

Abstract
Propionic acidemia (PA) and methylmalonic acidemia (MMA) are autosomal recessive disorders of propionyl-CoA (P-CoA) catabolism, which are caused by a deficiency in the enzyme propionyl-CoA carboxylase or the enzyme methylmalonyl-CoA (MM-CoA) mutase, respectively. The functional consequence of PA or MMA is the inability to catabolize P-CoA to MM-CoA or MM-CoA to succinyl-CoA, resulting in the accumulation of P-CoA and other metabolic intermediates, such as propionylcarnitine (C3), 3-hydroxypropionic acid, methylcitric acid (MCA), and methylmalonic acid (only in MMA). P-CoA and its metabolic intermediates, at high concentrations found in PA and MMA, inhibit enzymes in the first steps of the urea cycle as well as enzymes in the tricarboxylic acid (TCA) cycle, causing a reduction in mitochondrial energy production. We previously showed that metabolic defects of PA could be recapitulated using PA patient-derived primary hepatocytes in a novel organotypic system. Here, we sought to investigate whether treatment of normal human primary hepatocytes with propionate would recapitulate some of the biochemical features of PA and MMA in the same platform. We found that high levels of propionate resulted in high levels of intracellular P-CoA in normal hepatocytes. Analysis of TCA cycle intermediates by GC-MS/MS indicated that propionate may inhibit enzymes of the TCA cycle as shown in PA, but is also incorporated in the TCA cycle, which does not occur in PA. To better recapitulate the disease phenotype, we obtained hepatocytes derived from livers of PA and MMA patients. We characterized the PA and MMA donors by measuring key proximal biomarkers, including P-CoA, MM-CoA, as well as clinical biomarkers propionylcarnitine-to-acetylcarnitine ratios (C3/C2), MCA and methylmalonic acid. Additionally, we used isotopically labeled amino acids to investigate the contribution of relevant amino acids to production of P-CoA in models of metabolic stability or acute metabolic crisis. As observed clinically, we demonstrated that the isoleucine and valine catabolism pathways are the greatest sources of P-CoA in PA and MMA donor cells and that each donor showed differential sensitivity to isoleucine and valine. We also studied the effects of disodium citrate, an anaplerotic therapy, which resulted in a significant increase in the absolute concentration of TCA cycle intermediates, which is in agreement with the benefit observed clinically. Our human cell-based PA and MMA disease models can inform preclinical drug discovery and development where mouse models of these diseases are inaccurate, particularly in well-described species differences in branched-chain amino acid catabolism.
Keywords: organic acidemia, propionic acidemia, methylmalonic academia, TCA cycle, hepatocyte, hemodynamic flow
1. INTRODUCTION
Propionic acidemia (PA, MIM #606054) is an inborn error of metabolism caused by the dysfunction of propionyl-CoA carboxylase (PCC, EC 6.4.1.3) in the conversion of propionyl-CoA (P-CoA) to methylmalonyl-CoA (MM-CoA). Methylmalonic acidemia (MMA, MIM #251000, 251100, 251110) results from defects in MM-CoA mutase (MMUT, EC 5.4.99.2) or the enzymes involved in cofactor production (cblA, cblB, or cblD-MMA) that affect the conversion of MM-CoA to the tricarboxylic acid cycle (TCA) intermediate, succinyl-CoA. Abnormalities in these propionyl-CoA pathway enzymes lead to the accumulation of the metabolic intermediates P-CoA, propionyl-carnitine (C3), 3-hydroxypropionic acid, methylcitric acid (MCA), and, in MMA patients, methylmalonic acid (Fenton 2001).
PA affects approximately 1 in 250,000 individuals in the US and the prevalence can be as high as 1 in 1000-2000 in populations that are genetically at risk due to a higher incidence of consanguinity (e.g. Inuit, Greenland and specific tribes in Saudi Arabia; (Ravn et al. 2000; Carrillo-Carrasco and Venditti 1993; Ozand et al. 1994; Chapman et al. 2018). MMA impacts approximately 1 in 50,000-70,000 in the US (Chapman et al. 2018; Manoli and Venditti 1993).
Individuals with PA and MMA can present with life-limiting intermittent crises that are marked by metabolic acidosis and hyperammonemia in some cases. Long-term complications, including seizures, cardiomyopathies, metabolic stroke-like episodes and cardiac arrhythmias, severely impact the quality of life and cause progressive deterioration, sometimes ending in sudden death (Deodato et al. 2006; Pena 2012; Pena et al. 2012). Although seen occasionally in PA, MMA also has significant morbidity as a consequence of renal failure. There are no current definitive treatments for PA or MMA. Therapeutic options focus on dietary restriction of the precursors of P-CoA, such as odd-chain fatty acids and propiogenic amino acids (valine, methionine, isoleucine and threonine), while trying to maintain normal growth, scavenging excess P-CoA using carnitine, and symptomatically treating complications as they occur (Baumgartner et al. 2014; Sutton et al. 2012). Some patients with PA and MMA receive liver transplants to ameliorate symptoms primarily due to hyperammonemia or concomitant to a renal transplant (especially in MMA). However, in spite of the symptomatic relief, many of these patients still progress to the long-term sequelae of the disease. This highlights the unmet need to develop better therapies that would improve quality of life and lifespan of these patients.
The biochemical hallmark of PA and MMA is the accumulation of P-CoA and metabolites from the alternative oxidative pathways of P-CoA. P-CoA has been shown to inhibit NAGS, the obligate activator of carbamoyl phosphate synthetase I (CPS1), the rate-limiting enzyme of the urea cycle, and thereby affecting ureagenesis and inducing hyperammonemia (Coude, Sweetman, and Nyhan 1979). Additionally, P-CoA acts an alternative substrate or product inhibitor of multiple enzymes in the TCA cycle, further reducing the flux through the cycle and energy production (Cheema-Dhadli, Leznoff, and Halperin 1975; Fenton 2001; Gregersen 1981; Martin-Requero et al. 1983; Patel, DeBuysere, and Olson 1983; Schwab et al. 2006; Stumpf et al. 1980; Weidman and Drysdale 1979). This constellation of effects on the TCA cycle leads to progressive, severe derangement of mitochondrial energy production, reduced oxidative phosphorylation, increased oxidative stress, and ultimately, reduced mitochondrial DNA synthesis and mitochondrial structural derangement. These cumulative effects appear to also underlie the end-organ toxicity in PA, predominately observed in tissues with high energy demand (e.g. heart, muscle, kidney, brain).
Meaningful experimental models of PA and MMA are important to understand disease biochemistry and pathophysiology and test therapeutic approaches. A significant number of genetic animal models of PA and MMA have been developed in the past years to study PA and MMA (Buck et al. 2012; Chandler et al. 2007; Chandler et al. 2009; Forny et al. 2016; Guenzel et al. 2013; Manoli et al. 2013; Miyazaki et al. 2001; Peters et al. 2003; Peters et al. 2012). Homozygous knock-out mouse models of PA and MMA are neonatal lethal and pups typically die within 24 hours after birth. Hypomorphic animal models can survive for weeks and retain a phenotype with many of the features of human disease, including elevation of plasma C3/C2 ratio and MCA, variable hyperammonemia, and increased levels of tissue P-CoA, but suffer from a common weakness in that the models are designed to study a particular endpoint or therapeutic strategy and often demonstrate only a partial recapitulation of disease phenotype. In addition, mouse models do not adequately recapitulate disease biology due to inherent species-related differences with respect to both whole body amino acid composition and dietary amino acid requirements, especially during early development (Millward 1997; Wu et al. 2013). Also, differences in the tissue distribution of the endogenous enzymes, the regulation of branched-chain amino acid (BCAA) metabolism and the unpredictable expression and regulation of human proteins in transgenic mice make these models valuable but with some biochemical limitations to study human disease (Brosnan and Brosnan 2006; Suryawan et al. 1998; Sweatt et al. 2004). The use of patient cells for research has been limited to patient-derived fibroblasts. Although fibroblasts can be used to assess PCC and MMUT activity, these cells have considerably lower levels of P-CoA-related metabolites (Barash et al. 1989; Perez-Cerda et al. 2003; Ugarte et al. 1999; Varani et al. 1985; Berman, Tong, and Williams 1980). Previously, we described the recapitulation of PA disease biology in an organotypic model that utilized primary hepatocytes from a PA patient that were cultured in a three dimensional context in the presence of sinusoidal hemodynamic flow and transport (Chapman et al. 2016). PA patient-derived cells in this system showed restored hepatocyte morphology, organization and function, including the metabolic defects associated with PA.
Here, we further characterize patient-derived hepatocyte models as well as a chemically-induced PA hepatocyte model. In order to interrogate the biochemistry of these models, we developed methods to measure intracellular proximal biomarkers of the disease, including coenzyme A (CoA) esters, carnitine conjugates, and organic acids such as MCA and methylmalonic acid together with TCA cycle intermediates. We also utilized the patient-derived models to understand the biochemical basis behind clinical observations related to dietary formulations and beneficial effects of anaplerotic therapies. Our results indicate that PA and MMA patient-derived surrogate human liver models faithfully recapitulated the disease biochemistry and aligned with clinical observations, making them amenable for evaluation of therapies for the treatment of PA and MMA.
2. MATERIALS AND METHODS
2.1. Liver tissue procurement and isolation of hepatic cells
Primary hepatocytes were isolated from liver explants from 5 individuals with PA and 3 individuals with MMA (Table 1). Subjects were consented to the Children’s National Bio-repository for cells, tissues and DNA (CNHS IRB Pro0004911) following guidelines for the Federal Policy for the Protection of Human Subjects (the Common Rule, codified at 45 CFR Part 46). Hepatocyte isolation from liver tissue obtained from the patients were performed at either QPS Hepatic Biosciences (Research Triangle Park, NC) or at HemoShear Therapeutics, LLC (Charlottesville, VA) using a protocol previously described by LeCluyse (Lecluyse and Alexandre 2010). Control hepatocytes from healthy control livers were procured from QPS Hepatic Biosciences or isolated and cryopreserved at HemoShear Therapeutics. Genotypes of PA and MMA donors are listed in Table 1. Only donors that passed quality controls demonstrating a differentiated hepatocyte phenotype after cryopreservation were used in biomarker or flux experiments (PA1, PA3, PA5, MMA1, MMA2, MMA3).
Table 1:
List of PA and MMA donors with their genotypes used in these experiments.
| Patient ID | Disease | Genetic Mutation |
|---|---|---|
| PA1 | Propionic acidemia |
PCCA c.350G>A p.G117D homozygous |
| PA2 | Propionic acidemia |
PCCA c.350G>A p.G117D homozygous |
| PA3 | Propionic acidemia |
PCCA c.419A>G (p.H140R) homozygous |
| PA4 | Propionic acidemia |
PCCB c. 1218_1231del/insTAGAGCACAGGA (p. G406fs) homozygous |
| PA5 | Propionic acidemia |
PCCB c.1002C>A (p.Tyr334*); c.1219_1231del/insAGAGCACAGGA (p.Pro407Argfs*6) |
| MMA1 | Methylmalonic acidemia |
MMUT c.29dupT ( p.L11Tx38); c.1658delT (p.V553Gfsx17) |
| MMA2 | Methylmalonic acidemia |
MMUT c.406G>T (p.Val136Phe) homozygous |
| MMA3 | Methylmalonic acidemia |
MMUT c.2150G>T (p.Gly717Val); c.753+1G>A (Splice donor) |
2.2. Cell Culture and Device Operating Conditions
Primary hepatocytes were cultured in the HemoShear Therapeutics’ technology, referred to as the hepatocyte bioreactor, throughout the manuscript in presence of flow and transport to preserve the differentiated phenotype. Primary hepatocytes obtained as described above were plated in a collagen gel sandwich configuration on the undersurface of the membranes of 75 mm polycarbonate transwells (Corning) using previously described protocols (Figure S1; (Dash, Simmers, et al. 2013). The cultures were left overnight in maintenance medium (MM) that consisted of DMEM/F-12 supplemented with fetal bovine serum (10% at the time of plating). Additionally, the medium contained gentamycin (50 μg/ml), 0.2% ITS (Fisher/MediaTech MT-25–800CR), and dexamethasone (Cat# D4902, Sigma Aldrich, St. Louis, MO, 1 μM at plating and 250 nM thereafter). On the 2nd day, the transwells were set up within hepatocyte bioreactors in a configuration to allow for control of hemodynamics and transport as described previously (Dash, Simmers, et al. 2013). A proprietary hepatocyte flow medium, modified from MM but with significantly lower levels of key hormones and growth factors, was continuously perfused on both sides while shear stress was applied on the top surface based on the calculations described below. The devices were housed in a controlled environment at 37°C with 5% CO2 mixed with air. For all the flow experiments described in this study the shear stress of 0.6 dyn/cm2 was used derived from reference values for the pressure gradient across the sinusoid (ΔP), the radius of sinusoids (r), and the length of the sinusoids (I) obtained from the literature (Dash, Deering, et al. 2013). The hepatocytes were cultured for 7 days and harvested for measurement of both mitochondrial metabolites (acyl-CoAs) and other clinically relevant biomarkers (acyl-carnitines, MCA) in hepatocyte cell lysates.
For some experiments, primary hepatocytes from individual donors were plated in a collagen gel sandwich configuration on 24-well or 48-well plates (Corning) using previously described protocols. The cultures were incubated in MM. After day 2, and for the duration of the experiment, hepatocytes were cultured in normal or customized Hepatocyte Maintenance Medium purchased from Corning.
2.3. Treatments
Sodium propionate (Sigma) was added at concentrations ranging from 0.3-10 mM to cultured hepatocytes for 72hrs. After treatment, media was collected, and hepatocytes were harvested for the subsequent endpoints.
To recreate a milieu representative of a stable metabolic state, we used a low propiogenic custom media formulation designed based on amino acid levels measured in the plasma of PA and MMA patients that are metabolically stable and called this formulation ‘Baseline’ (Zwickler et al. 2012; Zwickler et al. 2014). To mimic a catabolic state or metabolic decompensation, we increased the levels of propiogenic amino acids and α-ketoacids by 5X or 10X based on the fold-changes observed in isoleucine, valine and leucine levels in Maple Syrup Urine Disease (MSUD) patients in crisis compared to stable metabolic states (Wajner et al. 2000). During metabolic crises, levels of propiogenic amino acids are not elevated in the plasma of PA and MMA patients, but P-CoA and MM-CoA accumulate in tissues instead. Therefore, we used data from MSUD patients, who accumulate BCAA in their plasma during decompensations due to a block more upstream in the BCAA catabolism pathway than PA and MMA, to simulate the amino acid load that occurs at a metabolic crisis. This formulation is referred to as ‘Catabolic’ or ‘MAX Catabolic’, respectively. Concentrations for each media formulation can be found in Table S1. All amino acid and α-keto acid supplements were purchased from Sigma or United State Biologicals.
To study the contribution of each individual amino acid and α-keto acid, each were replaced one at a time with an isotopically-labeled analog, and isotopically labeled P-CoA as well as unlabeled P-CoA were measured. 13C5-L-Valine and 13C5-Ketoisovalerate were purchased from Sigma; 13C6-L-Isoleucine, 13C5-L-Methionine, 13C4-L-Threonine were purchased from Cambridge Isotopes; and D8-ketomethylvalerate was purchased from Toronto Research.
In some donors, we titrated each P-CoA precursor amino acid and α-keto acid from 10 μM to 3000 μM, while keeping the remaining amino acids at low concentrations (equivalent to ‘baseline’ media). In those experiments, hepatocytes were treated for 5 days.
For TCA experiments, the cells were treated for 7 days with 1 mM disodium citrate (Sigma) or vehicle control in the presence of hemodynamic flow and transport. Hepatocytes were then harvested for measurement of TCA cycle intermediates by quantitative GC-MS/MS.
2.4. Immunostaining
At the prescribed time points in the experimental design, the device transwells were removed and washed gently with PBS followed by fixation of islands with 4% paraformaldehyde for 20 min. The samples were first permeabilized with 0.1% Triton-X for 20 min then washed with PBS and incubated with AlexaFluor-488 Phalloidin (Cat# A12379, Life Technologies), and TOPRO-3 (Cat# T3605, Life Technologies) or DAPI stains for 1 hr to label F-actin and nuclei, respectively. The samples were washed with PBS and then mounted for imaging on a Nikon C1+ Confocal System microscope.
2.5. Protein and Western Blots
Hepatocyte cell pellets were harvested using 100 μL of 2X Laemmli sample buffer (BioRad 161-0737) containing 2-β-mercaptoethanol (Biorad). Samples were boiled and sonicated on ice. Total protein lysates were resolved on a 10% SDS-PAGE gel and transferred to nitrocellulose. Blots were probed overnight with primary antibody and 30 min at room temperature with secondary antibody in blocking buffer (LI-COR 927-40000). Primary antibodies (1:1000) include: PCCA (Bethyl Laboratories), PCCB (Origene), MMUT (ThermoFisher) and GAPDH (Sigma). Secondary antibodies (1:15,000) include: IRDye 800CW Donkey Anti-Mouse (LI-COR 926-32212), IRDye 680LT Donkey Anti-Rabbit (LI-COR 926-68023), IRDye 800CW Donkey Anti-Rabbit (LI-COR 926-32213). The LI-COR Odyssey infrared imager was used for image acquisition and the LI-COR Odyssey Image Studio software used for densitometry analysis.
2.6. Acyl-CoAs and acyl-carnitines measurements
See Supplementary Methods for full description.
2.6.1. Extraction of acyl-CoAs from primary human hepatocytes.
Samples were placed on ice and lysed with 0.2 mL 70% acetonitrile, 0.5% TFA, 0.1 μM ethylmalonyl-CoA. Samples were dried under vacuum and centrifugation in the absence of heat and then re-suspended in 100 μL of 0.1% TFA containing 0.001% tween-20. Samples were subjected to final clean-up by vacuum filtration through a Millipore Multiscreen HTS GV 0.22 μm (MSGVN2250) plate. The samples were frozen at −80°C and shipped on dry ice to PureHoney Technologies (Billerica, MA).
2.6.2. Acylcarnitine extraction from primary human hepatocytes:
Samples were lysed with 70% acetonitrile containing 4 nM N-Methyl-MeD3-hexanoylcarnitine as the internal standard. Samples were centrifuged at 2500 x g for 25 min and the supernatants were subjected to vacuum drying with centrifugation. The resulting dried samples were dissolved in 150 μL of LCMS-grade water and subjected to filtration (Millipore MultiScreenHTS GV Filter Plate, 0.22 μm, clear, non-sterile: MSGVN2250). The samples were frozen at −80°C and shipped on dry ice to PureHoney Technologies (Billerica, MA).
2.6.3. Detection and quantitation of Acyl-CoAs and acylcarnitines by high-throughput mass-spectrometry.
At PureHoney Technologies (Billerica, MA), acyl-CoAs and acyl-carnitines extracted from primary hepatocytes were subjected to solid-phase extraction using an Agilent Rapidfire™ instrument with subsequent detection by mass-spectrometry using multi-reactant monitoring (MRM). Area under the curve integration was performed with the RapidFire™ Integrator (Agilent Technologies) for the desired analytes. Quantitation was completed using internal standards for normalization and external calibrants using a log-logistic regression model. Biomarker levels were normalized to cell counts and cell volume to account for differences in the number of cells plated for each donor.
2.7. Organic acid measurements
See Supplementary Methods for full description
2.7.1. Organic acid extraction from primary human hepatocytes:
Samples were lysed with 70% acetonitrile containing 500 nM 1,2,3-Propanetricarboxylic acid as the internal standard. Lysates were centrifuged at 2500 x g for 25 min and the supernatants were subjected to vacuum drying with centrifugation. The resulting dried samples were dissolved in 150 μL of LCMS-grade water and subjected to filtration (Millipore MultiScreenHTS GV Filter Plate, 0.22 μm, clear, non-sterile: MSGVN2250). The samples were frozen at −80°C and shipped on dry ice to PureHoney Technologies (Billerica, MA).
2.7.2. Detection and quantitation of defined organic acids by LC-MS/MS.
Primary human hepatocytes extracts were analyzed by LC-MS/MS using an Agilent 1100 system with binary pumps coupled to a Sciex 4000 mass spectrometer. Area under the curve integration was performed using Skyline Software (MacCross Lab Software) for the desired analytes. Quantitation was completed using internal standards for normalization and external calibrants using a log-logistic regression model.
2.8. Measurement of TCA cycle intermediates
Hepatocytes were scraped from transwells and lysed using ice-cold 80% methanol, transferred to a tube and flash-frozen until analysis. TCA cycle intermediates were measured by GC-MS/MS at the metabolomics core of the Goodman Cancer Research Center, McGill University using previously published methods (Mamer et al. 2013).
2.9. Statistics.
Linear models were fit to log-transformed data, and the statistical significance of treatment contrasts were assessed using the multiplicity-adjusted generalized linear hypothesis testing framework described by Hothorn et al (Hothorn, Bretz, and Westfall 2008). In cases where multiple donors were present within a given treatment condition, donors were included in the linear mixed-effects model as a random effect.
Dose response curves were analyzed using the ‘drc’ package available for the R programming language (Ritz et al. 2015). In particular, dose response parameters were derived from 4-parameter log-logistic curves that were fit to the data.
3. RESULTS.
3.1. Characterization of a chemically-induced human PA hepatocyte model
3.1.1. Treatment of normal human primary hepatocytes with propionate resulted in high levels of intracellular P-CoA as seen in hepatocytes from PA patients
The chemically-induced PA hepatocyte model was developed by treating normal primary hepatocytes with high concentrations of propionic acid. Treatment with propionic acid has been previous used in models of PA (Wilson et al. 2017). First, we tested whether propionate treatment of normal human primary hepatocytes would recapitulate some of the biochemical features of PA, such as elevation of the proximal key biomarker of the disease, P-CoA. Hepatocytes from normal donors cultured in 24-well plates and treated with increasing amounts of sodium propionate (0, 0.3, 1, 3, 10 mM) for 72 hours showed dose-dependent increases in intracellular P-CoA levels that range from 0.36 ± 0.03 μM to 19.57 ± 4.03 μM (Figure 1A). Although there is normal PCC activity in these hepatocytes, when propionate is given at a constant high dose P-CoA accumulates at high levels comparable to the levels reached in PA hepatocytes (Figure 1A and Figure 3A). The Km value of PCC for propionyl-CoA is 290 uM (Kalousek, Darigo, and Rosenberg 1980). Propionyl-CoA reaches an estimated steady state concentration of ~ 200 uM in the mitochondria at the highest PA dose, based on our measurements. Thus, intramitochondrial propionyl-CoA levels would be close to the Km value and the enzyme would be partially saturated.
Figure 1:
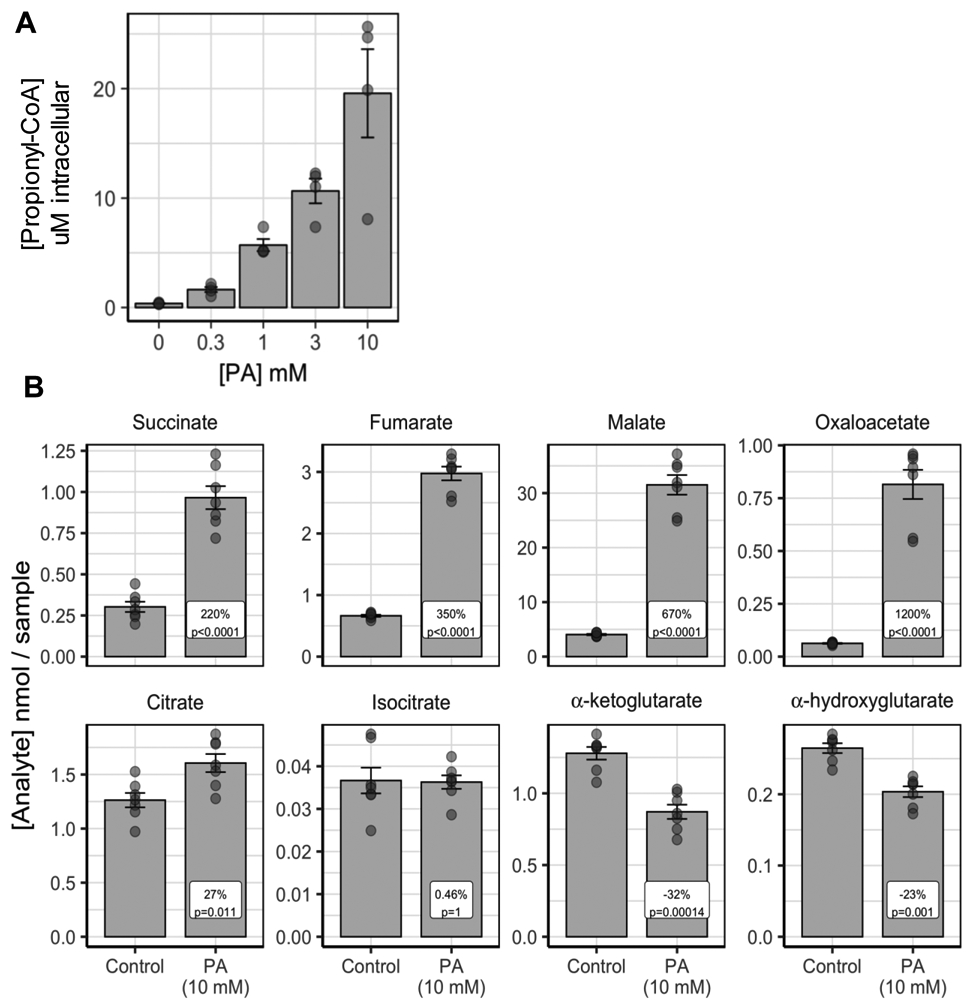
Propionic acid is converted to propionyl-CoA (P-CoA) in normal control hepatocytes. A. Propionic acid (PA) at 0, 0.3. 1, 3, 10 mM is converted to P-CoA in a dose-responsive fashion (1 donor, N=4 biological replicates/dose, N=1 technical replicates/biological replicate). B. Addition of 10 mM PA to normal hepatocytes increases the TCA cycle intermediates downstream of its entrance at succinyl-CoA (succinate, fumarate, malate, oxaloacetate, pyruvate and citrate). It does not increase the level of isocitrate, alpha-ketoglutarate or 2-hydroxyglutarate (1 donor, N=7 biological replicates, N=1 technical replicates/biological replicate). Error bars represent standard error of the mean.
Figure 3.

Elevated biochemical and disease biomarkers in propionic acidemia (PA) and methylmalonic acidemia (MMA) patient hepatocytes cultured under hemodynamics and flow. A. 12C-propionyl-CoA (P-CoA) is greatly elevated in the PA and MMA derived donor cells compared to normal donor cells. B. Schematic of P-CoA pathways and derived metabolites. C. Propionylcarnitine to acetylcarnitine ratio (C3/C2 ratio) is greatly increased in the PA and MMA derived donor cells. D. Methylcitric acid (MCA), which is derived from P-CoA and oxaloacetate, is elevated in the PA and MMA cells compared to controls. E. Intracellular methylmalonyl-CoA levels are elevated in MMA patients. F. 13C-methylmalonic acid (13C-MMA) is produced from 13C-propiogenic sources in MMA patients. Error bars represent the 95% confidence interval of the mean estimate. All data is shown. There are 2 technical replicates per biological replicate. Biological replicates range from 2 to 11 depending on the donor and measurement.
3.1.2. Propionate treatment increased TCA cycle intermediates, but showed some evidence of TCA inhibition
Propionic acid, P-CoA and its metabolites have been reported to reduce mitochondrial energy production by reducing acetyl-CoA levels and inhibiting some TCA cycle enzymes (Cheema-Dhadli, Leznoff, and Halperin 1975; Fenton 2001; Gregersen 1981; Martin-Requero et al. 1983; Patel, DeBuysere, and Olson 1983; Schwab et al. 2006; Stumpf et al. 1980; Weidman and Drysdale 1979). Thus, using normal primary hepatocytes cultured in presence of hemodynamics and flow, we tested the effect of 5-day exposure to high levels of propionate (10 mM) on TCA cycle intermediates. Analysis of TCA cycle intermediates indicated that propionate was incorporated into the TCA cycle as demonstrated by the significant increase seen in the levels of succinate (220%), fumarate (350%), malate (670%) and oxaloacetate (1200%) (Figure 1B). However, small changes were observed in citrate (27%), no change in isocitrate (0.5%), and a significant decrease in incorporation into α-ketoglutarate (−32%) and α-hydroxyglutarate (−23%) (Figure 1B). The pattern of the effect on TCA intermediates suggests that propionate enters into the TCA cycle through succinyl-CoA, as predicted, increasing intermediates immediately downstream. Small or no changes in citrate and isocitrate and a reduction in α-ketoglutarate levels are indicative of inhibition of citrate synthase and/or isocitrate dehydrogenase by propionic acid, P-CoA or a related metabolite. Alternatively, propionyl-CoA (made from propionic acid) may compete with acetyl-CoA for citrate synthase, resulting in conjugation to TCA cycle intermediate oxaloacetate to produce of methylcitric acid (MCA) and resulting in smaller changes in citric acid compared to oxaloacetate. This suggests that propionate treatment does not impact the TCA cycle in normal hepatocytes the same way as in PA patients and so the usefulness of a chemically-induced PA model to study TCA cycle and energy production is limited. Thus, we developed patient-derived PA and MMA human hepatocyte models, as described below.
3.2. Creation and characterization of PA and MMA models using patient-derived hepatocytes
3.2.1. Primary hepatocytes from multiple PA and MMA patients stably maintain differentiated morphology in a model with physiological hemodynamics and transport.
We first reported the recapitulation of the disease phenotype using primary hepatocytes from a single PA patient cultured in the hepatocyte bioreactor (Chapman et al. 2016). Since then, we have successfully isolated and cultured hepatocytes from five PA and three MMA patient explanted livers obtained through our IRB-approved transplant tissue procurement network (Table 1). Rapid isolation enabled by this coordinated effort and the unique ability of the hepatocyte bioreactor to keep the cells differentiated within the system using controlled hemodynamics and transport is critical to preserving hepatocyte function as well as maintaining the metabolic defect itself. After 7 days of culture in the system, the hepatocytes from the PA and MMA patients maintained a normal differentiated and polarized morphology as evidenced by the peripheral actin staining (green, Figure 2A).
Figure 2.

Characterization of the propionic acidemia (PA) and methylmalonic acidemia (MMA) donors. A. Hepatocytes isolated from PA and MMA patients and cultured in the hepatocyte biorreactor demonstrate retention of polarized morphology (Green – Phalloidin, Blue – Draq 5 or DAPI) that is stable over 7 days. B. Immunoblotting for propionyl-CoA carboxylase subunits, Pcca, Pccb demonstrates that PCCA mutants have not protein, but also have decreased Pccb. PCCB mutants make protein but it is decreased in quantity. C. Immunoblotting for Mmut demonstrate MMUT mutant MMA1 has not Mmut enzyme detected, but donor MMA2 does has a normal amount of enzyme present. +++ represents normal expression; ++ represents reduced expression; + represents low expression; +/− represents little to no expression, - represents no expression.
The genetic defect of each donor was further characterized by genotyping (Table 1) as well as by quantifying the expression of PCC or MMUT using western blots (Figure 2 B,C). PCC consists of two non-identical subunits, α and β, encoded by the PCCA and PCCB genes, respectively. The native enzyme is an α6β6 heterododecamer, where the α subunits are arranged as monomers decorating the central β6 hexameric core (Huang et al. 2010). The three PA patients with mutations in PCCA gene showed little or no expression of PCCA protein (Figure 2B). Importantly, these patients did not show expression of PCCB protein either, indicating that PCCA expression might be necessary for sustaining PCCB expression. This finding has not been previously reported so it is unclear whether this regulation occurs pre- or post-translationally. In contrast, two PA patients with mutations in the PCCB gene showed no expression of PCCB protein, but showed normal expression of PCCA.
Methylmalonic acidemia is caused either by a genetic defect in the MMUT gene itself or in one of the proteins (MMAA, MMAB, MMADHC) involved in the synthesis of its cofactor, 5’-deoxyadenosyl-cobalamin. Patients with no residual activity (MUT0) have the earliest and most severe disease presentation and poorest prognosis, followed by cblB mutations. Patients with MMUT mutations with residual enzyme activity (MUT−) or with clbA mutations are variably responsive to vitamin B12 therapy and are generally associated with less severe symptoms and a better prognosis. We analysed the genotype and MMUT expression of 3 MMA patients and found that only one patient (MMA1) showed no expression of MMUT and ,therefore, it is considered a MUT0. Patient MMA2 showed normal expression of MMUT and patient MMA3 showed reduced expression of MMUT. The MMUT protein expression profile is in complete alignment with the genotype of the patients (Table 1), such that MMA1 has a deletion and duplication in MMUT, MMA2 patient is homozygous for a missense mutation and MMA3 is a compound heterozygous for a missense mutation and a splice deletion. It has not been reported whether the missense mutation in MMA2 results in residual enzyme activity. The missense mutation c.2150G>T (p.Gly717Val) in exon 13 found in MMA3 has been identified as pathogenic mutation exhibiting a MUT − MMA phenotype (Crane et al. 1992). To summarize, we have a genotypic diverse group of patient-derived cells to create diverse models and characterize their biochemistry.
3.3. Recapitulation of biochemical features in the PA and MMA models from patient-derived hepatocytes
3.3.1. Elevated intracellular levels of CoA esters and carnitine esters in patient hepatocytes
To characterize the biochemical phenotype of the PA and MMA donors, we developed proprietary assays to assess intracellular proximal metabolites, such as the CoA esters, P-CoA, and MM-CoA, using high-throughput mass spectrometry (HT-MS/MS). We then used those methods to measure P-CoA and MM-CoA in hepatocytes from 3 PA and 3 MMA donors cultured for 7 days under physiological hemodynamics and transport. Levels of P-CoA were elevated by more than 10-fold in hepatocytes from all PA and MMA donors relative to normal donors cultured the same way (Figure 3A). Absolute levels of P-CoA varied from donor to donor but were always consistently higher than normal donors. Levels of acetyl-CoA varied between donors such that donors PA1 and MMA3 had levels similar to normal donors, while donors PA3, PA5, MMA1 and MMA2 showed a slight decrease relative to normal donors (Figure S2A). Free coenzyme A levels were similar between the normal and PA and MMA donors (Figure S2B). The levels of MM-CoA were elevated by 5-fold compared to normal donors in MMA donor cells only (Figure 3E). Overall, levels of MM-CoA have a higher baseline levels possibly due to the fact that this analytical method cannot separate MM-CoA from succinyl-CoA, resulting in a measurement that is the sum of both metabolites. Succinyl-CoA should be negligible in PA or MMA hepatocytes, but might explain the high baseline observed in normal donors. Regardless, it appears that MM-CoA accumulates at greater levels than P-CoA in this setting. P-CoA and MM-CoA are the most proximal and biochemically reliable biomarkers of the disease and thus, the utility of them as surrogates of disease state is made possible in these models (Figure 3B).
Elevation in P-CoA is not clinically assessed in patients because it is an intracellular metabolite. However, the levocarnitine ester derived from P-CoA, propionylcarnitine (C3), is readily detectable in plasma, serum, and urine (Kurczynski et al. 1989). Thus, we sought to measure clinically-relevant biomarkers, such as the intracellular ratio of propionyl-carnitine to acetyl-carnitine (C3:C2 ratio, Figure 3B), in patient hepatocytes and found that the C3:C2 ratio was significantly greater in PA and MMA hepatocytes as compared to normal (Figure 3C). Our estimated intracellular C3:C2 ratios are closely aligned with clinical data that considers blood C3:C2 ratios greater than 0.2 to be diagnostic of PA or MMA and with ratios in dried blood spots ranging from 0.39 to 2.55 (Al-Dirbashi et al. 2014).
3.3.2. High intracellular levels of organic acids in PA and MMA donors
P-CoA acts as an alternate substrate for citrate synthase, resulting in conjugation to TCA cycle intermediate oxaloacetate to produce of methylcitric acid (MCA), which is usually assayed from the urine clinically (Ando et al. 1972; Weidman and Drysdale 1979) (Figure 3B). We detected elevated intracellular MCA in hepatocytes from PA and MMA donors using an LC-MS/MS method. MCA was not detected in normal hepatocytes. We observed a greater variability in levels of MCA compared to P-CoA, suggesting donor differences in levels of TCA intermediates (oxaloacetate) and the propensity of citrate synthase to use P-CoA as an alternative substrate (Figure 3D). The intracellular levels of MCA in PA and MMA hepatocytes were comparable to the levels of observed in the plasma of patients that ranged from 8.8 to 34.7 μM (Fu et al. 2013).
Methylmalonic acid, formed by thioesterase-mediated hydrolysis of MM-CoA, is excreted and cleared in the urine of patients with MMA (Figure 3B). Although still controversial, methylmalonic acid is thought to be involved in the induction of chronic renal failure in MMA patients (Kolker and Okun 2005). We developed an LC-MS/MS method to detect levels of methylmalonic acid in patient hepatocytes using 13C-labeled propiogenic sources to allow for the precise measurement of 13C-methylmalonic acid. 12C-methymalonic acid has a high detection background, therefore it cannot be accurately measured in experiments without 13C-labeling. Using a special ‘catabolic’ media formulation with all propiogenic sources labeled as 13C (See section 3.5.1 and Table S1), we found elevated levels of 13C-methylmalonic acid in all 3 MMA hepatocyte donors (Figure 3E). 13C-methylmalonic acid was not detected in normal donors cultured under the same conditions. The PA and MMA patient-derived hepatocyte models developed herein provide a tool for relating proximal metabolites to clinically measurable biomarkers.
3.3.3. TCA cycle impact in a PA donor
To evaluate the impact of P-CoA and its metabolites to the TCA cycle, we measured levels of TCA cycle intermediates in normal and PA primary hepatocytes from donor PA1 cultured in the presence of hemodynamics and flow. Analysis of TCA cycle intermediates demonstrated significant decreases in levels of fumarate (−18%) and citrate (−20%) in donor PA1, compared to normal primary hepatocytes (Figure S3). This pattern of TCA intermediate levels suggests that in PA patient hepatocytes, P-CoA might be competing with acetyl-CoA for citrate synthase resulting in decreased citrate levels and increased MCA levels observed in the previous section (Figure 3D). Finally, the decreased levels of other intermediates, such as fumarate, in PA hepatocytes are in agreement with a lack of anaplerotic entry at the level of succinyl-CoA due to the block at PCC.
3.4. Contribution of P-CoA precursors in patient-derived PA and MMA models reveal different donor-dependent sensitivities to propiogenic sources
3.4.1. PA and MMA donors show unique responses to different propiogenic sources
Sources of P-CoA include the amino acids valine, isoleucine, threonine and methionine, odd chain fatty acids, and propionic acid derived from gut flora and the metabolism of the side chain of cholesterol. With appropriate diet and control of gut bacteria, nearly 100% of P-CoA is derived from amino acids, but their relative contributions have not been reliably determined. Here, we titrated each P-CoA precursor amino acid or its corresponding α-ketoacid from 10 μM to 3000 μM in 3 PA donors (PA1, PA3, PA5), while keeping the rest of the amino acids at low concentrations (baseline media formulation, see section 3.5.2) and measured P-CoA.
As shown before, isoleucine and its α-ketoacid, α-ketomethylvalerate (KMVA), efficiently produced P-CoA in all patient hepatocytes tested, with EC50 values varying by donor and ranging from 265 to 2350 μM (Figure 4). Valine and methionine did not significantly produce P-CoA in our dose range, thus it was not possible to calculate a reliable EC50 value. However, PA hepatocytes showed a significant response to the α-ketoacid of valine, α-ketoisovalerate (KIVA), with EC50 ranging from 190 to 1000 μM within PA donors. Threonine produced P-CoA only at high levels in some donors (EC50 > 1500 μM). Cells from each donor exhibited a different response to each individual propiogenic source. For example, cells from donor PA3 produced P-CoA at much lower concentrations of isoleucine, KMVA or KIVA than cells from other donors. It is possible that this biochemical phenomenon may underlie patient-to-patient differences in protein tolerance observed in the clinic.
Figure 4.
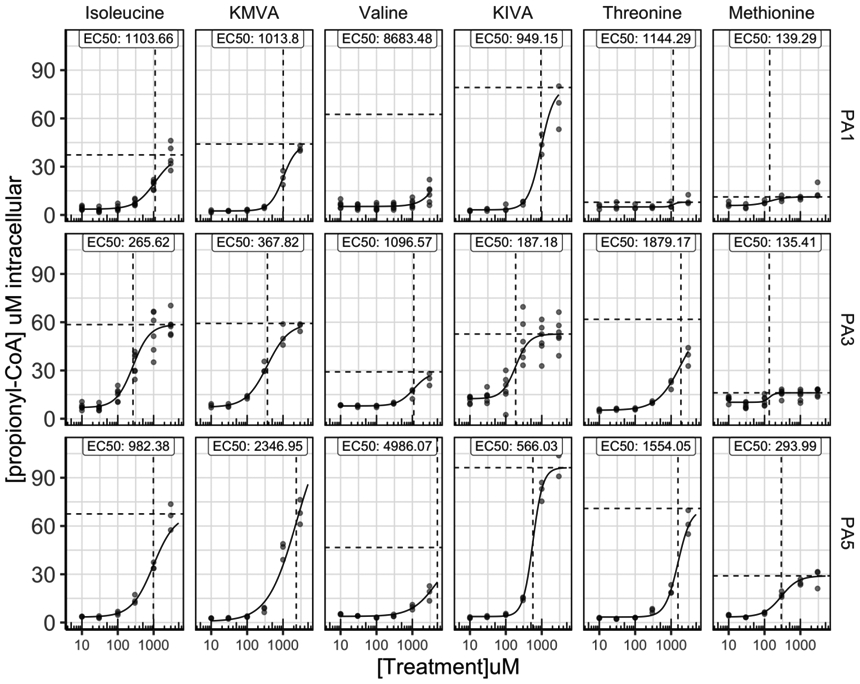
Donors show unique responses of propionyl-CoA (P-CoA) production from each amino acid source. Each P-CoA amino acid or α-ketoacid precursor was titrated from 10 μM to 3000 μM in multiple propionic acidemia (PA) donors, while keeping the rest of the amino acids at low concentrations and P-CoA levels were measured (N=3-6 biological replicates/dose; N=1 technical replicate/biological replicate).
3.4.2. P-CoA sources and their relative contribution in baseline and catabolic states
We sought to determine the contribution of each propiogenic source in the context of different disease states. By utilizing custom designed media formulations, we mimicked two disease states: 1) A ‘Baseline metabolic state’ by using a low propiogenic media based on propiogenic amino acid concentrations measured in the plasma of PA and MMA patients during periods of metabolic stability; or 2) A ‘Catabolic state’ by using a high propiogenic media based on projected load of propiogenic precursors during acute metabolic crises. During metabolic crises, amino acids from catabolism of muscle are used by tissues, including the liver, for energy production. Levels of propiogenic amino acids are not elevated in the plasma of PA and MMA patients, but P-CoA and MM-CoA accumulate in the tissues instead. Here, we simulated a metabolic crisis by increasing the levels of propiogenic amino acids in the culture media. In addition, we tested a “MAX catabolic state’ formulation using supra-physiological levels of propiogenic precursors (Table S1). To study the contribution of each P-CoA precursor in the different disease states, we replaced one precursor at a time with its 13C-labeled analog and measured 13C-P-CoA (blue) produced within 24 hrs from 13C-labeled precursors as well as endogenous 12C-P-CoA produced from unlabeled precursors (gray) (Figure 5). The study was performed using cells from 3 PA donors and 2 MMA donors.
Figure 5:

Propionyl-CoA (P-CoA) sources and their relative contribution in different disease states. Three different formulations were used to simulate different disease states: 1) ‘Baseline metabolic state’ 2) ‘Catabolic state’ or 3) “MAX catabolic state’ (Table S1). One P-CoA precursor at a time was replaced with its 13C-labeled analog within each media formulation and 13C-P-CoA (blue) was measured and % contribution was calculated. Endogenous 12C-P-CoA produced from the other unlabeled precursors is shown in gray bars. N= 4 biological replicates, N=1 technical replicates.
In the ‘baseline metabolic state’, levels of total intracellular P-CoA ranged from 2.5 to 27 μM depending on the donor. The contribution of each 13C-labeled precursor (blue bars) differed between donors, but for most donors the most effective propiogenic source was KMVA or isoleucine and the least effective propiogenic source was methionine (Figure 5). In the ‘catabolic state’, levels of total intracellular P-CoA ranged from 25 to 60 μM depending on the donor. Under these conditions, isoleucine and threonine efficiently produced P-CoA while valine and methionine contributed to only a small fraction of the P-CoA produced (<10%). When we used the α-ketoacid derivatives of valine and isoleucine, KIVA and KMVA, P-CoA was produced at significantly higher levels (>10% for most donors). Thus, α-ketoacids were more efficiently utilized by PA patient hepatocytes to produce P-CoA. Finally, with supra-physiological concentrations of propiogenic precursors (MAX catabolic), the P-CoA levels produced were similar to those produced in the catabolic condition, suggesting that propiogenic sources were already saturating in the catabolic condition. These results suggest that metabolites derived from the isoleucine and valine pathways represent the predominant source of P-CoA produced during metabolic decompensation in the patient-derived hepatocytes. Although, for some donors, such as MMA1, threonine was also major contributor to the production of P-CoA.
Additionally, we determined the contribution of each precursor to the production of MM-CoA using 2 MMA donors and found the same trends observed for the production of P-CoA by those MMA donor cells (Figure S4). We also observed differences in the donors’ response to precursors levels. Donor MMA1 was much more sensitive to precursor concentrations, showing saturation of MM-CoA production in the baseline condition, while donor MMA2 only displayed saturation in the catabolic condition (Figure S4).
3.5. Anaplerotic therapy in patient-derived PA hepatocytes increases the levels of TCA cycle intermediates
The conversion of P-CoA to succinyl-CoA is one of the major anaplerotic pathways of the TCA cycle. The lack of succinyl-CoA entry into the TCA cycle in PA and MMA patients results in deficiency of intermediates and reduced mitochondrial energy production. Longo et al. conducted a clinical trial to test the beneficial effects of different anaplerotic therapies in PA patients (NCT00645879). The most promising anaplerotic agent was citrate, a TCA cycle intermediate, that resulted in the increased urinary excretion of α-ketoglutarate, succinate, and fumarate. We sought to further understand the impact of citrate supplementation on TCA cycle mitochondrial energy production. Primary hepatocytes from donor PA1 in our PA disease model were treated for 7 days with either vehicle or 1 mM disodium citrate, and the levels of TCA cycle intermediates, fatty acids, and amino acids were quantified by GC-MS/MS. We observed a 250% increase in citrate levels, providing evidence that disodium citrate entered the TCA cycle (Figure 6A). Disodium citrate treatment also resulted in a significant increase in overall levels of other TCA cycle intermediates, including fumarate, malate, oxaloacetate, and α-ketoglutarate (Figure 6A). Additionally, disodium citrate treatment led to increased cataplerotic flux from two intermediates: (1) oxaloacetate flux to phosphoenolpyruvate (PEP) or aspartate, and (2) α-ketoglutarate flux to glutamate/glutamine and α-ketoglutarate interconverting to 2-hydroxyglutarate (Figure 6A, B). The levels of amino acids, fatty acids, and urea were measured by GC-MS/MS but not quantified. Figure 6B and Table S2 depict the % change relative to DMSO control calculated using the AUC measurements. There was an overall increase in amino acid and urea levels, and an overall decrease in fatty acid levels. The data support treatment with disodium citrate as having a positive anaplerotic effect on the TCA cycle with the potential for increased mitochondrial energy production, further supporting the clinical findings.
Figure 6:

Disodium citrate boost the TCA cycle in propionic acidemia (PA) hepatocytes A. TCA intermediate levels following treatment of PA donor hepatocytes with 1 mM disodium citrate (N=7 biological replicates; N=1 technical replicates). B. Cartoon illustrating TCA cycle and cataplerotic flux and showing percentage change for the different TCA intermediates and related pathways. Error bars represent standard error of the mean.
4. DISCUSSION
A major challenge in drug discovery is identifying cell-based models that best recapitulate the disease state. In this study, we developed two cellular approaches for modelling PA and MMA. The first approach consisted of a chemically-induced PA model created by adding propionic acid directly to normal human primary hepatocytes. Although we were able to partially recapitulate the PA disease-state biochemistry in this model, a key aspect of the disease, the TCA cycle dysfunction, was not fully recapitulated. The second approach utilized hepatocytes obtained from PA and MMA patient livers following liver transplant, where the relevant genetic mutation remains intact. Using this second approach we made several important observations that indicated a more complete recapitulation of the PA and MMA disease phenotypes as compared to the first approach. First, hepatocytes had elevated intracellular levels of key disease biomarkers, including P-CoA, the C3/C2 ratio, MCA and increased MM-CoA and methylmalonic acid for MMA donors, which aligned with the levels reported in the plasma or dried blood spots of PA and MMA patients (Al-Dirbashi et al. 2014; Fu et al. 2013). Second, using isotopically labeled propiogenic precursors, we demonstrated that the isoleucine and valine pathways are the major contributors to elevated levels of P-CoA, and that responses and susceptibilities to propiogenic precursors were donor-dependent, emphasizing the importance of customized diets and formulas. Third, we demonstrated that disodium citrate is an effective anaplerotic, increasing the levels of TCA cycle intermediates and cataplerosis in patient-derived hepatocytes, which is in agreement with clinical findings (Longo et al. 2017). Taken together, we have validated the utility and relevance of patient-derived PA and MMA organotypic models for the discovery and development of novel therapeutics.
Creating relevant in vitro human models of propionic and methylmalonic acidemia using patient cells.
Several models of PA and MMA have been developed in mice (Buck et al. 2012; Chandler et al. 2007; Chandler et al. 2009; Forny et al. 2016; Guenzel et al. 2013; Manoli et al. 2013; Miyazaki et al. 2001; Peters et al. 2003; Peters et al. 2012). Although these systems may be amenable to gene replacement therapy validation (Chandler et al. 2011; Chandler and Venditti 2010, 2012), the inherent differences in the tissue distribution of the endogenous enzymes and the regulation of BCAA metabolism compared to humans (Brosnan and Brosnan 2006; Suryawan et al. 1998; Sweatt et al. 2004), make them valuable but with some biochemical limitations for the human disease and challenging for novel small molecular drug approaches that regulate BCAA metabolism.
Research in rare diseases has been impacted by the limited availability of robust in vitro experimental models capable of recapitulating metabolic responses associated with the diseases. Until recently, in vitro human models relied primarily on patient-derived fibroblasts or lymphoblastoid cell lines (Anzmann et al. 2019; Chapman, Bush, and Zhang 2015; Darvish-Damavandi, Ho, and Kang 2016; Gallego-Villar et al. 2014). Although fibroblasts can be used to assess PCC and MMUT activity, these cells have considerably lower levels of activity of propionate pathways resulting in low metabolite levels and have little to no urea cycle activity or mitochondria function needed for in vitro disease modelling (Barash et al. 1989; Perez-Cerda et al. 2003; Ugarte et al. 1999; Varani et al. 1985; Berman, Tong, and Williams 1980). Inducible pluripotent stem cells (iPSCs) have been developed using fibroblasts from patients with PA and MMA cblB type, which may hold promise for tissue-specific model development in the future (Alonso-Barroso et al. 2017; Lopez-Marquez et al. 2019; Richard et al. 2018). More recently, models of PCCA-null and MMUT-null CRISPR edited HEK293 cells were developed that retained mitochondrial function and some of the relevant biochemical pathways related to MMA and PA (Anzmann et al. 2019).
To circumvent flaws inherent to these systems, we developed models using patient-derived primary hepatocytes. Although primary hepatocytes are challenging to procure and maintain in culture, primary hepatocytes afford the most faithful representation of liver metabolism and the patient’s original genotype. Success in this approach is dependent on three crucial factors: 1) access to patient tissue; 2) timely isolation of hepatocytes; and 3) the ability to maintain the hepatocytes in a functionally differentiated state in culture. In PA and MMA, liver transplantation is performed in patients with frequent metabolic decompensation. We have established a relationship with multiple pediatric liver transplantation centers in the US to gain IRB-approved access to livers from patients with PA and MMA. Livers identified for isolation are procured from the surgical team, perfused within 30 minutes of organ removal with concomitant timely isolation and cryopreservation of primary hepatocytes. Using the hepatocyte bioreactor, in vivo-like morphology and functionality is accurately reproduced and the metabolic defect observed in the donors is restored to be used for further experimentation. Most importantly, the clinically-relevant biochemical markers of disease state, including P-CoA, C3/C2 ratio, and MCA were maintained in cells from PA and MMA donors as compared to normal donors.
Variable responses to propiogenic sources shed light on the phenotypic spectrum in PA and MMA patients
Organic acidemias are a diverse group of genetic metabolic disorders, with extensive variation in the severity of the disease phenotypes associated with the specific enzyme defects and with unclear phenotype-genotype correlations (Kolker et al. 2015). This highlights the need to gain a deeper understanding of variability of key biomarkers as well as the most common triggers of disease severity. To reduce accumulation of P-CoA and MM-CoA and associated metabolites, restriction of natural protein and sometimes odd-chain fatty acids is used, as well as supplementation of a synthetic amino acid mixture devoid of valine, isoleucine, methionine, and threonine. Under this treatment regimen, disturbed plasma amino acid ratios and suboptimal growth has been observed (Manoli et al. 2016). Some patients suffered from severe acrodermatitis enteropathica or skin lesions due to isoleucine deficiency, which in some cases culminated in sepsis and death (Blecker et al. 1994; Bosch et al. 1998; Koopman and Happle 1990). Supplementation with isoleucine and valine to prevent essential amino acid deficiencies has become routine clinical practice in some centers, but defining an optimal dietary treatment for any given patient is difficult because of the need to balance essential amino acid intake needed for optimal growth against the production of toxic metabolites. Here, we showed that metabolites derived from the isoleucine and valine pathways are the major contributors to the production of P-CoA, followed next by those derived from threonine metabolism. In general, patient hepatocytes were more responsive to KIVA than to valine. This may be due to limitations of transport or branched-chain aminotransferase activity for valine. Although the relative contributions of the amino acids to the production of P-CoA were similar in most donors, each donor demonstrated a slightly different response to amino acid loading, which may explain why each patient responds differently during metabolic decompensation or to dietary supplementation. Our PA patient-derived surrogate human liver model may have potential for investigating the influence of precursors on the production of P-CoA and could inform medical food composition as well as sick-day formulas.
Anaplerosis impacts TCA cycle intermediates in PA and MMA
In PA patients, the absence of PCC prevents P-CoA from being converted into succinyl-CoA. This is a major anaplerotic pathway of the TCA cycle in some tissues during certain metabolic states, such as the brain and muscle during exercise or periods of low glucose (Davis, Spydevold, and Bremer 1980; Peuhkurinen 1984). Importantly, it is known that P-CoA itself reduces mitochondrial energy production by inhibition of the pyruvate dehydrogenase complex (Fenton 2001; Gregersen 1981; Martin-Requero et al. 1983; Patel, DeBuysere, and Olson 1983; Schwab et al. 2006), and that P-CoA can also act as an alternate substrate for citrate synthase, resulting in production of MCA which inhibits the TCA cycle enzymes, citrate synthase and isocitrate dehydrogenase (Cheema-Dhadli, Leznoff, and Halperin 1975; Fenton 2001; Weidman and Drysdale 1979). The chronic effect of inhibition of mitochondrial energy production leads to oxidative stress, mitochondrial DNA damage and altered mitochondrial morphology. The impact of this on organ biology is reflective of organ energy requirements; high-energy-demand organs such as the brain, muscle, liver and heart are primarily affected.
When we compared levels of TCA intermediates in PA hepatocytes to normal hepatocytes, we found small but significant reductions in fumarate and citrate, which is in agreement with a reduced succinyl-CoA-driven anaplerosis and citrate production from acetyl-CoA. The small magnitude of the changes in fumarate and citrate between PA and normal hepatocytes observed might be due to the culture conditions used that might not have favored a succinyl-CoA-driven anaplerosis. However, our system can be fine-tuned to allow for modeling metabolic conditions that would favor such state in the future.
We showed that supplementation with citrate in PA hepatocytes increased levels of intermediates and their flux within the TCA cycle. An increase in cataplerotic flux from the TCA cycle resulted in higher levels of amino acids, including glutamate and glutamine. Normally, glutamate dehydrogenase favors cataplerotic flux from the TCA cycle to produce glutamate. In patients with PA, low α-ketoglutarate levels result in the generation of α-ketoglutarate from glutamate and also replenishment of glutamate from glutamine (Filipowicz et al. 2006). Our data suggest that supplementation with citrate returns glutamate dehydrogenase to a cataplerotic flux. Overall, anaplerotic supplementation with citrate increases levels of TCA cycle intermediates. This provides evidence for a beneficial effect on mitochondrial energy production and supports clinical observations (Longo et al. 2017).
In summary, we have presented direct evidence that the PA and MMA disease phenotype can be retained in primary human hepatocytes derived from patients with PA and MMA when placed into the appropriate physiological context. We believe that these models have great potential for elucidating mechanisms that contribute to the disease pathophysiology and for identifying and testing new therapeutic interventions where animal models have limited utility. Finally, the paradigm deployed herein directly applies to other rare diseases with primary defects in the liver.
Supplementary Material
Acknowledgements:
The authors would like to express thanks to Christin Hamilton, Andrew Pryor, Justin Taylor, Diana Berry, Morgan Donovan, Crystal Passmore and Taylor Pourtaheri for technical assistance. We would also like thank our transplant network coordinator Jeanine Fogarty and collaborators Wanxing Cui and Nada Yazigi from Georgetown University Hospital, Washington, DC, Karl-Dimiter Bissig from Duke University, Durham, NC, Ira Fox and Alina Ostrowska from UPMC Children's Hospital of Pittsburgh and Lawrence Merrit from Seattle Children’s Hospital. We thank Brian Johns for his critical review of the manuscript. This research was supported in part by SBIR grant R44TR001407 and HemoShear Therapeutics, LLC.
Footnotes
Conflicts: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Al-Dirbashi OY, McIntosh N, McRoberts C, Fisher L, Rashed MS, Makhseed N, Geraghty MT, Santa T, and Chakraborty P. 2014. 'Analysis of methylcitrate in dried blood spots by liquid chromatography-tandem mass spectrometry', JIMD Rep, 16: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Barroso E, Brasil S, Briso-Montiano A, Navarrete R, Perez-Cerda C, Ugarte M, Perez B, Desviat LR, and Richard E. 2017. 'Generation and characterization of a human iPSC line from a patient with propionic acidemia due to defects in the PCCA gene', Stem Cell Res, 23: 173–77. [DOI] [PubMed] [Google Scholar]
- Ando T, Rasmussen K, Wright JM, and Nyhan WL. 1972. 'Isolation and identification of methylcitrate, a major metabolic product of propionate in patients with propionic acidemia', J Biol Chem, 247: 2200–4. [PubMed] [Google Scholar]
- Anzmann AF, Pinto S, Busa V, Carlson J, McRitchie S, Sumner S, Pandey A, and Vernon HJ. 2019. 'Multi-omics studies in cellular models of methylmalonic acidemia and propionic acidemia reveal dysregulation of serine metabolism', Biochim Biophys Acta Mol Basis Dis, 1865: 165538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash V, Elpeleg O, Amit R, Gottfried S, Yatziv S, and Gutman A. 1989. 'Propionic acidemia--biochemical studies', Isr J Med Sci, 25: 103–6. [PubMed] [Google Scholar]
- Baumgartner MR, Horster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, Burlina A, Fowler B, Grunert SC, Grunewald S, Honzik T, Merinero B, Perez-Cerda C, Scholl-Burgi S, Skovby F, Wijburg F, MacDonald A, Martinelli D, Sass JO, Valayannopoulos V, and Chakrapani A. 2014. 'Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia', Orphanet J Rare Dis, 9: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman JJ, Tong C, and Williams GM. 1980. 'Differences between rat liver epithelial and fibroblast cells in metabolism of purines', J Cell Physiol, 103: 393–8. [DOI] [PubMed] [Google Scholar]
- Blecker U, De Meirleir L, De Raeve L, Ramet J, and Vandenplas Y. 1994. 'Acrodermatitis-like syndrome in organic aciduria', Pediatrics, 93: 537. [PubMed] [Google Scholar]
- Bosch AM, Sillevis Smitt JH, Van Gennip AH, Abeling NG, Schutgens RB, Bakker HD, and Wijburg FA. 1998. 'Iatrogenic isolated isoleucine deficiency as the cause of an acrodermatitis enteropathica-like syndrome', Br J Dermatol, 139: 488–91. [DOI] [PubMed] [Google Scholar]
- Brosnan JT, and Brosnan ME. 2006. 'Branched-chain amino acids: enzyme and substrate regulation', J Nutr, 136: 207S–11S. [DOI] [PubMed] [Google Scholar]
- Buck NE, Dashnow H, Pitt JJ, Wood LR, and Peters HL. 2012. 'Development of transgenic mice containing an introduced stop codon on the human methylmalonyl-CoA mutase locus', PLoS One, 7: e44974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Carrasco N, and Venditti C. 1993. 'Propionic Acidemia' in Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Smith RJH and Stephens K (eds.), GeneReviews(R) (University of Washington, Seattle: [PubMed] [Google Scholar]
- University of Washington, Seattle: All rights reserved.: Seattle (WA)). [Google Scholar]
- Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr SE, Barry MA, and Venditti CP. 2011. 'Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia', Hum Gene Ther, 22: 477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Sloan J, Fu H, Tsai M, Stabler S, Allen R, Kaestner KH, Kazazian HH, and Venditti CP. 2007. 'Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle', BMC Med Genet, 8: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, and Venditti CP. 2010. 'Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy', Mol Ther, 18: 11–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, and Venditti CP. 2012. 'Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA)', Mol Genet Metab, 107: 617–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, and Venditti CP. 2009. 'Mitochondrial dysfunction in mut methylmalonic acidemia', FASEB J, 23: 1252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KA, Bush WS, and Zhang Z. 2015. 'Gene expression in cell lines from propionic acidemia patients, carrier parents, and controls', Mol Genet Metab, 115: 174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KA, Collado MS, Figler RA, Hoang SA, Armstrong AJ, Cui W, Purdy M, Simmers MB, Yazigi NA, Summar ML, Wamhoff BR, and Dash A. 2016. 'Recapitulation of metabolic defects in a model of propionic acidemia using patient-derived primary hepatocytes', Mol Genet Metab, 117: 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman Kimberly A., Gramer Gwendolyn, Viall Sarah, and Summar Marshall L.. 2018. 'Incidence of maple syrup urine disease, propionic acidemia, and methylmalonic aciduria from newborn screening data', Mol Genet Metab Rep, 15: 106–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema-Dhadli S, Leznoff CC, and Halperin ML. 1975. 'Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria', Pediatr Res, 9: 905–8. [DOI] [PubMed] [Google Scholar]
- Coude FX, Sweetman L, and Nyhan WL. 1979. 'Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia', J.Clin.Invest, 64: 1544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane AM, Jansen R, Andrews ER, and Ledley FD. 1992. 'Cloning and expression of a mutant methylmalonyl coenzyme A mutase with altered cobalamin affinity that causes mut-methylmalonic aciduria', J Clin Invest, 89: 385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvish-Damavandi M, Ho HK, and Kang TS. 2016. 'Towards the development of an enzyme replacement therapy for the metabolic disorder propionic acidemia', Mol Genet Metab Rep, 8: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash A, Deering TG, Marukian S, Thomas J, Desbans C, Alexandre E, Richert L, Blackman BR, and Wamhoff BR. 2013. "Human Primary Hepatocytes Under Controlled Hemodynamics Elicit Induction Responses to Drugs at Clinical Cmax Concentrations." In 52nd Annual Meeting of the Society of Toxicology, 10 San Antonio, Texas. . [Google Scholar]
- Dash A, Simmers MB, Deering TG, Berry DJ, Feaver RE, Hastings NE, Pruett TL, LeCluyse EL, Blackman BR, and Wamhoff BR. 2013. 'Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro', Am J Physiol Cell Physiol, 304: C1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EJ, Spydevold O, and Bremer J. 1980. 'Pyruvate carboxylase and propionyl-CoA carboxylase as anaplerotic enzymes in skeletal muscle mitochondria', Eur J Biochem, 110: 255–62. [DOI] [PubMed] [Google Scholar]
- Deodato F, Boenzi S, Santorelli FM, and Dionisi-Vici C. 2006. 'Methylmalonic and propionic aciduria', Am J Med Genet C Semin Med Genet, 142C: 104–12. [DOI] [PubMed] [Google Scholar]
- Fenton WA, Gravel RA, Rosenblatt DS 2001. 'Disorders of Propionate and Methylmalonate Metabolism' in Beaudert CR Sly William AL Valle S Scriver David (ed.), The Metabolic & Molecular Basis of Inherited Disease (McGraw-Hill: New York: ). [Google Scholar]
- Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, and Longo N. 2006. 'Metabolic changes associated with hyperammonemia in patients with propionic acidemia', Mol Genet Metab, 88: 123–30. [DOI] [PubMed] [Google Scholar]
- Forny P, Schumann A, Mustedanagic M, Mathis D, Wulf MA, Nagele N, Langhans CD, Zhakupova A, Heeren J, Scheja L, Fingerhut R, Peters HL, Hornemann T, Thony B, Kolker S, Burda P, Froese DS, Devuyst O, and Baumgartner MR. 2016. 'Novel Mouse Models of Methylmalonic Aciduria Recapitulate Phenotypic Traits with a Genetic Dosage Effect', J Biol Chem, 291: 20563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Xu YK, Chan P, and Pattengale PK. 2013. 'Simple, Fast, and Simultaneous Detection of Plasma Total Homocysteine, Methylmalonic Acid, Methionine, and 2-Methylcitric Acid Using Liquid Chromatography and Mass Spectrometry (LC/MS/MS)', JIMD Rep, 10: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Villar L, Perez B, Ugarte M, Desviat LR, and Richard E. 2014. 'Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts', Biochem Biophys Res Commun, 452: 457–61. [DOI] [PubMed] [Google Scholar]
- Gregersen N 1981. 'The specific inhibition of the pyruvate dehydrogenase complex from pig kidney by propionyl-CoA and isovaleryl-Co-A', Biochem Med, 26: 20–7. [DOI] [PubMed] [Google Scholar]
- Guenzel AJ, Hofherr SE, Hillestad M, Barry M, Weaver E, Venezia S, Kraus JP, Matern D, and Barry MA. 2013. 'Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing', Mol Ther, 21: 1316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Bretz F, and Westfall P. 2008. 'Simultaneous interference in general parametric models', Biom J., 50: 346–63. [DOI] [PubMed] [Google Scholar]
- Huang CS, Sadre-Bazzaz K, Shen Y, Deng B, Zhou ZH, and Tong L. 2010. 'Crystal structure of the alpha(6)beta(6) holoenzyme of propionyl-coenzyme A carboxylase', Nature, 466: 1001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalousek F, Darigo MD, and Rosenberg LE. 1980. 'Isolation and characterization of propionyl-CoA carboxylase from normal human liver. Evidence for a protomeric tetramer of nonidentical subunits', J Biol Chem, 255: 60–5. [PubMed] [Google Scholar]
- Kolker S, and Okun JG. 2005. 'Methylmalonic acid--an endogenous toxin?', Cell Mol Life Sci, 62: 621–4. [DOI] [PubMed] [Google Scholar]
- Kolker S, Valayannopoulos V, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Baric I, Karall D, Arnoux JB, Avram P, Baumgartner MR, Blasco-Alonso J, Boy SP, Rasmussen MB, Burgard P, Chabrol B, Chakrapani A, Chapman K, Cortes I. Saladelafont E., Couce ML, de Meirleir L, Dobbelaere D, Furlan F, Gleich F, Gonzalez MJ, Gradowska W, Grunewald S, Honzik T, Horster F, Ioannou H, Jalan A, Haberle J, Haege G, Langereis E, de Lonlay P, Martinelli D, Matsumoto S, Muhlhausen C, Murphy E, de Baulny HO, Ortez C, Pedron CC, Pintos-Morell G, Pena-Quintana L, Ramadza DP, Rodrigues E, Scholl-Burgi S, Sokal E, Summar ML, Thompson N, Vara R, Pinera IV, Walter JH, Williams M, Lund AM, and Garcia-Cazorla A. 2015. 'The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype', J Inherit Metab Dis, 38: 1059–74. [DOI] [PubMed] [Google Scholar]
- Koopman RJ, and Happle R. 1990. 'Cutaneous manifestations of methylmalonic acidemia', Arch Dermatol Res, 282: 272–3. [DOI] [PubMed] [Google Scholar]
- Kurczynski TW, Hoppel CL, Goldblatt PJ, and Gunning WT. 1989. 'Metabolic studies of carnitine in a child with propionic acidemia', Pediatr Res, 26: 63–6. [DOI] [PubMed] [Google Scholar]
- Lecluyse EL, and Alexandre E. 2010. 'Isolation and culture of primary hepatocytes from resected human liver tissue', Methods Mol Biol, 640: 57–82. [DOI] [PubMed] [Google Scholar]
- Longo N, Price LB, Gappmaier E, Cantor NL, Ernst SL, Bailey C, and Pasquali M. 2017. 'Anaplerotic therapy in propionic acidemia', Mol Genet Metab, 122: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Marquez A, Alonso-Barroso E, Cerro-Tello G, Bravo-Alonso I, Arribas-Carreira L, Briso-Montiano A, Navarrete R, Perez-Cerda C, Ugarte M, Perez B, Desviat LR, and Richard E. 2019. 'Generation and characterization of a human iPSC line (UAMi004-A) from a patient with propionic acidemia due to defects in the PCCB gene', Stem Cell Res, 38: 101469. [DOI] [PubMed] [Google Scholar]
- Mamer O, Gravel SP, Choiniere L, Chenard V, St-Pierre J, and Avizonis D. 2013. 'The complete targeted profile of the organic acid intermediates of the citric acid cycle using a single stable isotope dilution analysis, sodium borodeuteride reduction and selected ion monitoring GC/MS', Metabolomics, 9: 1019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Myles JG, Sloan JL, Shchelochkov OA, and Venditti CP. 2016. 'A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias', Genet Med, 18: 386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog K, Young S, Trivedi NS, Cheng J, Sloan JL, Chandler RJ, Abu-Asab M, Tsokos M, Elkahloun AG, Rosen S, Enns GM, Berry GT, Hoffmann V, DiMauro S, Schnermann J, and Venditti CP. 2013. 'Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia', Proc Natl Acad Sci U S A, 110: 13552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, and Venditti CP. 1993. 'Methylmalonic Acidemia'.
- Martin-Requero A, Corkey BE, Cerdan S, Walajtys-Rode E, Parrilla RL, and Williamson JR. 1983. 'Interactions between alpha-ketoisovalerate metabolism and the pathways of gluconeogenesis and urea synthesis in isolated hepatocytes', J Biol Chem, 258: 3673–81. [PubMed] [Google Scholar]
- Millward DJ 1997. 'Human amino acid requirements', J Nutr, 127: 1842–6. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Ohura T, Kobayashi M, Shigematsu Y, Yamaguchi S, Suzuki Y, Hata I, Aoki Y, Yang X, Minjares C, Haruta I, Uto H, Ito Y, and Muller U. 2001. 'Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene', J Biol Chem, 276: 35995–9. [DOI] [PubMed] [Google Scholar]
- Ozand PT, Rashed M, Gascon GG, Youssef NG, Harfi H, Rahbeeni Z, Garawi S. al, and Aqeel A. al. 1994. 'Unusual presentations of propionic acidemia', Brain Dev., 16 Suppl: 46–57. [DOI] [PubMed] [Google Scholar]
- Patel TB, DeBuysere MS, and Olson MS. 1983. 'The effect of propionate on the regulation of the pyruvate dehydrogenase complex in the rat liver', Arch Biochem Biophys, 220: 405–14. [DOI] [PubMed] [Google Scholar]
- Pena L, Burton BK 2012. 'Survey of health status and complications among propionic acidemia patients.', Am.J.Med.Genet.A. : 1641–46. [DOI] [PubMed] [Google Scholar]
- Pena L, Franks J, Chapman KA, Gropman A, Ah Mew N, Chakrapani A, Island E, MacLeod E, Matern D, Smith B, Stagni K, Sutton VR, Ueda K, Urv T, Venditti C, Enns GM, and Summar ML. 2012. 'Natural history of propionic acidemia', Mol Genet Metab, 105: 5–9. [DOI] [PubMed] [Google Scholar]
- Perez-Cerda C, Clavero S, Perez B, Rodriguez-Pombo P, Desviat LR, and Ugarte M. 2003. 'Functional analysis of PCCB mutations causing propionic acidemia based on expression studies in deficient human skin fibroblasts', Biochim Biophys Acta, 1638: 43–9. [DOI] [PubMed] [Google Scholar]
- Peters HL, Pitt JJ, Wood LR, Hamilton NJ, Sarsero JP, and Buck NE. 2012. 'Mouse models for methylmalonic aciduria', PLoS One, 7: e40609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H, Nefedov M, Sarsero J, Pitt J, Fowler KJ, Gazeas S, Kahler SG, and Ioannou PA. 2003. 'A knock-out mouse model for methylmalonic aciduria resulting in neonatal lethality', J Biol Chem, 278: 52909–13. [DOI] [PubMed] [Google Scholar]
- Peuhkurinen KJ 1984. 'Regulation of the tricarboxylic acid cycle pool size in heart muscle', J Mol Cell Cardiol, 16: 487–95. [DOI] [PubMed] [Google Scholar]
- Ravn K, Chloupkova M, Christensen E, Brandt NJ, Simonsen H, Kraus JP, Nielsen IM, Skovby F, and Schwartz M. 2000. 'High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase', Am J Hum Genet, 67: 203–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard E, Brasil S, Briso-Montiano A, Alonso-Barroso E, Gallardo ME, Merinero B, Ugarte M, Desviat LR, and Perez B. 2018. 'Generation and characterization of two human iPSC lines from patients with methylmalonic acidemia cblB type', Stem Cell Res, 29: 143–47. [DOI] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D. 2015. 'Dose-Response Analysis Using R', PLoS One, 10: e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Drose S, Brandt U, Hoffmann GF, Ter Laak H, Kolker S, and Smeitink JA. 2006. 'Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins', Biochem J, 398: 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf DA, McAfee J, Parks JK, and Eguren L. 1980. 'Propionate inhibition of succinate:CoA ligase (GDP) and the citric acid cycle in mitochondria', Pediatr Res, 14: 1127–31. [DOI] [PubMed] [Google Scholar]
- Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, and Hutson SM. 1998. 'A molecular model of human branched-chain amino acid metabolism', Am J Clin Nutr, 68: 72–81. [DOI] [PubMed] [Google Scholar]
- Sutton VR, Chapman KA, Gropman AL, MacLeod E, Stagni K, Summar ML, Ueda K, Ah Mew N, Franks J, Island E, Matern D, Pena L, Smith B, Urv T, Venditti C, and Chakarapani A. 2012. 'Chronic management and health supervision of individuals with propionic acidemia', Mol Genet Metab, 105: 26–33. [DOI] [PubMed] [Google Scholar]
- Sweatt AJ, Wood M, Suryawan A, Wallin R, Willingham MC, and Hutson SM. 2004. 'Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves', Am J Physiol Endocrinol Metab, 286: E64–76. [DOI] [PubMed] [Google Scholar]
- Ugarte M, Perez-Cerda C, Rodriguez-Pombo P, Desviat LR, Perez B, Richard E, Muro S, Campeau E, Ohura T, and Gravel RA. 1999. 'Overview of mutations in the PCCA and PCCB genes causing propionic acidemia', Hum Mutat, 14: 275–82. [DOI] [PubMed] [Google Scholar]
- Varani J, Dame M, Rediske J, Beals TF, and Hillegas W. 1985. 'Substrate-dependent differences in growth and biological properties of fibroblasts and epithelial cells grown in microcarrier culture', J Biol Stand, 13: 67–76. [DOI] [PubMed] [Google Scholar]
- Wajner M, Coelho DM, Barschak AG, Araujo PR, Pires RF, Lulhier FL, and Vargas CR. 2000. 'Reduction of large neutral amino acid concentrations in plasma and CSF of patients with maple syrup urine disease during crises', J Inherit Metab Dis, 23: 505–12. [DOI] [PubMed] [Google Scholar]
- Weidman SW, and Drysdale GR. 1979. 'The biosynthesis of methylcitrate', Biochem J, 177: 169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KA, Han Y, Zhang M, Hess JP, Chapman KA, Cline GW, Tochtrop GP, Brunengraber H, and Zhang GF. 2017. 'Inter-relations between 3-hydroxypropionate and propionate metabolism in rat liver: relevance to disorders of propionyl-CoA metabolism', Am J Physiol Endocrinol Metab, 313: E413–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Wu Z, Dai Z, Yang Y, Wang W, Liu C, Wang B, Wang J, and Yin Y. 2013. 'Dietary requirements of "nutritionally non-essential amino acids" by animals and humans', Amino Acids, 44: 1107–13. [DOI] [PubMed] [Google Scholar]
- Zwickler T, Haege G, Riderer A, Horster F, Hoffmann GF, Burgard P, and Kolker S. 2012. 'Metabolic decompensation in methylmalonic aciduria: which biochemical parameters are discriminative?', J Inherit Metab Dis, 35: 797–806. [DOI] [PubMed] [Google Scholar]
- Zwickler T, Riderer A, Haege G, Hoffmann GF, Kolker S, and Burgard P. 2014. 'Usefulness of biochemical parameters in decision-making on the start of emergency treatment in patients with propionic acidemia', J Inherit Metab Dis, 37: 31–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.