

Abstract
A major event in early embryo development is the awakening of the embryonic genome, a process of large-scale transcriptional induction termed zygotic genome activation (ZGA). To understand how ZGA is controlled temporally and spatially, tools are required to image and quantify nascent transcription in wholemount embryos. In this chapter, we describe a metabolic labeling approach that leverages 5-ethynyl uridine (5-EU) incorporation into newly transcribed RNAs. Subsequently, click chemistry is used to conjugate these nascent transcripts to fluorophores for wholemount confocal imaging or biotin for RNA sequencing. Such an approach facilitates direct visualization of the global transcriptional state of each cell during early embryogenesis and provides a spatial map of gene expression activity. We describe this procedure for imaging nascent transcription in a vertebrate embryo, and use as our model the onset of large-scale ZGA Xenopus laevis. Unlike cell culture systems in which 5-EU can be added to the media, metabolic labeling in Xenopus requires microinjection in 1-cell or 2-cell stage embryos. This method is a powerful tool to quantify the nascent transcriptome at a single-cell level and to dissect mechanisms that control ZGA. We propose that this methodology can be applied broadly in other embryonic systems, and demonstrate the feasibility using zebrafish cleavage stage embryos. Finally, we demonstrate how to sequence the nascent transcriptome via 5-EU incorporation and separation of zygotic versus maternal RNAs. Altogether, our generalizable methodology will facilitate new insights into gene regulation and spatial patterning of ZGA during early embryogenesis.
Keywords: nascent transcription, 5-ethynyl uridine (5-EU), zygotic genome activation; wholemount imaging; early embryogenesis; single-cell transcriptome
Introduction
Following fertilization, a vertebrate embryo undergoes autonomous cell divisions regulated by maternal factors loaded in the egg and in many systems the zygotic genome in dormant. After a define period the embryo initiates large-scale transcriptional activation as part of the maternal-to-zygotic transition (MZT) (Schier, 2007; Vastenhouw, Cao, & Lipshitz, 2019). The induction of the embryonic genome is termed zygotic genome activation (ZGA) which is essential for metazoan embryo development (Jukam, Shariati, & Skotheim, 2017). The timing for ZGA is tightly controlled within a species; however, it varies dramatically among species. The period following fertilization until ZGA onset ranges from several hours in zebrafish and Xenopus embryos to days for human embryos (M. T. Lee, Bonneau, & Giraldez, 2014). Despite extensive research on ZGA (Schulz & Harrison, 2019), how an embryo coordinates the spatial and temporal onset of ZGA in blastomeres remains a fundamental question in developmental biology.
A challenge in characterizing ZGA is quantitative measurement of nascent zygotic gene expression in wholemount embryos. Various methods have been successfully used to detect expression of zygotic genes during early development of multiple model organisms. Generally, these methods can be divided into two major categories – sequencing-based and imaging-based approaches. One major method is based on detecting zygotic gene expression via gene profiling. Traditionally, this has been carried out by sequencing RNA isolated from entire embryos at different stages of development for many species, including human (Gao et al., 2018; Liu et al., 2019), mouse (Lu et al., 2016), zebrafish (Aanes, Collas, & Alestrom, 2014; White et al., 2017), Xenopus (Gentsch, Owens, & Smith, 2019). An advantage of sequencing-based methods is that they are sensitive and can detect many, if not all, zygotic genes at the same time, which is helpful for characterizing gene expression patterns different stages of development. However, one significant shortcoming is that the expression level of each gene is derived from a population average of many cells and thus lacks information on cell-to-cell variation. Furthermore, these methods sacrifice a spatial view of ZGA across embryonic blastomeres. Separately, metabolic labeling of nascent transcripts can be performed using uridine analogs such as 4-thiouridine (4-SU) and 5-ethynyl uridine (5-EU), as has been done in Drosophila (Kwasnieski, Orr-Weaver, & Bartel, 2019), zebrafish (Chan et al., 2019; Heyn et al., 2014) and Xenopus (Gentsch et al., 2019). Moreover, single cell RNA-seq (scRNA-seq) has been performed on early embryos from human and mouse (Xue et al., 2013), and for embryos at relatively late stages of development from zebrafish (Farrell et al., 2018; Wagner et al., 2018) and Xenopus (Briggs et al., 2018). Although spatial relationships between individual cells can be inferred by computational reconstruction on known gene expression patterns, a spatial map cannot be built with high confidence for an embryo containing thousands of cells (Satija, Farrell, Gennert, Schier, & Regev, 2015). Additionally, the large sizes of blastomeres in early stages of embryos are poorly suited for scRNA-seq technologies.
The second major category of methods for detecting zygotic gene expression is based on imaging the expression of single gene or a handful of genes. Expression in fixed embryonic cells can be detected by single-molecule fluorescent in situ hybridization (smFISH) using gene-specific fluorescent probes and imaging. Such strategies have been successfully used in early embryos from Drosophila (Little & Gregor, 2018; Trcek, Lionnet, Shroff, & Lehmann, 2017), zebrafish (Stapel, Zechner, & Vastenhouw, 2017) and mouse (Ranisavljevic, Okamoto, Heard, & Ancelin, 2017; Xie, Timme, & Wood, 2018). However, such approaches are difficult to scale up to large, wholemount embryo. Separately, dynamics of gene transcription can be imaged live using RNA containing MS2 sequences that bind to MS2 coat protein-green fluorescent protein (GFP). Such strategies work well for single genes across narrow depths of cytoplasmic volume, such as in the syncytium of Drosophila embryos (Garcia, Tikhonov, Lin, & Gregor, 2013) and cortical cells in zebrafish (Campbell, Chao, Singer, & Marlow, 2015). In addition, live imaging using a catalytically dead Cas9 (dCas9) protein fused to GFP that recognizes target RNA through base pairing with a guide RNA (gRNA) has been used to detect transcription of zygotic genes expressed at a high level in early zebrafish embryos (Chan et al., 2019). In general, a limitation of these single-gene based methods is that they do not provide a global readout of transcriptional activity and live imaging can only be performed with a subset of cells and not whole embryos, particularly large vertebrate embryo models.
We developed a method to visualize and quantify large-scale zygotic transcription at the single-cell level across entire large vertebrate embryos using Xenopus laevis a model (Chen, Einstein, Little, & Good, 2019). This approach of metabolically labeling nascent zygotic transcripts using 5-ethynyl uridine (5-EU) leverages click chemistry to conjugate a fluorophore to EU-RNA for confocal imaging of wholemount embryos. The in situ imaging of transcripts in embryos maintains precise spatial information at cellular and tissue level and allows detecting the integrated transcriptional activity of all zygotic genes in each single cell. Such a wholemount strategy reveals spatial and temporal patterns of ZGA regulation that are often missed by other methods. We further verified this methodology for detecting nascent transcription to image large-scale ZGA in zebrafish embryos, suggesting the broad applicability of this method to other embryonic models. Importantly, our approach is also compatible with RNA-seq for characterization of the nascent transcriptome. Altogether, this methodology overcomes a number of limitations of previous techniques and offers a new tool to interrogate patterns of nascent transcription in embryos such as during embryonic genome activation.
Protocol
1. Labeling Nascent Transcripts in Early Embryogenesis Using 5-Ethynyl Uridine (5-EU)
Various studies across multiple model organisms have shown that uridine analogues, e.g., 5-bromouridine (BrU) (Aoki, Worrad, & Schultz, 1997; Kageyama, Nagata, & Aoki, 2004) and 4-thioluridine (4-SU) (Heyn et al., 2014; Heyn & Neugebauer, 2017), become phosphorylated upon delivery to embryonic blastomeres and subsequently incorporate into newly transcribed RNAs Thus, these metabolic labeling approaches have been used to measure nascent transcripts during early development. However, the requirement of an antibody to detect BrU and the presence of endogenous thiols in embryos limit the utility of 5-BrU and 4-SU for imaging embryonic genome activation at the single-cell level in large wholemount embryos.
Recently, another uridine analog 5-ethynyl uridine (5-EU), was shown to overcome the limitations noted above for labeling newly transcribed RNA in cultured cells (Jao & Salic, 2008). 5-EU is readily taken up from the medium by living cells and after phosphorylation becomes 5-ethynyl uridine triphosphate (5-EUTP), which is specifically incorporated into newly made RNAs but not DNA. The alkyne group in 5-EU is not naturally occurring and thus it can be very selectively and efficiently reacted with azides by a copper (I)-catalyzed alkyne-azide cycloaddition reaction, termed a click reaction (Jao & Salic, 2008). Additionally, this specific reaction ensures that background reactivity is very low. Nascent EU-RNA can be conjugated to fluorescent azides for imaging transcriptional activity at the onset of ZGA in individual cells of wholemount embryos via confocal imaging (Chan et al., 2019; Chen et al., 2019; Jao & Salic, 2008). Alternatively, the nascent transcriptome can be sequenced via biotinylation of EU-RNA and streptavidin bead purification (Chan et al., 2019; Chen et al., 2019).
We have developed a methodology to detect nascent zygotic transcripts in single cells of wholemount Xenopus laevis cleavage stage embryos (Figure 1A) (Chen et al., 2019). The steps are broken down as follows, corresponding to the subsections of this protocol: (1A) microinjection of 5-EU into 1-cell embryos and (1B) fixing blastula embryos at developmental times corresponding to the onset of large-scale, (2) performing a click chemistry reaction to conjugate a fluorophore to nascent EU-RNA, (3) additional DNA dye and antibody staining, and (4) confocal imaging of cleared wholemount embryos. The use of metabolic labeling with 5-EU allows for selective detection of nascent transcripts within a larger pool of maternal RNAs, which are highly abundant in the early embryo (Figure 1B). Although embryos are fixed to allow for conjugating fluorophores with click chemistry, the rapid divisions of embryonic blastomeres enable the transcriptional output of a cell to be approximated by EU-RNA signal in the nucleus. Upon synthesis, this nascent EU-RNA enriches in the interphase nucleus (Figure 1C) due to insufficient time; the interphase duration is approximately 20 minutes during cleavage stage. At mitosis and nuclear envelop breakdown this RNA is dispersed throughout cell (Figure 1C). This feature enables quantitation the transcriptional activity in the current cell cycle by integrating total EU-RNA signal in the nucleus. Separately, transcriptional history can be calculated by measuring the total accumulated EU-RNA in the cell. In the case of Xenopus laevis, wholemount embryos fixed at 5 hours post-fertilization (hpf) (around embryonic cleavage 10 or C10), 7 hpf (around C13), 9 hpf (around C16) display a spatially graded pattern of single-cell transcriptional activation (Figure 1D) (Chen et al., 2019).
Figure. 1. Method overview for EU-RNA imaging.
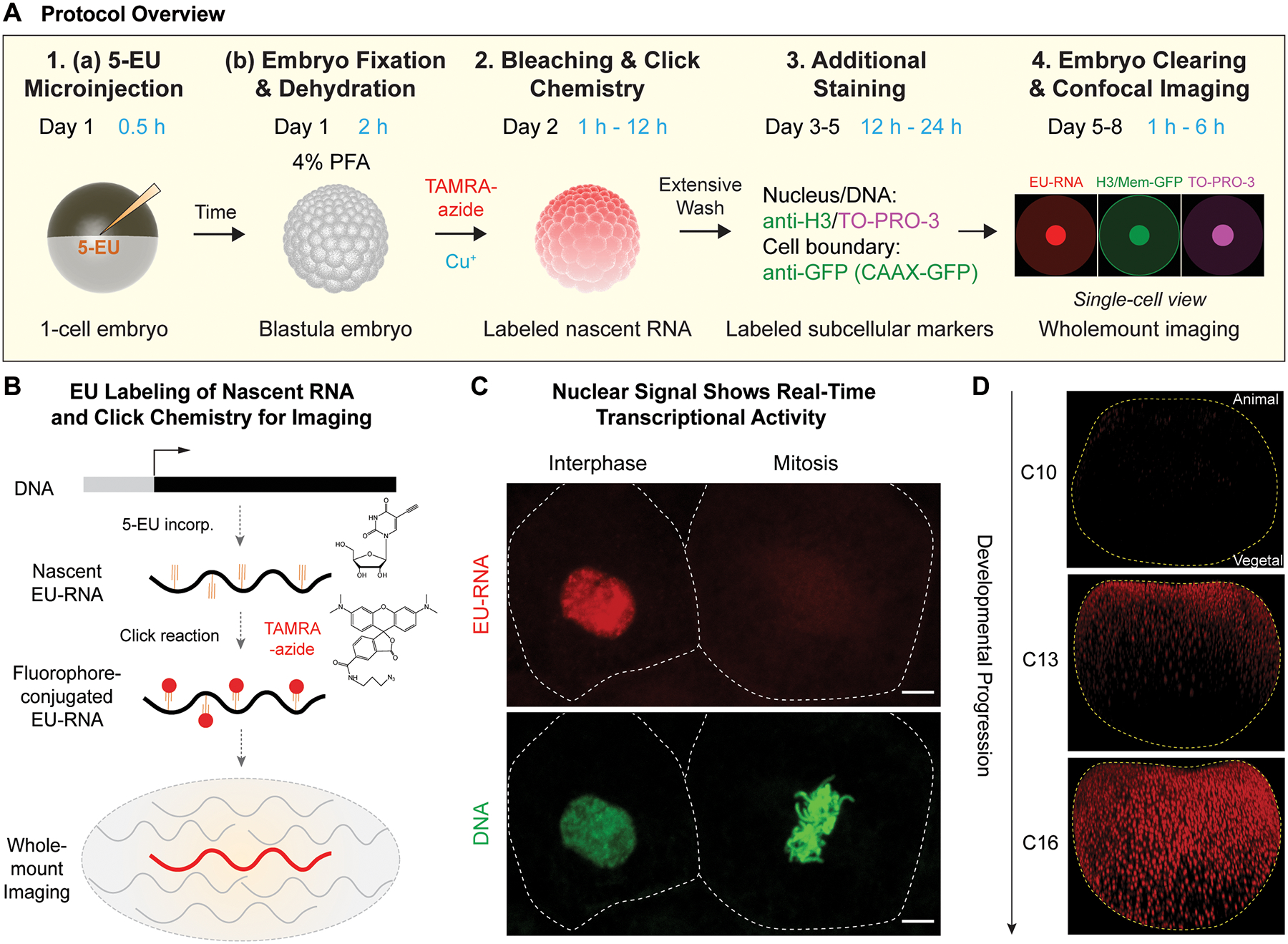
(A) Schematic summary for labeling and visualizing nascent RNA transcripts in wholemount embryos during early development. Numbering correspond to methods sections in main text. 1A: microinjection of 1-cell embryos with 5-ethynyl uridine (5-EU); 1B: fixation of blastula embryos in 4% paraformaldehyde and dehydration in methanol for long-term storage; 2: following bleaching, nascent RNAs in an embryo conjugated to a fluorescent azide using a click reaction; 3: Staining DNA is required for image segmentation, which can be further augmented with additional immunostaining; 4: Clearing embryos for confocal imaging. Example nascent EU-RNA (red), nucleus/cell boundary (green) and DNA (magenta). (B) Schematic of EU-RNA labeling of early embryogenesis: injected 5-EU converts to 5-EUTP and incorporates into nascent but not maternal transcripts; TAMRA-azide is conjugated to EU-RNA and click chemistry. Nascent zygotic transcripts can be visualized; maternal RNAs are unlabelled. (C) Transcriptional output is measured by accumulated EU-RNA in nucleus. Example of neighboring interphase cell and a mitotic cells, in an embryo at 8–9 hours post-fertilization (hpf). Scale bar, 5 μm. Dashed white lines indicate cell boundary. (D) Nascent EU-RNA accumulation as a function of embryo development for wholemount Xenopus laevis embryos. Embryonic cleavage 10 (C10) at ~ 5 hrs p.f., prior to large-scale ZGA, embryonic cleavage C13 at ~ 7–7.5 hrs p.f. with partial onset of large-scale ZGA, and embryonic cleavage C16 at 9 hrs p.f. showing complete spatial onset of ZGA across embryo. Orientation: animal pole up; vegetal pole down.
For EU-RNA imaging in any embryonic system, controls must be performed to demonstrate the sensitivity and specificity of the nascent gene expression. Although 5-EU can be taken up by cultured cells in media, it is not readily absorbed by Xenopus laevis embryos and therefore microinjection is required to deliver 5-EU. The final concentration of 5-EU inside the embryo is extremely critical as low concentrations of 5-EU may reduce the sensitivity of fluorescent labeling for imaging (Figure 2A and 2B) and high concentrations of 5-EU are toxic to normal developmental progression. We recommend always performing a control with co-microinjection of 5-EU with α-amanitin, an inhibitor of RNA polymerase II, to ensure that nuclear EU-RNA signal is selective, representing bona-fide nascent transcripts (Figure 2C and 2D). The EU-labeling method is sensitive enough to image burst of transcription – seen as spots - at early stages of ZGA onset (Figure 3). For analysis of large-scale genome activation, we often set a threshold minimum amount of transcript accumulated in the blastomere nucleus that corresponds to more widespread zygotic transcription (e.g., see nuclei at C12 in Figure 3).
Figure 2. Sensitivity and specificity of nascent EU-RNA imaging in early embryogenesis.
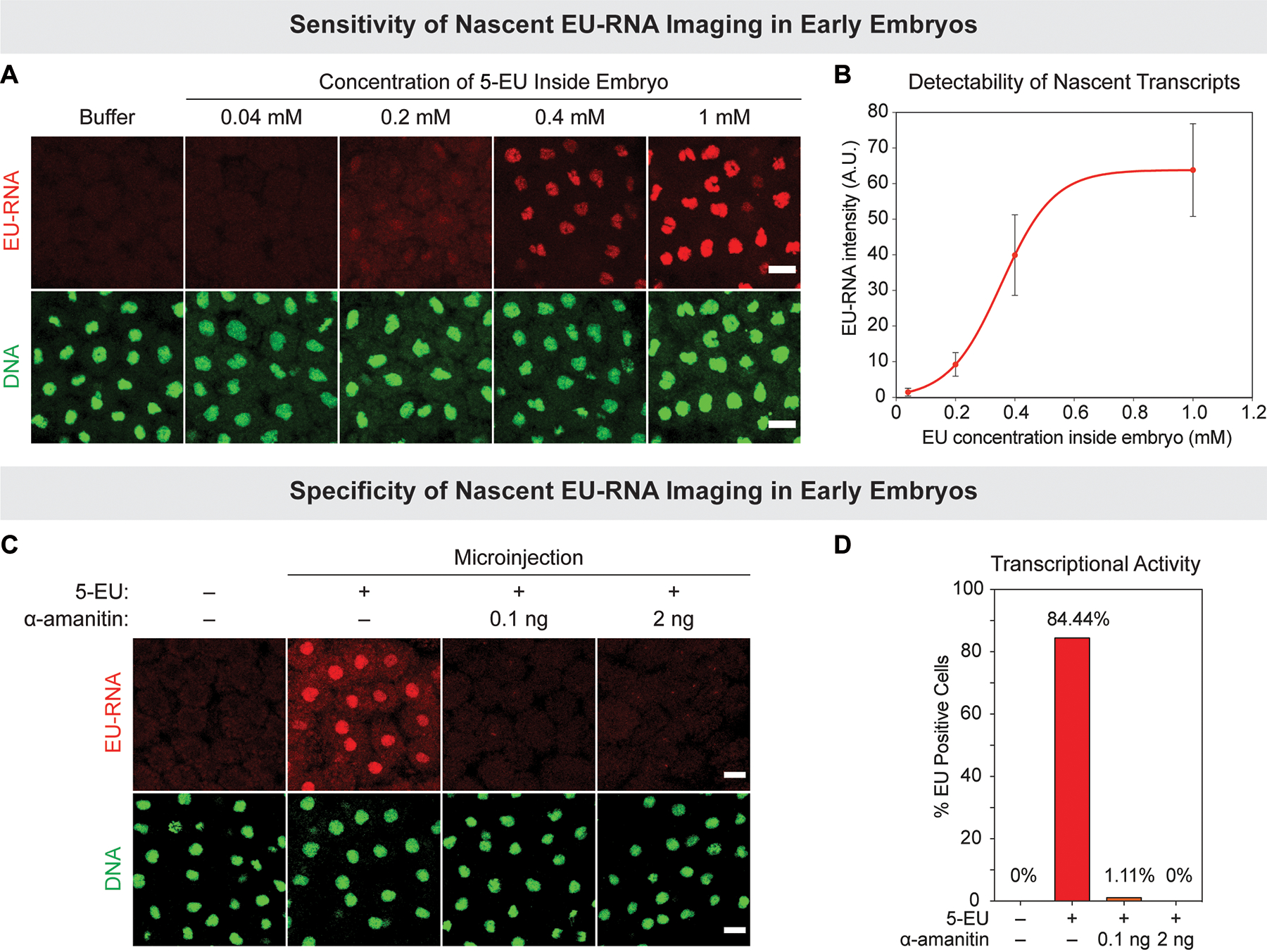
(A and B) Sensitivity: dose-dependence of EU-RNA signal intensity of 5-EU concentration inside embryo. (A) Xenopus embryos were microinjected with buffer or indicated final concentrations of 5-EU, fixed at 10 hpf and processed to image nascent EU-RNA (red) and DNA staining (green). (B) Quantification of nuclear EU-RNA intensity (A.U., arbitrary units) after cytoplasmic background subtraction. Mean ± standard deviation, with a nonlinear fit for visualization purposes. N > 20 cells for each group. (C and D) Evaluating specificity of EU-RNA signal via α-amanitin co-injection. (C) Xenopus embryos microinjected with 5-EU alone or co-microinjected with 5-EU and 0.1 ng (to inhibit RNAPII) or 2 ng of α-amanitin (to inhibit RNAPII and RNAPIII). Embryos fixed at 8 hrs and 45 min pf. (D) Quantification of percentage of EU-RNA positive cells whose nuclear EU-RNA intensity exceeds am empirical threshold. N > 60 cells for each group. Scale bars, 20 μm.
Figure 3. Temporal onset of ZGA and levels of nascent transcription.
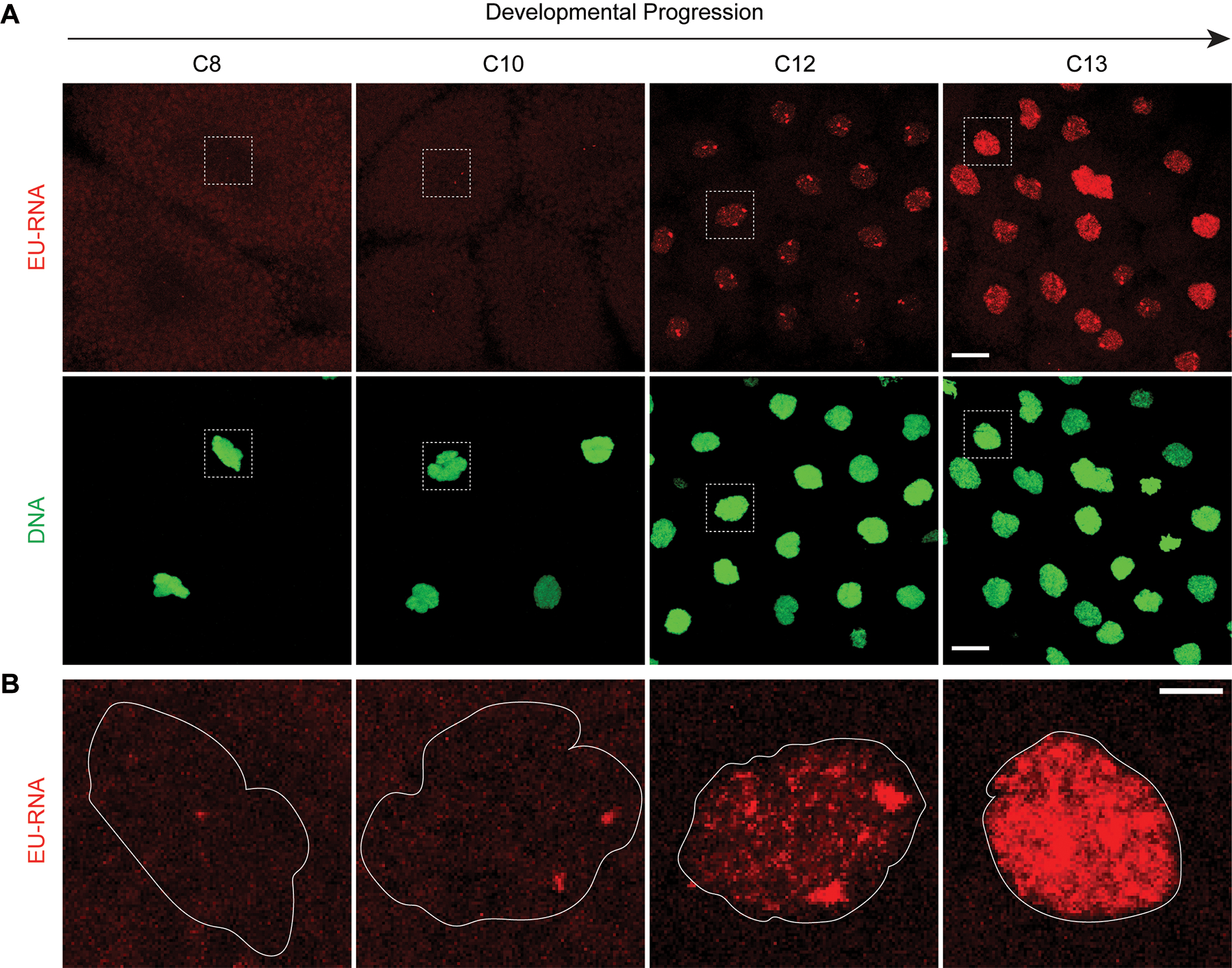
(A) Xenopus embryos microinjected with 5-EU and fixed at timepoints before and surrounding ZGA. EU-RNA (red) and DNA (green). Maximum intensity projections of confocal stacks at 63× magnification visualizing the embryo animal pole. Scale bar 20 μm. (B) Zoomed, enlarged view of EU-RNA in nuclei in the dashed white boxes from panels in A. Distinct levels of nascent transcription are apparent from weak diffuse signal early, to transcriptional foci, to nascent transcripts filling nucleus. Outline of nucleus in white. Scale bar 5 μm.
In this protocol, we followed previous methods for fixing and bleaching Xenopus embryos (C. Lee, Kieserman, Gray, Park, & Wallingford, 2008; Wallingford, 2010). Fixation is required to preserve the integrity of RNAs in embryos. The choice of fixative and buffer is critical because they can impact the variability of the results. We found 4% PFA in 1× MEM, a common fixative solution used in in situ hybridization (IHS), works most efficiently for downstream click reaction, DNA staining and immunostaining for Xenopus laevis blastula embryos. Bleaching is critical for eliminating the light-absorbing pigments present in the animal pole. Lastly, clearing embryos in BABB, a mixture of benzyl alcohol (BA) and benzyl benzoate (BB), drastically improves image quality for many types of embryos.
1.1. Materials
Fertilized Xenopus laevis eggs. We obtained Xenopus embryos by in vitro fertilization (IVF). All animal experiments were performed according to the Animal Use Protocol approved by the University of Pennsylvania Animal Care and Use Committee.
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (Sigma-Aldrich, cat. no. H3375)
Ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific, cat. no. BP120–500)
Ethylenebis(oxyethylenenitrilo)tetraacetic acid (EGTA) (Thermo Fisher Scientific, cat. no. AC409915000)
Potassium chloride (KCl) (Thermo Fisher Scientific, cat. no. P217500)
Magnesium chloride (MgCl2) (Thermo Fisher Scientific, cat. no. M33–500)
Magnesium sulfate (MgSO4) (Thermo Fisher Scientific, cat. no. M65–500)
Calcium chloride (CaCl2) (Sigma-Aldrich, cat. no. C5080)
MOPS (Thermo Fisher Scientific, cat. no. AC172631000)
Sodium hydroxide (NaOH) (Thermo Fisher Scientific, cat. no. S318-1)
Hydrochloric acid (HCl) (Thermo Fisher Scientific, cat. no. A144SI212)
Bovine serum albumin (BSA) (Sigma-Aldrich, cat. no. A3059)
Triton X-100 (Sigma-Aldrich, cat. no. 10789704001)
L-cysteine (Sigma-Aldrich, cat. no. C7352)
Ficoll® 400 (Sigma-Aldrich, cat. no. F4375)
RNase-free water (Thermo Fisher Scientific, cat. no. BP561-1)
Methanol (Thermo Fisher Scientific, cat. no. A4524)
Methanol, anhydrous (Sigma-Aldrich, cat. no. 322415)
Dimethyl sulfoxide (DMSO), anhydrous (Thermo Fisher Scientific, cat. no. D12345)
Hydrogen peroxide (Sigma-Aldrich, cat. no. H1009)
Formamide (Thermo Fisher Scientific, cat. no. AC181090010)
20× SSC (Mediatech, cat. no. MT46-020-CM)
5-ethynyl uridine (5-EU) (Thermo Fisher Scientific, cat. no. E10345)
16% Paraformaldehyde (PFA) (EMS, cat. no. 15710-S)
1.2. Equipment
Dissection stereomicroscope (Leica, model no. MZ6)
PLI-100 picoliter microinjector (Medical Systems Corp., Greenvale, NY)
Glass needle (Sutter Instrument, cat. no. BF100-58-10)
Micropipette puller (Sutter Instrument, model no. P-87)
Glass Petri dish, 6 cm (Corning, cat. no. 3160-60)
Plastic transfer pipette (Thermo Fisher Scientific, cat. no. 13-711-9AM)
Glass chamber for microinjection
Glass vials with caps, 2 ml (Thermo Fisher Scientific, cat. no. C4010-17)
Tube rotator (Thermo Fisher Scientific, cat. no. 88881001)
1.3. Protocol
Before starting the protocol, prepare the following solutions required for this section.
0.1× Marc’s Modified Ringer’s (MMR): To prepare 0.1× MMR, dilute 1 part of 20× MMR (100 mM HEPES pH 7.8, 2 mM EDTA, 2 M NaCl, 40 mM KCl, 20 mM MgCl2, and 40 mM CaCl2) in 199 parts of Milli-Q® water. Store at room temperature up to 1 month.
Dejelly solution: To prepare dejelly solution, mix 1 g of L-cysteine, 200 μl of 10 N NaOH and bring up to 30 ml with Milli-Q® water. Prepare fresh solution before experiment.
3% Ficoll (wt/vol) in 0.5× MMR: To prepare 3% Ficoll/0.5× MMR, mix 10 ml of 15% Ficoll (15% Ficoll® is prepared by dissolving 37.5 g of Ficoll in 250 ml Milli-Q water followed by autoclave) and 1.25 ml of 20× MMR and bring up the final volume to 50 ml with Milli-Q water. Store at 4 ℃ for up to 1 month.
1× TBS, pH 7.6: To prepare 10× TBS, dissolve 2.42 g of Tris base and 8.77 g of NaCl in 50 ml of RNase-free water. Adjust pH to 7.6 with HCl, filter sterilize and store at 4 ℃. To prepare 1× TBS (pH 7.6), add 1 part of 10× TBS (pH 7.6) and 9 parts of RNase-free water. Store at 4 ℃ for up to 6 months.
1× TBST, pH 7.6: To prepare 1× TBST, add 50 μl Triton X-100 to 50 ml 1× TBS (pH 7.6) prepared by RNase-free water to generate a 0.1% (vol/vol) detergent solution. Store at 4 ℃ for up to 2 weeks.
4% PFA/1× MEM: To prepare 4% PFA/1× MEM, mix 1 vial (10 ml) of 16% PFA stock and 4 ml of 10× MEM (1000 mM MOPS pH 7.4, 20 mM EGTA, and 10 mM MgSO4) and add RNase-free water to 40 ml. Prepare fresh fixative before experiment.
Bleaching solution: Bleaching solution is 0.5× SSC containing 5% (vol/vol) formamide and 2% (vol/vol) H2O2. To prepare 10 ml of bleaching solution, mix 250 μl of 20× SSC, 500 μl of formamide, 670 μl of H2O2 and 8.58 ml of RNase-free water. Prepare fresh solution right before use.
5-ethynyl uridine (5-EU) (100 mM stock): To prepare 100 mM 5-EU stock, dissolve 5 mg of 5-EU in 186.4 μl of 1× TBS (pH7.6) prepared by RNase-free water. Mix thoroughly and make aliquots of 2 μl. Store at −80 ℃ for up to 12 months.
1.3.1. Microinjection of 5-EU in Embryos
-
1
Preparation of embryos (see Note 1): Follow a standard protocol for in vitro fertilization (IVF) of eggs in a glass dish and subsequent development in 0.1× MMR (Sive, Grainger, & Harland, 2007). At ~ 30 minutes post-fertilization (mpf), dejelly the embryos. Pour off 0.1× MMR and add the freshly prepared dejelly solution to cover the embryos. Rinse briefly and discard the dejelly solution. Add the dejelly solution again and incubate for 3–5 min at room temperature; gently swirling several times. Stop the incubation once the jelly coats are obviously detached from embryos by pouring off the dejelly solution and replacing with 0.1× MMR. Wash the embryos with 0.1× MMR five times. Keep the dejellied embryos in 0.1× MMR for further development.
-
2
Transfer ~ 50 embryos (see Note 2) using plastic transfer pipettes to a glass chamber containing 3% Ficoll/0.5× MMR, in preparation for microinjection. Place the embryos in the middle of the chamber.
-
3
EU microinjection calculation: Thaw an aliquot of 100 mM 5-EU stock on ice. Dilute the 100 mM 5-EU stock in 1× TBS (pH 7.6) or any other buffer suitable for microinjection, based on three considerations: i) embryo volume, ii) desired EU concentration, iii) stage. For example, the typical desirable maximum volume for microinjection in embryos of Xenopus laevis and zebrafish is 10 nl and 2 nl, respectively. Further, the average embryo volume for Xenopus laevis and zebrafish is ~ 1 μl and ~ 0.3 μl, respectively. The final concentration of 5-EU inside an embryo significantly affects the sensitivity of labeling nascent RNA (Figure 2A and 2B). While increasing 5-EU concentration increases the detectability of EU-RNA in cells, microinjection of high concentrations of 5-EU is toxic to embryos and thus should be avoided. In our experience, the balance the detectability and toxicity the optimal range of 5-EU concentration inside embryo is 0.4–0.6 mM. A typical experiment for Xenopus is to microinject 5-EU to achieve a final concentration of 0.5 mM inside the embryo (see Note 3).
-
4
Fill needle and microinject embryos: Fill glass needle ~ 1 μl of appropriate 5-EU dilution, and microinject 10 nl into Xenopus laevis 1-cell embryos using a picoliter microinjector. For more details follow a standard protocol for performing microinjections (Sive, Grainger, & Harland, 2010).
-
5
Incubate embryos: After microinjection, carefully transfer the embryos to a new glass dish containing 3% Ficoll/0.5× MMR to stabilize injected embryos. Incubate for 1–2 h, then carefully transfer the embryos to a new glass dish with 0.1× MMR to continue embryonic development. Note that elongated incubation with Ficoll may interfere with normal developmental timing.
1.3.2. Embryo Fixation
-
6
Collect and fix 5-EU-microinjected embryos using a plastic transfer pipette to select ~ 10–20 embryos at appropriate stages. For Xenopus laevis we collect from 5–9 hpf at room temperature. To prevent excess buffer transfer, let embryos settle down to the bottom of tip via gravity (see Note 4). Gently transfer the embryos dropwise into 2-ml glass vials filled with 4% PFA/1× MEM solution in a chemical hood. Cap vials tightly.
-
7
To complete fixation, rotate the glass vials at a low speed of 10 rotations per minute (rpm) for a minimum of 2 h at room temperature or for overnight at 4 °C.
-
8
Dehydrate embryos: After fixation, carefully remove the fixative in a chemical hood without disturbing the embryos. Fill the glass vials with 100% methanol and close the caps tightly. Rotate the glass vials for 5 min on a rotator and then replace with fresh methanol. Repeat for a total of at least 3 times to completely dehydrate the embryos.
-
9
Store the embryos at −20 °C for up to six months.
1.3.3. Bleaching Embryos
-
10
Rehydrate embryos by replacing methanol in the glass vials sequentially with solutions of 75% methanol, 50% methanol, 25% methanol and 0% methanol in 0.5× SSC. For each new solution, rotate the glass vials for 5 min at low speed. Wash embryos with 0.5× SSC at least two more times. Note that embryos must be completely rehydrated before proceeding.
-
11
Bleach embryos: Carefully remove 0.5× SSC in the glass vials from the previous step. Add 2 ml of the bleaching solution, i.e., 5% (vol/vol) formamide and 2% (vol/vol) H2O2 in 0.5× SSC, and close the caps of glass vials tightly. Lay down the glass vials in a box the surface of which is coated with aluminum foil. Place the box under bright light source from a bulb. Adjust the distance between the bulb and glass vials to ensure embryos receive sufficient light without overheating. Bleach embryos for 5–6 h (see Note 5).
-
12
SSC Wash: Carefully remove the bleaching buffer from the glass vials by using pipettes (see Note 6) and make sure to avoid touching the embryos. Fill up the glass vials with 0.5× SSC. Rotate for 5 min at low speed. Repeat 0.5× SSC wash and rotation.
-
13
TBST Wash: Carefully remove 0.5× SSC and fill up glass vials with 1× TBST (pH7.6). Rotate for 30 min at low speed. Carefully remove 1× TBST (pH7.6) and repeat washing embryos by rotation with 1× TBST (pH7.6) for a minimum total of 4 h; replacing 1× TBST (pH7.6) every 30 min. Note that the embryos must be thoroughly washed because residue from the bleaching solution can interfere with the efficiency of the click chemistry reaction.
-
14
TBS Wash: Carefully remove 1× TBST (pH7.6) and fill up the glass vials with 1× TBS (pH7.6). Rotate the glass vials for 5 min at low speed. Carefully remove 1× TBS (pH7.6) and repeat wash. Finally, completely remove any residual 1× TBS (pH7.6) first by pouring and then using a P10 pipette. Do so carefully to avoid causing damage to embryos. The embryos are ready for use in next steps but they can also be dehydrated in methanol and stored for later processing (see Note 7).
2. Click Chemistry: Conjugation of Fluorophores to Nascent Transcripts, EU-RNAs,
In the following steps, the nascent EU-RNA transcripts are conjugated with fluorescent azide, such as tetramethylrhodamine (TAMRA)-azide, by a copper (I)-catalyzed alkyne-azide cycloaddition reaction, also known as a click reaction (Jao & Salic, 2008). Before starting this reaction, it is important that any residual bleaching solution should be completely removed by extensive washing. Although click reactions are usually fast (e.g., 30 minutes) for fixed culture cells, the large size of Xenopus embryos requires extensive reaction time (e.g., within 12 hours) to completely label RNAs. Short incubation time of 1 hour lead to labeling only a cortical layer of cells. Alternatively, extended incubation of over 12 hours can lead to higher background due to nonspecific labeling. Thus, the optimal timing click reaction should be optimized for your specific embryo and will likely scale with embryo size. Note that wash times after click reaction should also be extended in large embryos.
2.1. Materials
Tetramethylrhodamine (TAMRA)-azide (Abcam, cat. no. ab146486)
Copper sulfate (CuSO4) (Sigma-Aldrich, cat. no. 61230)
Ascorbic acid (Sigma-Aldrich, cat. no. A7506)
2.2. Equipment
Variable speed nutator (Thermo Fisher Scientific, cat. no. S06622)
2.3. Protocol
Before starting the protocol, prepare the following solutions required in this section. Note that all following solutions and reagents should be ideally prepared using RNase-free conditions.
Tris-HCl, pH8.5 (1 M stock): To prepare Tris-HCl, pH8.5 (1 M stock), dissolve 6 g of Tris base in 50 ml of RNase-free water. Adjust pH to 8.5 with HCl, filter sterilize and store at 4 ℃ for up to 6 months.
TAMRA-azide (10 mM stock): To prepare 10 mM TAMRA-azide stock, dissolve 5 mg of TAMRA-azide in 975 μl anhydrous DMSO. Mix thoroughly and make aliquots of 10 μl. Store at −80 ℃ for up to 12 months.
CuSO4 (1 M stock): To prepare 1 ml of CuSO4 (1 M stock), dissolve 160 mg of CuSO4 in 1 ml RNase-free water. Mix thoroughly. Store at 4 ℃ for up to 12 months.
Ascorbic acid (1 M stock): To prepare 1 ml of ascorbic acid (1 M stock), dissolve 176 mg of ascorbic acid in 1 ml RNase-free water. Mix thoroughly. Prepare fresh solution right before use.
2.3.1. Conjugating EU-RNA with Fluorophore by Click Reaction
Prepare click reaction mix: Sequentially add the stock solutions of the following reagents to a 1.5 ml microcentrifuge tube at final concentrations of 100 mM Tris-HCl (pH8.5), 1 mM CuSO4, 25 μM TAMRA-azide, and 100 mM ascorbic acid. It is important that this reaction mix be made fresh each time; always add ascorbic acid in last step (see Note 8). For example, to prepare a reaction mix of 2 ml, sequentially add 1593 μl of RNase-free water, 200 μl of 1 M Tris-HCl (pH8.5), 2 μl of 1 M CuSO4, 5 μl of TAMRA-azide, and 200 μl of 1 M ascorbic acid. Gently mix well by pipetting up and down several times.
Click reaction: Add 200–500 μl click reaction mix to each glass vial containing embryos from the last step of previous section. Wrap each glass vial with aluminum foil to prevent light exposure. Incubate embryos for 12 h at room temperature on a nutator in the dark (see Note 9).
Stop reaction and briefly wash embryos. Carefully remove the click reaction mix and fill up the glass vials with 1× TBST (pH7.6). Wrap vials with aluminum foil and rotate embryos for 5 min at low speed in the dark. Repeat washing embryos with 1× TBST (pH7.6) once more.
Extensively wash embryos: Wash embryos with 1× TBST (pH7.6) for entire day at room temperature by rotating the glass vials on a rotator at low speed in the dark. Change 1× TBST (pH7.6) for every 1–2 h. A minimum of six washes is required (see Note 10). Extent of washing can be gauged by decreasing coloration of embryo. For larger embryos, additionally wash with 1× TBST (pH7.6) overnight at 4 °C at low speed in the dark, and then wash again for an entire day, changing 1× TBST (pH7.6) for every 1–2 h.
3. Staining: Nuclear Staining using DNA Dyes and Optional Immunostaining
In this section, we describe staining of DNA and general steps for immunocytochemistry following click reaction for labeling EU-RNA in embryos. We adapt the methods from a previous described protocol (C. Lee et al., 2008). Staining of nuclear DNA with TO-PRO-3 facilitates nucleus segmentation in later steps. Note that complete washing from click reaction step is required before immunostaining and/or DNA staining with dyes; inadequate washing often leads poor staining.
3.1. Materials
Goat serum (Abcam, cat. no. ab7481)
Anti-Histone H3 antibody (Abcam, cat. no. ab1791)
Goat anti-rabbit Alex Fluor® 488 secondary antibody (Thermo Fisher Scientific, cat. no. A27034)
TO-PRO-3 (Thermo Fisher Scientific, cat. no. T3605)
3.2. Equipment
Variable speed nutator (Thermo Fisher Scientific, cat. no. S06622)
3.3. Protocol
Before starting the protocol, prepare the following solution required in this section.
Blocking solution: Blocking solution is 10% (vol/vol) goat serum and 0.2% (wt/vol) BSA in 1× TBST (pH7.6). To prepare 10 ml of blocking solution, add 1 ml of goat serum and 20 mg of BSA in 9 ml of 1× TBST (pH7.6). Prepare fresh solution right before use or store at −20 ℃ for up to 1 month.
If there is no immunostaining, jump to Step 5 for DNA staining.
Block embryos: Carefully remove 1× TBST (pH7.6) from the glass vials and add 200–500 μl of the blocking solution (10%(vol/vol) goat serum and 0.2% (wt/vol) BSA in 1× TBST, pH7.6). Wrap the vials with aluminum foil and incubate for 2–3 h at room temperature on a nutator at low speed in the dark (see Note 11).
Primary antibody incubation: carefully remove the blocking solution from the glass vials using pipettes. Dilute an antibody (such as anti-histone H3 antibody at 1:1000) in the blocking solution. Add 200–500 μl of the diluted anti-histone H3 antibody to each glass vial for 10–20 embryos. Wrap the glass vials with foil and incubate overnight at 4 °C on a nutator at low speed in the dark.
Washing embryos: carefully remove the antibody solution from the glass vials and fill up with 1× TBST (pH7.6). Rotate embryos for 5 min then carefully remove 1× TBST (pH7.6). Repeat washing embryos with 1× TBST (pH7.6) once more. Wash embryos with 1× TBST (pH7.6) for all day at room temperature by rotating the glass vials on a rotator. Change 1× TBST (pH7.6) every 1–2 h. A minimum of six washes is required.
Secondary antibody incubation and/or DNA staining: Dilute a secondary antibody such as goat anti-rabbit Alex Fluor 488 secondary antibody and/or TO-PRO-3 DNA Stain at 1:500 in the blocking solution. Add 200–500 μl of the secondary antibody and/or TO-PRO-3 mix to each glass vial. Wrap the glass vials with aluminum foil and incubate embryos for overnight at 4 °C on at low speed in the dark.
Final extensive wash: carefully remove the secondary antibody and/or TO-PRO-3 mix from the glass vials and fill up with 1× TBST (pH7.6) to quickly rinse by rotating embryos for 5 min; repeat rinsing embryos with 1× TBST (pH7.6) once more. Make sure to wash embryos with 1× TBST (pH7.6) all day via rotation at room temperature. Change 1× TBST (pH7.6) every 1–2 h. A minimum of six washes is required.
For general troubleshooting of subsections on click chemistry and staining, see Note 12.
4. Confocal Imaging of Nascent Transcription in Cleared, Wholemount Embryos
The preparation of Xenopus embryos for confocal imaging has been described in detail (Wallingford, 2010). Due to their large size and yolk, Xenopus embryos are difficult to image without first clearing the embryos. The clearing reagent BABB, 1 part of benzyl alcohol (BA) mixed with 2 parts of benzyl benzoate (BB), is used for clearing thick tissues and works broadly for many types of embryos. To obtain best results, it is recommended to clear embryos for 24–48 h before imaging. Inadequate clearing may result in uneven signals in different regions of embryos while excessive clearing may cause the embryo to fragment or diminish fluorescent signals.
To image the same embryo from different sides, e.g., the animal pole and the vegetal pole of a Xenopus embryo, it is preferable that imaging chamber be able to be flipped without turning internal contents. To build such a chamber, we stick hollowed 3M VHB™ tape on glass covers. The thickness of 3M VHB™ tape is ~ 1.14 mm, which is very close to the average diameter of a Xenopus embryo (~ 1.2 mm). Embryos can be situated properly inside the chamber with desired orientation, i.e., animal pole on the top and vegetal pole at the bottom. After imaging one side, the chamber can be flipped by 180° and the embryo can be imaged again from the other side. For Xenopus laevis wholemount imaging, we use a laser scanning confocal microscope and 10x air objective to fully image the entire embryo or a 25×oil objective and zoom 0.6x combined with image stitching.
4.1. Materials
Methanol (Thermo Fisher Scientific, cat. no. A4524)
Methanol, anhydrous (Sigma-Aldrich, cat. no. 322415)
Benzyl alcohol (Sigma-Aldrich, cat. no. 305197)
Benzyl benzoate (ACROS Organics, cat. no. 105860010)
Clearing reagent (BABB): To prepare clearing reagent BABB, mix benzyl alcohol (BA) and benzyl benzoate (BB) at a ratio of 1:2 using glass transfer pipets. Store at room temperature for up to 2 weeks.
4.2. Equipment
Cover glass, No. 1, 22×40 mm (VWR, cat. no. 48393048)
VHB™ Tape (3M, cat. no. GPH-110GF)
Glass Pasteur pipets (VWR, cat. no. 14673-010)
Razor blade (Thermo Fisher Scientific, cat. no. 18-100-970)
Dissecting forceps (Thermo Fisher Scientific, cat. no. S08096)
Confocal microscopes (Zeiss LSM 710, Plan-Apochromat 10× / numerical aperture (NA) 0.45 objective, Plan-Apochromat 25× / NA 0.8 and 63× / NA 1.4 immersion oil objectives)
4.3. Protocol
4.3.1. Clearing Embryos
-
1
Dehydrate embryos: carefully remove 1× TBST (pH7.6) from the glass vials and fill up with 100% methanol. Rotate embryos for 5 min, then carefully remove methanol and repeat washing embryos with methanol for a total of at least 3 times.
-
2
Completely dehydrate embryos: Further dehydrate embryos with anhydrous methanol at least three times following procedures in the previous step. Note that embryos must be completely dehydrated before clearing. Embryos can be stored in methanol at −20 °C overnight or for longer time before further processing.
-
3
Clear embryos: completely remove anhydrous methanol from the glass vials. Add 500 μl of the clearing reagent (benzyl alcohol (BA): benzyl benzoate (BB) = 1:2) into each glass vial. Wait for ~ 10 min and let embryos settle down to the bottom of the vial. Carefully remove the clearing reagent and replace with fresh clearing agent. Note: embryos are nearly invisible after clearing so extra care must be taken to avoid losing them during pipetting. Repeat replacement of clearing reagent 2–3 times until no schlieren lines are visible. For complete clearing, keep embryos in the clearing reagent for 24–48 h before imaging, changing the clearing reagent for multiple times. Note that embryos in clearing reagent can be stored for short term at 4 °C; however, long-time storage may result in a reduction in fluorescence signal and fragmentation of embryos.
4.3.2. Preparing Imaging Chambers and Confocal Imaging
-
4
To make tape chambers, cut the 3M VHB™ tape into squares of 10 mm × 10 mm. Draw a square of 0.8 cm × 0.8 cm at the center of the tape using a razor blade and remove the center piece using dissecting forceps, which leaves a frame of tape. Transfer the frame of tape and let it adhere to the surface of a 20 mm × 40 mm cover-glass. Gently press the corners to make sure that the tape tightly adheres to the cover-glass. Peel off the thin plastic film on the tape using dissecting forceps.
-
5
Carefully transfer embryos from the glass vials in Step 3 into tape chambers. Disperse embryos evenly inside the frame of tape. Gently place a second cover-glass on top to assemble the chamber to be a sandwich with embryos between two cover-glasses. Avoid air bubbles during this process.
-
6
Confocal microscopy: Under typical imaging conditions on a Zeiss LSM 710 we use lasers 561 nm (0.15% power), 633 nm (10% power) and 488 nm (10% power), without saturating signals. To image whole embryos, use the Plan-Apochromat 10× / numerical aperture (NA) 0.45 objective. To image embryos at high resolution, use the Plan-Apochromat 25× / NA 0.8 or 63× / NA 1.4 immersion oil objective. Use the following settings to acquire image stacks: scan mode = frame, frame size = 1,024 × 1,024, speed = 9, averaging number = 2, bit depth = 8-bit, direction = bidirectional, method = mean, zoom = 0.6. To collect z-stacks, adjust the positions of objective and set the first (bottom) and last (top) images of the embryo. Set interval = 5 μm (for 10×), 2 μm (for 25× immersion oil) or 1 μm (for 63× immersion oil).
5. Image Processing and Analysis
Image stacks collected from confocal microscopy can be used for wholemount three-dimensional (3D) image segmentation to quantify levels of nascent transcription. We found that nucleus segmentation with background subtraction gave the most direct readout of transcriptional activity in blastomeres during cleavage stages. We found it most convenient to process and segment images using the Imaris 9.2 software, but similar approaches can be carried out using ImageJ or Matlab. The corrections we perform for large embryos include, signal attenuation due to thick and an optical correction for the 10× objective due to refractive index mismatches (~ 1.5 vs 1, BABB vs air). Nuclei are easily segmented based on DNA stain or immunstaining with nuclear protein such as antibody to histone H3. Cell diameter of relatively round blastomeres was estimated using inner nuclear distances between object centroids. Cells at interphase can be separated from mitotic cells using the sphericity parameter, the threshold of which should be empirically determined.
6. Broader Impacts & Future Directions
6.1. Generalizability to Embryonic Systems: Imaging ZGA in Zebrafish Blastula Embryos
To determine whether our method for imaging zygotic genome activation can be applied to other embryonic systems we tested two approaches in blastula embryos from the zebrafish, Danio rerio. We microinjected 5-EU in 1-cell stage zebrafish embryo and fixed them at 3–4 hpf when large-scale genome activation occurs (Figure 4A). After processing using the protocol described above, we found that nascent transcripts could be detected in single cells of wholemount embryos similar to what we observed for Xenopus (Figure 4B). Second, we tested whether zebrafish embryos can be incubated with 5-EU in the medium, instead of microinjection, as works for cell culture (Jao & Salic, 2008). We removed the chorion of zebrafish embryos at 1-cell stage, incubated them with 1 mM EU in the medium starting from 1 hpf to 4 hpf (Figure 4C), and then fixed them. We found that nascent transcripts could be detected in single cells in wholemount embryos, the signal and pattern of which was similar to embryos with EU microinjection (Figure 4D). This and other studies (Chan et al., 2019; Kwasnieski et al., 2019) show that 5-EU can used for tracking zygotic genome activation in a variety of embryonic systems. The choice of 5-EU microinjection or incubation should be determined empirically for any new embryo of interest.
Figure 4. Labeling and imaging nascent transcripts in zebrafish embryos.
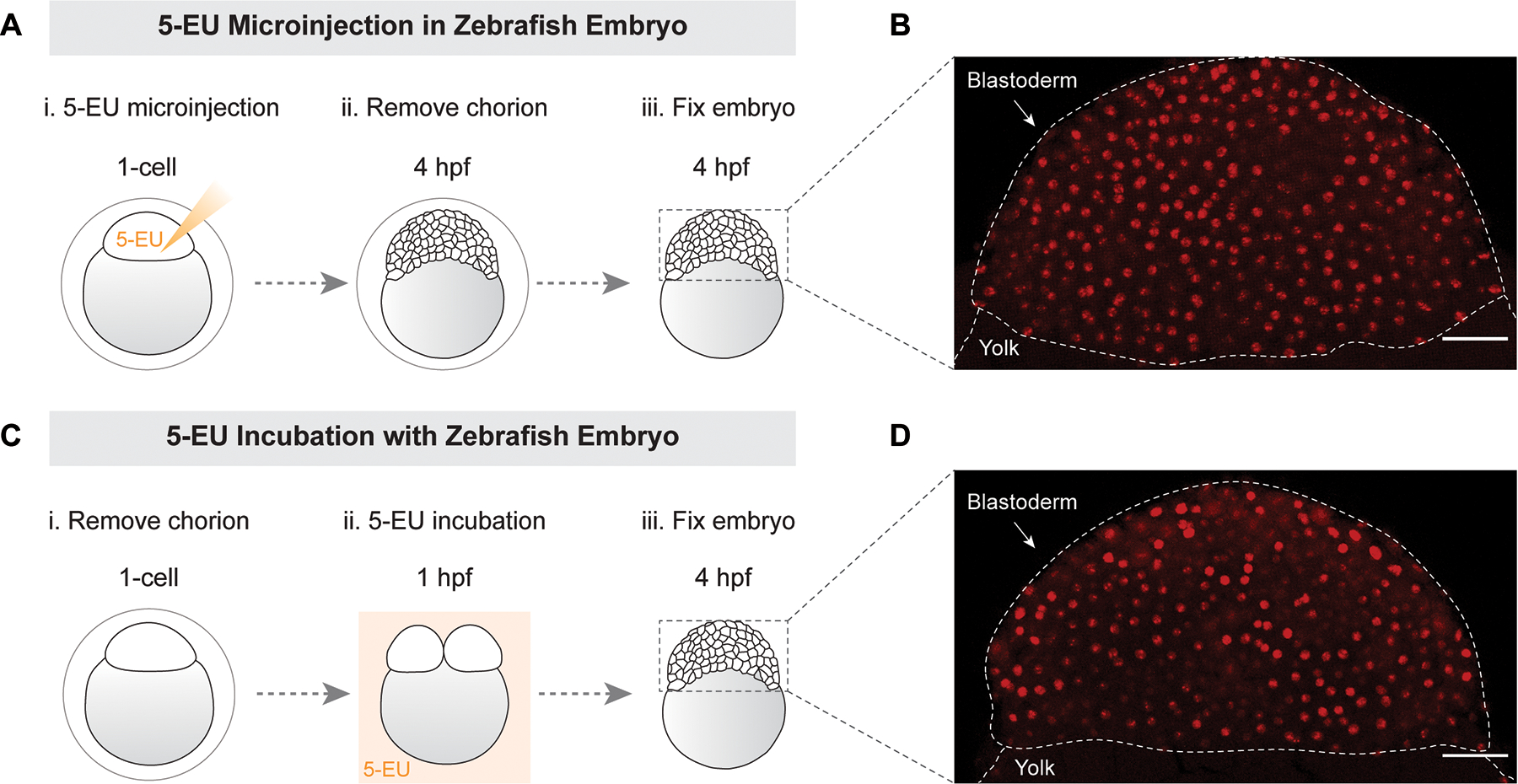
(A) schematic of 5-EU microinjection in the blastodisc at 1-cell stage. (B) chorion was removed prior to fixation at 4 hpf. Image of 5-EU in blastoderm from 5-EU injection. (C) schematic of zebrafish embryos incubated with 5-EU following chorion removal. (D) image of 5-EU in blastoderm from 5-EU incubation. Confocal imaging at 25× magnification. Maximum intensity projections of Z-stacks for embryos (only blastoderms are shown). The dashed lines indicate the shape and position of blastoderm relative to yolk. Scale bar 50 μm.
6.2. Sequencing the Nascent Transcriptome via Biotinylation of EU-RNA
EU-RNA imaging provides a single-cell and spatial perspective of large-scale genome activation during early embryogenesis. However, this approach lacks information on the identify of transcripts produced. Importantly, metabolic labeling with 5-EU can be directed towards (1) in vivo imaging via conjugation to an azide-flourophore and (2) in vitro characterization of the nascent transcriptome via RNA-seq via conjugation with azide-biotin (Figure 5) (Chan et al., 2019; Chen et al., 2019; Kwasnieski et al., 2019). To generate and analyze the nascent transcriptome, total RNA is isolated from EU-microinjected embryos, and EU-RNA is biotinylated and then purified using streptavidin beads (Figure 5). Nascent EU-RNA pulled down by beads is reversed transcribed and the cDNA is used for making nascent transcriptome libraries for RNAseq. Conversely, the flowthrough RNA that does not bind to beads is reversed transcribed and used to make maternal transcriptome libraries (Kwasnieski et al., 2019). Thus, using 5-EU metabolic labeling, in addition to wholemount imaging, the dynamics of both maternal and zygotic transcripts can be measured during early embryo development.
Figure 5. Sequencing EU-RNA to characterize the nascent transcriptome.
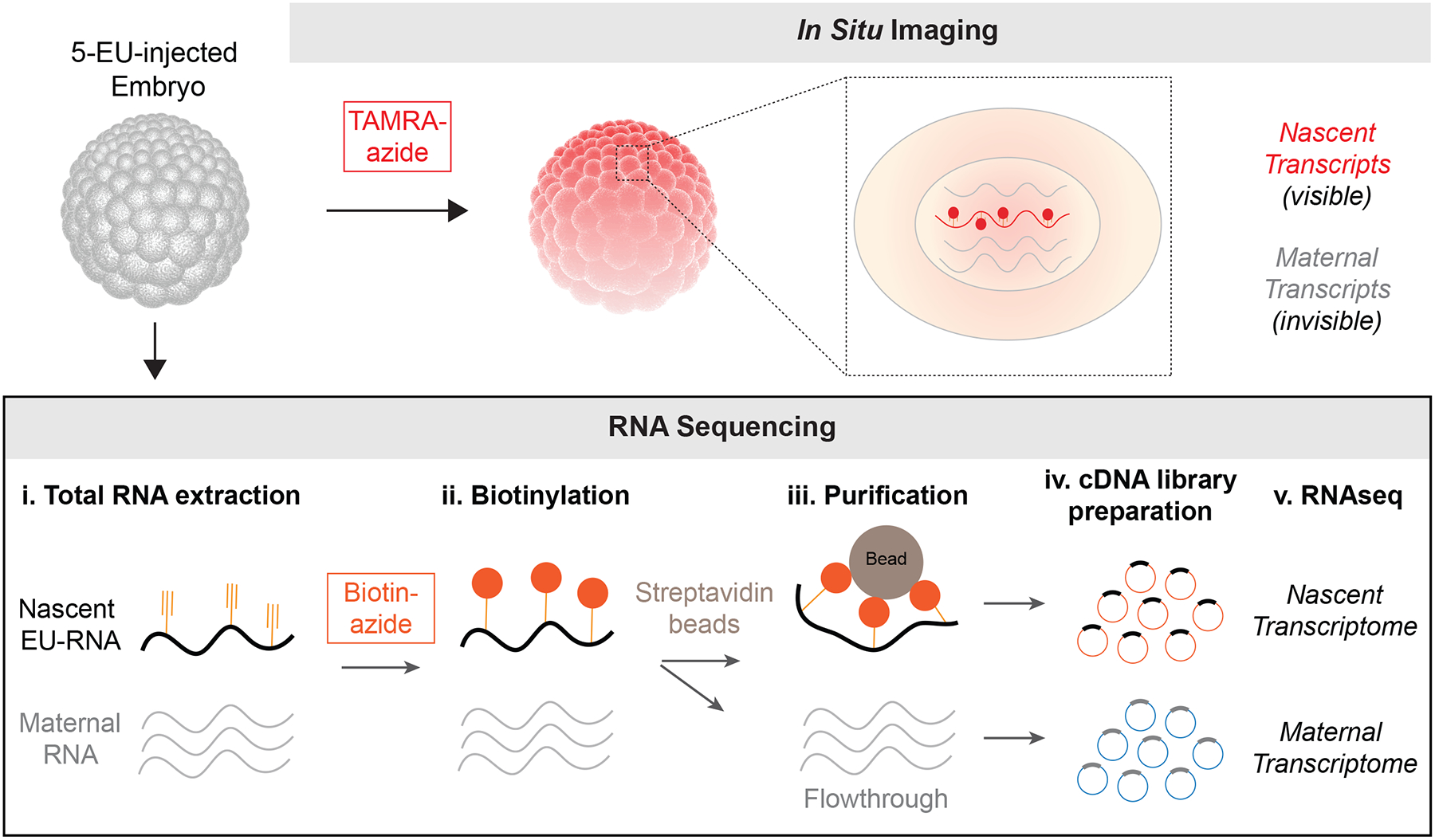
5-EU injected embryos can either be fixed and labeled for wholemount imaging of nascent transcription at a single-cell level, or RNA can be isolated from embryos, biotinylated and separated from unlabeled RNA to generate nascent transcriptome libraries for sequencing. Unbound RNA flowthrough defines the maternal transcriptome.
Summary
How the zygotic genome is activated during embryogenesis is a fundamental question in biology. This chapter describes a novel method to image nascent transcription in single cells of wholemount early embryos and to characterize their nascent transcriptomes. This approach provides temporal and spatial information on the pattern of ZGA, and it can be applied to a variety of model embryonic systems. This method will help to advance our understanding of gene regulation in complex tissues such as the blastula early embryo.
Notes
Egg quality and IVF: The quality of eggs and embryos is extremely important for all downstream processing. Ideally, embryos from IVFs with over 90% efficiency should be used in this protocol. To achieve high efficiency, freshly prepare buffers and sperm solutions used for IVF. Additionally, we recommend screening eggs from multiple frogs to identify clutch with highest quality of IVF.
Embryo budgeting: The number of embryos microinjected is dependent on the purpose of the experiment. As a rule of thumb for imaging, a minimum of 5–6 embryos should be collected for each stage in treatment and experimental groups. To achieve this we recommend microinjecting at least 10 embryos to image 5–6 of highest quality. Some embryos can be damaged or lost during later processing.
5-EU intracellular concentration: When first identifying the optical level of 5-EU for embryos from a new species, two factors should be determined empirically: sensitivity and toxicity. A final concentration of 5-EU at 0.2–0.6 mM often works for most embryos, including those from zebrafish, Xenopus tropicalis and Xenopus laevis. Higher concentrations produce higher signals for imaging, but concentrations above 1 mM often leads to toxicity and slow embryo development.
To avoid diluting the fixative, add embryos to the glass vial, while transferring only a minimal volume of media. Embryos will enrich in the tip of plastic transfer pipette due to gravity by waiting for 5–10 second. Excess buffer transferred will slow down the rate of fixation. If necessary, one can replace the fixative with fresh fixative to ensure rapid and complete fixation.
Bleaching: Use bleaching solution prepared freshly, immediately prior to the experiment. When adding the bleaching solution to the glass vials, be careful not to fill to very top as exposure of H2O2 to light for long periods of time will generate air bubbles that can cause vials to explode. Be careful not to overheat embryos because it may cause damage that worsens image quality. The duration of bleaching depends on the visibility of pigments in the animal pole of embryos. Bleaching can be ended when all pigments are whitened. Insufficient bleaching can lead to light absorption during fluorescence imaging.
Floating embryos: Following bleaching, take extra care when replacing buffer surrounding the embryos. Small air bubbles generated during bleaching can cause the embryos to float which can make buffer removal more challenging. Work slowly and carefully. Keep in mind that embryos may become softened after extended bleaching and can be easily destroyed by shear forces during pipetting.
Pause step: Completely dehydrated embryos in 100% methanol can be stored long-term at −20 °C. This enables one to process separate sets of embryos at different times and then to image them all at one.
Click reaction details: Always freshly prepare the click reaction mix right before use. Also, avoid using previously used aliquots of reagents. Add ascorbic acid last to the reaction mix. We store 5-EU aliquots in aqueous solution of TBS or water at 100 mM stock concentration at −80 ℃. Use of DMSO as solvent can slow embryos development (e.g., 0.5% injected DMSO may extend cleavage stage cell cycle or lead to reduced embryogenesis efficiency).
Volume of click reaction: The volume of click reaction mix to be added in each vial depends on the number of embryos inside. Minimally, one wants a volume sufficient for submerging all embryos inside the vial. The incubation time should be empirically determined for embryos from different species. Generally, the size of embryos affects the timing for click reaction, i.e., large embryos requires longer incubation than small embryos. Insufficient incubation may result in no or little labeling of EU-RNA inside the embryo, while over-incubation may cause higher nonspecific background. In our experience with Xenopus laevis, ~ 1.2 mm diameter embryos, we used 200–500 μl of reaction volume in a glass vial for 10–20 embryos, and incubated the reaction for ~ 12 h. For larger embryos we would perhaps scale up reaction volumes and times; for smaller embryos we would shorten reaction times.
Importance of washing: Complete washing of embryos following the click reaction is required for successful downstream processing, including immunostaining and DNA staining. An important indicator of complete washing is that no color remains in the embryos from fluorescent azides, e.g., red from TAMRA-azide. However, we still wash embryos for an extended period such as another day once red TAMRA-azide signal is gone. Insufficient washing may result in high background. Therefore, embryos must be extensively and adequately washed before further processing. Expect to extend washing times listed in this protocol for embryos > 2 mm in diameter.
Blocking of embryos: The volume of the blocking buffer to be added in each vial should be sufficient to submerge all embryos inside the vial. For large-sized embryos, e.g., > 1 mm in diameter, the incubation can also be performed overnight at 4 °C.
Troubleshooting. Always prepare buffers for processing fixed embryos with RNase-free water. If DNA staining is weak or non-specific, try making a fresh preparation of MEMFA for fixation step. If EU or DNA staining appear somewhat nonspecific, particularly in cortical layers of embryo, it may be helpful to increase washing time (particularly if embryos are larger than those of Xenopus laevis). If embryos are being damaged during processing make sure to always fill glass tubes to the very top so that large air pockets are not present during rotation.
Acknowledgements
We thank the Cell and Developmental Biology Microscopy Core at the University of Pennsylvania for technical support in confocal imaging, National Xenopus Resource (NXR) for training and guidance on frogs and the Dr. Mary Mullins lab for providing zebrafish embryos. This work was supported in part by Burroughs Wellcome Fund, Charles E. Kaufman Foundation, the March of Dimes and the National Institute of General Medical Sciences (R35GM12874) (M.C.G.).
References
- Aanes H, Collas P, & Alestrom P (2014). Transcriptome dynamics and diversity in the early zebrafish embryo. Brief Funct Genomics, 13(2), 95–105. doi: 10.1093/bfgp/elt049 [DOI] [PubMed] [Google Scholar]
- Aoki F, Worrad DM, & Schultz RM (1997). Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo. Dev Biol, 181(2), 296–307. doi: 10.1006/dbio.1996.8466 [DOI] [PubMed] [Google Scholar]
- Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW, & Klein AM (2018). The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science, 360(6392). doi: 10.1126/science.aar5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PD, Chao JA, Singer RH, & Marlow FL (2015). Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development, 142(7), 1368–1374. doi: 10.1242/dev.118968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Tang Y, Miao L, Darwich-Codore H, Vejnar CE, Beaudoin JD, … Giraldez AJ (2019). Brd4 and P300 Confer Transcriptional Competency during Zygotic Genome Activation. Dev Cell, 49(6), 867–881 e868. doi: 10.1016/j.devcel.2019.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Einstein LC, Little SC, & Good MC (2019). Spatiotemporal Patterning of Zygotic Genome Activation in a Model Vertebrate Embryo. Dev Cell, 49(6), 852–866 e857. doi: 10.1016/j.devcel.2019.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, & Schier AF (2018). Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 360(6392). doi: 10.1126/science.aar3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wu K, Liu Z, Yao X, Yuan S, Tao W, … Liu J (2018). Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell, 173(1), 248–259 e215. doi: 10.1016/j.cell.2018.02.028 [DOI] [PubMed] [Google Scholar]
- Garcia HG, Tikhonov M, Lin A, & Gregor T (2013). Quantitative imaging of transcription in living Drosophila embryos links polymerase activity to patterning. Curr Biol, 23(21), 2140–2145. doi: 10.1016/j.cub.2013.08.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch GE, Owens NDL, & Smith JC (2019). The Spatiotemporal Control of Zygotic Genome Activation. iScience, 16, 485–498. doi: 10.1016/j.isci.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, & Neugebauer KM (2014). The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep, 6(2), 285–292. doi: 10.1016/j.celrep.2013.12.030 [DOI] [PubMed] [Google Scholar]
- Heyn P, & Neugebauer KM (2017). Purification of Zygotically Transcribed RNA through Metabolic Labeling of Early Zebrafish Embryos. Methods Mol Biol, 1605, 121–131. doi: 10.1007/978-1-4939-6988-3_8 [DOI] [PubMed] [Google Scholar]
- Jao CY, & Salic A (2008). Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A, 105(41), 15779–15784. doi: 10.1073/pnas.0808480105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukam D, Shariati SAM, & Skotheim JM (2017). Zygotic Genome Activation in Vertebrates. Dev Cell, 42(4), 316–332. doi: 10.1016/j.devcel.2017.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama S, Nagata M, & Aoki F (2004). Isolation of nascent messenger RNA from mouse preimplantation embryos. Biol Reprod, 71(6), 1948–1955. doi: 10.1095/biolreprod.104.031906 [DOI] [PubMed] [Google Scholar]
- Kwasnieski JC, Orr-Weaver TL, & Bartel DP (2019). Early genome activation in Drosophila is extensive with an initial tendency for aborted transcripts and retained introns. Genome Res, 29(7), 1188–1197. doi: 10.1101/gr.242164.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kieserman E, Gray RS, Park TJ, & Wallingford J (2008). Whole-mount fluorescence immunocytochemistry on Xenopus embryos. CSH Protoc, 2008, pdb prot4957. doi: 10.1101/pdb.prot4957 [DOI] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, & Giraldez AJ (2014). Zygotic genome activation during the maternal-to-zygotic transition. Annu Rev Cell Dev Biol, 30, 581–613. doi: 10.1146/annurev-cellbio-100913-013027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SC, & Gregor T (2018). Single mRNA Molecule Detection in Drosophila. Methods Mol Biol, 1649, 127–142. doi: 10.1007/978-1-4939-7213-5_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Leng L, Liu C, Lu C, Yuan Y, Wu L, … Lin G (2019). An integrated chromatin accessibility and transcriptome landscape of human pre-implantation embryos. Nat Commun, 10(1), 364. doi: 10.1038/s41467-018-08244-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Inoue A, Suzuki T, Zhao K, & Zhang Y (2016). Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell, 165(6), 1375–1388. doi: 10.1016/j.cell.2016.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranisavljevic N, Okamoto I, Heard E, & Ancelin K (2017). RNA FISH to Study Zygotic Genome Activation in Early Mouse Embryos. Methods Mol Biol, 1605, 133–145. doi: 10.1007/978-1-4939-6988-3_9 [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, & Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol, 33(5), 495–502. doi: 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AF (2007). The maternal-zygotic transition: death and birth of RNAs. Science, 316(5823), 406–407. doi: 10.1126/science.1140693 [DOI] [PubMed] [Google Scholar]
- Schulz KN, & Harrison MM (2019). Mechanisms regulating zygotic genome activation. Nat Rev Genet, 20(4), 221–234. doi: 10.1038/s41576-018-0087-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, & Harland RM (2007). Xenopus laevis In Vitro Fertilization and Natural Mating Methods. CSH Protoc, 2007, pdb prot4737. doi: 10.1101/pdb.prot4737 [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, & Harland RM (2010). Microinjection of Xenopus embryos. Cold Spring Harb Protoc, 2010(12), pdb ip81. doi: 10.1101/pdb.ip81 [DOI] [PubMed] [Google Scholar]
- Stapel LC, Zechner C, & Vastenhouw NL (2017). Uniform gene expression in embryos is achieved by temporal averaging of transcription noise. Genes Dev, 31(16), 1635–1640. doi: 10.1101/gad.302935.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T, Lionnet T, Shroff H, & Lehmann R (2017). mRNA quantification using single-molecule FISH in Drosophila embryos. Nat Protoc, 12(7), 1326–1348. doi: 10.1038/nprot.2017.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Cao WX, & Lipshitz HD (2019). The maternal-to-zygotic transition revisited. Development, 146(11). doi: 10.1242/dev.161471 [DOI] [PubMed] [Google Scholar]
- Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, & Klein AM (2018). Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science, 360(6392), 981–987. doi: 10.1126/science.aar4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB (2010). Preparation of fixed Xenopus embryos for confocal imaging. Cold Spring Harb Protoc, 2010(5), pdb prot5426. doi: 10.1101/pdb.prot5426 [DOI] [PubMed] [Google Scholar]
- White RJ, Collins JE, Sealy IM, Wali N, Dooley CM, Digby Z, … Busch-Nentwich EM (2017). A high-resolution mRNA expression time course of embryonic development in zebrafish. Elife, 6. doi: 10.7554/eLife.30860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Timme KA, & Wood JR (2018). Using Single Molecule mRNA Fluorescent in Situ Hybridization (RNA-FISH) to Quantify mRNAs in Individual Murine Oocytes and Embryos. Sci Rep, 8(1), 7930. doi: 10.1038/s41598-018-26345-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Huang K, Cai C, Cai L, Jiang CY, Feng Y, … Fan G (2013). Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature, 500(7464), 593–597. doi: 10.1038/nature12364 [DOI] [PMC free article] [PubMed] [Google Scholar]