

Abstract
Advances in bioconjugation, the ability to link biomolecules to each other, small molecules, surfaces, and more, can spur the development of advanced materials and therapeutics. We have discovered that pyrocinchonimide, the dimethylated analog of maleimide, undergoes a surprising transformation with biomolecules. The reaction targets amines and involves an imide transfer, which has not been previously reported for bioconjugation purposes. Despite their similarity to maleimides, pyrocinchonimides do not react with free thiols. Though both lysine residues and the N-termini of proteins can receive the transferred imide, the reaction also exhibits a marked preference for certain amines that cannot solely be ascribed to solvent accessibility. This property is peculiar among amine-targeting reactions and can reduce combinatorial diversity when many available reactive amines are available, such as in the formation of antibody-drug conjugates. Unlike amides, the modification undergoes very slow reversion under high pH conditions. The reaction offers a thermodynamically controlled route to single or multiple modifications of proteins for a wide range of applications.
Keywords: drug delivery, slow-release, lysine modification, antibody-drug conjugates, next-generation maleimides
Graphical Abstract
Introduction
The thiol-maleimide click reaction is an important mainstay in bioconjugation chemistry. Typically, the maleimide nitrogen atom carries a payload molecule chosen for experimental or therapeutic purposes. The reaction proceeds via nucleophilic attack by a sulfur atom at the maleimide alkene in a classical 1,4-conjugate addition (or ‘thio-Michael’) mechanism. This reaction covalently links the payload to the target under mild conditions in aqueous buffers.1,2 The product of the thiol-maleimide reaction is an unstable thiosuccinimide, which can undergo reversion via a retro-Michael process. In situ, the regenerated maleimide can then react with other thiols, shuffling the payload to off-target sites, introducing a potential source of toxicity, and decreasing selectivity for therapeutic bioconjugates.3–5 Thiosuccinimides are also susceptible to irreversible hydrolytic ring-opening reactions in aqueous conditions, forming long-lived succinamic acid thioethers.3,4,6–10 These and other impish behaviors have inspired detailed investigations,11,12 especially in the context of maleimides used in preparing antibody-drug conjugates (ADCs).13,14
Next-generation maleimides (NGMs) are maleimide derivatives designed to attenuate the issues described above. The archetypal NGMs, halomaleimides, feature a halogen substituent at one or both alkene carbons of the maleimide.15–17 Halomaleimides undergo an overall addition-elimination reaction; after the canonical thio-Michael step, the intermediate halosuccinimide rapidly eliminates HX, reforms the hydrolytically stable maleimide heterocycle, and generates a vinylic S-C covalent bond. Dihalo-, dithio-, and diaryloxymaleimides have also been developed for cross-linking reactions.18–21 This technology has been successfully employed for re-bridging reduced interchain antibody disulfides, and arming antibodies with the anticancer drug, monomethyl auristatin E22 or the photodynamic, theranostic fluorophore IRDye 800 CW.23
The mono-methylated analog of maleimide (‘citraconimide’) has also been reported for the assembly of doxorubicin-Fab′ ADCs,24 and may be considered a member of the NGM family. The citraconimide thio-Michael addition reaction exhibits excellent regioselectivity for nucleophilic attack at the methylated alkene carbon,25,26 and the product methylsuccinimides exhibit improved hydrolytic stability compared to standard succinimides.24 5-Methylene pyrrolones are maleimide =O → =CH2 isosteres that exhibit soft electrophilic character at the exocyclic γ/δ-alkene, and can participate in 1,6-conjugate addition reactions with thiols. The adduct gains no additional stereocenters and can tracelessly revert to starting materials at elevated pH, thus providing a pH-controlled catch-and-release process.27 Another class of maleimide isosteres, 1,2-dihydropyridazine-3,6-diones undergo a similar retro-Michael reaction for cargo release.28
Examined herein is an ostensibly overlooked NGM reagent, pyrocinchonimide (Pci), the dimethylated analog of maleimide. The Pci motif (also referred to as dimethylmaleimide or N-dimethylmaleoyl) was previously established as an N-protecting group for aminosugar synthesis by Richard R. Schmidt.29 Prominent examples of Pci compounds in the patent literature include antimicrobials,30–33 water soluble34 and photochemically cross-linked polymers,35 silicon-based adhesives,36 herbicides and other plant growth regulators and their synthetic intermediates,37–44 pharmaceuticals and prophylactics for treatment of obesity45 and urinary incontinence,46 liquid crystal thermoset monomers,47 synthetic intermediates of allosteric glucokinase modulators48 and alpha-2C adrenergic receptor antagonists,49 internal ribosome entry site-mediated protein synthesis inhibitors,50 and in oligosaccharide manufacturing.51 Despite this considerable precedent, the reactivity of Pci compounds with proteins has never been specifically addressed.
The investigation revealed that the reactivity of Pci towards thiols is profoundly diminished compared to maleimide and citraconimide. Fluorescence and MS labeling experiments with proteins revealed only trace quantities of the expected thio-Michael adduct. Instead, a previously unreported imide transfer reaction was observed (i.e., a transpyrocinchonimidation); both acyl groups are moved from the electron-poor nitrogen atoms of select Pci derivatives, to the solvent-exposed amines of lysine side chains and N-termini on target proteins. This imide transfer was then leveraged for direct bioconjugation of a fluorescent label to a target protein under mild conditions using a modified pyrocinchonimide derivative. Additionally, we have explored the possibility for protein-bound Pci groups to participate in UV-activated intermolecular cycloadditions with terminal alkenes. The results establish Pci compounds as a new species of amine-targeting bioconjugation reagents, with qualities that make it distinct from NHS esters, maleimides, and other NGMs (Figure 1).
Figure 1. The unique modality of pyrocinchonimides.
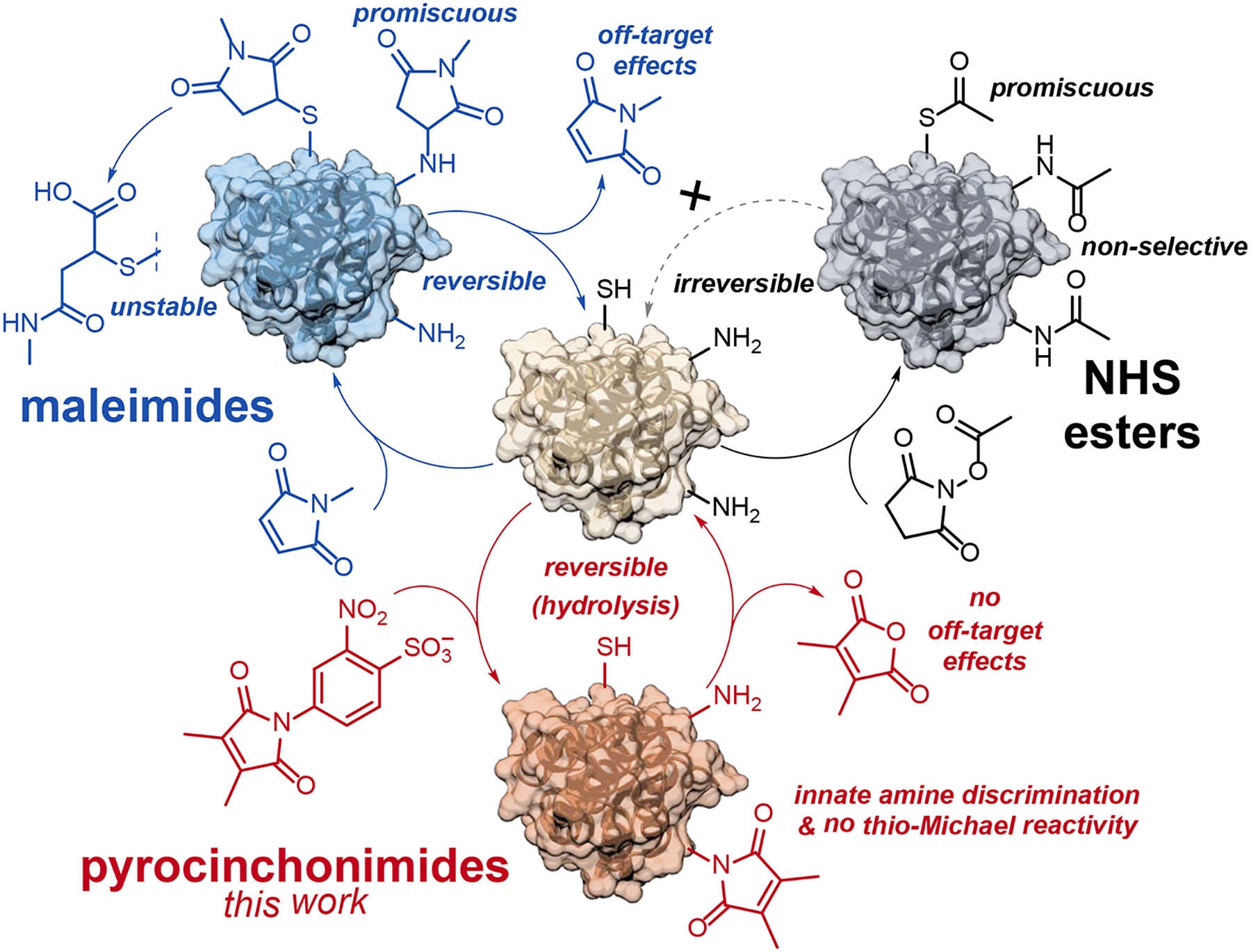
The behavior of the Pci bioconjugation is compared with its two closest analogs, maleimides and NHS esters.
Results and Discussion
Synthesis of Pyrocinchonimides
The most common method for preparing N-substituted maleimides is via the reaction of 1° amines with maleic anhydride. Ring-opening acylation of the amine results in an isolable maleamic acid intermediate, which can be cyclized under dehydrating conditions. This two-step sequence is equally appropriate for citraconimides, but not Pci compounds. At least two independent sources have reported that the condensation of 1° amines with pyrocinchonic anhydride furnishes the cyclic imide without observation of the intermediate pyrocinchonamic acid (Scheme 1).52,53 However, the existence of the pyrocinchonamic acid intermediate is well-established spectroscopically, and the species can be trapped as the carboxylate triethylammonium salt.40,54
Scheme 1. Facile cyclization of pyrocinchonimides.
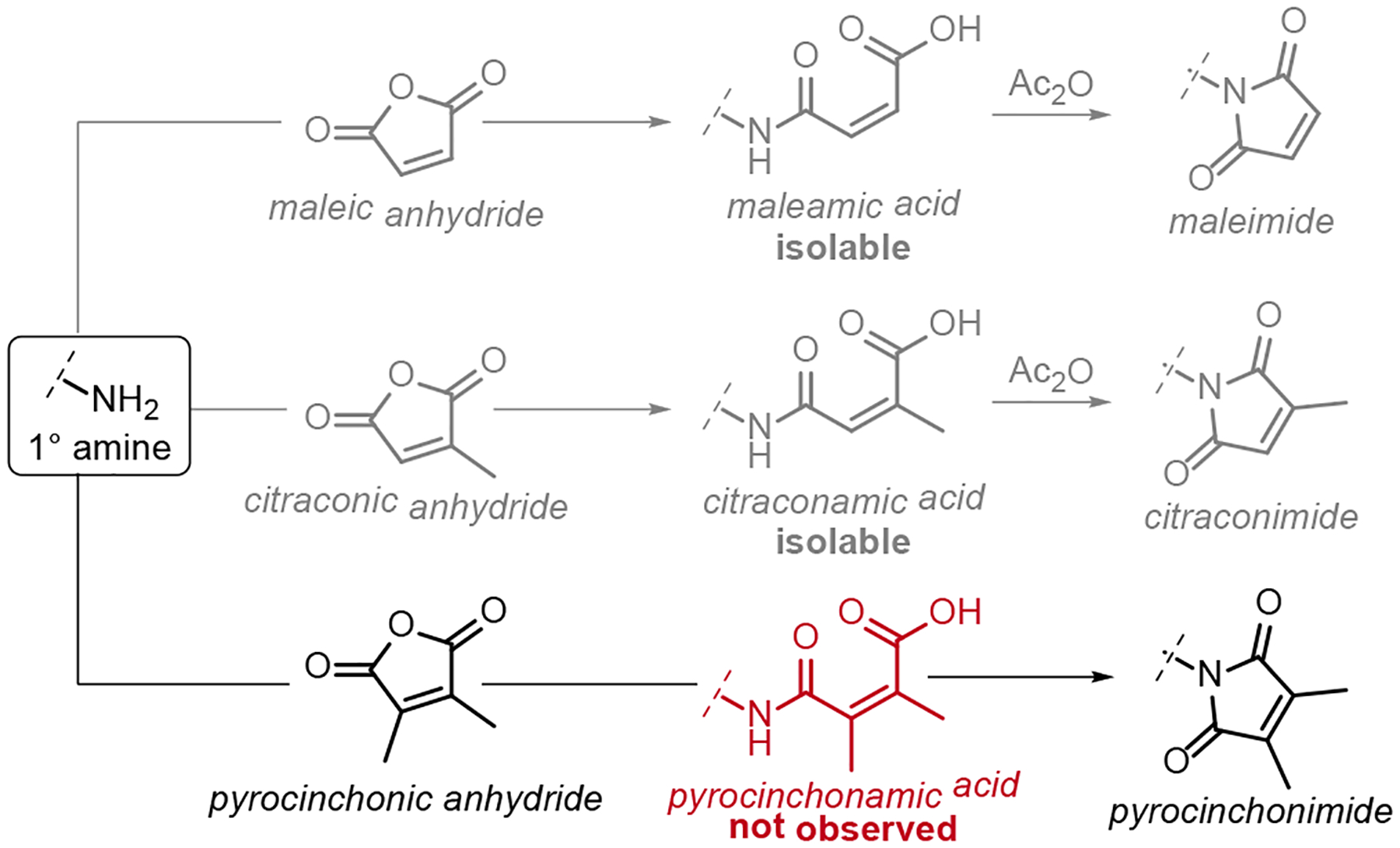
In contrast to the reactions of 1° amines with maleic and citraconic anhydrides, the ring-opened pyrocinchonamic acid intermediate spontaneously cyclizes to afford the imide at ambient temperatures without requiring dehydrating conditions.
The disparity in cyclization efficiency between citraconimide and pyrocinchonimide is consistent with the behaviors of their parent diacids. Both maleic (CAS: 110–16-7) and citraconic (CAS: 498–23-7) acids have been isolated and characterized as pure compounds. However, pyrocinchonic acid undergoes rapid cyclization to its anhydride during attempted isolation, and a sample of the purified acid has never been described.55 The formation of pyrocinchonic anhydride exhibits immeasurably fast ring-closing kinetics in organic solvents, reflecting a profound entropic preference for the cyclized state.56,57 This behavior has been ascribed to angle compression (the Thorpe-Ingold effect); the mutual repulsion of the vinylic methyl groups drives the carboxylic acids close together, and accelerates the ring closing step.58,59 The same stereoelectronic and steric effects could operate in the amic acid, resulting in the apparent absence of this intermediate during the synthesis of Pci derivatives. Thus, mild conditions should suffice for Pci synthesis, despite many reports applying the strongly dehydrating conditions typical for citraconimide and maleimide formation.
Guided by the reports described above, a general methodology provided relatively effortless access to a wide variety of Pci derivatives in satisfactory-to-excellent yields with respect to the amine starting material (Scheme 2). Briefly, a 1° aryl- or alkylamine was combined with catalytic 4-dimethylaminopyridine (DMAP) and excess pyrocinchonic anhydride in a mixture of CH2Cl2 and DMF. The reaction was typically judged complete by thin layer chromatography (TLC) after stirring at rt for 6–72 h (condition i). Anhydrous DMF is not required for these syntheses. In general, the components of the product mixture were easily separated by flash chromatography, and unreacted pyrocinchonic anhydride can be recovered for use in future reactions. Alcohol substituents are tolerated (i.e., ester formation does not occur), as are the sodium salts of carboxylic and sulfonic acids. Electron poor and sterically demanding amines can result in very slow reactions; however, Pci derivatives for challenging starting materials could still be achieved in reasonable time periods by omitting the CH2Cl2 and increasing the reaction temperature (condition ii at 50 °C and condition iii at 90 °C).
Scheme 2. Synthesis of N-substituted Pci compounds.
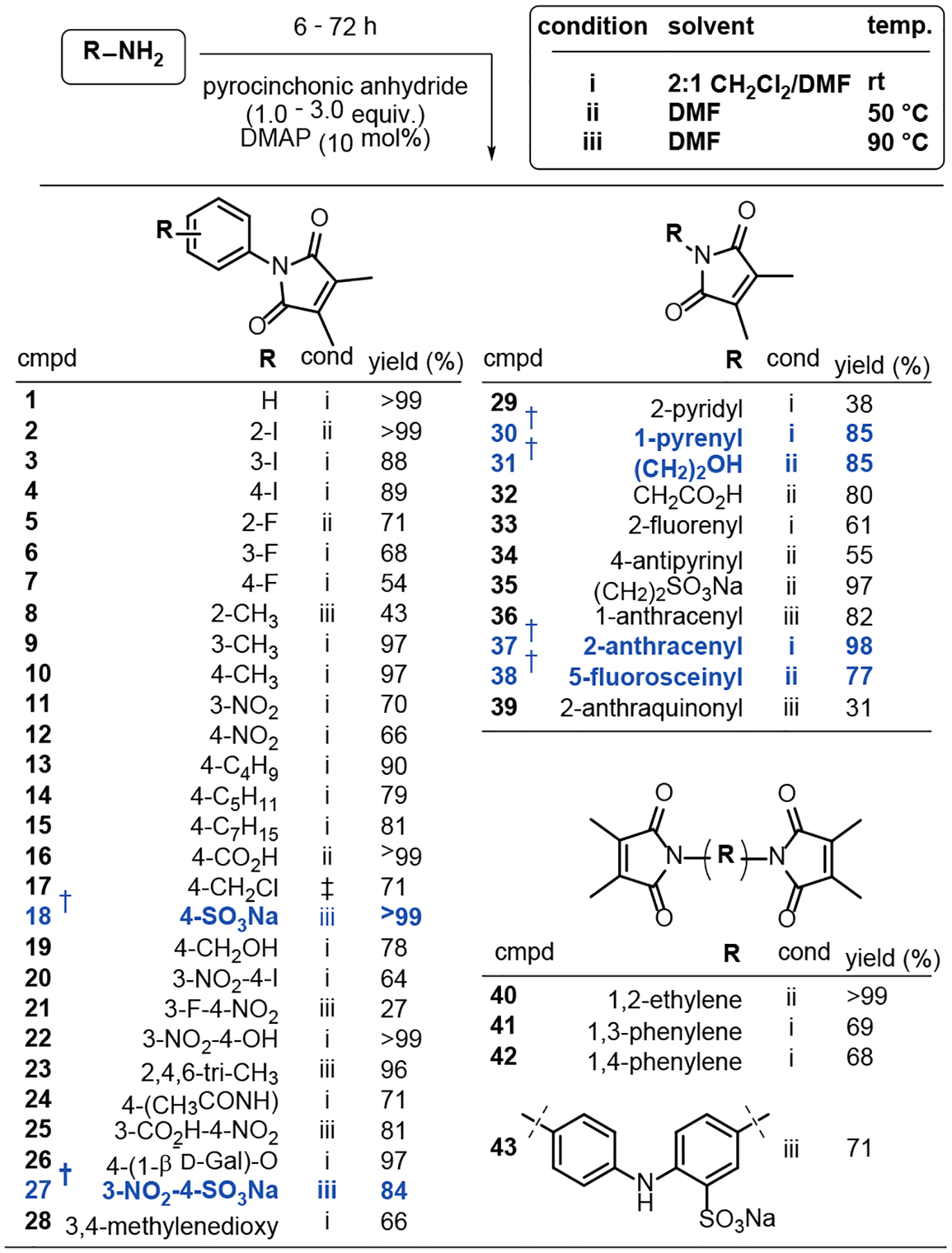
†Compounds 18, 27, 30, 31, 37, and 38 (in blue) are the subject of bioconjugation experiments reported in the following sections. ‡Synthesized from 19 via reaction with MsCl/Et3N. The identities of 18 and 27 were unambiguously confirmed by single crystal X-ray diffraction analysis (Supporting Information Section A).
In the following sections, the ethanolamine derivative 31, polyaromatic derivatives 30 and 37, and the fluorescein derivative 38 were used as probes to study the surprisingly reluctant thio-Michael addition of pyrocinchonimides. Then, experiments involving the sulfanilic acid derivatives 18 and 27 establish a previously unreported transpyrocinchonimidation reaction with proteins.
Pyrocinchonimides Do Not Bioconjugate to Free Thiols
Pci participation in thio-Michael reactions appears sporadically in the prior literature. The Nε-Pci derivative of Nα-acetyl lysine was reported to undergo a thio-Michael reaction with methyl 3-mercaptopropionate and Nα-acetylcysteine methyl ester.60 Reactions of glutathione (GSH) with N-(2-hydroxyethyl)pyrocinchonimide (32) and N-phenylpyrocinchonimide (1) have also been reported.26,61 However, in at least one counter example, no reaction was observed after treatment of a Pci derivative with GSH.62 No spectra are provided for the thio-Michael Pci adducts in these prior reports.
In our hands Pci compounds are not reactive towards thiols. NMR experiments failed to detect any transformation of the Pci groups of 30 and 37 after prolonged exposure (3 h) to 10 mol% excess ß-mercaptoethanol (BME) at elevated temperatures (50 °C) in DMSO-d6. In contrast, conventional maleimides were consumed within minutes under identical conditions (Scheme 3 and Figure S1 in the Supporting Information Section B). Further NMR experiments with GSH and water-soluble Pci derivatives 17 and 31 established the general lack of reactivity to be independent of the Pci N-substituent or choice of solvent. The rate constant for pyrocinchonimide reactions with thiols has been previously shown to be slowed by a factor of 3.15×105 compared to maleimides.26 However, we have failed to observe any appreciable formation of thiol-Pci adducts, even in the presence of large excess of free thiol for extended periods. It is possible that the conjugate addition could occur at extreme temperatures or pH, outside of acceptable ranges for bioconjugation.
Scheme 3. Attempted small molecule thio-Michael reactions. Reactions of Pci compounds 18, 31, 30 and 37 with GSH and BME.
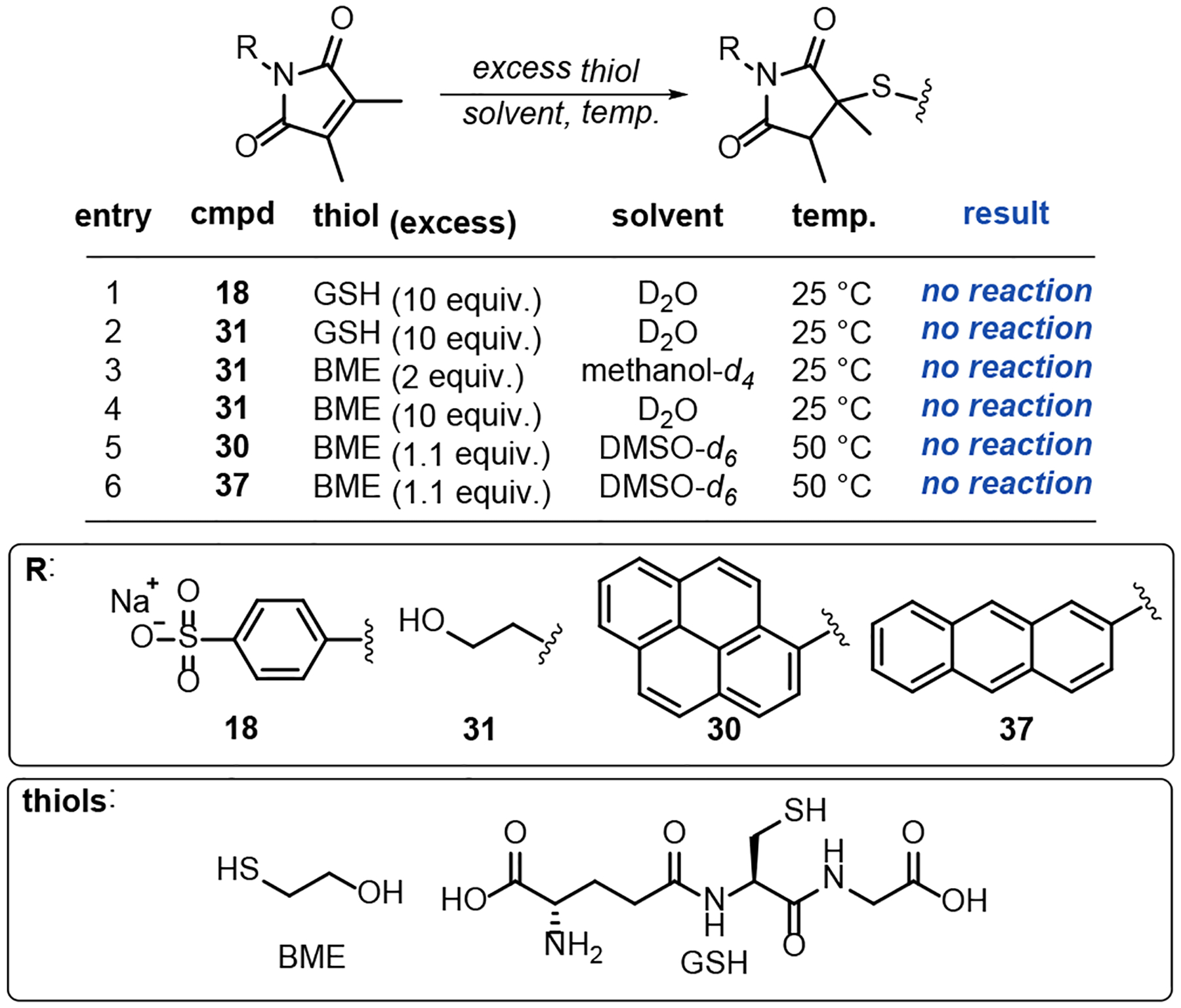
Additional reactions and positive controls appear in the Supporting Information Section B. No reaction indicates undetectable thio- or aza- Michael addition products in an NMR experiment with a cryoprobe-equipped 600 MHz spectrometer.
The reaction of Pci groups with cysteine residues on proteins was also investigated using single cysteine and cysteine-free variants of dihydrofolate reductase (DHFR-Cys and DHFR) and an exonuclease deficient Klenow fragment of DNA polymerase I (KF-Cys and KF). Fluoroscein derivatives 5-F-Pci (38) and its standard maleimide analog 5-F-Mal were chosen as fluorescent reporters, and the bioconjugation result was evaluated by SDS-PAGE (Figure 2 and Figure S2 in the Supporting Information Section B). The two KF variants readily reacted with 5-F-Mal, and no difference in fluorescence intensity for the Cys and Cys-free variants was observed. The apparent unimportance of a free cysteine residue in the bioconjugation of KF with 5-F-Mal is striking, but not entirely unexpected. Despite their reputation for exclusive thiol reactivity, maleimides are known to react with other nucleophiles, and especially amines.63–67 5-F-Pci (38) also reacted to a small extent with both KF variants, suggesting a Cys-independent addition occurs at a low level for KF. These 5-F-Pci (38) conjugates cannot be detected or identified by mass spectrometry (MS), indicative of their vanishingly small quantities. The low fluorescence of the reaction with 5-F-Pci (38), compared to its maleimide analog, is commensurate with its greatly attenuated thiol-maleimide bioconjugation efficiency, and corroborates the results obtained from the small molecule studies.
Figure 2. Probing the elusive Pci thio-Michael bioconjugation.
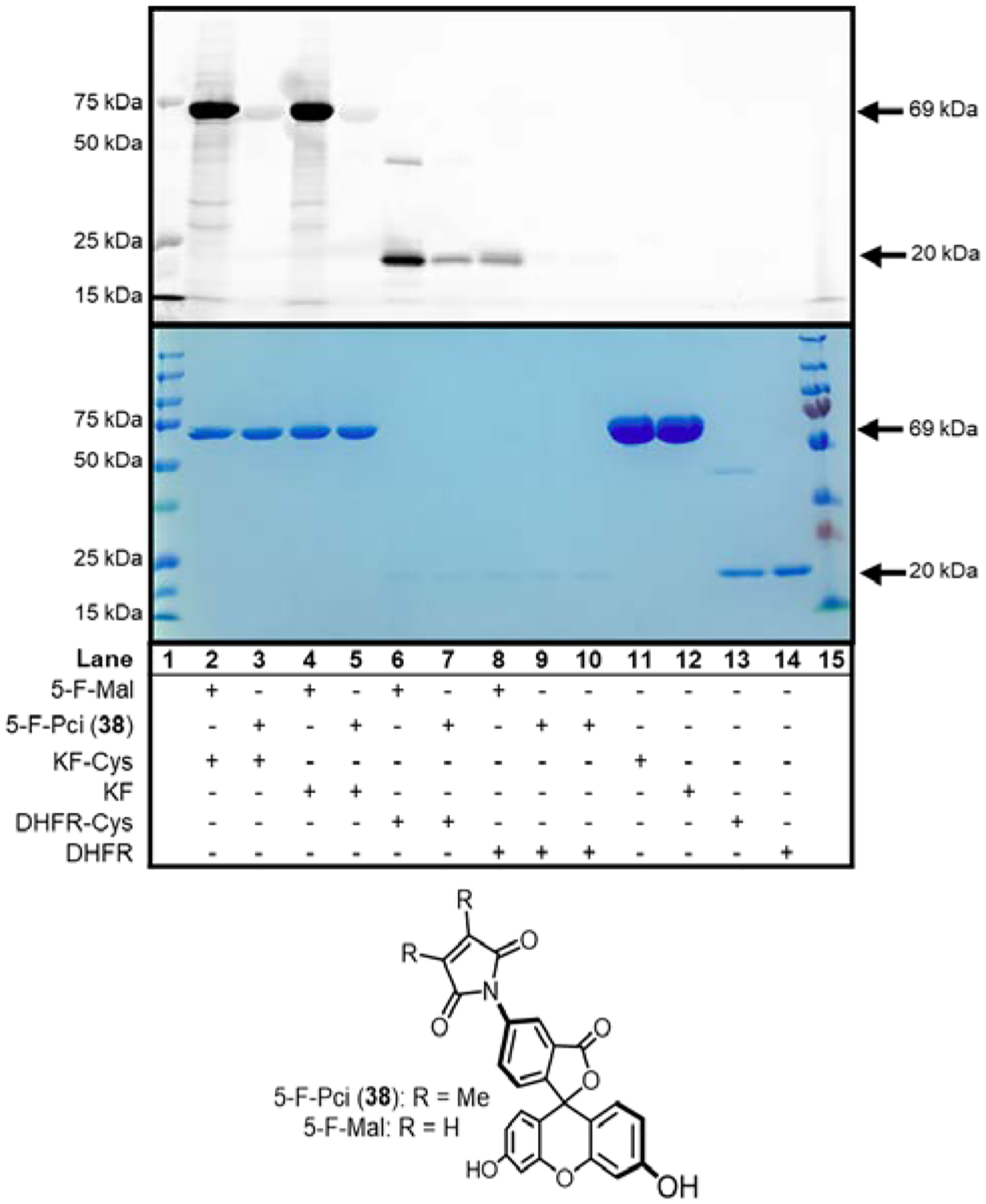
In this representative 15% SDS-PAGE, KF and DHFR are analyzed after treatment with 5-F-Pci (38) or 5-F-Mal for 2 h at pH 7.2 and 25 °C. The protein gels were visualized by a Typhoon scanner with fluorescence excitation at 256 nm (top gel), then stained with Coomassie brilliant blue as a control for loading and protein purity (lower gel). The reaction of single cysteine proteins with 5-F-Pci (38) uncovered Cys-dependent fluorescence for DHFR-Cys (lanes 7 and 9–10), but not KF-Cys (lanes 3 and 5). Conventional 5-F-Mal yielded efficient fluorescent labeling of single cysteine proteins as expected (lanes 2 and 6), but also extensive Cys independent labeling (lanes 4 and 8). Negative control experiments featured identical conditions without 5-F-Pci (38) or 5-F-Mal treatment (lanes 11–14). The precision plus Kaleidoscope standards fluorescent ladder (lane 1) and PageRuler plus prestained protein ladder (lane 15) were used as references.
The reactions of 5-F-Pci (38) and 5-F-Mal with DHFR are equally informative. As with KF, the presence of a free cysteine is unnecessary for the formation of bioconjugates with 5-F-Mal, although DHFR presents less non-specific labeling than KF. Remarkably, bioconjugation of DHFR with 5-F-Pci (38) appears cysteine-dependent, but the dimness of the band permits only a conservative estimate for this reaction’s efficiency, which again failed to yield products detectable by MS. Taken together, the results with both proteins demonstrate Pci compounds, unlike conventional maleimides, are not good candidates for bioconjugation via the canonical thio-Michael reaction. This may not be a surprising result; the two methyl substituents on the Pci heterocycle are expected to contribute approximately −12 kcal·mol−1 of stability to the alkene, relative to the unsubstituted maleimide, through hyperconjugative effects alone.68 This inference can explain the lack of both thio- and aza-Michael addition reactions by Pci compounds.
The Transpyrocinchonimidation Reaction
In aqueous conditions, pyrocinchonimides equilibrate between their imide and non-isolable amic acid forms.54–57 The mechanism likely proceeds via nucleophilic attack of a water molecule or hydroxide ion at the imide carbonyl groups, followed by elimination of the amide, resulting in the ring-opened structure. The product amic acid then rapidly recyclizes via the reverse process, reaching an equilibrium position that is strongly influenced by pH.54 This reactivity forms the mechanistic rationale for the following observations.
In addition to oxygen-based nucleophiles, nitrogen atoms can attack Pci carbonyls. In the presence of free amines, and presumably operating by the same mechanism, transient pyrocinchonic diamides can be expected to form. In this situation, the ring-closing step associated with Pci re-formation must occur with the ejection of a constituent nitrogen atom. Thermodynamic considerations dictate that the amine with the lowest pKa offers the best potential leaving group during this step. Therefore, leveraging the established mechanism for pyrocinchonimide equilibration, it should be possible to achieve an overall imide-transfer reaction from an electron-depleted nitrogen atom to an electron-rich one (Scheme 4).
Scheme 4. Mechanism for transpyrocinchonimidation.
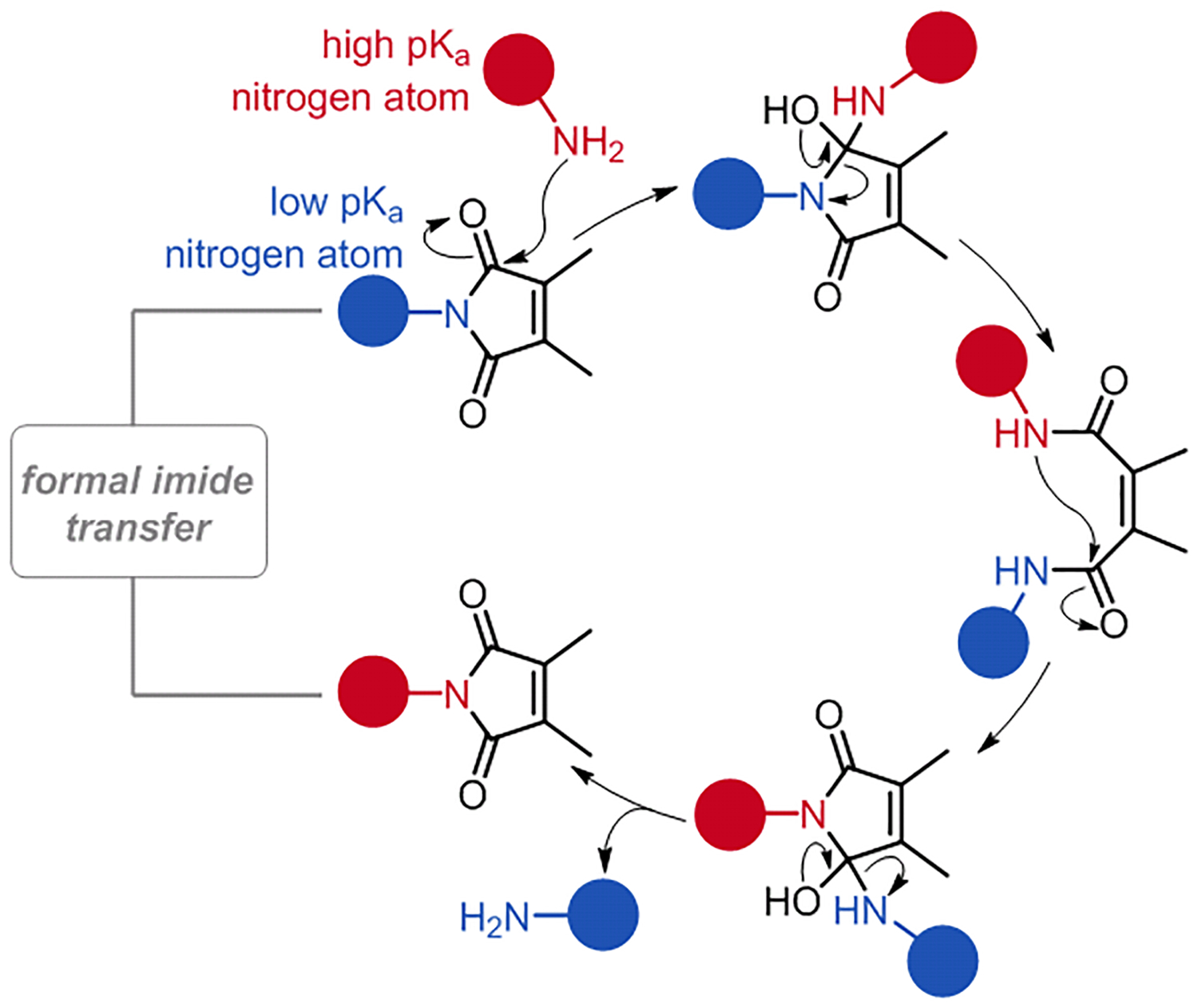
Attack of the pyrocinchonimide carbonyl groups by an electron-rich 1° amine can result in an overall imide transfer reaction (proton transfers omitted for simplicity). The depicted process is reversible and thermodynamically controlled.
The operability of this concept is now demonstrated. Compound 18 was originally synthesized for its ease of use in bioconjugation experiments owing to its excellent stability and solubility in water. Compound 18 is derived from sulfanilic acid, which features an ammonium ion >10-fold more acidic than acetic acid (pKa 3.23 vs 4.75).69 The electron deficiency of the nitrogen atom is further frustrated by the withdrawing effects of the twin acyl substituents of the Pci motif in 18. In the presence of the considerably more basic ε-NH2 of lysine residues (pKa ≈ 10.5) on target biomolecules, a formal imide transfer (transpyrocinchonimidation) was consistently observed. The reaction is a close analogue of a reported phthaloyl (Phth) protecting group installation method, where Phth-transfer also occurs from an acidic (carbamate) nitrogen atom.70 Compound 27, which carries an additional electron-withdrawing nitro substituent on the sulfanilic acid portion of the molecule, was designed to further lower the pKa for the pyrocinchonimide donor nitrogen atom and accelerate the imide transfer.
To further investigate the imide transfer reaction, compounds 18 and 27 were reacted with human insulin which provides a small, readily characterized peptide hormone to receive the imide. First, 20 μM insulin solutions were incubated with 100 mM 18 or 27 at 25 °C for 2 h; imide-transfer products were quantified by whole-protein ESI LC-MS, and the modified residues were subsequently identified by digest analysis (Figure 3a). The deconvoluted mass spectra were integrated over the regions corresponding to the calculated masses of the starting material and bioconjugates within a ± 0.5 Da range, and the relative intensities were calculated as a proportion of the sum total. Consistent with the proposed mechanism, insulin samples treated with 27 exhibited the highest proportions of Pci modification (Figure 3b). Additionally, insulin modified with 18 resulted exclusively in a single modification (1×Pci) of the peptide (within noise levels), whereas 27 delivered high proportions of the doubly-modified species (2×Pci).
Figure 3. Transpyrocinchonimidation of insulin.
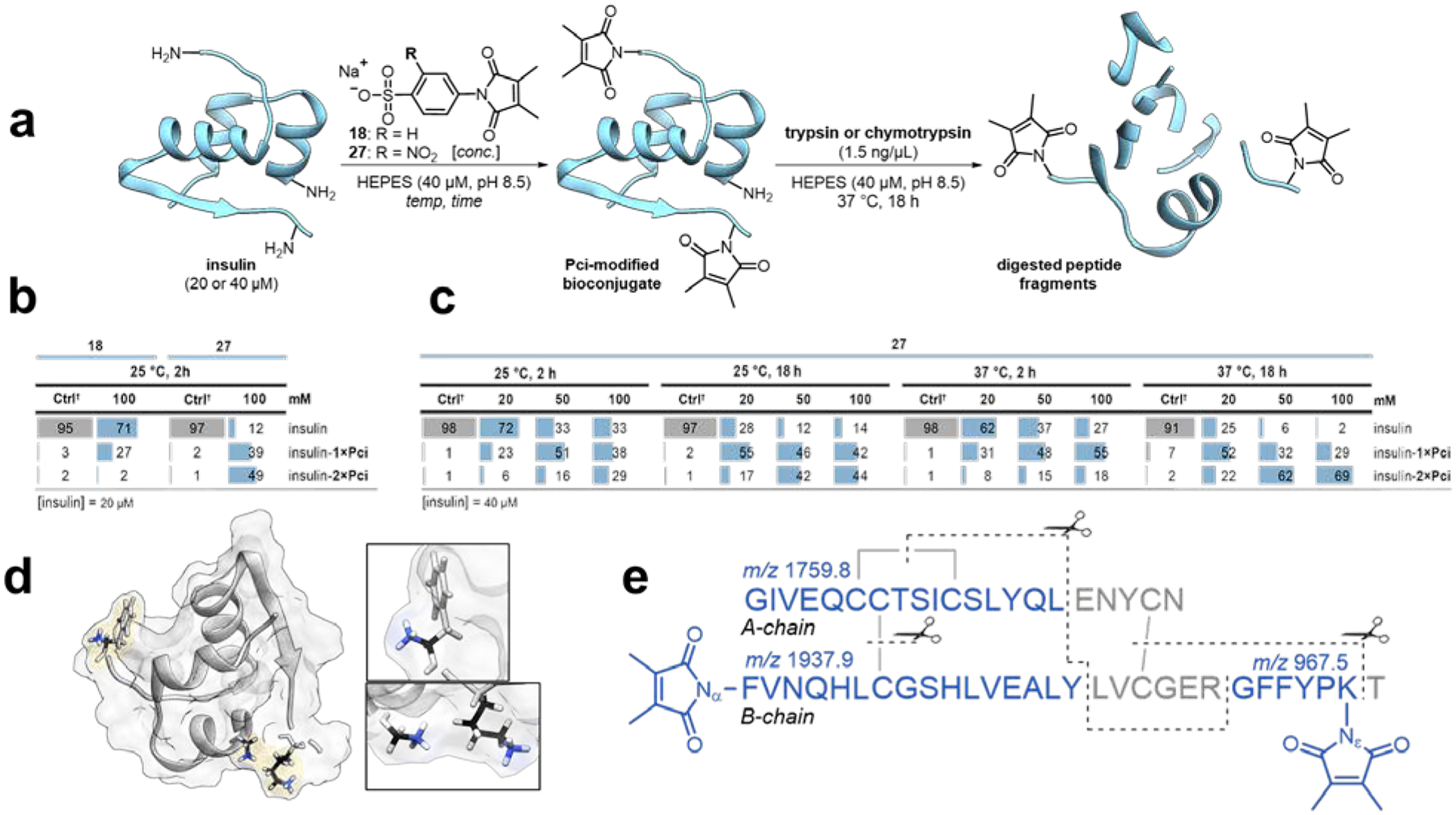
a) Schematic for Pci transfer followed by intact protein ESI LC-MS and tandem digest MALDI-TOF of insulin-Pci bioconjugates. b-c) Estimated proportions (%) of Pci bioconjugates detected in reaction mixtures, calculated as the quotient of the signal intensities ascribed to the starting human, monomeric insulin and its bioconjugates. Peak integration values obtained from the deconvoluted spectra were used to perform this analysis, which assumes equal ionizability for the insulin starting material and the Pci-modified products. d) This view of insulin’s structure highlights the two N-terminal α-NH2 groups and the ε-NH2 of B-chain K29 (PDB: 1MSO). e) Two modification sites were identified on the insulin B-chain by trypsin or chymotrypsin digest analysis: the N-terminal α-NH2, and the K29 ε-NH2. No modification was detected on the A chain’s α-NH2. The detected fragment masses, and corresponding cleavage sites are indicated. †Control samples were not exposed to the Pci-transfer reagents; this measurement can assess background noise in the mass ranges of the detected bioconjugates and helps to benchmark confidence in product detection.
Next, compound 27 (20 to 100 mM) was incubated with insulin (40 μM) in pH 8.5 solution at 25 and 37 °C for 2 and 18 h, and the full matrix of these conditions was explored (Figure 3c). At the most aggressive conditions examined (37 °C, 18 h, 100 mM 27), the unmodified insulin levels were undetectable above the noise, and 69% of the bioconjugates carried two Pci groups. Notably, only two of the three available amines in insulin were modified; the ε-NH2 nitrogen atom of insulin’s one lysine side chain, and one of the two N-termini participated in the imide-transfer with either reagent, despite all three atoms being solvent-exposed. For comparison, bioconjugation with an NHS ester modified all three amines. (Figure 3d–e and Figure S3 in the Supporting Information Section B). Thus, imide transfer by compound 27 to insulin reaction exhibits an innate selectivity for certain amines.
The imide transfer reaction was then evaluated in a more challenging context. Trastuzumab, also known as Herceptin (Genentech), is an antibody that binds to human epithelial growth factor receptor 2 (HER2), and is a biotherapeutic treatment for HER2-positive breast cancer.71 Methods for preparing well-defined trastuzumab bioconjugates are in high demand for antibody-mediated delivery of chemo- and radiotherapeutic agents to tumor sites.72,73 Two 3.5 μM trastuzumab solutions were incubated with 3.5 and 8.75 mM 27 at 25 °C for 2 and 18 h (conditions I and II, respectively) in buffered 40 mM HEPES (pH 8.5). Prior to LC-MS analysis, the product mixtures were stripped of their N-linked surface glycans via digestion with PNGase F at 37 °C for 18 h.74 ESI-MS analysis of the intact antibodies revealed average drug-to-antibody ratios (DAR, with Pci representing the ‘drug’)75,76 of 0.9 and 2.3 for conditions I and II, respectively. In the product mixture obtained from I, the trastuzumab-1×Pci bioconjugate was found to be the most dominant signal, save for signal from the unmodified antibody. The trastuzumab-4×Pci bioconjugate was the highest level of modification observed for this condition (Figure 4). In contrast, condition II yielded a more extensively modified antibody product mixture, with the lowest ion count observed for the unmodified antibody. The modification density was also increased, with at most eight Pci-moieties detected per antibody, and the most dominant signal was assigned to the trastuzumab-2×Pci bioconjugate.
Figure 4. Transpyrocinchonimidation of trastuzumab.
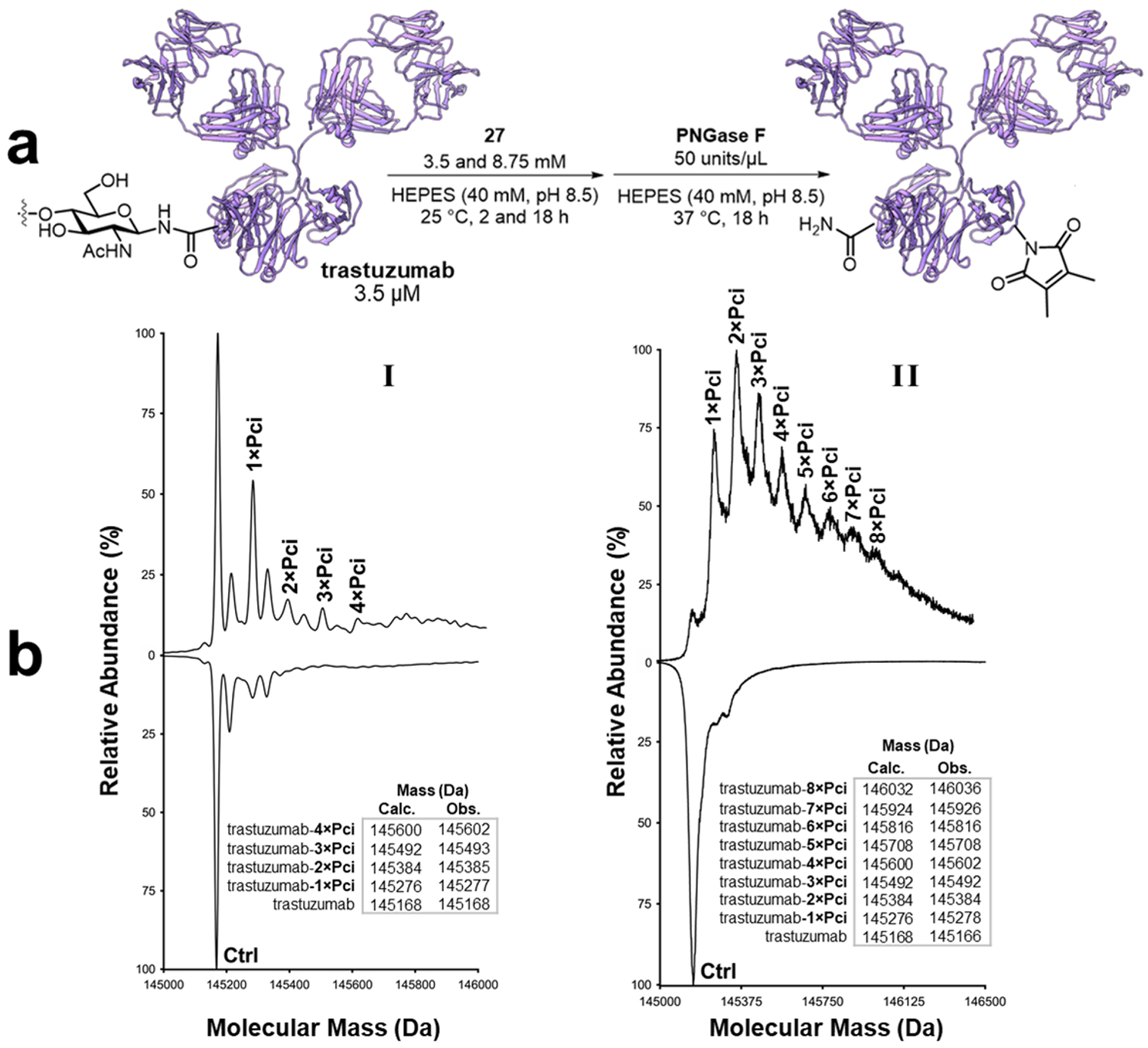
a) Schematic depiction of Pci bioconjugation of trastuzumab, followed by deglycosylation prior to ESI LC-MS analysis. b) Deconvoluted mass spectra of deglycosylated trastuzumab Pci-bioconjugates after treatment with (I) 3.5 mM 27 for 2 h, or (II) 8.75 mM 27 for 18 h at 25 °C. Traces on the top-half of the graph were obtained for the Pci-modified samples; traces on the bottom-half of the graph were obtained from the unmodified control samples of trastuzumab.
The cytotoxic trastuzumab ADC, ‘emtansine’ (Kadcyla), exhibits an average DAR of 3.5, also with as many as eight modifications per antibody.77 Even small increases in DAR values can dramatically increase product heterogeneity. Lysine-modified ADCs with DARs in the range 0–6, with approximately 40–45 lysine residues available for modification, can generate a combinatorial library with over four million unique members.73,78,79 Such heterogeneity can result in unwanted behavior, including toxicity and loss of efficacy.80,81 The challenge of addressing the combinatorial diversity of lysine-targeted bioconjugates has only recently started to receive a reply.82–85 The low DAR values obtained with the Pci reagent 27 suggest that transpyrocinchonimidation may be a viable addition to the NHS alternatives toolkit, particularly in situations where bioconjugate heterogeneity is a persistent problem.
Protein-bound Pci groups could feasibly be expected to migrate from the bioconjugate to off-target amines. To examine this possibility, Pci-modified insulin bioconjugates were incubated with a series of potential small molecule-, peptide-, and protein-based Pci-scavengers for periods of up to 22 days (Figures S4–S5 in the Supporting Information Section B). The insulin bioconjugate was observed to undergo a slow, pH-dependent reversion to its unmodified form; however, no evidence for Pci-exchange to amine groups on scavenger molecules was found. This behavior is typical of a class of bioconjugates intended for extended release of therapeutic payloads from targeted delivery systems, such as hydrogels and nanoparticles.86–88
Our attention then turned to developing a Pci reagent with a covalently attached payload. Our laboratory uses bioconjugation for construction of single molecule devices based on single-walled carbon nanotube field-effect transistors (SWCNT-FETs).89–91 These devices rely on the quasi-irreversible binding of aromatic hydrocarbon-based anchors to the SWCNT side-wall.92 Our previously reported strategies for achieving protein-nanotube bioconjugates exclusively applied N-(1-pyrenyl)maleimide, reacting with a single-cysteine mutant of the target protein. Here, a Pci reagent was designed to target amines, not Cys thiols, and the pyrene substituent is carried by the Pci ß-carbon, rather than its heterocyclic N-atom. The key step in the synthesis of the target is a frustratingly stubborn Wittig condensation, involving the phosphorus ylide derived from citraconimide. (Scheme 5). The poor performance of this process is likely the result of a combination of ylide stabilization via its betaine resonance, and the poor reactivity of the aryl aldehyde (compared to the alkyl aldehyde precedents).93–96 Improved procedures for introducing cargo to the Pci motif are currently in active development.
Scheme 5.
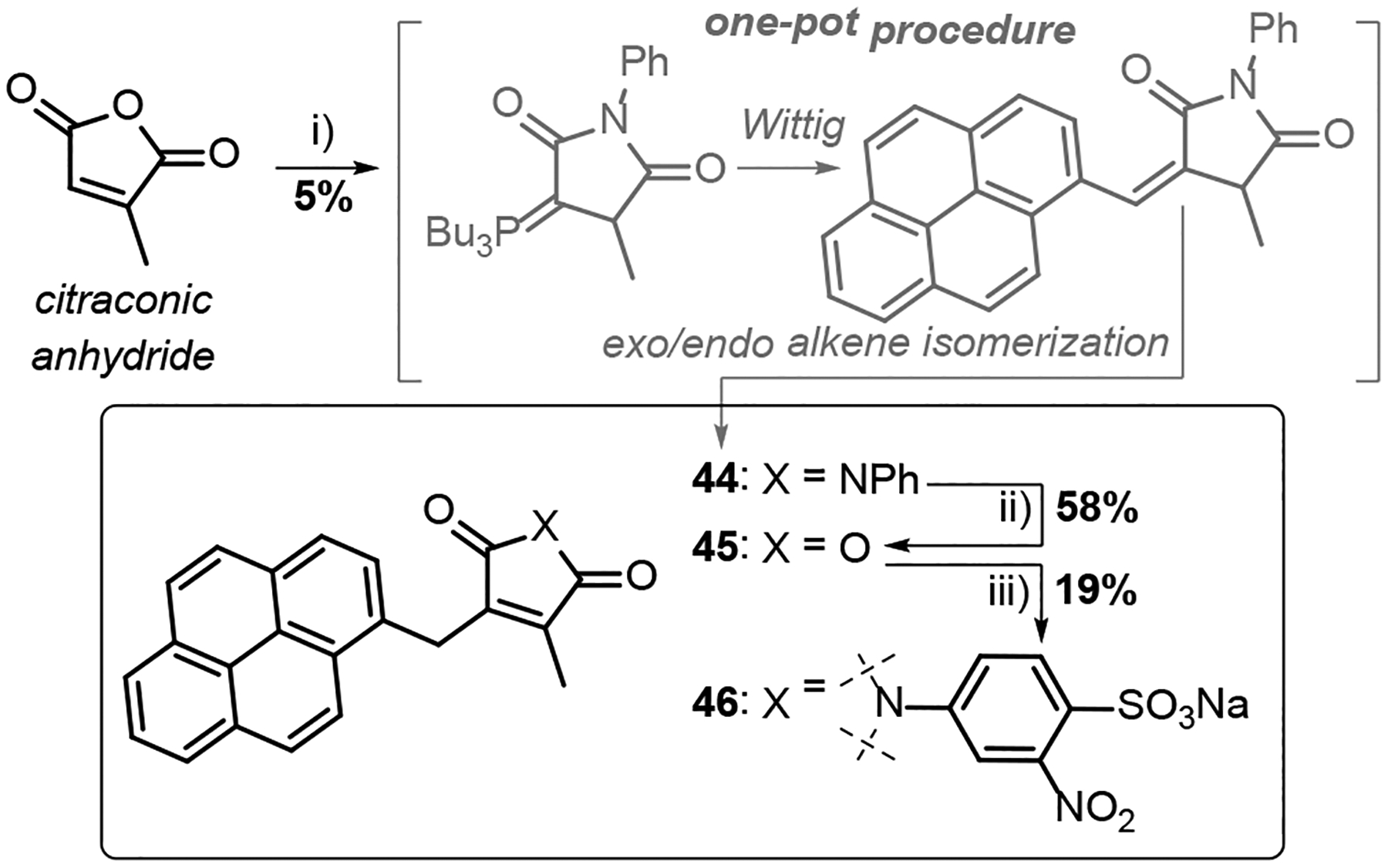
Reagents and conditions i) PhNH2, AcOH, 160 °C, 1 h, then Ac2O, 160 °C, 0.5 h, then PBu3, pyrene-1-carboxaldehyde, NaOAc 160 °C, 48 h, then aq. HCl, 180 °C, 16 h. ii) NaOH, MeOH, CH2Cl2, rt, 30 min;97 iii) sodium 4-sulfonato-3-nitraniline, DMF, 90 °C, 48 h. Compound 44, the product of the multistep one-pot procedure, was unambiguously identified by single-crystal X-ray diffraction analysis (Supporting Information Section A).
The reactivity of the pyrene-modified Pci reagent 46 was first investigated with insulin. Unsurprisingly, the cumbersome steric character and greatly reduced solubility of compound 46 compared to 27 resulted in lower transformation efficiency (Figure S6 in the Supporting Information Section B). Furthermore, the more sterically crowded molecule 46 also affects the intrinsic selectivity of the bioconjugation; exactly one modification of insulin was observed in this example. Compound 46 was then employed for modification of Taq polymerase (Taq). Taq was incubated with compound 46 at 37 °C for 18 h (pH 8.5). After removal of the excess Pci reagent by dialysis, the activity of the modified enzyme was verified by high resolution agarose gel assays to resolve dsDNA from ssDNA.91 The Taq bioconjugate mixture retained 95% of the activity observed for the unmodified enzyme (Figure S7 in the Supporting Information Section B). The bioconjugate mixture was incubated with pristine SWCNTs using standard procedures and single-molecule attachments were investigated by atomic-force microscopy (AFM). AFM imaging revealed a single 1 nm feature, consistent with previous observations of similar proteins (Figure 5).
Figure 5. Applying the imide transfer to pyrene bioconjugation of Taq. A schematic diagram of a SWCNT-FET noncovalently bioconjugated to a single molecule of Taq.
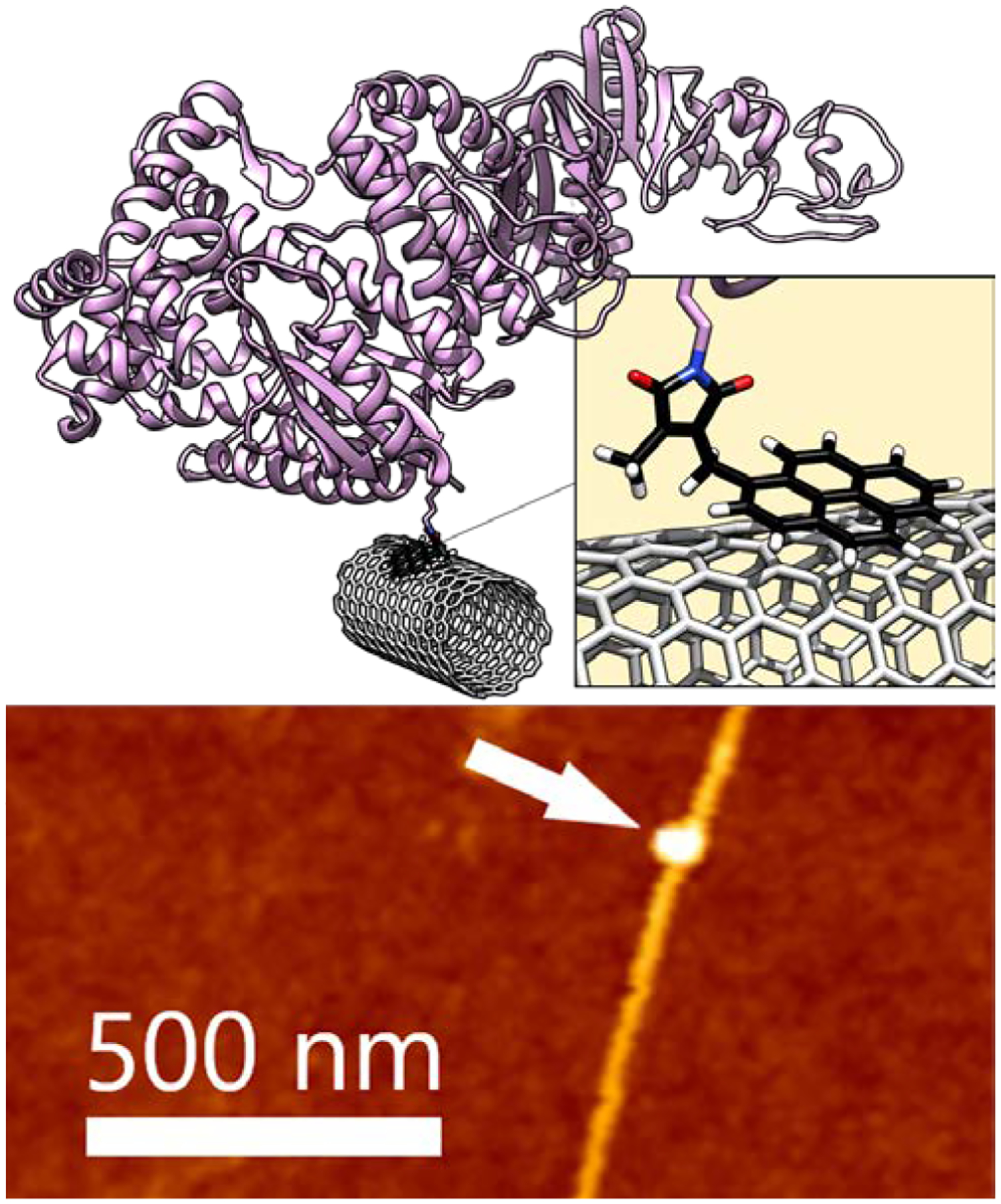
A pyrene-Pci molecule, 46, (black) is adhered to the SWCNT-FET through π−π stacking. Atomic force microscopy shows the expected 1−2 nm diameter of the SWCNT-FET with a single Taq attachment obtained using linker 46 (1 nm, white arrow).
UV-Activated Cycloadditions
Motivated by the efficiency of 27 to introduce Pci into proteins, we sought reactions capable of further elaborating the Pci bioconjugates. Cycloadditions of alkenes with maleimide derivatives have been extensively examined in the literature. The synthesis of cyclobutane-containing fused di-, tri- and tetracyclic scaffolds via UV-initiated [2+2] cycloadditions of maleimide-like compounds are especially well-documented. In 2001, an unfamiliar [5+2] cycloaddition of Pci was reported,98 and was then applied to a challenging total synthesis.99 The reaction involves a formal insertion of two alkene carbons into the Pci ring between the nitrogen atom and one of its carbonyls, resulting in an overall two-carbon ring-expansion, and the formation of a seven-membered dihydroazepinedione (Dhzd). Initial mechanistic proposals for this unusual transformation have since been succeeded by robust, experimentally validated models.100–102
Two hypothesized photochemical processes compete to support the [2+2] or the [5+2] cycloaddition pathways. Sensitized irradiation in the presence of an appropriate chromophore populates the C=C triplet state and facilitates the [2+2] pathway. Direct irradiation, which operates in the absence of a photosensitizer, populates the C-N singlet state, causing homolytic cleavage, and results in an amide/acyl diradical intermediate. While energy transfer from the singlet to the triplet state can occur through intersystem crossing, the presence of electron-donating methyl groups at the alkene weakens the C-N bond and supports diradical formation.102 The diradical intermediate undergoes a formal [5+2] cycloaddition in the presence of alkenes, or else spontaneously recombines to regenerate the starting Pci molecule.
These UV-activated processes offer a bioorthogonal route to further functionalization of Pci-modified proteins (Scheme 6a). Two alkene-based secondary modifiers were chosen to investigate this approach: a commercially available PEG derivative and an EDTA-derived metal ligand (47), which was synthesized using a one-pot method inspired by existing procedures103 (Scheme 6b). The PEG derivative demonstrates a typical solubilization scheme for biotherapeutics. The introduction of metal complexes to proteins has a wide scope of utility, including the chelation of radioisotopes for targeted radiotherapy, as contrast and imaging agents in nuclear medicine, and as luminescent probes, serving as alternatives to traditional organic fluorophores. EDTA is a general-purpose ligand that can form stable complexes with a variety of different metal atoms with many potential applications. Solutions of Pci-modified insulin (~40 μM) were incubated with mPEG-alkene-1K (40 mM, MW ~1000 Da) or 47, under direct irradiation from a 450 W medium-pressure Hg lamp (228–420 nm), and temperature regulation below 8 °C was achieved with a high-velocity flow of chilled water (Figure S8 in the Supporting Information Section B).
Scheme 6.
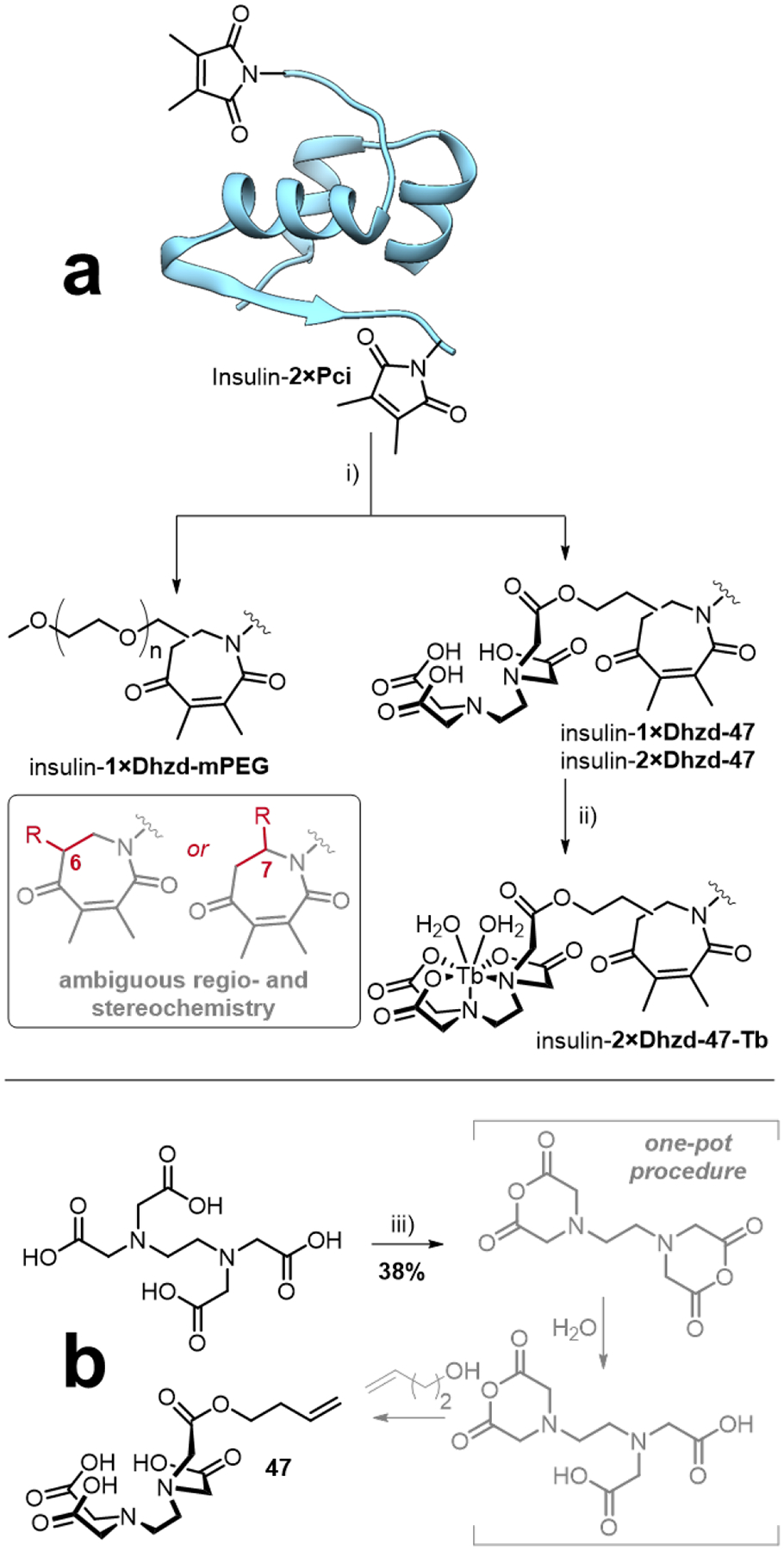
Reagents and conditions i) 40 mM mPEG-alkene 1K or 40 mM 47, hν 228–420 nm, 40 mM HEPES (pH 8.5), 5–10 °C, 4 h; ii) 10 mM terbium(III) chloride, 40 mM HEPES (pH 8.5), 25 °C, 18 h; iii) Ac2O, DMF, 60 °C, 24 h, then H2O (1 equiv.), 75 °C, 2 h, then 3-buten-1-ol, DMAP, 80 °C, 24 h.
Analysis of the product mixtures by ESI LC-MS revealed masses consistent with the target cycloadducts (Figure S8 in the Supporting Information Section B). The insulin-1×Dhzd-mPEG cycloadduct presented a range of m/z signals separated by ~44 Da, a typical polymeric PEG MS fingerprint. Only the 1×Dhzd-mPEG attachment was detected, with a second Pci group remaining unmodified. In the cycloaddition reaction with 47, both the 1× and 2×Dhzd-47 insulin adducts were observed. After incubation with terbium(III) chloride prior to further analysis, only the insulin-2×Dhzd-47-Tb complex was observed (as the dihydrate). The increased background noise of the mass spectra precluded estimation of product yields; photolytic decomposition of the protein likely occurred, resulting in additional mass fragments. Furthermore, the structures in Scheme 6 are drawn for the [5+2] product Dhzd compounds, which is concordant with the published mechanism. However, the [2+2] cyclobutane product with the same mass could also form, and thus the exact identity of the cycloadducts remains unclear. Resolving this uncertainty will likely require a comprehensive X-ray crystallography experiment beyond the scope of the present study.
Conclusion
The Pci bioconjugation reported here offers useful capabilities and a reaction modality that is distinctive from maleimides and NHS esters. The investigation demonstrates that Pci compounds react very poorly with thiols via the conventional thio-Michael pathway, despite scattered reports to the contrary. Therefore, Pci reagents are likely to be compatible with thiols, including Cys residues on protein targets and bystanders, and the reducing agents BME and GSH. The imide transfer reaction may also be helpful in addressing frontier challenges in bioconjugation chemistry. We demonstrate effective imide transfer can result in low conjugation loadings and DARs compared to NHS methods, even when very large excesses of Pci reagent are employed (Figures S9–10 in the Supporting Information Section B). The Pci modification of proteins is slowly lost to hydrolysis at neutral-to-basic pH; however, no evidence for protein-protein imide exchange was found (Figures S4–5 in the Supporting Information Section B). The fact that the hydrolyzed anhydride/diacid is unreactive towards amine groups on neighboring proteins also suggests that prior activation (e.g. 18 and 27) is essential. In contrast to maleimide reversion, the terminal nature of Pci hydrolysis may protect against unwanted off-target modification in situ.104 We expect the Pci group may be of value in situations where ADC development is confounded by product heterogeneity issues, or where a slow release of the payload is desirable. Lastly, evidence has been collected that suggests tandem Pci-transfer/UV-cycloaddition procedures may be a viable approach to the future development of Pci capabilities.
Although Pci is a structural analogue of maleimide, the Pci-transfer is more functionally aligned with an emerging class of alternative amine-targeting methods and reagents,105 including the 6 π -azaelectrocyclization reagents,106–110 squarimides,111–113 iminoboronates,114 TAK-242 derivatives,115,116 diazonium terephthalate esters,117 two-component formaldehyde/indole couplings,118 phthalaldehydes,119 acylfluorides,120 palladium-catalyzed arylations,121 sulfonyl acrylates,122,123 fluorophenyl esters,85 DSH-mediated acylations,124 heteroaryl methylsulfones,125 dinitroimidazoles,126 and linchpin-directed modification strategies.127,128 The Pci-transfer reagents 18 and 27 were used in unusually high concentrations in the present study, compared with typical concentration ranges employed in bioconjugate chemistry. However, these are only the first generation of these reagents. Exploration of other water-soluble variants with alternative substitution patterns around the aniline portion of the Pci transfer reagent is fertile ground for further research. We anticipate that new iterations of these reagents with improved reaction kinetics and defined site-selectivity profiles will be the subjects of future studies.
General Experimental Methods
Small molecule high-resolution mass spectra (HRMS) were obtained by electrospray ionization (ESI) on a Waters (Micromass) LCT Premier equipped with a time-of-flight (TOF) mass analyzer. Proton (1H, 400 and 600 MHz), carbon (13C, 100 and 150 MHz), and fluorine (19F, 564 MHz) nuclear magnetic resonance (NMR) spectra were obtained on Bruker instruments equipped with a switchable QNP or BBFO probe. NMR samples were prepared in CDCl3 and DMSO-d6, and residual protonated solvent was used as an internal chemical shift standard. 1H and 13C assignments were determined using HSQC and 10 Hz optimized HMBC 2D-NMR analyses. Optical rotatory powers were measured in the sodium D band (589 nm) on a Jasco P-1020 polarimeter. Fourier transform infrared (FTIR) spectra were obtained as neat samples on a Jasco 4700 attenuated total reflectance (ATR) instrument using a diamond-coated zinc selenide sample accessory. X-ray data was collected on a Bruker SMART APEX II diffractometer. Flash chromatography was carried out on silica gel 60 according to the method established by Still et al.,129 or using a Teledyne Isco CombiFlash preparative chromatography system. Analytical thin layer chromatography (tlc) was conducted on aluminum-backed 2 mm thick silica gel 60 GF254, and chromatograms were visualized under a UV lamp (254 and 365 nm) or by chemical staining with ceric ammonium molybdate (Hanessian’s stain) or potassium permanganate (KMnO4).
Transpyrocinchonimidation products were quantified by ESI LC-MS (ACQUITY UPLC H-class system, Xevo G2-XS QTof, Waters Corporation). The protein was separated from reaction buffer using a phenyl guard column at 45 °C (ACQUITY UPLC BEH Phenyl VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 mm X 5 mm, Waters). The 5 min analytical method used 0.2 mL/min flow rate of a gradient of Buffer A consisting of 0.1% formic acid in water and Buffer B, 100% acetonitrile (Table S1). The Xevo Z-spray source was operated in positive MS resolution mode, 400–4000 Da, with a capillary voltage of 3000 V and a cone voltage of 40 V (NaI / CsI and Leu-enkephalin lock-mass calibration).130 Nitrogen was used as the desolvation gas at 350 °C at a flow-rate of 800 L/h. Total average mass spectra were reconstructed from the charge state ion series using the MaxEnt1 algorithm from Waters MassLynx software V4.1 SCN949 according to the manufacturer’s instructions. The parameters for the deconvolution of insulin and insulin-Pci were 0.05 Da peak separation, and the combined ion series for 650–2300 m/z was processed under MaxEnt1 using an acquisition range of 5700–7300 Da, 0.5 Da/channel resolution, Uniform Gaussian with 0.5 Da width at half height, minimum intensity ratios of 33% for left and right, and 12 maximum number of iterations. The deconvolution of trastuzumab and trastuzumab-Pci applied the following modifications to the parameters mentioned above: combined ion series for m/z 1800–4000 processed under MaxEnt1 using 145,000–150,000 Da range and 1.0 Da/channel resolution. A representative total ion chromatogram (TIC), combined m/z spectrum, and deconvoluted spectrum for insulin and trastuzumab can be found in the Supporting Information Section B, Figure S12 and Figure S18. Data files were exported in .txt format for further processing (GraphPad Prism, v. 8.0) and quantification (MS-Excel). A typical semi-quantitative assessment of the relative abundances (%) as measured by the ESI LC-MS analysis is illustrated in Supporting Information Section B, Figure S19. This method was applied to the analysis of Pci bioconjugates with both insulin and trastuzumab.
Supplementary Material
Acknowledgements
We gratefully acknowledge the support of the NHGRI of the NIH (1R01HG009188-01). M.B.R. thanks the American Australian Association and the Dow Chemical Company for a Dow Chemical Company Fellowship. K.N.G. was supported by a training grant from NIH (1R01HG009188-01) and a National Science Foundation Graduate Research Fellowship Program (DGE-1839285). J.A.G. thanks the Maximizing Access to Research Careers (MARC) Program, funded by the NIH (T34GM069337). R.P.D. was supported by a training grant from the NIH (5T32CA009054-37). This work would not have been possible without the assistance and support of Dmitry Fishman and the UCI Laser Spectroscopy Laboratory, and Felix Grun and the UCI Mass Spectrometry Facility. We would also like to thank Donald Laudicina for assistance with the ESI LC-MS analysis of trastuzumab-Pci bioconjugates, and Samantha Beasley and Robert Spitale for use of the flash chromatography system, and Edward Nelson for the generous gift of the Trastuzumab antibody. Research was supported by an Anti-Cancer Challenge research grant from the University of California, Irvine Chao Family Comprehensive Cancer Center.
Footnotes
Supporting Information. NMR and IR spectra for compounds 1−47, crystallographic data for 18, 27 and 44 (CIF), additional bioconjugate MS and gel electrophoresis experiments, bioconjugate stability/exchange assays, detailed materials and protocols.
References
- (1).Ravasco JMJM; Faustino H; Trindade A; Gois PMP Bioconjugation with Maleimides: A Useful Tool for Chemical Biology Chem. - Eur. J 2019, 25, 43–59. [DOI] [PubMed] [Google Scholar]
- (2).Nair DP; Podgórski M; Chatani S; Gong T; Xi W; Fenoli CR; Bowman CN The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry Chem. Mater 2014, 26, 724–744. [Google Scholar]
- (3).Baldwin AD; Kiick KL Tunable Degradation of Maleimide–Thiol Adducts in Reducing Environments Bioconjugate Chem 2011, 22, 1946–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fontaine SD; Reid R; Robinson L; Ashley GW; Santi DV Long-Term Stabilization of Maleimide–Thiol Conjugates Bioconjugate Chem 2015, 26, 145–152. [DOI] [PubMed] [Google Scholar]
- (5).Szijj PA; Bahou C; Chudasama V Minireview: Addressing the Retro-Michael Instability of Maleimide Bioconjugates Drug Discov. Today Technol 2018, 30, 27–34. [DOI] [PubMed] [Google Scholar]
- (6).Gregory JD The Stability of N-Ethylmaleimide and Its Reaction with Sulfhydryl Groups J. Am. Chem. Soc 1955, 77, 3922–3923. [Google Scholar]
- (7).Finnegan RA; Mueller WH Base-Catalyzed Addition and Solvolysis Reactions of N-Phenylmaleimide in Methanol J. Pharm. Sci 1965, 54, 1257–1260. [DOI] [PubMed] [Google Scholar]
- (8).Machida M; Machida MI; Kanaoka Y Hydrolysis of N-Substituted Maleimides: Stability of Fluorescence Thiol Reagents in Aqueous Media Chem. Pharm. Bull 1977, 25, 2739–2743. [Google Scholar]
- (9).Kalia J; Raines RT Catalysis of Imido Group Hydrolysis in a Maleimide Conjugate Bioorg. Med. Chem. Lett 2007, 17, 6286–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tan X-J; Cheng S-S; Shi Y; Xing D-X; Liu Y; Li H; Feng W-Q; Yang J-B Hydrolytic Degradation of N,N′-Ethylenedimaleimide: Crystal Structures of Key Intermediates and Proposed Mechanisms J. Mol. Struct 2016, 1125, 514–521. [Google Scholar]
- (11).Shafer DE; Inman JK; Lees A Reaction of Tris(2-Carboxyethyl)Phosphine (Tcep) with Maleimide and A-Haloacyl Groups: Anomalous Elution of Tcep by Gel Filtration Anal. Biochem 2000, 282, 161–164. [DOI] [PubMed] [Google Scholar]
- (12).Kantner T; Watts AG Characterization of Reactions between Water-Soluble Trialkylphosphines and Thiol Alkylating Reagents: Implications for Protein-Conjugation Reactions Bioconjugate Chem 2016, 27, 2400–2406. [DOI] [PubMed] [Google Scholar]
- (13).Wei C; Zhang G; Clark T; Barletta F; Tumey LN; Rago B; Hansel S; Han X Where Did the Linker-Payload Go? A Quantitative Investigation on the Destination of the Released Linker-Payload from an Antibody-Drug Conjugate with a Maleimide Linker in Plasma Anal. Chem 2016, 88, 4979–4986. [DOI] [PubMed] [Google Scholar]
- (14).Ohri R; Bhakta S; Fourie-O’donohue A; Dela Cruz-Chuh J; Tsai SP; Cook R; Wei B; Ng C; Wong AW; Bos AB et al. High-Throughput Cysteine Scanning to Identify Stable Antibody Conjugation Sites for Maleimide- and Disulfide-Based Linkers Bioconjugate Chem 2018, 29, 473–485. [DOI] [PubMed] [Google Scholar]
- (15).Nunes JPM; Vassileva V; Robinson E; Morais M; Smith MEB; Pedley RB; Caddick S; Baker JR; Chudasama V Use of a Next Generation Maleimide in Combination with Thiomab Antibody Technology Delivers a Highly Stable, Potent and near Homogeneous Thiomab Antibody-Drug Conjugate (Tdc) RSC Adv 2017, 7, 24828–24832. [Google Scholar]
- (16).Schumacher FF; Nunes JPM; Maruani A; Chudasama V; Smith MEB; Chester KA; Baker JR; Caddick S Next Generation Maleimides Enable the Controlled Assembly of Antibody-Drug Conjugates Via Native Disulfide Bond Bridging Org. Biomol. Chem 2014, 12, 7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Forte N; Livanos M; Miranda E; Morais M; Yang X; Rajkumar VS; Chester KA; Chudasama V; Baker JR Tuning the Hydrolytic Stability of Next Generation Maleimide Cross-Linkers Enables Access to Albumin-Antibody Fragment Conjugates and Tri-Scfvs Bioconjugate Chem 2018, 29, 486–492. [DOI] [PubMed] [Google Scholar]
- (18).Hull EA; Livanos M; Miranda E; Smith MEB; Chester KA; Baker JR Homogeneous Bispecifics by Disulfide Bridging Bioconjugate Chem 2014, 25, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee MTW; Maruani A; Baker JR; Caddick S; Chudasama V Next-Generation Disulfide Stapling: Reduction and Functional Re-Bridging All in One Chem. Sci 2016, 7, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Marculescu C; Kossen H; Morgan RE; Mayer P; Fletcher SA; Tolner B; Chester KA; Jones LH; Baker JR Aryloxymaleimides for Cysteine Modification, Disulfide Bridging and the Dual Functionalization of Disulfide Bonds Chem. Commun 2014, 50, 7139–7142. [DOI] [PubMed] [Google Scholar]
- (21).Morais M; Nunes JPM; Karu K; Forte N; Benni I; Smith MEB; Caddick S; Chudasama V; Baker JR Optimisation of the Dibromomaleimide (Dbm) Platform for Native Antibody Conjugation by Accelerated Post-Conjugation Hydrolysis Org. Biomol. Chem 2017, 15, 2947–2952. [DOI] [PubMed] [Google Scholar]
- (22).Nunes JPM; Morais M; Vassileva V; Robinson E; Rajkumar VS; Smith MEB; Pedley RB; Caddick S; Baker JR; Chudasama V Functional Native Disulfide Bridging Enables Delivery of a Potent, Stable and Targeted Antibody-Drug Conjugate (Adc) Chem. Commun 2015, 51, 10624–10627. [DOI] [PubMed] [Google Scholar]
- (23).Pye H; Butt MA; Reinert HW; Maruani A; Nunes JPM; Marklew JS; Qurashi M; Funnell L; May A; Stamati I et al. A Her2 Selective Theranostic Agent for Surgical Resection Guidance and Photodynamic Therapy Photochem. Photobiol. Sci 2016, 15, 1227–1238. [DOI] [PubMed] [Google Scholar]
- (24).Hong LPT; Scoble JA; Doughty L; Coia G; Williams CC Cancer-Targeting Antibody–Drug Conjugates: Site-Specific Conjugation of Doxorubicin to Anti-Egfr 528 Fab′ through a Polyethylene Glycol Linker Aust. J. Chem 2011, 64, 779–789. [Google Scholar]
- (25).Earl RA; Clough FW; Townsend LB Chemical Investigations of Citraconimide J. Heterocycl. Chem 1978, 15, 1479–1483. [Google Scholar]
- (26).Miyadera T; Kosower EM Receptor Site Labeling through Functional Groups. 2. Reactivity of Maleimide Groups J. Med. Chem 1972, 15, 534–537. [DOI] [PubMed] [Google Scholar]
- (27).Zhang Y; Zhou X; Xie Y; Greenberg MM; Xi Z; Zhou C Thiol Specific and Tracelessly Removable Bioconjugation Via Michael Addition to 5-Methylene Pyrrolones J. Am. Chem. Soc 2017, 139, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bahou C; Spears RJ; Aliev AE; Maruani A; Fernandez M; Javaid F; Szijj PA; Baker JR; Chudasama V Use of Pyridazinediones as Extracellular Cleavable Linkers through Reversible Cysteine Conjugation Chem. Commun 2019, 55, 14829–14832. [DOI] [PubMed] [Google Scholar]
- (29).Aly MRE; Castro-Palomino JC; Ibrahim E-SI; El-Ashry E-SH; Schmidt RR The Dimethylmaleoyl Group as Amino Protective Group. Application to the Synthesis of Glucosamine-Containing Oligosaccharides Eur. J. Org. Chem 1998, 2305–2316. [Google Scholar]
- (30).耿 雪敏; 朱 英红; 陈 万里. The Synthetic Method of 3- Amino Pyrrolidine Hydrochloride 2018. CN109369493A.
- (31).Acharya S; Panda SK; Das P; Agarwal B Novel Compositions and Therapeutic Methods 2017. WO2017173024A1.
- (32).陈 小龙; 陆 跃乐; 李 忠; 范 永仙; 张 丽君; 李 延娟; 沈 寅初. N-Substituted Cis-Butene Imidodicarbonic Diamide Compound, Preparation Method Thereof and Antibacterial Application 2020. CN107033060B.
- (33).陈 小龙; 陆 跃乐; 范 永仙; 李 延娟; 沈 寅初. N Substitution Maleimide Compounds and Its Preparation Based on Pharmaceutical Intermediate Are Studied with Antibacterial Activity 2017. CN107162950A.
- (34).Roth M 2,3-Dimethylmaleimido-Alkyl Haloacetates 1987. US4656292A.
- (35).Baumann M; Kvita V; Roth M; Waterhouse JS Imidyl Compounds 1978. US4107174A.
- (36).Lohmann D; Wyler S Silanes Containing Imide Groups 1981. US4271074A.
- (37).Kume T; Goto T; Kamochi A; Yamaguchi N; Yanagi A; Hayakawa H; Yagi S 1-(1,4-Benzoxazin-3-on-6-Yl)-Dialkylmaleimides and Use as Herbicides 1988. US4729784A.
- (38).Kume T; Goto T; Kamochi A; Yanagi A; Yagi S; Miyauchi H 2,5-Dihydropyrroles 1989. US4828604A.
- (39).Moser H; Pissiotas G; Brunner H-G; Bohner B; Baumann M N-Phenyl-Maleimides and Herbicidal and Plant Growth Regulating Methods of Use Thereof 1989. US4804400A.
- (40).Kohsaka H; Takase M Butenoic Acid Derivatives and Use as Herbicides 1991. US5059237A.
- (41).Arabori H; Yamazaki S; Arahira M; Murakami A N-Substituted-3-(Nitrogen-Containing 5-Membered Ring)-Benzenesulfonamide Derivatives, and Herbicidal Compositions 1992. US5127937A.
- (42).Wepplo PJ Substituted Benzisoxazole and Benzisothiazole Herbicidal Agents 1996. US5484763A.
- (43).Lachia MD; Screpanti C; De Mesmaeker A; Lumbroso AFJC; Rendine S Plant Growth Regulating Compounds 2017. US9751867B2.
- (44).Morris JA; Hennessy AJ; Boehmer JE Herbicidal Pyrrolone Derivatives 2016. WO2016071360A1.
- (45).Suzuki N; Nihei Y; Ichinose H; Tanaka H; Yasa N; Hatanaka T; Masuzawa Y; Nakanishi E; Kondo N Benzene Compounds 2007. US20070105899A1.
- (46).Izumimoto N; Kawai K; Kawamura K; Fujimura M; Komagata T Remedies or Preventives for Urinary Frequency or Urinary Incontinence and Morphinan Derivatives Having Nitrogen-Containing Heterocyclic Group 2008. US7320984B2.
- (47).Jung MS; Cho CK; Kim MJ; Park CW; Ahn YH; Jang SW; Chang JH Liquid Crystal Thermoset Monomer or Oligomer, Thermosetting Liquid Crystal Polymer Composition Comprising the Same and Printed Circuit Board Using the Same 2010. US7655155B2.
- (48).Urbanski M; Xiang A; Zeck R Substituted Pyrrolones as Allosteric Modulators of Glucokinase 2014. US8680122B2.
- (49).Haikarainen A; Kumpulainen E; Pohjakallio A; Pystynen J; Wang S Benzodioxane Derivatives and Their Pharmaceutical Use 2018. WO2018002437A1.
- (50).Geraa JF; Lichtenstein L; Jung ME; Lee J; Holmes B; Benavides-Serrato A Inhibitors of Ires-Mediated Protein Synthesis 2017. WO2017192665A1.
- (51).岡崎 宏紀; 湯浅 宏昭; 落合 洋文; 赤木 博; 雄大 佐藤; 空處 弘一. Oligosaccharide Compound, Manufacturing Method Therefor, and Intermediate Thereof 2018. JPWO2013141350A1.
- (52).Fleš D; Vuković R; Kuzmić AE; Bogdanić G; Piližota V; Karlović D; Markuš K; Wolsperger K; Vikić-Topić D Synthesis and Spectroscopic Evidences of N-Arylmaleimides and N-Aryl-2,3-Dimethylmaleimides Croat. Chem. Acta 2003, 76, 69–74. [Google Scholar]
- (53).Kshirsagar UA; Argade NP Facile Approach to Diverse Range of 1,3-Diaza-Heterocycles: Angular/Linear Selectivity Paradigm and a Remarkable Intramolecular Methyl Migration Tetrahedron 2009, 65, 5244–5250. [Google Scholar]
- (54).Su S; Du F-S; Li Z-C Synthesis and Ph-Dependent Hydrolysis Profiles of Mono- and Dialkyl Substituted Maleamic Acids Org. Biomol. Chem 2017, 15, 8384–8392. [DOI] [PubMed] [Google Scholar]
- (55).Bartlett PD The Chemical Properties of the Methyl Group J. Chem. Ed 1953, 30, 22–31. [Google Scholar]
- (56).Koskikallio J Dimethylmaleic Acid-Dimethylmaleic Anhydride Equilibrium Suom. Kemistil. B 1956, 29B, 5–7. [Google Scholar]
- (57).Koskikallio J Kinetics of the Hydrolysis and Formation of Dimethylmaleic Anhydride in Solvent Mixtures Acta Chem. Scand 1956, 10, 822–830. [Google Scholar]
- (58).Allinger NL; Zalkow V Conformational Analysis. Ix. The Gem-Dimethyl Effect J. Org. Chem 1960, 25, 701–704. [Google Scholar]
- (59).Wheeler OH; Rodríguez EEG Solvolysis of Methylmaleic Anhydrides J. Org. Chem 1961, 26, 4763–4764. [Google Scholar]
- (60).Tressl R; Wondrak G; Kersten E; Rewicki D Structure and Potential Crosslinking Reactivity of a New Pentose-Specific Maillard Product J. Agr. Food Chem 1994, 42, 2692–2697. [Google Scholar]
- (61).Palanki MSS; Bhat A; Lappe RW; Liu B; Oates B; Rizzo J; Stankovic N; Bradshaw C Development of Novel Linkers to Conjugate Pharmacophores to a Carrier Antibody Bioorg. Med. Chem. Lett 2012, 22, 4249–4253. [DOI] [PubMed] [Google Scholar]
- (62).Koechel DA; Tarloff JB; Rankin GO Acute Effects of Alkylating Agents on Canine Renal Function: Specifically Designed Synthetic Maleimides J. Med. Chem 1983, 26, 85–90. [DOI] [PubMed] [Google Scholar]
- (63).Uno BE; Dicken RD; Redfern LR; Stern Charlotte m.; Krzywicki GG; Scheidt KA Calcium(Ii)-Catalyzed Enantioselective Conjugate Additions of Amines Chemical Sci 2018, 9, 1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Uno BE; Deibler KK; Villa C; Raghuraman A; Scheidt KA Conjugate Additions of Amines to Maleimides Via Cooperative Catalysis Adv. Syn. Cat 2018, 360, 1719–1725. [Google Scholar]
- (65).Zubkov FI; Kvyatkovskaya EA; Nikitina EV; Amoyaw PNA; Kouznetsov VV; Lazarenko VA; Khrustalev VN Comment on “an Unexpected Formation of the Novel 7-Oxa-2-Azabicyclo[2.2.1]Hept-5-Ene Skeleton During the Reaction of Furfurylamine with Maleimides and Their Bioprospection Using a Zebrafish Embryo Model” by C. E. Puerto Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 407 Org. Biomol. Chem 2017, 15, 6447–6450. [DOI] [PubMed] [Google Scholar]
- (66).Alonso CMA; Palumbo A; Bullous AJ; Pretto F; Neri D; Boyle RW Site-Specific and Stoichiometric Conjugation of Cationic Porphyrins to Antiangiogenic Monoclonal Antibodies Bioconjugate Chemistry 2010, 21, 302–313. [DOI] [PubMed] [Google Scholar]
- (67).Mustafa A; Asker W; Khattab S; Zayed S. M. a. D. On the Reactivity of the Unsaturated System in N-Arylmaleimides J. Org. Chem 1961, 26, 787–789. [Google Scholar]
- (68).Braida B; Prana V; Hiberty PC The Physical Origin of Saytzeff’s Rule Angew. Chem. Int. Ed 2009, 48, 5724–5728. [DOI] [PubMed] [Google Scholar]
- (69).Serjeant EP; Dempsey B Ionisation Constants of Organic Acids in Aqueous Solution IUPAC Chemical Data Series No. 23 1979, New York: Pergamon Press, Inc., 251. [Google Scholar]
- (70).Nefkens GHL Synthesis of Phthaloyl Amino Acids under Mild Conditions Nature 1960, 185, 309. [DOI] [PubMed] [Google Scholar]
- (71).Wilson FR; Coombes ME; Brezden-Masley C; Yurchenko M; Wylie Q; Douma R; Varu A; Hutton B; Skidmore B; Cameron C Herceptin (Trastuzumab) in Her2-Positive Early Breast Cancer: A Systematic Review and Cumulative Network Meta-Analysis Syst. Rev 2018, 7, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Scott AM; Wolchok JD; Old LJ Antibody Therapy of Cancer Nat. Rev. Cancer 2012, 12, 278. [DOI] [PubMed] [Google Scholar]
- (73).Agarwal P; Bertozzi CR Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development Bioconjugate Chem 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Tarentino AL; Plummer TH In Methods Enzymol; Academic Press: 1994; Vol. 230, p 44–57. [DOI] [PubMed] [Google Scholar]
- (75).Wakankar A; Chen Y; Gokarn Y; Jacobson FS Analytical Methods for Physicochemical Characterization of Antibody Drug Conjugates mAbs 2011, 3, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Basa L In Antibody-Drug Conjugates; Ducry L, Ed.; Humana Press: Totowa, NJ, 2013, p 285–293. [Google Scholar]
- (77).Kim MT; Chen Y; Marhoul J; Jacobson F Statistical Modeling of the Drug Load Distribution on Trastuzumab Emtansine (Kadcyla), a Lysine-Linked Antibody Drug Conjugate Bioconjugate Chem 2014, 25, 1223–1232. [DOI] [PubMed] [Google Scholar]
- (78).Wang L; Amphlett G; Blättler WA; Lambert JM; Zhang W Structural Characterization of the Maytansinoid–Monoclonal Antibody Immunoconjugate, Hun901–Dm1, by Mass Spectrometry Protein Sci 2005, 14, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ponte JF; Sun X; Yoder NC; Fishkin N; Laleau R; Coccia J; Lanieri L; Bogalhas M; Wang L; Wilhelm S et al. Understanding How the Stability of the Thiol-Maleimide Linkage Impacts the Pharmacokinetics of Lysine-Linked Antibody–Maytansinoid Conjugates Bioconjugate Chem 2016, 27, 1588–1598. [DOI] [PubMed] [Google Scholar]
- (80).Hamblett KJ; Senter PD; Chace DF; Sun MMC; Lenox J; Cerveny CG; Kissler KM; Bernhardt SX; Kopcha AK; Zabinski RF et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate Clin. Cancer Res 2004, 10, 7063–7070. [DOI] [PubMed] [Google Scholar]
- (81).Tsuchikama K; An Z Antibody-Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries Protein & Cell 2018, 9, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Chen X; Muthoosamy K; Pfisterer A; Neumann B; Weil T Site-Selective Lysine Modification of Native Proteins and Peptides Via Kinetically Controlled Labeling Bioconjugate Chem 2012, 23, 500–508. [DOI] [PubMed] [Google Scholar]
- (83).Forte N; Benni I; Karu K; Chudasama V; Baker JR Cysteine-to-Lysine Transfer Antibody Fragment Conjugation Chem. Sci 2019, 10, 10919–10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Matos MJ; Oliveira BL; Martínez-Sáez N; Guerreiro A; Cal PMSD; Bertoldo J; Maneiro M; Perkins E; Howard J; Deery MJ et al. Chemo- and Regioselective Lysine Modification on Native Proteins J. Am. Chem. Soc 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Pham GH; Ou W; Bursulaya B; Didonato M; Herath A; Jin Y; Hao X; Loren J; Spraggon G; Brock A et al. Tuning a Protein-Labeling Reaction to Achieve Highly Site Selective Lysine Conjugation ChemBioChem 2018, 19, 799–804. [DOI] [PubMed] [Google Scholar]
- (86).Baldwin AD; Kiick KL Reversible Maleimide–Thiol Adducts Yield Glutathione-Sensitive Poly(Ethylene Glycol)–Heparin Hydrogels Polym. Chem 2013, 4, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Bandyopadhyay A; Gao J Targeting Biomolecules with Reversible Covalent Chemistry Curr. Opin. Chem. Biol 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Cao Z; Li W; Liu R; Li X; Li H; Liu L; Chen Y; Lv C; Liu Y Ph- and Enzyme-Triggered Drug Release as an Important Process in the Design of Anti-Tumor Drug Delivery Systems Biomed. Pharmacother 2019, 118, 109340. [DOI] [PubMed] [Google Scholar]
- (89).Choi Y; Moody IS; Sims PC; Hunt SR; Corso BL; Perez I; Weiss GA; Collins PG Single-Molecule Lysozyme Dynamics Monitored by an Electronic Circuit Science 2012, 335, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Olsen TJ; Choi Y; Sims PC; Gul OT; Corso BL; Dong C; Brown WA; Collins PG; Weiss GA Electronic Measurements of Single-Molecule Processing by DNA Polymerase I (Klenow Fragment) J. Am. Chem. Soc 2013, 135, 7855–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Pugliese KM; Gul OT; Choi Y; Olsen TJ; Sims PC; Collins PG; Weiss GA Processive Incorporation of Deoxynucleoside Triphosphate Analogs by Single-Molecule DNA Polymerase I (Klenow Fragment) Nanocircuits J. Am. Chem. Soc 2015, 137, 9587–9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Chen RJ; Zhang Y; Wang D; Dai H Noncovalent Sidewall Functionalization of Single-Walled Carbon Nanotubes for Protein Immobilization J. Am. Chem. Soc 2001, 123, 3838–3839. [DOI] [PubMed] [Google Scholar]
- (93).Hedaya E; Theodoropulos S The Preparation and Reactions of Stable Phosphorus Ylides Derived from Maleic Anhydrides, Maleimides or Isomaleimides Tetrahedron 1968, 24, 2241–2254. [Google Scholar]
- (94).Mangaleswaran S; Argade NP An Efficient Synthesis of Dimethylmaleic Anhydride Synthesis 2002, 2002, 0865–0868. [Google Scholar]
- (95).Easwar S; Argade NP A Facile Synthesis and Enzymatic Resolution of Naturally Occurring Remotely Functionalized Alkylmethylmaleic Anhydrides from Aspergillus Wentii: Aspergillus Acids a-D Synthesis 2006, 2006, 831–838. [Google Scholar]
- (96).Desai SB; Argade NP A Facile Synthesis of Ras Farnesyl-Protein Transferase Inhibitor Chaetomellic Acid A J. Org. Chem 1997, 62, 4862–4863. [Google Scholar]
- (97).Theodorou V; Skobridis K; Tzakos AG; Ragoussis V A Simple Method for the Alkaline Hydrolysis of Esters Tetrahedron Lett 2007, 48, 8230–8233. [Google Scholar]
- (98).Booker-Milburn Kevin i.; Anson Christopher e.; Clissold C; Costin Nicola j.; Dainty Richard f.; Murray M; Patel D; Sharpe A Intramolecular Photocycloaddition of N-Alkenyl Substituted Maleimides: A Potential Tool for the Rapid Construction of Perhydroazaazulene Alkaloids Eur. J. Org. Chem 2001, 2001, 1473–1482. [Google Scholar]
- (99).Booker-Milburn KI; Dudin LF; Anson CE; Guile SD Formal Intramolecular [5 + 2] Photocycloaddition Reactions of Maleimides: A Novel Approach to the Cde Ring Skeleton of (−)-Cephalotaxine Org. Lett 2001, 3, 3005–3008. [DOI] [PubMed] [Google Scholar]
- (100).Roscini C; Cubbage KL; Berry M; Orr-Ewing AJ; Booker-Milburn KI Reaction Control in Synthetic Organic Photochemistry: Switching between [5+2] and [2+2] Modes of Cycloaddition Angew. Chem. Int. Ed 2009, 48, 8716–8720. [DOI] [PubMed] [Google Scholar]
- (101).Kumarasamy E; Raghunathan R; Jockusch S; Ugrinov A; Sivaguru J Tailoring Atropisomeric Maleimides for Stereospecific [2 + 2] Photocycloaddition—Photochemical and Photophysical Investigations Leading to Visible-Light Photocatalysis J. Am. Chem. Soc 2014, 136, 8729–8737. [DOI] [PubMed] [Google Scholar]
- (102).Raghunathan R; Kumarasamy E; Jockusch S; Ugrinov A; Sivaguru J Engaging Electronic Effects for Atropselective [5+2]-Photocycloaddition of Maleimides Chem. Commun 2016, 52, 8305–8308. [DOI] [PubMed] [Google Scholar]
- (103).Takeshita T; Shimohara TA; Maeda S Synthesis of Edta‐Monoalkylamide Chelates and Evaluation of the Surface‐Active Properties J. Am. Oil Chem. Soc 1982, 59, 104–107. [Google Scholar]
- (104).Shen B-Q; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu S-F; Mai E et al. Conjugation Site Modulates the in Vivo Stability and Therapeutic Activity of Antibody-Drug Conjugates Nat. Biotechnol 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- (105).Chaubet G; Thoreau F; Wagner A Recent, Non-Classical, Approaches to Antibody Lysine Modification Drug Discov Today Technol 2018, 30, 21–26. [DOI] [PubMed] [Google Scholar]
- (106).Degruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: A Chemist’s Guide Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Koniev O; Wagner A Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation Chem. Soc. Rev 2015, 44, 5495–5551. [DOI] [PubMed] [Google Scholar]
- (108).Tanaka K; Kitadani M; Fukase K Target-Selective Fluorescent “Switch-on” Protein Labeling by 6π-Azaelectrocyclization Org. Biomol. Chem 2011, 9, 5346–5349. [DOI] [PubMed] [Google Scholar]
- (109).Tanaka K; Masuyama T; Hasegawa K; Tahara T; Mizuma H; Wada Y; Watanabe Y; Fukase K A Submicrogram-Scale Protocol for Biomolecule-Based Pet Imaging by Rapid 6π-Azaelectrocyclization: Visualization of Sialic Acid Dependent Circulatory Residence of Glycoproteins Angew. Chem. Int. Ed 2008, 47, 102–105. [DOI] [PubMed] [Google Scholar]
- (110).Tanaka K; Fujii Y; Fukase K Site-Selective and Nondestructive Protein Labeling through Azaelectrocyclization-Induced Cascade Reactions ChemBioChem 2008, 9, 2392–2397. [DOI] [PubMed] [Google Scholar]
- (111).Rudd SE; Roselt P; Cullinane C; Hicks RJ; Donnelly PS A Desferrioxamine B Squaramide Ester for the Incorporation of Zirconium-89 into Antibodies Chem. Commun 2016, 52, 11889–11892. [DOI] [PubMed] [Google Scholar]
- (112).Ian Storer R; Aciro C; Jones LH Squaramides: Physical Properties, Synthesis and Applications Chem. Soc. Rev 2011, 40, 2330–2346. [DOI] [PubMed] [Google Scholar]
- (113).Marchetti LA; Kumawat LK; Mao N; Stephens JC; Elmes RBP The Versatility of Squaramides: From Supramolecular Chemistry to Chemical Biology Chem 2019, 5, 1398–1485. [Google Scholar]
- (114).Cal PMSD; Vicente JB; Pires E; Coelho AV; Veiros LSF; Cordeiro C; Gois PMP Iminoboronates: A New Strategy for Reversible Protein Modification Journal of the American Chemical Society 2012, 134, 10299–10305. [DOI] [PubMed] [Google Scholar]
- (115).Patterson JT; Wilson HD; Asano S; Nilchan N; Fuller RP; Roush WR; Rader C; Barbas CF Human Serum Albumin Domain I Fusion Protein for Antibody Conjugation Bioconjugate Chem 2016, 27, 2271–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Asano S; Patterson JT; Gaj T; Barbas CF Iii. Site-Selective Labeling of a Lysine Residue in Human Serum Albumin Angew. Chem., Int. Ed 2014, 53, 11783–11786. [DOI] [PubMed] [Google Scholar]
- (117).Diethelm S; Schafroth MA; Carreira EM Amine-Selective Bioconjugation Using Arene Diazonium Salts Org. Lett 2014, 16, 3908–3911. [DOI] [PubMed] [Google Scholar]
- (118).Larda ST; Pichugin D; Prosser RS Site-Specific Labeling of Protein Lysine Residues and N-Terminal Amino Groups with Indoles and Indole-Derivatives Bioconjugate Chem 2015, 26, 2376–2383. [DOI] [PubMed] [Google Scholar]
- (119).Tung CL; Wong CTT; Fung EYM; Li X Traceless and Chemoselective Amine Bioconjugation Via Phthalimidine Formation in Native Protein Modification Org. Lett 2016, 18, 2600–2603. [DOI] [PubMed] [Google Scholar]
- (120).Dovgan I; Ursuegui S; Erb S; Michel C; Kolodych S; Cianférani S; Wagner A Acyl Fluorides: Fast, Efficient, and Versatile Lysine-Based Protein Conjugation Via Plug-and-Play Strategy Bioconjugate Chem 2017, 28, 1452–1457. [DOI] [PubMed] [Google Scholar]
- (121).Lee HG; Lautrette G; Pentelute BL; Buchwald SL Palladium-Mediated Arylation of Lysine in Unprotected Peptides Angew. Chem. Int. Ed 2017, 56, 3177–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Matos MJ; Jiménez-Osés G; Bernardes GJL In Bioconjugation: Methods and Protocols; Massa S, Devoogdt N, Eds.; Springer New York: New York, NY, 2019, p 25–37. [Google Scholar]
- (123).Matos MJ; Oliveira BL; Martinez-Saez N; Bertoldo J; Bernardes GJL; Guerreiro A; Cal PMSD; Bernardes GJL; Maneiro M; Perkins E et al. Chemo- and Regioselective Lysine Modification on Native Proteins J. Am. Chem. Soc 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Yamatsugu K; Furuta M; Xi S; Amamoto Y; Liu J; Kawashima SA; Kanai M Kinetic Analyses and Structure-Activity Relationship Studies of Synthetic Lysine Acetylation Catalysts Biorg. Med. Chem 2018, 26, 5359–5367. [DOI] [PubMed] [Google Scholar]
- (125).Hwang D; Tsuji K; Park H; Burke TR; Rader C Site-Specific Lysine Arylation as an Alternative Bioconjugation Strategy for Chemically Programmed Antibodies and Antibody-Drug Conjugates Bioconjugate Chem 2019, 30, 2889–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Luo Q; Tao Y; Sheng W; Lu J; Wang H Dinitroimidazoles as Bifunctional Bioconjugation Reagents for Protein Functionalization and Peptide Macrocyclization Nat. Commun 2019, 10, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Adusumalli SR; Rawale DG; Thakur K; Purushottam L; Reddy NC; Kalra N; Shukla S; Rai V Chemoselective and Site-Selective Lysine-Directed Lysine Modification Enables Single-Site Labeling of Native Proteins Angew Chem Int Ed 2020, ASAP. [DOI] [PubMed] [Google Scholar]
- (128).Adusumalli SR; Rawale DG; Singh U; Tripathi P; Paul R; Kalra N; Mishra RK; Shukla S; Rai V Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology J. Am. Chem. Soc 2018, 140, 15114–15123. [DOI] [PubMed] [Google Scholar]
- (129).Still WC; Kahn M; Mitra A Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution J. Org. Chem 1978, 43, 2923–2925. [Google Scholar]
- (130).Sztáray J; Memboeuf A; Drahos L; Vékey K Leucine Enkephalin—a Mass Spectrometry Standard Mass Spectrom. Rev 2011, 30, 298–320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.