

Abstract
Noncanoncial inflammasome activation by cytosolic lipopolysaccharide (LPS) causes pyroptotic cell death facilitated by gasdermin D (GSDMD) pore formation. In this issue of Immunity, Huang et al. describe how cytosolic LPS in endothelial cells does not cause cell death but restrains endothelial cell proliferation.
Bacterial lipopolysaccharide (LPS) is a potent pathogen-associated molecular pattern (PAMP) that triggers robust inflammation during sepsis. While plasma membrane-localized TLR4 recognizes extracellular LPS, intracellular LPS induces noncanonical inflammasome formation through an interaction between LPS and murine caspase-11 (Shi et al., 2017). This interaction promotes cleavage of gasdermin D (GSDMD), and upon cleavage, the N terminus of GSDMD (GSDMD-NT) forms pores in the plasma membrane, facilitating pyroptosis, a form of inflammatory cell death (Shi et al., 2017). Activities of GSDMD outside of its role in cell death are not well understood, and its relationship with other innate immune sensing pathways remains unclear. In this issue of Immunity, Huang et al. (2020) describe how LPS-stimulated GSDMD cleavage leads to the formation of GSDMD-NT pores on the mitochondria, causing the release mitochondrial DNA (mtDNA) to activate the cyclic GMP AMP synthase (cGAS)-Stimulator of Interferon Genes (STING) pathway to suppress cellular proliferation (Huang et al., 2020) (Figure 1).
Figure 1. LPS in the Cytosol of Endothelial Cells Activates cGAS-STING Suppression of YAP1.
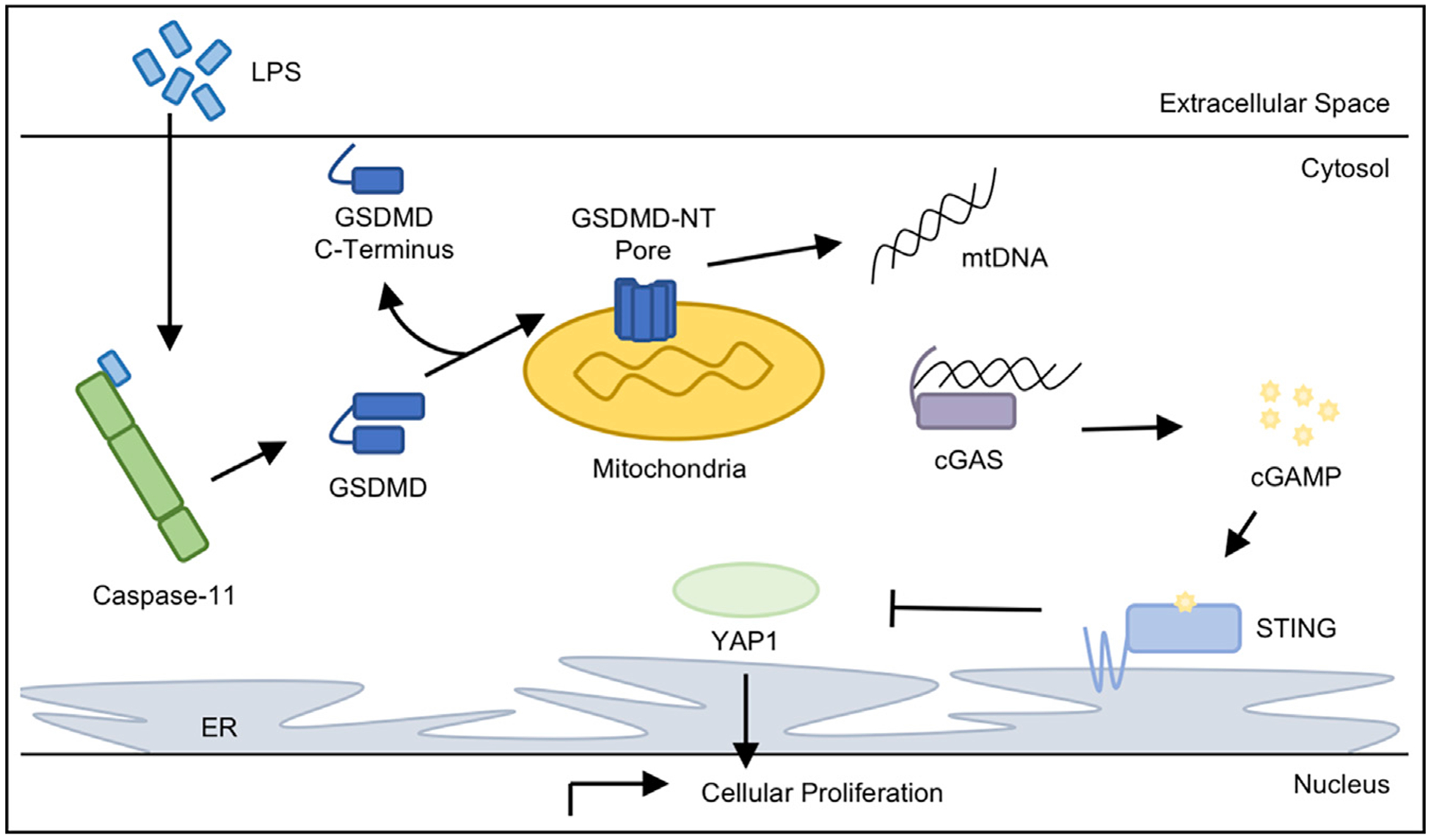
Intracellular LPS stimulates murine caspase-11 to form the noncanonical inflammasome and cleave GSDMD. Canonically, the cleaved GSDMD-NT forms a pore in the plasma membrane to cause cellular ion gradient depolarization and the release of inflammatory cytokines to facilitate pyroptotic cell death. However, Huang and colleagues discovered that LPS-stimulated cleavage of GSDMD in murine endothelial cells leads to the concentration of the GSDMD-NT on the mitochondria, presumably perforating the mitochondrial membrane. GSDMD-NT pore formation on the mitochondria releases mtDNA into the cytosol, where it is detected by the cytosolic DNA sensor cGAS. Upon binding mtDNA, cGAS produces the secondary messenger cGAMP, which is detected by the adaptor protein STING. Here, STING activation through cGAMP binding inhibited YAP1 translocation into the nucleus, preventing transcriptional upregulation of genes promoting cellular proliferation.
To study the role of intracellular LPS in the endothelium, the authors first studied the effects of transfected LPS on cultured endothelial cells. Only a small percentage of endothelial cells died upon LPS transfection, and instead, the authors observed significant decreases in mitochondrial membrane potential (MMP) and increases in intracellular DNA content. Next, the authors found that GSDMD-NT was present in the mitochondrial fraction endothelial cells and that the intracellular DNA was of mitochondrial origin. Notably, both the drop in MMP and the presence of mtDNA in the cytosol were decreased by caspase inhibition and dependent on caspase-11 and GSDMD. While the relative distribution of GSDMD on the mitochondria versus the plasma membrane was not addressed, these data suggest GSDMD-NT may preferentially perforate mitochondria upon LPS stimulation in endothelial cells.
Next, the authors considered the cecal ligation puncture (CLP) model, which induces polymicrobial sepsis, to study intracellular LPS in vivo. Upon CLP, both wild-type (WT) and Casp11fl/fl mice had increased cytosolic double-stranded DNA (dsDNA) content in endothelial cells and sustained injury to the endothelial barrier. However, mice that lacked caspase-11 in the endothelium or totally lacked GSDMD restored endothelial barrier integrity more rapidly, exhibited increased cellular proliferation, and lacked increases in cytosolic DNA. These in vivo data corroborate the observations in vitro and imply that the GSDMD-NT mediates the release of mtDNA, causing sustained injury to the endothelium.
During canonical pyroptosis, the GSDMD-NT fragment localizes to the plasma membrane to form a pore, leading to cellular depolarization and secretion of pyroptotic cytokines, including interleukin (IL)-1β and IL-18 (Shi et al., 2017). GSDMD targets plasma membrane through an interaction with lipids on the inner leaflet of the plasma membrane, phosphatidylinositol-4-phosphate (PI(4) P) and PI(4,5)P2 (Barnett and Kagan, 2020). How the GSDMD-NT preferentially perforates the mitochondria over the plasma membrane in endothelial cells is unclear. However, GSDMD-NT also binds cardiolipin, a lipid present on the inner membrane of the mitochondria. Cardiolipin is exposed on mitochondrial surface following stress (Dudek, 2017), and perhaps such stress within LPS transfected endothelial cells redirects GSDMD to the surface of the mitochondria over the plasma membrane. While others noted the GSDMD-NT can perforate the mitochondria (Platnich et al., 2018), this activity was not observed in the absence of cell death, as Huang and colleagues demonstrated here. Further research will clarify this potential alternative mechanism of action for cleaved GSDMD-NT, and further characterization of GSDMD-NT localization in this model and others will illuminate how this switch of cell-fate decisions occurs.
Following mitochondrial membrane permeabilization via GSDMD, Huang and colleagues observed cytosolic mtDNA. Cytosolic DNA is detected by the intracellular DNA sensor cGAS, stimulating the production of 2′3′-cyclic GMP AMP (cGAMP), a secondary messenger detected by the adaptor protein STING (Li and Chen, 2018). Upon binding cGAMP, STING promotes cytokine production, including anti-viral type I interferons (IFNs) (Li and Chen, 2018). To identify the role of cGAS-STING signaling in their model, Huang and colleagues determined if cGAMP was produced upon LPS exposure in vivo. Indeed, cGAMP was found in the lungs of in mice subjected to LPS stimulation in a caspase-11-dependent manner. Additionally, upregulation of IFN and IFN-stimulated gene production, hallmarks of STING activation, were dependent on caspase-11 and GSDMD. Together, these experiments point toward a model in which intracellular LPS activates caspase-11, leading to GSDMD cleavage into GSDMD-NT, which perforates mitochondrial membrane. Mitochondrial DNA is then released through GSDMD-NT mitochondrial pores, causing cGAS-STING activation in endothelial cells.
As noted earlier, Huang and colleagues found that murine endothelial cells lacking caspase-11 and GSDMD were proliferative upon CLP, while WT cells were not. This suggested LPS sensing by caspase-11, and subsequent GSDMD cleavage suppressed endothelial cell proliferation. Because these activities culminated in mtDNA release and cGAS-STING activation, the investigators next found that transfection of either mtDNA or cGAMP was sufficient to suppress endothelial-cell proliferation. Similarly, mice lacking cGAS demonstrated increased cellular proliferation and recovery from barrier injury in the lung endothelium following CLP as compared to WT animals. In a reciprocal experiment, mice treated with a STING agonist following LPS exposure suppressed cellular proliferation in the endothelium and led to sustained barrier injury. These data suggested a role for the cGAS-STING pathway in suppressing cellular proliferation.
cGAS-STING signaling influences the cell cycle, as this pathway promotes cellular senescence (Li and Chen, 2018). However, outside of this role, little is known about the relationship between cGAS-STING signaling and the cell cycle. Therefore, the observation that LPS exposure led to decreased endothelial cell proliferation in a cGAS-STING-dependent manner warranted further mechanistic study. To address this, the authors investigated the involvement of the YAP and/or TAZ transcriptional regulators, which are components of the Hippo signaling pathway that promotes cellular proliferation and tissue repair in the endothelium (Zanconato et al., 2019). To this end, the investigators found that cGAMP or mtDNA transfection into endothelial cells caused YAP1 phosphorylation, suppressing its nuclear translocation and presumably the transcription of proliferative genes. This included cyclin D, which showed decreased expression by immunoblot upon cGAMP or mtDNA transfection. The authors also investigated potential kinases that might mediate YAP1 phosphorylation, including LATS1, IKKε, and TBK1. Finally, upon CLP, endothelial cells lacking YAP1 had lower rates of proliferation and greater endothelial barrier injury when compared to Yap1fl/fl endothelial cells, suggestive of a role for YAP1 in these processes.
cGAS-STING signaling is implicated in many disease states, including various pathogenic infections and cancers. More recently, the role of this pathway in promoting cellular senescence was observed, linking cGAS-STING signaling to the control of the cell cycle (Li and Chen, 2018). In this study, the authors found that sensing of mtDNA by cGAS restrained cellular proliferation following cytosolic LPS exposure, suggesting that this pathway controls the cell cycle beyond senescence induction. While the authors implicate YAP1 signaling, it is unclear if other pathways contribute to the antiproliferative effect of cGAS-STING activation. Additional research will determine if this occurrence is observed broadly or is limited to specific cell types. Also, these data mandate further study into the association between cGAS-STING activation and YAP and/or TAZ signal transduction. Through this work and others, the relationship between cGAS-mediated DNA sensing and the cell cycle is emerging as a key component of this innate immune sensing pathway.
Altogether, this study by Huang and colleagues describes a phenomenon by which inflammasome activation promotes cGAS-STING activation in the absence of cell death. Other groups have reported that inflammasome activation can suppress cGAS-STING signaling during infection (Banerjee et al., 2018), while others demonstrated that the STING pathway can activate the inflammasome (Gaidt et al., 2017; Swanson et al., 2017), highlighting the complex relationship between these two pathways. Redirecting PAMP-mediated pyroptotic cell death to promote the production of mtDNA, a danger-associated molecular pattern (DAMP), to activate a different signal transduction cascade is a stark example of intracellular crosstalk. The physiological benefit of such crosstalk following septic injury is unclear. While cell death was restrained in this context, the endothelial barrier remained compromised and unable to repair through cellular proliferation. This suggests that the activity of LPS in sepsis goes beyond canonical sensing mechanisms and further drives its pathology through the induction of DAMPs. More research is required to determine why and in what contexts GSDMD acts to DAMPen pyroptosis.
Footnotes
DECLARATION OF INTERESTS
J.P.-Y.T. is the co-founder of IMMvention Therapeutix and a member of the Burroughs Wellcome Fund Board of Directors.
REFERENCES
- Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, et al. (2018). Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49, 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett KC, and Kagan JC (2020). Lipids that directly regulate innate immune signal transduction. Innate Immun. 26, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J (2017). Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol 5, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, and Malik AB (2020). Mitochondrial DNA Activates cGAS Signaling and Suppresses YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 52, this issue, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, Komada T, Trotman-Grant AC, Brandelli JR, Chun J, et al. (2018). Shiga Toxin/Lipopolysaccharide Activates Caspase- 4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 25, 1525–1536. [DOI] [PubMed] [Google Scholar]
- Shi J, Gao W, and Shao F (2017). Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci 42, 245–254. [DOI] [PubMed] [Google Scholar]
- Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, Petrucelli A, Barber GN, Benedict CA, and Ting JP (2017). A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med 214, 3611–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2019). YAP and TAZ: a signalling hub of the tumour microenvironment. Nat. Rev. Cancer 19, 454–464. [DOI] [PubMed] [Google Scholar]