H. Targeting the metastatic bone microenvironment by MicroRNAs. Front Endocrinol (Lausanne). 2018;9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Yumoto K, Eber MR, Wang J, et al. Axl is required for TGF‐beta2‐induced dormancy of prostate cancer cells in the bone marrow. Sci Rep. 2016;6(1):36520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Taichman RS, Patel LR, Bedenis R, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One. 2013;8(4):e61873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Martin TJ. Parathyroid hormone‐related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol Rev. 2016;96(3):831‐871. [DOI] [PubMed] [Google Scholar]
- 175. Ruppender NS, Merkel AR, Martin TJ, Mundy GR, Sterling JA, Guelcher SA. Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells. PLoS One. 2010;5(11):e15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol. 2013;61 Chapter 14: p. Unit:14‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S a. 2003;100(7):3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451(7176):345‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ricci‐Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature. 2007;445(7123):111‐115. [DOI] [PubMed] [Google Scholar]
- 180. Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci. 2004;117(Pt 16):3539‐3545. [DOI] [PubMed] [Google Scholar]
- 181. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030‐1037. [DOI] [PubMed] [Google Scholar]
- 182. Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140(1):62‐73. [DOI] [PubMed] [Google Scholar]
- 183. Sikandar SS, Pate KT, Anderson S, et al. NOTCH signaling is required for formation and self‐renewal of tumor‐initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70(4):1469‐1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi‐1 regulate self‐renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063‐6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL. Cell signaling by urokinase‐type plasminogen activator receptor induces stem cell‐like properties in breast cancer cells. Cancer Res. 2010;70(21):8948‐8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor‐positive cells. Dev Biol. 2005;277(2):443‐456. [DOI] [PubMed] [Google Scholar]
- 187. Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Kritikou EA, Sharkey A, Abell K, et al. A dual, non‐redundant, role for LIF as a regulator of development and STAT3‐mediated cell death in mammary gland. Development. 2003;130(15):3459‐3468. [DOI] [PubMed] [Google Scholar]
- 189. Dieter SM, Ball CR, Hoffmann CM, et al. Distinct types of tumor‐initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011;9(4):357‐365. [DOI] [PubMed] [Google Scholar]
- 190. Ranganathan AC, Zhang L, Adam AP, Aguirre‐Ghiso JA. Functional coupling of p38‐induced up‐regulation of BiP and activation of RNA‐dependent protein kinase‐like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66(3):1702‐1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Schewe DM, Aguirre‐Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009;69(4):1545‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10(12):871‐877. [DOI] [PubMed] [Google Scholar]
- 193. Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood. 2003;101(8):3142‐3149. [DOI] [PubMed] [Google Scholar]
- 194. Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS‐354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532‐4539. [DOI] [PubMed] [Google Scholar]
- 195. Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016‐4019. [DOI] [PubMed] [Google Scholar]
- 196. Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672‐679. [DOI] [PubMed] [Google Scholar]
- 197. Butler SJ, Richardson L, Farias N, Morrison J, Coomber BL. Characterization of cancer stem cell drug resistance in the human colorectal cancer cell lines HCT116 and SW480. Biochem Biophys Res Commun. 2017;490(1):29‐35. [DOI] [PubMed] [Google Scholar]
- 198. Todaro M, Alea MP, di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin‐4. Cell Stem Cell. 2007;1(4):389‐402. [DOI] [PubMed] [Google Scholar]
- 199. Ding XW, Wu JH, Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010;86(17–18):631‐637. [DOI] [PubMed] [Google Scholar]
- 200. Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89(4):491‐502. [DOI] [PubMed] [Google Scholar]
- 201. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756‐760. [DOI] [PubMed] [Google Scholar]
- 202. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova‐Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22(4):436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Balic M, Lin H, Young L, et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12(19):5615‐5621. [DOI] [PubMed] [Google Scholar]
- 205. Wang N, Docherty F, Brown HK, et al. Mitotic quiescence, but not unique "stemness," marks the phenotype of bone metastasis‐initiating cells in prostate cancer. FASEB J. 2015;29(8):3141‐3150. [DOI] [PubMed] [Google Scholar]