

Upon stress, JNK stimulates microtubule dynamics in human epithelial cell lines. Henrie et al. show that JNK phosphorylates the rescue factor CLIP-170, increasing its retention frequency on the lattice behind comets, at locations that correspond to potential future rescue sites.
Abstract
The stress-induced c-Jun N-terminal kinase (JNK) controls microtubule dynamics by enhancing both microtubule growth and rescues. Here, we show that upon cell stress, JNK directly phosphorylates the microtubule rescue factor CLIP-170 in its microtubule-binding domain to increase its rescue-promoting activity. Phosphomimetic versions of CLIP-170 enhance its ability to promote rescue events in vitro and in cells. Furthermore, while phosphomimetic mutations do not alter CLIP-170’s capability to form comets at growing microtubule ends, both phosphomimetic mutations and JNK activation increase the occurrence of CLIP-170 remnants on the microtubule lattice at the rear of comets. As the CLIP-170 remnants, which are potential sites of microtubule rescue, display a shorter lifetime when CLIP-170 is phosphorylated, we propose that instead of acting at the time of rescue occurrence, CLIP-170 would rather contribute in preparing the microtubule lattice for future rescues at these predetermined sites.
Graphical Abstract

Introduction
Microtubules (MTs) are dynamic and polarized nanotubes constituted from αβ-tubulin dimers. MT minus ends are often blocked at organizing centers, while MT plus ends explore the cytoplasmic space, thanks to dynamic instability (Mitchison and Kirschner, 1984). The latter consists of alternative phases of MT polymerization and depolymerization, with transitions called catastrophes (from growth or pause to a phase of shrinking) and rescues (from shrinking to pause or to growth).
In cells, the intrinsic properties of MTs are regulated by a variety of microtubule-associated proteins (MAPs). Among them, the plus end–tracking proteins (+TIPs) accumulate at growing plus ends, where they appear as comets (for review, see Akhmanova and Steinmetz, 2015). Cytoplasmic Linker Protein-170 (CLIP-170) was the first +TIP identified (Diamantopoulos et al., 1999; Perez et al., 1999). CLIP-170 functions as a linker for dynein-based MT transport. Its C-terminal motif recruits the p150GLUED subunit of dynactin in neurons to initiate dynein-mediated transport (Moughamian et al., 2013; Nirschl et al., 2016). In nonneuronal cells, CLIP-170 and p150GLUED allow endocytic vesicle transport (Pierre et al., 1992; Kedashiro et al., 2015) and initiate retrograde transport of herpes virus particles (Jovasevic et al., 2015). The N-terminal head domain of CLIP-170 also links the plus ends of MTs to the cortical actin meshwork by interacting with IQGAP1, leading to cell polarization (Fukata et al., 2002). CLIP-170 may also contribute to actin assembly by interacting through its coiled-coil domain with the nucleation and elongation factor mDia (Henty-Ridilla et al., 2016). During mitosis, CLIP-170 is recruited to kinetochores by p150GLUED, where it facilitates chromosome attachment to MT plus ends (Tanenbaum et al., 2006).
Besides these linker functions, CLIP-170 was characterized as a rescue factor in vitro, since its dimerized N-terminal head domain (termed H2) was shown to promote MT nucleation, elongation, and rescues and could do so by interacting with tubulin oligomers (Diamantopoulos et al., 1999; Arnal et al., 2004). In cells, coinhibition of CLIP-170 and of its paralog CLIP-115 using a dominant-negative abolished ∼80% of the rescue events (Komarova et al., 2002), which were restored by the expression of CLIP-170 monomeric N-terminal head domain (termed H1). CLASP is a CLIP-170 partner that also promotes rescues (Mimori-Kiyosue et al., 2005; Al-Bassam et al., 2010) and displays a strong ability to prevent catastrophes (Aher et al., 2018; Lawrence et al., 2018; Lindeboom et al., 2019). CLASP2β/γ activation requires interaction with the coiled-coil domain of CLIP-170 (this is dispensable for CLASP1/2α long isoforms). Activated CLASP mediates rescues in vitro and prevents catastrophes by forming rear comets that protect lagging protofilaments, allowing them to catch the tip of growing MTs (Aher et al., 2018). In contrast, CLIP-170 forms transient immobile remnants on the MT lattice at the trailing end of +TIP comets (Perez et al., 1999). Such remnants can overlap GTP-like islands, which comprise architectural defects and small repaired lattice domains, especially at MT crossings (de Forges et al., 2016). GTP-like islands function as potential rescue hotspots along MTs (Dimitrov et al., 2008; Aumeier et al., 2016; Vemu et al., 2018).
CLIP-170 binding to MTs, which relies on the H1 domain of the protein (Pierre et al., 1992), was reported to be regulated by phosphorylation (Rickard and Kreis, 1991). H1 contains two cytoskeleton-associated protein glycine-rich (CAP-Gly) domains flanked by serine-rich regions and by canonical SxIP and SxIP-like motifs (Chen et al., 2019). These domains participate in targeting CLIP-170 to comets (Pierre et al., 1992), mainly via interactions with α-tubulin and with the End-Binding (EB) +TIP family (Peris et al., 2006; Mishima et al., 2007; Weisbrich et al., 2007; Bieling et al., 2008; Dixit et al., 2009; Chen et al., 2019). The N-terminal domain of CLIP-170 can also associate with the C-terminal region of the protein to allow auto-inhibitory folding (Weisbrich et al., 2007; Goodson et al., 2003; Lansbergen et al., 2004). Phosphorylations in the third serine-rich cluster (which includes Ser312) of the H1 domain promote such intramolecular folding, reducing comet length at growing MT plus ends (Lee et al., 2010). This can be achieved by several protein kinases such as AMP-activated protein kinase (AMPK; Nakano et al., 2010), protein kinase A (Lee et al., 2010), protein kinase N (Collazos et al., 2011), and polo-like kinase 1 (PLK1; Kakeno et al., 2014). Conversely, the open conformation of CLIP-170 makes its C-terminal region free to bind other CAP-Gly–containing proteins such as p150GLUED (Weisbrich et al., 2007; Goodson et al., 2003; Lansbergen et al., 2004). Hence, in an open conformation, CLIP-170 can initiate dynein/dynactin transport in neurons (Nirschl et al., 2016). Independently, some phosphorylations were shown to increase CLIP-170 binding to MTs, as proposed for mammalian target of rapamycin (Choi et al., 2002) or for Cdc2/Cdk1 in the G2/M phase of the cell cycle (at T287; Yang et al., 2009). This T287 phosphorylation is important for CLIP-170–mediated recruitment of PLK1 at kinetochores (Amin et al., 2014). Furthermore, the priming of CLIP-170 by PLK1 (at S195) controls the phosphorylation by casein kinase 2 of S1318 to recruit dynactin (Li et al., 2010). Similarly, the phosphorylation of S1384 by leucin-rich repeat kinase 1 regulates CLIP-170’s capacity to interact with dynein/dynactin complexes (Kedashiro et al., 2015). However, whether the rescue factor activity of CLIP-170 is regulated by phosphorylation is currently unknown.
We showed earlier that the MT molecular motor kinesin-1 regulates interphase MT dynamics in epithelial cells in a way that involves the activation of JNK to accelerate MT polymerization and promote rescues (Daire et al., 2009). JNKs, extracellular signal-regulated kinases (ERKs), and p38 belong to the mitogen activated protein kinase (MAPK) superfamily, which is activated during development and upon various stresses (for reviews, see Bogoyevitch and Kobe, 2006; Zeke et al., 2016). JNK1–3 are serine/threonine kinases that phosphorylate consensus SP or TP motifs. They are activated by dual threonine and tyrosine phosphorylation mediated by upstream MAPK kinases (MKK4 or MKK7), thanks to scaffolding proteins like those of the JNK-interacting protein (JIP) family (for reviews, see Zeke et al., 2016; Hotamisligil and Davis, 2016). As cargoes of kinesin-1, JIPs favor local JNK activation via a kinesin–JIP–JNK pathway (Verhey et al., 2001; Daire et al., 2009). Many neuronal MAPs (Doublecortin, MAP2, MAP1B, Stathmin, SCG10, SCLIP, Tau, and WDR62), which control MT growth or stabilization, are known as JNK substrates (for review, see Zeke et al., 2016). In most cases, MAP phosphorylation by JNK stabilizes MTs to sustain neuron development and function and to protect MTs upon stress. JNK also regulates MT-based transport, especially in neurons, by directly phosphorylating motors such as KIF5C (Padzik et al., 2016) as well as cargoes such as Bim (Lei and Davis, 2003) or synaptotagmin-4 (Bharat et al., 2017). However, the actors that contribute to the effectiveness of rescues downstream of JNK remain unknown.
Here, we show that upon cell stress, CLIP-170 is directly phosphorylated by JNK in its H1 domain to promote MT rescues. Phosphorylation occurs in the two serine-rich regions that flank its first CAP-Gly domain, at three independent sites (T25, T45, and S147) that are phosphorylated with different kinetics and efficacy. Phosphomimetic mutants of the CLIP-170 H1 domain increase its rescue-promoting activity in cells and in vitro. In cells, phosphomimetic mutants of the full-length protein are still recruited into +TIP comets, but they form remnants at the trailing end of comets at higher frequency. Furthermore, the residence time of CLIP-170 phosphomimetic mutants is shortened in the remnants. Similar results on CLIP-170 WT remnants were obtained upon JNK-activating stress. As the transient CLIP-170 trailing spots overlap future rescue hotspots, our results suggest that phospho–CLIP-170 functions more rapidly and more efficiently to prepare the MT lattice for future rescues.
Results
CLIP-170 is a new substrate of JNK in epithelial cells
To analyze the phosphorylation of endogenous CLIP-170 (Fig. 1 A) by JNK, we acutely stressed HeLa cells by exposure to anisomycin (1 h), to ultraviolet C (UVC; 10 min followed by 50 min without exposure), or to excess NaCl (1 h). We tried to detect changes in the phosphorylation status of the endogenous protein by SDS-PAGE. Our results revealed no clear shift in CLIP-170 electrophoretic mobility (Fig. 1 B and Fig. S1 A for expression levels and JNK activation controls). We thus repeated these experiments after 2 h of preincubation of cells with specific inhibitors of the MAP kinases MAP/ERK-kinase (MEK)/ERK1/2 (U0126), p38α/β (SB203580), or JNK1/2 (SP600125). Only JNK inhibition slightly accelerated the migration of CLIP-170, while the inhibition of ERK or p38 had no effect (Fig. 1 B and Fig. S1, B and C for expression levels, JNK activation controls, and data in other cell lines). These observations suggested that CLIP-170 might be a substrate for JNK in living cells. They also revealed that measuring gel shifts of the full-length protein in conventional SDS-PAGE was not satisfying to properly detect phosphorylation.
Figure 1.

The phosphorylations of CLIP-170 at T25 and S147 residues are mediated by JNK upon various acute cell stresses. (A) Sketches of human CLIP-170 organization, with the fluorescent tags used and the five amino acids mutated in this study. The asterisks indicate the location of the SxIP/SxIP-like motifs. The five last amino acids of CLIP-170 are indicated. (B) Western blot analyses of endogenous CLIP-170 migration variation in HeLa cells after exposure to acute stresses (1.2 µM anisomycin, 1 h; 200 J/m2 UVC 10 min followed by 50 min without UV; 350 mM NaCl, 1 h), with or without 2-h pretreatment with inhibitors of MEK/ERK1/2 (10 µM U0126), p38α/β (10 µM SB203580), or JNK1/2 (20 µM SP600125). (C) Phos-tag Western blots of GFPT186A-H1 showing the phosphorylations at T25, S147, and S312 upon various HeLa cell stresses combined or not with MAPK inhibitions. Each phosphorylated form was identified as a single band (P) above the nonphosphorylated form (np) when cells expressed mutants that could not bear any other detectable phosphorylation (GFPT186A-H1 mutations indicated on the left). The percentages of each phospho-form to total GFPT186A-H1 are indicated below each gel (P%). GFPT186A-H1 was probed using anti–CLIP-170 antibody. Western blots with anti–phospho-JNK and anti–β-actin antibodies show JNK activation levels (bottom of the panel). The histograms show the mean phosphorylation levels ± SD, with individual values shown as black dots. Statistical analyses were done using Kruskal-Wallis nonparametric analysis of variance, followed when appropriate by Mann-Whitney U test comparisons with controls. n indicates the number of independent experiments in B and C. *, P < 0.05. Ani., anisomycin; inh., inhibitoin; P-JNK, phospho-JNK.
Figure S1.

JNK-mediated phosphorylation of CLIP-170 at T25 and S147 in various cell lines (related to Figs. 1 and 2). (A and B) CLIP-170 expression in HeLa cells subjected to acute stresses and to MAPK inhibition. (A) Western blots of cell lysates obtained with the acute stress conditions depicted in Fig. 1 B. (B) Western blots of cell lysates obtained after acute MAPK inhibition coupled with anisomycin stress, as described in Fig. 1 B. The histograms (A and B) show the mean ratio between CLIP-170 and β-actin expression levels ± SD, with individual values shown as black dots. Statistics were done using Kruskal-Wallis nonparametric analysis of variance. (C) Western blot analyses of endogenous CLIP-170 migration in lysates of MEF, HuH7, and PtK2 cells subjected to the indicated acute stresses. CLIP-170 motility was increased upon JNK inhibition, except for PtK2 cells (note the absence of T25 in rat-kangaroo CLIP-170 sequence shown in Fig. S2). (D) Western blots of GFPT186A-H1 from HeLa and PtK2 cell lysates to show its expression levels in experiments corresponding to Fig. 1 C, Fig. 2, and E of this figure. In the 3 h–DMSO condition, “25” means that the GFP-H1 mutant expressed is suitable for the detection of T25 phosphorylation (therefore, it stands for T147A-S312A). Accordingly, “147” stands for T25A-S312A, “312” for T25A-S147A, and “AAA” for T25A-S147A-S312A GFPT186A-H1 mutants. For cotransfected Flag-MKK7-JNK1APF, “25” stands for T45A-S147A-S312A, “45” for T25A-S147A-S312A, “147” for T25A-T45A-S312A, “312” for T25A-T45A-S147A, and AAAA for T25A-T45A-S147A-S312A GFPT186A-H1 mutants. (E) Phos-tag Western blots of GFPT186A-H1 showing the phosphorylations at T25, S147, and S312 (P = phosphorylated; np = nonphosphorylated) in PtK2 and HeLa cells, stressed as indicated. The percentages of each phospho-form to total-GFPT186A-H1 are indicated below each gel (P%). (F) Phosphorylation levels were measured as in E in RPE-1, HuH7, and MDA-MB-231 cells. GFPT186A-H1 proteins were probed using the anti–CLIP-170 antibody. The histograms (E and F) show the mean phosphorylation levels ± SD, with individual values shown as black dots. Statistical analyses were done using Kruskal-Wallis nonparametric analysis of variance followed by Mann-Whitney nonparametric comparisons with controls. n indicates the number of independent experiments. *, P < 0.05. n.s., not significant. Ani., anisomycin; P-JNK, phospho-JNK; inh., inhibition.
In humans, CLIP-170 displays nine putative sites (SP or TP) for phosphorylation by JNK, all located in the N-terminal H1 domain of the protein (Fig. 1 A). These sites flank the two CAP-Gly domains, and four of them (S147, T182, T287, and S310) are well conserved between species (Fig. S2). To increase the detection sensitivity of phosphorylation, we moved to the study of GFP-H1 mutants using Phos-tag SDS-PAGE, which allows the retardation of phosphoprotein migration. Being a substrate of JNK at T186 (unpublished observation), GFP was mutated into GFPT186A throughout the study (Fig. 1 A). In unstressed HeLa cells, we detected basal phosphorylation at only three sites (T25, S147, and S312), as revealed by the migration of the corresponding nonphosphorylatable AAA mutants into a single band and by the presence of a single additional retarded band when two mutations were combined (Fig. 1 C, left panel). Upon acute (1 h) anisomycin-mediated stress (resulting in JNK1 and JNK2 phospho-activation), S312, which does not belong to an SP motif, was not up-regulated by JNK and kept a mean proportion of phosphorylation around 20%. In contrast, phosphorylations at T25 and S147 (two consensus sites for JNK located in the serine-rich regions 1 and 2, respectively) were both enhanced by stress. T25 phosphorylation was low in the basal condition (mean of 5%) and rapidly increased upon anisomycin exposure (5 min) to reach a mean proportion of 25% after 30 min. The phosphorylation at S147 was readily detected (43%) before the treatment and increased up to 61% after 30 min of stress (Fig. 1 C, left panel; and Fig. S1 D for expression levels).
Figure S2.

Conservation of CLIP-170 N-terminal domain (H1 domain) in eight species (related to Fig. 1 A). Identical residues are indicated with stars, while similar residues are marked with one or two dots. Consensus sites for MAPK/JNK are highlighted in yellow, with the T25P, T45P, and S147P sites characterized in this paper highlighted in magenta. The two CAP-Gly domains are underlined, and the SxIP and SxIP-like domains are framed and highlighted in cyan. The known phosphorylation sites for PLK1 (S195) and AMPK/PKA/PLK1 (S312) are indicated in red font. The known phosphorylation site for Cdc2 (T287) is a TP motif (yellow).
To confirm that H1 phosphorylations at T25 and S147 were mediated by JNK, 2-h MAPK inhibitions were started before the 1-h anisomycin treatments. The proportions of phospho-GFPT186A-H1 at T25 and S147 returned to basal levels only upon JNK1/2 inhibition, not upon MEK/ERK1/2 or p38α/β inhibition (Fig. 1 C, middle panels). The substantial level of basal S147 phosphorylation that remained after JNK inhibition indicates that this site is also targeted by another kinase(s). In other epithelial cell lines (PtK2, RPE-1, HuH7, and MDA-MB-231), anisomycin exposure also triggered JNK-mediated phosphorylations of GFPT186A-H1 at T25 and S147 (Fig. S1, D–F). Furthermore, HeLa cells subjected to UVC or excess NaCl also exhibited phosphorylated GFPT186A-H1 at T25 (15%–18%) and at S147 (52%–55%) through the JNK pathway (Fig. 1 C, right panels).
To perform sustained (24 h) JNK activation, we overexpressed constitutively active constructs encoding two JNK isoforms (the short isoform JNK1α1 and the C-terminally extended JNK2α2) fused to FLAG-MKK7β2, one of their upstream MAPK kinases (Lei et al., 2002). The negative control consisted of overexpressing JNK1α1 in which the TPY motif phosphorylated by MKK7β2 was mutated into APF. As expected, this long-term JNK activation in HeLa cells induced high levels of phosphorylation of GFPT186A-H1 at T25 (mean proportions of 58% and 50% for JNK1 and 2, respectively) and at S147 (73%–65%), without affecting the S312 phosphorylation level. It also phosphorylated two additional sites, one identified as T45 (38%–30%) and another one still uncharacterized (X; 27%–21%; Fig. 2 and Fig. S1 D for expression levels).
Figure 2.
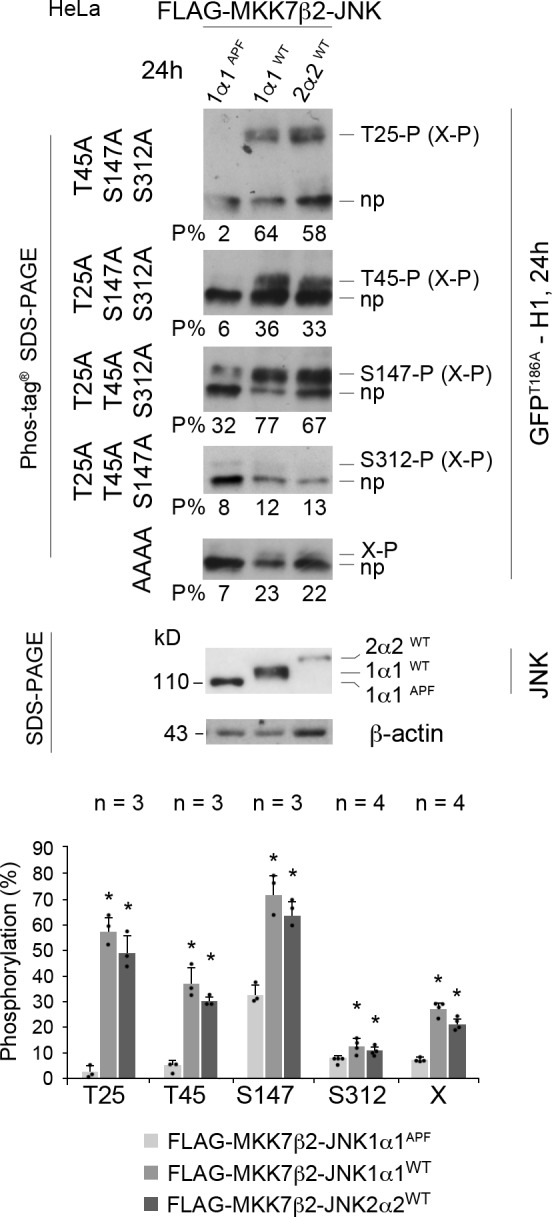
Long-term expression of active JNK phosphorylates CLIP-170 at T25, S147, and T45. Phos-tag Western blots of GFPT186A-H1 showing phosphorylations at T25, T45, S147, S312, and at an additional unidentified (X) residue. These phosphorylated bands (P) were retarded above the nonphosphorylated form of each protein (np). HeLa cells were cotransfected 24 h with the GFPT186A-H1 phospho mutants indicated on the left and the FLAG-MKK7β2-JNK isoforms indicated on the top before phosphorylation analysis. Phos-tag Western blots were revealed with an anti–CLIP-170 antibody. The percentages of each phospho-form to total-GFPT186A-H1 are indicated below each gel (P%). In the two bottom blots, the expression levels of JNK constructs were detected with an anti-FLAG antibody and anti–β-actin as a loading control (size difference is due to the phosphorylation of JNK1α1WT and JNK2α2WT versus JNK1α1APF and to an extended C-terminal region in JNK2α2WT). Note that X-P may occur in combination with the other phosphorylations. The histograms show the mean % phosphorylation levels ± SD, with individual values shown as black dots. Statistical analyses were done using Kruskal-Wallis nonparametric analysis of variance, followed when appropriate by Mann-Whitney U test comparisons with the control (APF mutant). n indicates the number of independent experiments. *, P < 0.05.
Altogether, these experiments indicate that in multiple cell lines, JNK (without isoform specificity) phosphorylates consensus SP and TP motifs in the serine-rich regions that flank the first CAP-Gly domain of CLIP-170. S147, which is highly conserved in evolution, displayed a detectable basal phosphorylation level. Acute (1 h) JNK activation phosphorylates CLIP-170 at T25 and at S147, while sustained (24 h) JNK activation further phosphorylates T45 and at least one unidentified site.
Phosphorylation of CLIP-170 (H1) by JNK in vitro
To test whether the H1 domain of CLIP-170 is directly phosphorylated by JNK, we incubated recombinant active GST-JNK1α1 with mutated GFPT186A-H1, purified from HeLa cells by GFP-Trap. We found that phosphorylations at T25 and at S147 reached high levels (mean proportions of 75% and 63%, respectively; Fig. 3), consistent with the high JNK-mediated phosphorylation they exhibited in cells (Fig. 1 and Fig. 2). T45, which was less sensitive to JNK activity in cells, was only partially phosphorylated (at 41%) in vitro. It is noteworthy that, as in cells that exhibited sustained JNK activity, recombinant GST-JNK1α1 phosphorylated at least one additional unidentified residue X by 22%. As expected, recombinant JNK did not phosphorylate GFPT186A-H1 at S312.
Figure 3.
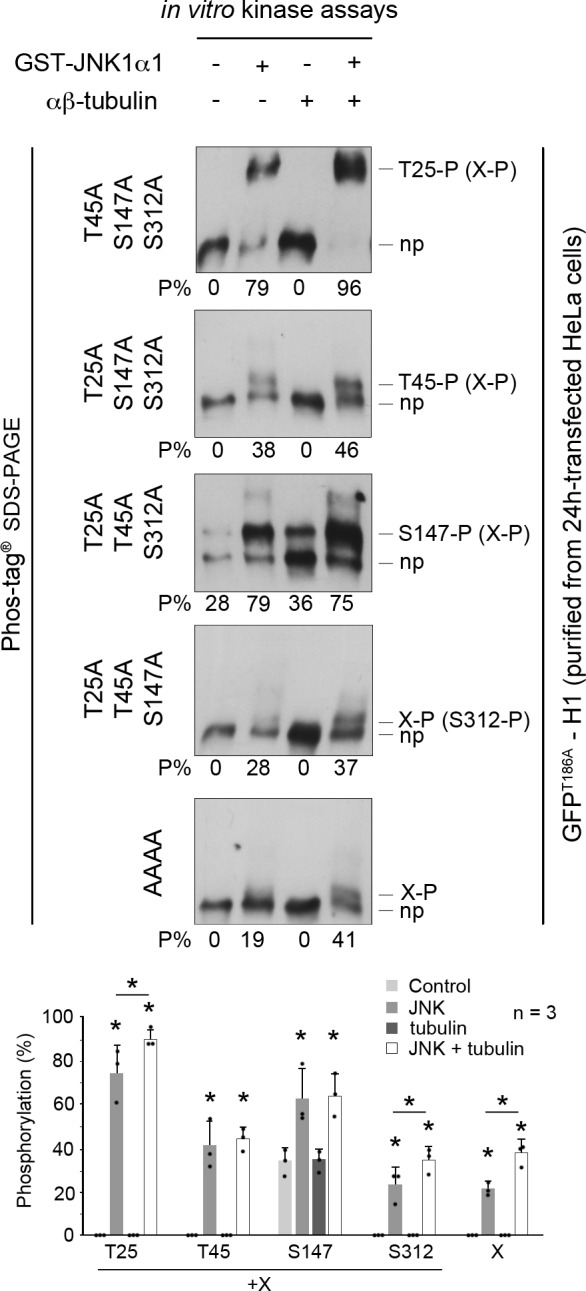
JNK phosphorylates the head domain of CLIP-170 at T25, T45, and S147 in vitro. In vitro kinase assays were performed for 1 h using mouse recombinant activated GST-JNK1α1 and mutated GFPT186A-H1 purified from HeLa cells, with or without porcine brain αβ-tubulin loaded with GTP. Phosphorylations at T25, T45, S147, S312, and X are detected as the upper bands (P), while the bottom bands correspond to the nonphosphorylated forms (np). The Phos-tag Western-blots were probed using an anti–CLIP-170 antibody. The percentages of each phospho-form to total-GFPT186A-H1 are indicated below each gel (P%). Note that X-P may occur in combination with the other phosphorylations. Individual values are shown as black dots. The histograms show the mean % phosphorylation values ± SD, with individual values shown as black dots. Statistical analyses were done using Mann-Whitney U test comparisons with controls (each JNK condition was compared with the corresponding control without JNK; the JNK conditions with and without tubulin were also compared and noted with a line underneath the * sign when significant). n indicates the number of independent experiments. *, P < 0.05.
As the main three amino acids phosphorylated by JNK flank the first CAP-Gly domain of CLIP-170, we further sought to determine if their phosphorylation could be affected by the binding of H1 to tubulin. Repeating the same in vitro kinase assays with purified porcine αβ-tubulin (using a stoichiometry of two tubulin dimers for one H1) revealed a slight increase in phosphorylation efficacy at T25 (from 75% to 91%) but also at X (from 22% to 38%; Fig. 3).
Together, these data confirm that the phosphorylations at T25, T45, and S147 (and at X) observed in cells are directly regulated by JNK. Furthermore, we found that tubulin can be a minor enhancer of phosphorylation at T25 (and at X).
CLIP-170 phosphomimetic mutants stimulate MT rescues in cells
We then focused on the functional relevance of JNK-dependent phosphorylations of CLIP-170, especially on their capacity to stimulate MT rescues. To this end, we studied the effects of both nonphosphorylatable (A) and phosphomimetic (E) mutants of full-length mCherry–CLIP-170 in PtK2 cells stably expressing GFP–α-tubulin (Fig. 4 C and Fig. S3 for expression levels). MT dynamic instability at plus ends was recorded with GFP-tubulin, and the mean values of the four main parameters obtained were plotted in diamond graphs after normalization relative to WT values (Lacroix et al., 2014). Note that the expression of WT mCherry–CLIP-170 did not dramatically change MT dynamics parameters compared with those of untransfected cells (Fig. S3 C, Table S1, and Table S2). Furthermore, MT growth speed was roughly unaffected by CLIP-170 phosphorylations. The mean shrinking speed and the catastrophe frequency happened to be more variable, but they showed no consistent change relative to controls. In contrast, the overall rescue frequency was doubled compared with WT when E, EE, EEE mutants were expressed (0.091 s−1 on average for the E mutants vs. 0.043 s−1 for the WT experiments), while it remained unchanged with the corresponding A mutants (0.041 s−1 on average; Fig. 4 A, Table S3, and Table S4 for mean values).
Figure 4.

Full-length CLIP-170 phosphomimetics increase rescue frequency in cells. MT dynamics parameters were determined from time-lapse imaging of living PtK2 cells stably expressing GFP–α-tubulin and transiently expressing WT, nonphosphorylatable (A), or phosphomimetic (E) mCherry–CLIP-170 transgenes. (A) Diamond graphs represent mean values of MT dynamic instability parameters after normalization relative to the WT. (B) The values (from the same experiments as in A) of rescue frequencies in each of the depicted classes are reported in box plots showing representative percentiles and outliers. (C) Sample images of the MT network of living PtK2 GFP–α-tubulin cells expressing mCherry–CLIP-170 WT (inset zooms on a comet). Note that the CLIP-170 patches are due to its Zn finger domains and did not interfere with MT dynamics. Scale bars correspond to 5 µm. The numerical mean values ± SD of each parameter are shown in A and B, but also the numbers of cells, MTs, and rescues are detailed in Table S3. The statistical comparisons were performed using one-factor ANOVA followed by Fisher's protected t tests for pairwise comparisons (Table S4). n indicates the number of independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Figure S3.

mCherry–CLIP-170 expression levels used to record MT dynamics and CLIP-170 comet/remnant parameters (related to Figs. 4 and 7). (A) Western blots of CLIP-170 were performed to compare the expression levels of the endogenous CLIP-170 with that of the mCherry–CLIP-170 transgenes used in Figs. 4 and 7 and in B–D. Note that the endogenous PtK2 CLIP-170 is not well recognized by the human antibody. Since acrylamide SDS-PAGE was migrated for a long time to separate endogenous and transgenic CLIP-170, low-molecular mass proteins were lost from the gel, and this precluded the use of β-actin as loading control. (B) Sample images of the MT network (GFP–α-tubulin) and mCherry–CLIP-170 comets in living PtK2 (left panels) and MEF (right panels) cells (insets zoom on comets). mCherry–CLIP-170 patches/aggregates are due to the presence of the C-terminal Zn-finger domain of CLIP-170 and do not interfere with MT dynamics. Scale bars correspond to 5 µm. (C) MT dynamics parameters were determined from time-lapse imaging of living PtK2 cells stably expressing GFP–α-tubulin and transiently expressing or not the WT mCherry–CLIP-170 transgene. The same analysis was done in WT MEF cells transfected with GFP–α-tubulin. Diamond graphs represent mean values of MT dynamic instability parameters after normalization relative to the mCherry–CLIP-170 WT. The values of the overall rescue frequencies are reported in box plots showing representative percentiles and outliers. (D) Full-length CLIP-170 phosphomimetics increase the frequency of rescues with immediate or delayed regrowth in cells. MT dynamics parameters were determined from time-lapse imaging of living PtK2 cells stably expressing GFP-α-tubulin and transiently expressing the indicated mCherry–CLIP-170 transgenes (related to Fig. 4 B). The values of the rescue frequencies in each class are reported in box plots showing representative percentiles and outliers. The numerical mean values ± SD of each parameter are shown in C and D, but also the numbers of cells, MTs, and transitions are detailed in Table S1. The statistical comparisons were performed using one-factor ANOVA followed by Fisher's protected t tests for pairwise comparisons (Table S2). n indicates the number of independent experiments. ****, P < 0.0001. n.s., not significant.
These results prompted us to explore more in detail how these mutants affected the different classes of rescue. In the literature, rescue events were initially defined as shrinkage-to-growth transitions (Walker et al., 1988). However, shrink-to-pause transitions are also considered rescues (Dhamodharan and Wadsworth, 1995; Moriwaki and Goshima, 2016). Therefore, we considered rescues as interruptions of MT shrinking, followed either by a regrowth phase (immediately or after a pause) or by a resumption of shrinking after a pause (Fig. 4 B, diagrams). We observed that the E, EE, or EEE mutants selectively stimulated rescues with regrowth (0.062 s−1 on average) compared with A, AA, or AAA mutants (0.023 s−1) and with the WT control (0.025 s−1) in PtK2 cells (Fig. 4 B, Table S3, and Table S4). Note that rescue stimulation was very similar when rescues were directly followed by a regrowth phase and when the shrinking and regrowth phases were separated by a pause (Fig. S3 D, Table S1, and Table S2). In contrast, we observed that rescues without regrowth (corresponding to transient pauses in depolymerization) were not affected by the expression of CLIP-170 mutants (Fig. 4 B).
We then tested if GFPT186A-H1 phosphomimetic mutants are also better rescue factors than their nonphosphorylatable versions. Compared with full-size CLIP-170, the comets formed by H1 at the tip of growing MTs are less contrasted due to additional binding all along the MT body (Fig. 5 D and Fig. S4 for expression levels; Pierre et al., 1994). Therefore, expressing fluorescent H1 alone in PtK2 cells allowed us to follow MT dynamics without the need for GFP–α-tubulin expression. The overall rescue frequency was again the most responsive parameter to the phosphomimetic mutations (EE: 0.100 s−1 vs. WT: 0.054 s−1 in Ptk2 cells). The frequency of rescues with regrowth doubled compared with WT (0.086 s−1 vs. 0.039 s−1 for WT), while rescues without regrowth (the transient pauses in depolymerization) remained unchanged. These results were also observed in HeLa cells (Fig. 5, Table S5, and Table S6).
Figure 5.

CLIP-170 H1 phosphomimetics increase rescue frequency in cells. MT dynamics parameters were determined from time-lapse imaging of living PtK2, HeLa, or Clip1/Clip2 KO MEF cells expressing GFPT186A-H1 T25-S147 WT, AA, or EE transgenes. (A) Diamond graphs represent mean values of MT dynamic instability parameters after normalization relative to WT in PtK2 and HeLa cells. (B) The values (from the same experiments as in A) of rescue frequencies in each of the depicted classes are reported in box plots showing representative percentiles and outliers. (C) The overall rescue frequency values for KO MEF cells treated or not with anisomycin 1.2 µM (for ∼30 min) are reported in box plots showing representative percentiles and outliers. (D) Sample images of the MT network of living PtK2 cells expressing GFPT186A-H1 WT (inset zooms on comets). This type of GFP fluorescent signal was used in A, B, and C to follow MT dynamics. Scale bars correspond to 5 µm. The numerical mean values ± SD of each parameter are shown in A, B, and C, but also the numbers of cells, MTs, and rescues are detailed in Table S5. The statistical comparisons were performed using one-factor ANOVA followed by Fisher's protected t tests for pairwise comparisons (Table S6). n indicates the numbers of independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. n.s., not significant. Ani., anisomycin.
Figure S4.

GFPT186A-H1 expression levels used to record MT dynamic parameters (related to Fig. 5). (A) Western blots of CLIP-170 were performed to compare the expression levels of the endogenous CLIP-170 with that of the GFPT186A-H1 transgenes used in Fig. 5 and in panels B and C. Due to the low detection of endogenous CLIP-170 in PtK2 and the absence of endogenous CLIP in Clip1/Clip2 KO MEF cells, we could not estimate precisely the level of expression of H1 in these lines. Instead, we show normalized ratios to β-actin. n indicates the number of independent experiments. (B) Sample images of the MT network and comets highlighted by GFPT186A-H1 in living PtK2, HeLa, and MEF cells (insets zoom on comets). Scale bars correspond to 5 µm. (C) Sample event showing a GFPT186A-H1 remnant (cyan arrowheads) left behind a comet before an MT rescue occurs at the same site (magenta arrowheads) in HeLa cells. The recorded sequence is shown both in the form of relevant frames and in a kymograph. The length scale bar is indicated both for the images and for the kymograph.
To directly demonstrate that an acute stress exerts a positive effect on MT rescue frequency through the phosphorylation of the T25 and S147 sites by JNK, we used Clip1/Clip2 knockout (KO) mouse embryonic fibroblast (MEF) cells, depleted of both endogenous CLIP-170 and of its paralog CLIP-115 (Goldspink et al., 2017). As expected in these KO cells, anisomycin enhanced MT rescue frequencies only in the presence of WT GFPT186A-H1 transgenes, but not with the AA or EE versions (Fig. 5 C, Table S5, Table S6, and Fig. S4 for expression levels).
Our results reveal that CLIP-170 phosphorylation(s) at T25, T45, or S147 increase(s) the rescue frequency by a mechanism that selectively involves its head domain. Both T25 and S147 are responsive to acute stress response involving JNK to elicit the observed increase in rescue frequency. This enhancement operates specifically on immediate or delayed MT regrowth. Thus, phospho–CLIP-170 appears as a potent rescue factor that specifically functions to elicit MT regrowth rather than stopping MT disassembly.
H1 phosphomimetic mutants promote MT rescues in vitro
To reconstitute in a minimal system the effects of CLIP-170 phosphorylations on its rescue activity, we used in vitro–grown MTs from guanylyl (α,β) methylenediphosphonate (GMPCPP) seeds, with or without recombinant His-superfolderGFPT187A-H1 (His-sfGFPT187A-H1 WT or EEE). As expected due to the absence of EB1, H1 distributed along the MT lattice without comet formation. However, we noticed that H1 EEE displayed lower binding to the GMPCPP seeds than to the rest of the MTs, while H1 WT showed an equal binding (Fig. 6 A). Concerning MT dynamic parameters at plus ends, rescues (mostly occurring without pause in vitro) that were rare with tubulin alone (mean frequency of 0.075 s−1) did not statistically increase with WT H1 (0.088 s−1) but raised up to 2.4 times with EEE (0.21 s−1; Fig. 6 B, Table S7, and Table S8). We also observed a decreased catastrophe frequency in the presence of both H1 forms compared with assay without H1. This could reveal a mild anti-catastrophe effect of H1 in vitro, which was not observed in living cells with mCherry–CLIP-170 (Fig. S3 C). Overall, mutations of the H1 domain of CLIP-170 that mimic its phosphorylation by JNK appear to be sufficient to autonomously promote MT rescues.
Figure 6.

CLIP-170 H1 phosphomimetics promote MT rescues in vitro. Recombinant His-sfGFPT187A-H1 in its WT or in its T25E-T45E-S147E (EEE) versions was added or not to purified tubulin in in vitro MT dynamics assays monitored by TIRF microscopy. (A) The sketch at the top indicates that MT (yellow, tubulin-TAMRA and His-sfGFPT187A-H1) elongation and dynamics were initiated at GMPCPP seeds (cyan, tubulin-HiLyte 647) and took place on kinesin-1 heavy chain (KHC) kept in nonmotile conformation in the presence of AMPPNP. In the assay, His-sfGFPT187-H1 phosphomimetic (EEE) binding was weaker than that of the WT form to GMPCPP seeds. Intensity profile of the images shown are plotted below, and the normalized values of GFP fluorescence/micrometer ± SD are shown on the right panel. (B) Sample kymographs derived from recordings of the assay in A show the occurrence of rescues at MT plus ends (magenta arrows). The cyan dashed lines correspond to the end of the GMPCPP seeds. The diamond graph shows the mean values of each dynamic instability parameter after normalization relative to WT. The values of overall rescue frequencies are reported in box plots showing representative percentiles and outliers. The numerical mean values ± SD of each parameter are shown, but also the numbers of MTs and rescues are detailed in Table S7. The statistical comparisons were performed using one-factor ANOVA followed by Fisher's protected t tests for pairwise comparisons (Table S8). n indicates the number of independent experiments. ****, P < 0.0001. Scale bars are indicated for images and kymographs. ns, not significant.
Remnant behavior of CLIP-170 phosphomimetics in cells
Given the surprising finding that phosphomimetic H1 mutants displayed a lower affinity for GMPCPP MTs in vitro, we decided to address carefully how JNK-mediated phosphorylations affect CLIP-170 binding to cellular MTs. In PtK2 cells expressing GFP–α-tubulin, the overexpression of A, AA, or AAA or of E, EE, or EEE mutants of mCherry–CLIP-170 did not globally affect the MT network (Fig. S3 B). Our data are in accordance with those of Goldspink et al. (2017), reporting a lack of strong MT network defect of Clip1/Clip2 double KO cells. Also, mCherry–CLIP-170 recruitment into comets was not affected, since comet lengths remained similar in PtK2 cells expressing mutants or WT controls (Fig. 7, A and B; Table S9 and Table S10). This indicates that phosphorylations do not trigger CLIP-170 self-inhibition and detachment from MT growing plus ends, contrasting with the phosphorylations in the third serine stretch that includes S312 (Lee et al., 2010). As expected, CLIP-170 comets leave transient trailing dots on MTs (CLIP-170 remnants) after their passage (Fig. 7 A). However, the quantifications revealed that the phosphomimetic mutations significantly increased the frequency of comet fragmentations (mean values of 0.131 s−1 for EEE vs. 0.058 s−1 for WT), while the lifetime of the remnants was significantly shorter (4.4 s for EEE vs. 10.7 s for WT). This result was confirmed using mCherry–CLIP-170 EE and AA mutants, both in PtK2 (Fig. 7 B, Table S9, and Table S10) and in Clip1/Clip2 KO MEF cells (Fig. S5, Table S11, and Table S12).
Figure 7.

Full-length CLIP-170 phosphomimetics frequently form short-lived remnants, highlighting potential future rescue sites. Parameters of comets and remnants were measured from time-lapse imaging of PtK2 cells stably expressing GFP–α-tubulin and transiently expressing WT, nonphosphorylatable (A), or phosphomimetic (E) mCherry–CLIP-170 transgenes. (A) Sample image sequences showing the progression of mCherry–CLIP-170 comets (perpendicular dashed lines at the front tips) in cells treated or not with 1.2 µM anisomycin or 20 µM SP600125 (30 min to 1 h). CLIP-170 remnants are left behind comets (white arrows). (B) From the same experiments as in A, the values of comet and remnant parameters are reported in box plots showing representative percentiles and outliers. The histogram on the right shows the ratios (means normalized to the WT values ± SD) between the frequencies of MT regrowth and those of remnant occurrence. The mean values ± SD of each parameter are shown, but also the numbers of cells, comets, and remnants are detailed in Table S9. The statistical comparisons were performed using one-factor ANOVA followed by Fisher's protected t tests for pairwise comparisons (Table S10). (C) Sample event showing an mCherry–CLIP-170 remnant (cyan arrowheads) left behind a comet before an MT rescue occurred at the same site (magenta arrowheads). The recorded sequence is shown in both the form of relevant frames and in a kymograph. n indicates the number of independent experiments. ***, P < 0.001; ****, P < 0.0001. Scale bars are indicated for images and kymographs.
Figure S5.

Full-length CLIP-170 phosphomimetic frequently forms short-lived remnants, highlighting potential future rescue sites in MEF cells (related to Fig. 7). (A) Sample images of the MT network (GFP–α-tubulin) and mCherry–CLIP-170 comets in living Clip1/Clip2 KO MEF cells (insets zoom on comets). mCherry–CLIP-170 patches/aggregates are due to the presence of the C-terminal Zn-finger domain of CLIP-170 and do not interfere with MT dynamics. Scale bars correspond to 5 µm. Arrows point to remnants behind comets. (B) The comet lengths and remnant parameters (from the same experiments as in A) are reported in box plots showing representative percentiles and outliers. The numerical mean values ± SD of each parameter are shown, but also the numbers of cells, comets, and remnants are detailed in Table S11. The statistical comparisons were performed using one-factor ANOVA (Table S12). n indicates the number of independent experiments. ****, P < 0.0001. n.s., not significant.
We estimated earlier that ∼35% of the GTP-like islands actually function to promote rescues (Dimitrov et al., 2008) and that CLIP-170 remnants could highlight future rescue hotspots given the good coincidence they showed with GTP-like islands (de Forges et al., 2016). Accordingly, we currently show events in which the location of remnants of WT mCherry–CLIP-170 (Fig. 7 C) or GFPT186A-H1 (Fig. S4 C) is actually predictive of future MT rescue sites. Given the relationship between the occurrence of a remnant and that of a rescue, one could derive a parameter representing the rescue effectiveness of remnants (i.e., the ratio between the frequency of rescues and the frequency of remnant occurrence). These ratios were consistently higher for EE or EEE mutants than for WT or for AA or AAA mutants in PtK2 cells (Fig. 7 B and Table S9).
To assess whether JNK-mediated cell stress can similarly affect CLIP-170 remnant behavior, we modulated JNK activity in PtK2 cells expressing mCherry–CLIP-170 WT. JNK inhibition by SP600125 caused uniform retention of CLIP-170 behind comets. This phenotype precluded a reliable quantification of remnant occurrence frequency and of their persistence. Conversely, as with the phosphomimetic mutant, when JNK was activated by anisomycin, the frequency of CLIP-170 WT remnant occurrence increased (mean values of 0.088 s−1 for anisomycin vs. 0.059 s−1 for control) and remnant persistence decreased (5.95 s for anisomycin vs. 8.19 s for control; Fig. 7 B, Table S9, and Table S10).
Therefore, we propose that when CLIP-170 is phosphorylated by JNK, it is more frequently retained at the trailing end of comets, allowing frequent rescues with consecutive MT regrowth. Since the time spent by CLIP-170 at remnants is opposite to its efficiency in promoting rescue, this suggests that the CLIP-170 retained in these remnants is necessary to prepare future rescues before their occurrence.
Discussion
The regulation of MT dynamics is essential for cells to adapt to their environment. Among the events that drive MT dynamics, rescues result from the intrinsic properties of tubulin and their modulation by MAPs. CLIP-170 and its partner CLASPs are major MT rescue factors. CLASPs have also been shown to prevent catastrophe events; probably due to this additional function, CLASP depletion reduces interphase MT density, while CLIP-170 is dispensable for the maintenance of the network (Komarova et al., 2002; Mimori-Kiyosue et al., 2005; Goldspink et al., 2017). Given that rescue frequency is stimulated by the kinesin-1–JNK signaling pathway (Daire et al., 2009), we report here for the first time a direct phosphorylation by the stress kinase JNK of CLIP-170 at T25, T45, and S147 (Figs. 1, 2, and 3). Strikingly, phosphomimetic versions of CLIP-170 approximately doubled the occurrence of rescues in cells by promoting immediate or delayed MT regrowth (Fig. 4, Fig. 5, and Fig. S3 D). We validated CLIP-170 T25 and S147 amino acids as being determinant in the response to acute JNK-induced stress (anisomycin), thanks to the use of Clip1-Clip2 KO MEF cells (Fig. 5 C). We also reproduced rescue stimulation in vitro using a phosphomimetic H1 domain (Fig. 6). These findings show that the positive effect of JNK on MT rescue activity in response to changes in cell environment (Daire et al., 2009) can be mediated by the rescue factor CLIP-170.
Rescue events frequently occur at predetermined GTP-islands/MT defects (Dimitrov et al., 2008), which are generated along the MT body by mechanical constraints, lattice defect, or repair (Vemu et al., 2018), especially at MT crossings (de Forges et al., 2016; Aumeier et al., 2016). GTP-islands could function as depolymerizing stop signs, as observed with artificial islands made of GMPCPP-bound tubulin (Tropini et al., 2012). Consistent with its rescue factor function, mCherry–CLIP-170 is found at discrete spots termed remnants, which are left behind the comets (Fig. 7), and endogenous CLIP-170 is found at MT crossovers, where GTP-like islands are enriched (de Forges et al., 2016). We measured earlier that CLIP-170 depletion did not affect the overall distribution of GTP-like islands (de Forges et al., 2016). However, our phosphomimetic CLIP-170 proteins approximately doubled the frequency of remnant occurrences (Fig. 7 and Fig. S5), suggesting that phosphorylated CLIP-170 may be more efficiently interacting with these particular hotspots during MT elongation (in comets) and soon after it (in the remnants). We also found that the dwell time of CLIP-170 phosphomimetic mutants at remnants was decreased by ∼60%, which means that after retention, most of the CLIP-170 is released in a way that depends on its phosphorylation state. This result was confirmed upon anisomycin-mediated stress, as we observed a 50% increase in the frequency of remnant occurrence associated with a 40% decrease of remnant dwell time (Fig. 7). We also established that CLIP-170 remnants could highlight future rescue hotspots before the actual rescues take place (Fig. 7 C and Fig. S4 C). The exact state of tubulin in these hotspots is still unknown and may vary during the occurrence of MT defects and, subsequently, during their possible reparation/preparation for future rescue. As suggested by the lower affinity of the EEE forms in vitro for the GMPCPP lattice (Fig. 6 A), a faster detachment of EEE CLIP-170 from the remnants could reflect a lower binding to the hotspots, when they become actual GTP-islands. Together, these results support the hypothesis that CLIP-170 actively participates in preparing the MT lattice for future rescues.
Our in vitro data clearly indicate that the phosphoform of CLIP-170 is a strong autonomous promoter of rescues (Fig. 6). However, in cells, we cannot exclude that phosphorylation of CLIP-170 by JNK might modulate binding to its partners in order to further control MT rescue efficiency. For example, CLIP-170 phosphorylation nearby its SxIP/SxIP-like motifs could reduce its binding to EB1 (Honnappa et al., 2009). Interestingly, CLIP-170 partners also leave trailing dots behind comets, as revealed in published videos or immunofluorescent images for CLIP-115 (Hoogenraad et al., 2000), EBs (Aumeier et al., 2016; Mustyatsa et al., 2019), CLASP2 (Lawrence et al., 2018; Wittmann and Waterman-Storer, 2005), p150GLUED (Vaughan et al., 2002), or SLAIN2 (van Haren et al., 2018). Accordingly, EB3 localizes to self-repair sites (Aumeier et al., 2016) and EB1 colocalizes with newly incorporated tubulin at repaired sites (Vemu et al., 2018). More directly, phospho–CLIP-170 at rescue hotspots could help to recruit small tubulin oligomers (Arnal et al., 2004). If phosphorylations could trigger conformational changes in the N-terminal unstructured domain of CLIP-170 (S1 domain; Fig. 1 A) or in the domain between its two CAP-Gly, it could allow easier tubulin dimer/oligomer incorporation into the lattice. In that case, CLIP-170 would resemble CLASPs, which are able to bind to tubulin oligomers (Maki et al., 2015) and to simultaneously bind to polymerized and soluble tubulin (Al-Bassam et al., 2010; Lawrence et al., 2018). CLASP capacity to bring tubulin near lagging protofilaments allows them to catch the front comet of growing MTs (Aher et al., 2018) and mostly serves to prevent catastrophes. Our in vitro experiments suggested that H1 (either the WT or the EEE mutant) can also prevent catastrophes (Fig. 6), but whether such effect occurs in cells and resembles that of CLASPs deserves further exploration in the future. Regarding other potential mechanisms, von Hippel-Lindau protein and MT severing enzymes have also been shown to indirectly promote MT regrowth by inhibition of tubulin GTPase activity (Thoma et al., 2010) or by induction of MT architectural defects followed by MT repair, thus causing the occurrence of GTP-islands (Vemu et al., 2018; Kuo et al., 2019). To what extent CLIP-170 is implicated in these processes still remains unknown.
Whether JNK-mediated phosphorylation of CLIP-170 occurs anywhere in the cytoplasm or locally (on CLIP-170 surrounding MTs, at the comets or even at the remnants left behind comets) remains to be determined. In cells, JNK activation generally depends on specific scaffolding proteins such as JIPs (Zeke et al., 2016; Yasuda et al., 1999), which can associate with MTs as they are cargos of the kinesin-1 and dynein/dynactin motors (Verhey et al., 2001; Fu and Holzbaur, 2013; Fu et al., 2014). Indeed, depletion of kinesin-1 reduces basal JNK activation and MT rescue frequency (Daire et al., 2009), suggesting local JNK activation near the growing end of MTs. This local JNK activation could affect other +TIPs, such as CLIP-115/CLIP2/CYLN2, p150GLUED, CLIPR-59/CLIP3, RSNL2/CLIP4, CYLD, CAP350, KIF13B, or TBCB, which all harbor SP/TP JNK-consensus sites adjacent to their CAP-Gly domain(s) and putative SxIP/SxIP-like motifs. In addition, we found several putative JNK sites in SLAIN2, CLASPs, and IQGAP1. Thus, it still remains to be discovered if JNK could orchestrate a special dynamic protein network to control rescue events, as well as other parameters of MT dynamics.
Here, we show that CLIP-170 phosphorylation by JNK links cell sensing of strong stresses to the control of MT rescues and that this pathway may be finely tuned. Indeed, T25, T45, and S147 residues on exogenous H1 exhibit differential sensitivity to JNK-mediated phosphorylation in cells, and S147 already displays a detectable level of basal phosphorylation (Fig. 1 C and Fig. 2). The latter finding is consistent with a proteomic study (Beausoleil et al., 2004) showing basal phosphorylation of CLIP-170 at S147. Hence, JNK can probably phosphorylate CLIP-170 (and possibly other MAPs) at low levels to sustain MT dynamics in basal cell conditions and further increase its control upon acute or sustained stress.
Altogether, our findings shed new light on the regulation of MT rescues and point out the importance of CLIP-170 and JNK, which may function as part of a preparation machinery for future rescue hotspots during MT elongation or soon after it. This is in agreement with accumulating data showing the importance of MTs in controlling their own maintenance and life span. MT dynamics more generally appear as a sensor of stress (Mackeh et al., 2014), and it will be worth investigating the role of this new JNK–CLIP-170 pathway in diverse pathologies, including cancer, diabetes, and neuropathies, as well as in liver regeneration.
Materials and methods
Plasmids, antibodies, cell culture, and treatment
The sequence of human CLIP-170 N-terminal domain (NP_002947; National Center for Biotechnology Information) from amino acids 1–347 (H1 domain) was aligned with corresponding CLIP-170 sequences of mouse (Mus musculus; D3Z3M7), rat (Rattus norvegicus; Q9JK25), rat-kangaroo (Dipodomys ordii; XP_012878519), chicken (Gallus gallus; O42184), danio (Danio rerio; XP_021335153), Xenopus (Xenopus tropicalis; XP_012818562), and drosophila (Drosophila melanogaster; NP_724047, CG5020) using the software Clustal Omega (EMBL-EBI).
PtK2 (rat-kangaroo kidney epithelium) cells stably expressing GFP-tubulin were a gift of Dr. Alexey Khodjakov (Wadsworth Center, Albany, NY), and immortalized MEF cells (WT line) were obtained from Dr. Neil D. Perkins (Newcastle University, Newcastle, UK) and from Dr. Niels Galjart (Erasmus MC, Rotterdam, Netherlands) Clip1/Clip2 KO line (Goldspink et al., 2017). Initial PtK2 cells are from (American Type Culture Collection [ATCC]; CCL-56). HeLa (human cervical cancer, ATCC; CCL2), RPE-1 (immortalized retina human epithelium, human telomerase reverse transcriptase; ATCC; CRL-4000), HuH7 (human hepatocarcinoma; kind gift of Dr. Ama Gassama, UMR 1193, Centre Hépato-Biliaire, Villejuif, France), and MDA-MB-231 (human metastatic breast cancer; ATCC HTB-26) cell lines were cultured in DMEM (Gibco) and supplemented with 10% fetal calf serum (Sigma) and 1% antibiotic-antifungal mixture (Gibco). PtK2 cells were grown without antibiotics, and MEF cells were supplemented with 20% fetal calf serum. Cells were regularly checked for mycoplasma by DAPI staining.
Human GFP-H1 (Pierre et al., 1992; corresponding amino acid sequence indicated in Fig. S2), mCherry–CLIP-170 (de Forges et al., 2016; corresponding amino acid sequence in Uniprot #30622 isoform 3) mutated for this study (see below), GFP–α−tubulin (from Dr. Roger Y. Tsien, Howard Hughes Medical Center, University of San Diego, San Diego, CA), and Flag-MKK7β2-JNK1α1WT, Flag-MKK7β2-JNK1α1APF, and Flag-MKK7β2-JNK2α2WT (Lei et al., 2002; Addgene; #19726, #19730, and #19727; from Dr. Roger Davis, University of Massachusetts, Worcester, MA) plasmids were transfected with Turbofect (Thermo Fisher Scientific) or JetPEI (Polyplus transfection) for 24 h (1 µg GFPT186A-H1, 1 µg Flag-MKK7-JNK) or 48 h (0.25 µg mCherry-CLIP, 0.6 µg GFP–α−tubulin, or 1 µg GFPT186A-H1) before analysis (Phos-tag Western blots and whole cell imaging, respectively). Plasmid pET28a-His-sfGFP-Nterm-T7 (Novagen; #69864) was used to clone H1 in order to express and purify recombinant His-sfGFPT187A-H1 proteins for in vitro MT dynamic assays (see sections below).
The rabbit polyclonal anti–CLIP-170 (#16234 directed to the N-terminal region) was a gift from Dr. Franck Perez (CNRS UMR 144, Institut Curie, Paris, France). The rabbit anti-Flag DYKDDDK (PA1-984B) was from Thermo Fisher Scientific, and the mouse anti–β-actin-HRP (C4) was from Santa Cruz Biotechnology. The rabbit anti–phospho-JNK Thr183/Tyr185 (81E11) and HRP-mouse and -rabbit secondary antibodies were from Cell Signaling.
Cell treatments were performed by adding 0.1% DMSO (control), 350 mM NaCl, 1.2 µM anisomycin, 20 µM SP600125 (Enzo), 10 µM U0126, or 10 µM SB203580 (Sigma) to the culture medium (1 h) or by exposition to 200 J/m2 UVC (10 min followed by 50 min with no treatment).
Site-directed mutagenesis
Human GFP-H1 and mCherry–CLIP-170 mutants were produced by PCR using a site-directed mutagenesis kit (QuickChange Lightning Site–directed, Agilent Technologies; #210519). For GFP mutations, a T186A mutation was made in the GFP-H1 plasmid, with the following primer pairs: for-T186A 5′-TCAACTAGCAGACCATTA-TCAACAAAATGCTCCAATTGGCGATG-3′ and rev-T186A 5′-CATCGCCAATT-GGAGCATTTTGTTGATAATGGTCTGCTAGTTGA-3′ (underlined nucleotides indicate mutated sites), and a T187A mutation in the His-sfGFP-H1 plasmid, with the following primer pairs: for-T187A 5′-CTACCAGCAGAACGCCCCCATCGGCGA-3′ and rev-T187A 5′-TCGCCGATGGGGGCGTTCTGCTGGTAG-3′. For CLIP-170 mutations, primer pairs were as follows: for-T25A 5′-AGCACAGCTCTGAAGGCACCTA-CGGCTGTTG-3′ and rev-T25A 5′-CAACAGCCGTAGGTGCCTTCAGAGCTGTGCT-3′; for-T25E 5′-GGAAGCACAGCTCTGAAGGAACCTACGGCTGTTGTAGC-3′ and rev-T25E 5′-GCTACAACAGCCGTAGGTTCCTTCAGAGCTGTGCTTCC-3′; for-T45A 5′-AGTGAAAAAGCATCAAGCGCTCCATCATCTGAGACTC-3′ and rev-T45A 5′-GAGTCTCAGATGATGGAGCGCTTGATGCTTTTTCACT-3′; for-T45E 5′-TCCAGTGAAAAAGCATCAAGCGAGCCATCATCTGAGACTCAGGAG-3′ and rev-T45E 5′-CTCCTGAGTCTCAGATGATGGCTCGCTTGATGCTTTTTCACTGGA-3′; for-S147A 5′-CTCCCGAGCTACTGCACCGCTGTGCAC-3′ and rev-S147A 5′-GTGCACAGCGGTGCAGTAGCTCGGGAG-3′; for-S147E 5′-CTCCCGAGCTACT-GAACCGCTGTGCACTTC-3′ and rev-S147E 5′-GAAGTGCACAGCGGTTCAG-TAGCTCGGGAG-3′; and for-S312A 5′-GAAGCGCAGCCCTGCTGCCTCTTCCCT-3′ and rev-S312A 5′-AGGGAAGAGGCAGCAGGGCTGCGCTTC-3′. All mutageneses were verified by DNA sequencing (Eurofins).
Immunoprecipitation and in vitro kinase assay
For the in vitro kinase assays, GFPT186A-H1 proteins were purified using GFP-Trap magnetic beads (Chromotek; ACT-CM-GFM0250) from transfected HeLa cells (1 µg, 24 h) and were resuspended 80 mM Pipes, pH 6.9, 1 mM EGTA, and 2 mM MgCl2, pH 6.9 (PEM) buffer, supplemented with 150 mM NaCl, 0.25 mM DTT, 0.1 mM EGTA, 5 mM ATP (Sigma), 25 mM MgCl2, 25 mM β-glycero-phosphate, and 1 mM Na3VO4. In the kinase assay, immunoprecipitated GFPT186A-H1 protein (1 µg/µl) bound to GFP-Trap beads was incubated at 30°C for 60 min with or without mouse recombinant active GST-JNK1α1 according to the manufacturer's instructions (100 ng; Precisio J2455; Sigma). αβ-tubulin purified from porcine brain (3.7 µg/µl) as described below was added or not to the assays in GTP-loaded forms.
Tubulin purification
Porcine brain MAP-free tubulin was purified as described in Walker et al. (1988). Briefly, brains from freshly slaughtered pigs were immediately put into ice and, after removal of meninges, homogenized by blending in 0.5 volume of PEM buffer (100 mM Pipes, 2 mM MgSO4, and 1 mM EGTA, pH 6.9) supplemented with 1 mM PMSF, 10 mg/ml leupeptin, 1 µg/ml pepstatin A, and 10 µg/ml TAME (all from Sigma). After centrifugation (100,000 g, 60 min, 4°C), the supernatants (cytosol) were diluted 1:1 with PEM-Glycerol buffer (66% [vol/vol] glycerol). Then, they were subjected to MT polymerization (45 min, 37°C) after the addition of 0.2 mM GTP (Sigma). After MT sedimentation (100,000 g, 45 min, 25°C), the MT pellets were resuspended in 20% of the homogenate volume with PEM buffer containing 0.2 mM GTP and dissolved by gentle homogenization every 5 min using a Dounce homogenizer (pestle “A,” on ice, 30 min). After clarification by centrifugation (100,000 g, 45 min, 4°C), the supernatants were subjected to another cycle of polymerization-depolymerization before tubulin purification by ion exchange chromatography on a column of phosphocellulose (Sigma) equilibrated with 25 mM Pipes, 1 mM EGTA, and 0.5 mM MgSO4, pH 6.7, containing 0.5 mM GTP and 1 mM DTT (Sigma). The fractions containing the peak of tubulin were identified by Bradford protein assay (Bio-Rad), then pooled and subjected to MT polymerization (30 min, 37°C) after dissolution of sodium L-glutamate (Sigma) at a final concentration of 1 M. After a final sedimentation (100,000 g, 30 min, 25°C) and pellet dissolution on ice, tubulin was assayed for protein concentration, frozen in liquid nitrogen, and stored at −80°C until use.
Live cell imaging of MT and CLIP-170 dynamics
Parameters of MT dynamics at MT plus ends and of remnants were determined on living PtK2 (stably expressing GFP–α-tubulin or not), HeLa, or MEF cells transfected for 48 h with 1 µg of GFPT186A-H1 or 0.25 µg of mCherry–CLIP-170 (±0.6 µg GFP–α-tubulin). Cells were grown on coverslips and treated or not with 1.2 µM anisomycin or 20 µM SP600125. Living cells were filmed at room temperature (RT; 20°C–25°C) in their culture medium by mounting the coverslips on a microscope slide above adhesive tape strips to make a small chamber. mCherry–CLIP-170 patches/aggregates are due to the presence of the C-terminal Zn-finger domain of CLIP-170 and do not interfere with MT dynamics. All the cells presenting a too-high level of transfection (CLIP-170 relocalization on MT lattice, MT bundling) were excluded from the analysis. Time-lapse sequences (1 image every 2.5 s, 80 frames) were acquired on a Leica DMLB microscope through a 100× 1.3 NA objective, using a Scion CFW1312M chanrge-coupled device camera and homemade software.
Image processing and analyses
For time-lapse imaging, MTs were tracked manually in ImageJ using the segmented line tool and a homemade multikymograph macro command. Briefly, for each selection recorded in the selection manager and for each image of a time-lapse sequence, the command performed the straightening of an image of configurable width along the selection. The images obtained were directly assembled to build an “extended” kymograph (as shown in Fig. 7 C). Alternatively, the straightened images underwent pixel maximum intensity projection or arithmetic mean calculation across their width (perpendicular to the selection) to generate classical kymographs in which each straightened image yielded a single line of pixels (as shown in Fig. 6 B).
Dynamic parameters were calculated according to Dhamodharan and Wadsworth (1995), using ImageJ and the above kymograph macro to outline the extremity of MTs with the segment line and obtain the coordinates of each point. All subsequent calculations were made in Microsoft Excel. Each phase in which the MT length varied by <0.5 µm was considered as a pause (p). The following dynamic instability parameters were calculated for each MT. For polymerizing and depolymerizing phases, growth (g) and shrinkage (s) rates are the extent of MT elongation or shortening (in micrometers) divided by the duration of the phase (in seconds). As catastrophes can occur only in growing or paused MTs, their frequency was calculated by dividing the number of catastrophes (“g to s” + “p to s” transitions) by the total time spent in growing and paused phases (temporal frequency). Rescues can only occur on depolymerizing MTs. Thus, the overall rescue frequency was calculated by dividing the number of rescues (“s to g” + “s to p” transitions) by the total time spent in the shortening phase (temporal frequency). The calculation of rescue frequencies with or without regrowth took into account the subsequent phase to label each rescue event (“s to g,” and “s to p” then “g” for rescues with regrowth; “s to p” then “s” again for rescues without regrowth). Of note, a few rescue events that consisted of “s to p” transitions identified at the very end of the time-lapse sequences could not be categorized as “with regrowth” or “without regrowth.” This explains why the overall rescue frequency is most often higher than the sum of the frequencies of rescues with regrowth and without regrowth. In all the figures, the diamond graphs (Lacroix et al., 2014) recapitulate the mean parameter values, and the box plots display the 10th, 25th, 50th, 75th, and 90th percentiles as well as the outlier points, as indicated.
For the comet and remnant parameters, a segmented line (in ImageJ) was drawn manually along each comet (without remnants) from a single frame (= comet length). During tracking, the remnants left at the trailing end of each comet were counted and their life span measured (= remnant persistence). The frequency of remnant occurrence for each comet was determined by dividing the number of remnants by the comet’s lifetime. Comet tracks were always chosen following three criteria: (1) comets should not overlap during the entire growth cycle; (2) the moment of the comet’s appearance and disappearance should be clearly identified; and (3) comets should not reach the periphery of the cell where they were indistinguishable. The values are presented in box plots displaying the 10th, 25th, 50th, 75th, and 90th percentiles as well as the outlier points.
The cell and comet images were processed as follows using ImageJ. After dead pixel removal by applying a despeckle filter, and after optional registration and cropping for time-lapse sequences, the images were subjected to a slight smoothing using a 0.5-pixel radius Gaussian filter and finally to image dynamics adjustment by automatic contrast enhancement with no more than 0.3% of saturated pixels.
Expression and purification of recombinant His-GFPT187A-H1 proteins
We cloned H1 in the modified plasmid pET28a-His-sfGFPT187A-Nterm-T7 (see mutagenesis GFP) using circular polymerase extension cloning technology (In-Fusion HD; Clontech). We used our WT and EEE mCherry–CLIP-170 plasmids for H1 PCR amplification with the primer pairs: for 5′-GCTGTACAAGGGATCCATGAGTATGCTAAAGCCAAGTGGG-3′ and rev 5′-GTGCGGCCGCAAGCTTTCATATCTTCCTGGCGTAACGGG-3′, with BamH1 and HindIII restriction sites indicated in bold. We linearized pET28a-His-sfGFPT187A-Nterm-T7 with BamH1 and HindIII enzymes and performed clonings following Clontech instructions.
For recombinant protein expression pET28a-His-sfGFPT187A-H1 (WT and EEE) plasmids growing BL21 cells were induced with 1 mM IPTG for 2.5 h at 37°C. After 20-min centrifugation (4,000 g, 4°C), pelleted cells were washed in 40 mM Tris-HCl and 10 mM imidazole, pH 8.0, and snap frozen in liquid nitrogen. Cells were lysed by the addition of 25 µg/ml DNaseI, 5 mM MgCl2, and 0.2% TritonX100 for 20 min on ice. After dilution with 1 volume of 1 M NaCl and centrifugation (15,000 rpm, 30 min, 4°C), proteins in the supernatant were purified on Ni-nitrilo-triacetic acid agarose matrix (Qiagen). For this purpose, Ni-nitrilo-triacetic acid was equilibrated in 5 mM imidazole, 0.5 M NaCl, and 20 mM Tris-HCl, pH 7.9, loaded with the protein samples, washed, and eluted with the same buffer containing 30 mM and 600 mM imidazole. For the fractions containing His-sfGFPT187A-H1, the buffer was exchanged with 80 mM Pipes, 1 mM EGTA, and 1 mM MgCl2, pH 6.8, using centricons (10 kD). Protein concentrations were determined by BCA (Pierce), and integrity and purity of proteins were confirmed by SDS-PAGE.
In vitro MT dynamics assays
GMPCPP MT seeds were assembled according to Mohan et al. (2013). A mixture (1:10 wt/wt) of HiLyte647-labeled tubulin (Cytoskeleton Inc.) and of phosphocellulose-purified porcine brain tubulin (see tubulin purification above) was polymerized with 1 mM GMPCPP (Jena Biosciences) at 37°C for 30 min. MTs were centrifuged at 120,000 g for 10 min in an Airfuge Centrifuge (Beckman). After supernatant removal and replacement by the same volume of PEM buffer, MTs were depolymerized (20 min by gentle pipetting on ice) and repolymerized with 1 mM GMPCPP (30 min, 37°C). After centrifugation (120,000 g, 10 min), the pellet was resuspended in 2.5 times the initial volume of PEM containing 10% glycerol, aliquoted, frozen in liquid nitrogen, and kept at −80°C until use.
Total internal reflection fluorescence microscopy assays were performed as described in Campanacci et al. (2019). Briefly, MT dynamic assays were performed in microchambers made of two glass coverslips assembled with gently melted parallel parafilm spacers. After extensive washing with ice-cold methanol and air drying, the chambers were coated (5 min, RT) using a solution of PEM supplemented with casein at 0.2 mg/ml, 10 µg/ml recombinant kinesin-1 heavy chain (Cytoskeleton Inc.), and 1 mM adenylyl-imidodiphosphate (AMPPNP; Sigma), as described in de Forges et al. (2016). After blocking with PEM containing 0.5 mg/ml casein (5 min) and three rinses with PEM containing 0.2 mg/ml casein and 1 mM AMPPNP, a suspension of GMPCPP seeds (1:200 vol/vol in PEM containing 1 mM AMPPNP, 0.5% β-mercaptoethanol, and an oxygen-scavenging mixture: 0.8 mg/ml catalase [Sigma], 2 mg/ml glucose oxidase [Sigma], and 2 M glucose [Euromedex]) was infused into the chamber.
MTs were elongated off GMPCPP seeds using a mixture of 12 µM phosphocellulose-purified tubulin containing 5% wt/wt of TAMRA-labeled tubulin (Cytoskeleton Inc.), with or without the addition of 300 nM recombinant His-sfGFPT187A-H1 and 1 mM GTP in PEM buffer supplemented with the oxygen-scavenging mixture. Three-color TIRF recordings (one image every 4 s, at least 200 frames) were performed at 37°C using a Nikon inverted microscope Eclipse Ti equipped with a Nikon Perfect Focus System, a Nikon APO TIRF ×100 1.49 NA oil immersion objective lens, and a Hamamatsu ORCA FLASH camera (Dr. Sandrine Lévêque-Fort, Institut des Sciences Moléculaires d’Orsay, Université Paris-Saclay, Paris, France). Dynamic parameters were measured from kymographs derived from the image sequences (ImageJ), as described in the Image processing and analyses section.
Phos-tag and Western blotting
To visualize the migration shifts of GFPT186A-H1 mutants, we used 6% polyacrylamide SDS gels containing 20 µM Phos-tag (Wako; AAL-107) and 40 µM MnCl2. Gels were run for 90 min at 150 V in a BIO-RAD Mini-Protean II system kept in the dark at RT. PageRuler prestained protein ladder (Thermo Fisher Scientific; 10–170 kD) was used to follow migration but was not appropriate to accurately determine any molecular mass due to Phos-tag effect. Tris/HCl and PEM buffers used in the in vitro kinase assay were diluted five times in water before loading on Phos-tag gels. After migration, gels were incubated for 15 min in transfer buffer containing 1 mM EDTA to chelate Mn2+ cations before transfer. To visualize the shifts of endogenous CLIP-170, we used classical 6% acrylamide SDS gels that ran for 2 h at 150 V at RT to keep only the proteins of apparent molecular mass >110 kD (noted as “long migration” in the figures). To quantify mCherry–CLIP-170 and GFPT186A-H1 transfection levels, we compared transgene accumulation levels with that of endogenous CLIP-170, β-actin, and P-JNK on classical 6% (mCherry) or 9% (GFPT186A) gels that ran for 1 h at 150 V at RT. Quantifications of scanned Amersham Hyperfilm ECL (GE Healthcare) were performed with ImageJ, and Microsoft Excel software was used for plotting.
Statistical analyses
All statistical comparisons were performed using StatView 5.0, SAS Software. Comparisons of the Western blot quantifications were performed using Kruskal-Wallis nonparametric analysis of variance followed, if appropriate, by Mann-Whitney U test for comparisons with the indicated controls. Comparisons of dynamic instability parameters in cells or in vitro as well as those of the remnant parameters were made using one-factor ANOVA tests followed, if appropriate, by Fisher’s protected t tests for subsequent pairwise comparisons. The following symbols were used: * for P < 0.05, ** for P < 0.01, *** for P < 0.001, and **** for P < 0.0001.
Online supplemental material
Fig. S1 shows JNK-mediated phosphorylation of CLIP-170 at T25 and S147 in various cell lines (related to Fig. 1 and Fig. 2). Fig. S2 shows sequence alignment in various species of CLIP-170 N-terminal domain (H1; related to Fig. 1 A). Fig. S3 shows mCherry–CLIP-170 expression levels for live cell imaging (related to Fig. 4 and Fig. 7). Fig. S4 shows GFPT186A-H1 expression levels for live cell imaging (related to Fig. 5). Fig. S5 shows CLIP-170 comet and remnant parameters in Clip1/Clip2 KO MEF cells (related to Fig. 7). Table S1 presents parameters of MT dynamic instability related to Fig. S3. Table S2 shows statistical values related to Table S1. Table S3 presents parameters of MT dynamic instability related to Fig. 4. Table S4 shows statistical values related to Table S3. Table S5 presents parameters of MT dynamic instability related to Fig. 5. Table S6 shows statistical values related to Table S5. Table S7 presents parameters of MT dynamic instability related to Fig. 6. Table S8 shows statistical values related to Table S7. Table S9 presents CLIP-170 comet and remnant parameters related to Fig. 7 B. Table S10 shows statistical values related to Table S9. Table S11 presents CLIP-170 comet and remnant parameters related to Fig. S5. Table S12 shows statistical values related to Table S11.
Supplementary Material
presents parameters of MT dynamic instability related to Fig. S3.
shows statistical values related to Table S1.
presents parameters of MT dynamic instability related to Fig. 4.
shows statistical values related to Table S3.
presents parameters of MT dynamic instability related to Fig. 5.
shows statistical values related to Table S5.
presents parameters of MT dynamic instability related to Fig. 6.
shows statistical values related to Table S7.
presents CLIP-170 comet and remnant parameters related to Fig. 7 B.
shows statistical values related to Table S9.
presents CLIP-170 comet and remnant parameters related to Fig. S5.
shows statistical values related to Table S11.
Acknowledgments
The authors are very grateful to Drs. Franck Perez, Roger Davis, Ama Gassama, Alexey Khodjakov, Roger Y. Tsien, Neil D. Perkins, and Niels Galjart for sharing materials. We also thank Dr. Sandrine Levêque-Fort for hosting at the Institut des Sciences Moléculaires d’Orsay and her help in acquiring images on the TIRF microscope, and the reviewers for their helpful comments.
This work was supported by the Fondation ARC pour la Recherche sur le Cancer (PJA2019 1209695).
The authors declare no competing financial interests.
Author contributions: Conception, design of the work, and funding acquisition were done by C. Poüs and B. Benoit. Clonings were done by H. Henrie, I. Cantaloube, M. Talantikite, V. Stoppin-Mellet, and B. Benoit. Western blots were done by H. Henrie and B. Benoit. Live cell imaging was done by H. Henrie, M. Talantikite, D. Bakhos-Douaihy, C. Poüs, A. Pilon, and B. Benoit. The comet and remnant parameters were determined by D. Bakhos-Douaihy. The MT dynamics parameters in cells were determined by H. Henrie, D. Bakhos-Douaihy, C. Poüs, A. Pilon, and B. Benoit. M. Talantikite purified the proteins. In vitro assays were done by A. Pilon, D. Bakhos-Douaihy, and C. Poüs. Interpretation of data was done by D. Bakhos-Douaihy, A. Baillet, C. Poüs, and B. Benoit. The manuscript was drafted by H. Henrie, C. Poüs, and B. Benoit and revised by A. Baillet, A. Pilon, and D. Bakhos-Douaihy. Figures were mounted by B. Benoit and C. Poüs. All supplementary tables, statistical analyses, schematic drawings, and software development were performed by C. Poüs.
References
- Aher A., Kok M., Sharma A., Rai A., Olieric N., Rodríguez-García R., Katrukha E.A., Weinert T., Olieric V., Kapitein L.C., et al. 2018. CLASP Suppresses Microtubule Catastrophes through a Single TOG Domain. Dev. Cell. 46:40–58.E8. 10.1016/j.devcel.2018.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A., and Steinmetz M.O.. 2015. Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol. 16:711–726. 10.1038/nrm4084 [DOI] [PubMed] [Google Scholar]
- Al-Bassam J., Kim H., Brouhard G., van Oijen A., Harrison S.C., and Chang F.. 2010. CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev. Cell. 19:245–258. 10.1016/j.devcel.2010.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin M.A., Itoh G., Iemura K., Ikeda M., and Tanaka K.. 2014. CLIP-170 recruits PLK1 to kinetochores during early mitosis for chromosome alignment. J. Cell Sci. 127:2818–2824. 10.1242/jcs.150755 [DOI] [PubMed] [Google Scholar]
- Arnal I., Heichette C., Diamantopoulos G.S., and Chrétien D.. 2004. CLIP-170/tubulin-curved oligomers coassemble at microtubule ends and promote rescues. Curr. Biol. 14:2086–2095. 10.1016/j.cub.2004.11.055 [DOI] [PubMed] [Google Scholar]
- Aumeier C., Schaedel L., Gaillard J., John K., Blanchoin L., and Théry M.. 2016. Self-repair promotes microtubule rescue. Nat. Cell Biol. 18:1054–1064. 10.1038/ncb3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S.A., Jedrychowski M., Schwartz D., Elias J.E., Villén J., Li J., Cohn M.A., Cantley L.C., and Gygi S.P.. 2004. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA. 101:12130–12135. 10.1073/pnas.0404720101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat V., Siebrecht M., Burk K., Ahmed S., Reissner C., Kohansal-Nodehi M., Steubler V., Zweckstetter M., Ting J.T., and Dean C.. 2017. Capture of Dense Core Vesicles at Synapses by JNK-Dependent Phosphorylation of Synaptotagmin-4. Cell Rep. 21:2118–2133. 10.1016/j.celrep.2017.10.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieling P., Kandels-Lewis S., Telley I.A., van Dijk J., Janke C., and Surrey T.. 2008. CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol. 183:1223–1233. 10.1083/jcb.200809190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch M.A., and Kobe B.. 2006. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70:1061–1095. 10.1128/MMBR.00025-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanacci V., Urvoas A., Cantos-Fernandes S., Aumont-Nicaise M., Arteni A.-A., Velours C., Valerio-Lepiniec M., Dreier B., Plückthun A., Pilon A., et al. 2019. Insight into microtubule nucleation from tubulin-capping proteins. Proc. Natl. Acad. Sci. USA. 116:9859–9864. 10.1073/pnas.1813559116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wang P., and Slep K.C.. 2019. Mapping multivalency in the CLIP-170-EB1 microtubule plus-end complex. J. Biol. Chem. 294:918–931. 10.1074/jbc.RA118.006125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.H., Bertram P.G., Drenan R., Carvalho J., Zhou H.H., and Zheng X.F.S.. 2002. The FKBP12-rapamycin-associated protein (FRAP) is a CLIP-170 kinase. EMBO Rep. 3:988–994. 10.1093/embo-reports/kvf197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collazos A., Michael N., Whelan R.D.H., Kelly G., Mellor H., Pang L.C.H., Totty N., and Parker P.J.. 2011. Site recognition and substrate screens for PKN family proteins. Biochem. J. 438:535–543. 10.1042/BJ20110521 [DOI] [PubMed] [Google Scholar]
- Daire V., Giustiniani J., Leroy-Gori I., Quesnoit M., Drevensek S., Dimitrov A., Perez F., and Poüs C.. 2009. Kinesin-1 regulates microtubule dynamics via a c-Jun N-terminal kinase-dependent mechanism. J. Biol. Chem. 284:31992–32001. 10.1074/jbc.M109.007906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Forges H., Pilon A., Cantaloube I., Pallandre A., Haghiri-Gosnet A.-M., Perez F., and Poüs C.. 2016. Localized Mechanical Stress Promotes Microtubule Rescue. Curr. Biol. 26:3399–3406. 10.1016/j.cub.2016.10.048 [DOI] [PubMed] [Google Scholar]
- Dhamodharan R., and Wadsworth P.. 1995. Modulation of microtubule dynamic instability in vivo by brain microtubule associated proteins. J. Cell Sci. 108:1679–1689. [DOI] [PubMed] [Google Scholar]
- Diamantopoulos G.S., Perez F., Goodson H.V., Batelier G., Melki R., Kreis T.E., and Rickard J.E.. 1999. Dynamic localization of CLIP-170 to microtubule plus ends is coupled to microtubule assembly. J. Cell Biol. 144:99–112. 10.1083/jcb.144.1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov A., Quesnoit M., Moutel S., Cantaloube I., Poüs C., and Perez F.. 2008. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science. 322:1353–1356. 10.1126/science.1165401 [DOI] [PubMed] [Google Scholar]
- Dixit R., Barnett B., Lazarus J.E., Tokito M., Goldman Y.E., and Holzbaur E.L.. 2009. Microtubule plus-end tracking by CLIP-170 requires EB1. Proc. Natl. Acad. Sci. USA. 106:492–497. 10.1073/pnas.0807614106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M.-M., and Holzbaur E.L.F.. 2013. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol. 202:495–508. 10.1083/jcb.201302078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M.-M., Nirschl J.J., and Holzbaur E.L.F.. 2014. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell. 29:577–590. 10.1016/j.devcel.2014.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M., Watanabe T., Noritake J., Nakagawa M., Yamaga M., Kuroda S., Matsuura Y., Iwamatsu A., Perez F., and Kaibuchi K.. 2002. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 109:873–885. 10.1016/S0092-8674(02)00800-0 [DOI] [PubMed] [Google Scholar]
- Goldspink D.A., Rookyard C., Tyrrell B.J., Gadsby J., Perkins J., Lund E.K., Galjart N., Thomas P., Wileman T., and Mogensen M.M.. 2017. Ninein is essential for apico-basal microtubule formation and CLIP-170 facilitates its redeployment to non-centrosomal microtubule organizing centres. Open Biol. 7 160274 10.1098/rsob.160274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson H.V., Skube S.B., Stalder R., Valetti C., Kreis T.E., Morrison E.E., and Schroer T.A.. 2003. CLIP-170 interacts with dynactin complex and the APC-binding protein EB1 by different mechanisms. Cell Motil. Cytoskeleton. 55:156–173. 10.1002/cm.10114 [DOI] [PubMed] [Google Scholar]
- Henty-Ridilla J.L., Rankova A., Eskin J.A., Kenny K., and Goode B.L.. 2016. Accelerated actin filament polymerization from microtubule plus ends. Science. 352:1004–1009. 10.1126/science.aaf1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnappa S., Gouveia S.M., Weisbrich A., Damberger F.F., Bhavesh N.S., Jawhari H., Grigoriev I., van Rijssel F.J., Buey R.M., Lawera A., et al. 2009. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 138:366–376. 10.1016/j.cell.2009.04.065 [DOI] [PubMed] [Google Scholar]
- Hoogenraad C.C., Akhmanova A., Grosveld F., De Zeeuw C.I., and Galjart N.. 2000. Functional analysis of CLIP-115 and its binding to microtubules. J. Cell Sci. 113:2285–2297. [DOI] [PubMed] [Google Scholar]
- Hotamisligil G.S., and Davis R.J.. 2016. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 8 a006072 10.1101/cshperspect.a006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovasevic V., Naghavi M.H., and Walsh D.. 2015. Microtubule plus end-associated CLIP-170 initiates HSV-1 retrograde transport in primary human cells. J. Cell Biol. 211:323–337. 10.1083/jcb.201505123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakeno M., Matsuzawa K., Matsui T., Akita H., Sugiyama I., Ishidate F., Nakano A., Takashima S., Goto H., Inagaki M., et al. 2014. Plk1 phosphorylates CLIP-170 and regulates its binding to microtubules for chromosome alignment. Cell Struct. Funct. 39:45–59. 10.1247/csf.14001 [DOI] [PubMed] [Google Scholar]
- Kedashiro S., Pastuhov S.I., Nishioka T., Watanabe T., Kaibuchi K., Matsumoto K., and Hanafusa H.. 2015. LRRK1-phosphorylated CLIP-170 regulates EGFR trafficking by recruiting p150Glued to microtubule plus ends. J. Cell Sci. 128:385–396. 10.1242/jcs.161547 [DOI] [PubMed] [Google Scholar]
- Komarova Y.A., Akhmanova A.S., Kojima S., Galjart N., and Borisy G.G.. 2002. Cytoplasmic linker proteins promote microtubule rescue in vivo. J. Cell Biol. 159:589–599. 10.1083/jcb.200208058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y.-W., Trottier O., Mahamdeh M., and Howard J.. 2019. Spastin is a dual-function enzyme that severs microtubules and promotes their regrowth to increase the number and mass of microtubules. Proc. Natl. Acad. Sci. USA. 116:5533–5541. 10.1073/pnas.1818824116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix B., Bourdages K.G., Dorn J.F., Ihara S., Sherwood D.R., Maddox P.S., and Maddox A.S.. 2014. In situ imaging in C. elegans reveals developmental regulation of microtubule dynamics. Dev. Cell. 29:203–216. 10.1016/j.devcel.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbergen G., Komarova Y., Modesti M., Wyman C., Hoogenraad C.C., Goodson H.V., Lemaitre R.P., Drechsel D.N., van Munster E., Gadella T.W. Jr., et al. 2004. Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J. Cell Biol. 166:1003–1014. 10.1083/jcb.200402082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence E.J., Arpag G., Norris S.R., and Zanic M.. 2018. Human CLASP2 specifically regulates microtubule catastrophe and rescue. Mol. Biol. Cell. 29:1168–1177. 10.1091/mbc.E18-01-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-S., Komarova Y.A., Nadezhdina E.S., Anjum R., Peloquin J.G., Schober J.M., Danciu O., van Haren J., Galjart N., Gygi S.P., et al. 2010. Phosphorylation controls autoinhibition of cytoplasmic linker protein-170. Mol. Biol. Cell. 21:2661–2673. 10.1091/mbc.e09-12-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K., and Davis R.J.. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA. 100:2432–2437. 10.1073/pnas.0438011100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K., Nimnual A., Zong W.-X., Kennedy N.J., Flavell R.A., Thompson C.B., Bar-Sagi D., and Davis R.J.. 2002. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22:4929–4942. 10.1128/MCB.22.13.4929-4942.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Liu X.S., Yang X., Wang Y., Wang Y., Turner J.R., and Liu X.. 2010. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments. EMBO J. 29:2953–2965. 10.1038/emboj.2010.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom J.J., Nakamura M., Saltini M., Hibbel A., Walia A., Ketelaar T., Emons A.M.C., Sedbrook J.C., Kirik V., Mulder B.M., et al. 2019. CLASP stabilization of plus ends created by severing promotes microtubule creation and reorientation. J. Cell Biol. 218:190–205. 10.1083/jcb.201805047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeh R., Lorin S., Ratier A., Mejdoubi-Charef N., Baillet A., Bruneel A., Hamaï A., Codogno P., Poüs C., and Perdiz D.. 2014. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate α-tubulin acetyltransferase-1 (αTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J. Biol. Chem. 289:11816–11828. 10.1074/jbc.M113.507400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki T., Grimaldi A.D., Fuchigami S., Kaverina I., and Hayashi I.. 2015. CLASP2 Has Two Distinct TOG Domains That Contribute Differently to Microtubule Dynamics. J. Mol. Biol. 427:2379–2395. 10.1016/j.jmb.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y., Grigoriev I., Lansbergen G., Sasaki H., Matsui C., Severin F., Galjart N., Grosveld F., Vorobjev I., Tsukita S., et al. 2005. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 168:141–153. 10.1083/jcb.200405094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima M., Maesaki R., Kasa M., Watanabe T., Fukata M., Kaibuchi K., and Hakoshima T.. 2007. Structural basis for tubulin recognition by cytoplasmic linker protein 170 and its autoinhibition. Proc. Natl. Acad. Sci. USA. 104:10346–10351. 10.1073/pnas.0703876104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T., and Kirschner M.. 1984. Dynamic instability of microtubule growth. Nature. 312:237–242. 10.1038/312237a0 [DOI] [PubMed] [Google Scholar]
- Mohan R., Katrukha E.A., Doodhi H., Smal I., Meijering E., Kapitein L.C., Steinmetz M.O., and Akhmanova A.. 2013. End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc. Natl. Acad. Sci. USA. 110:8900–8905. 10.1073/pnas.1300395110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki T., and Goshima G.. 2016. Five factors can reconstitute all three phases of microtubule polymerization dynamics. J. Cell Biol. 215:357–368. 10.1083/jcb.201604118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moughamian A.J., Osborn G.E., Lazarus J.E., Maday S., and Holzbaur E.L.F.. 2013. Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J. Neurosci. 33:13190–13203. 10.1523/JNEUROSCI.0935-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustyatsa V.V., Kostarev A.V., Tvorogova A.V., Ataullakhanov F.I., Gudimchuk N.B., and Vorobjev I.A.. 2019. Fine structure and dynamics of EB3 binding zones on microtubules in fibroblast cells. Mol. Biol. Cell. 30:2105–2114. 10.1091/mbc.E18-11-0723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A., Kato H., Watanabe T., Min K.-D., Yamazaki S., Asano Y., Seguchi O., Higo S., Shintani Y., Asanuma H., et al. 2010. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat. Cell Biol. 12:583–590. 10.1038/ncb2060 [DOI] [PubMed] [Google Scholar]
- Nirschl J.J., Magiera M.M., Lazarus J.E., Janke C., and Holzbaur E.L.F.. 2016. α-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 14:2637–2652. 10.1016/j.celrep.2016.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padzik A., Deshpande P., Hollos P., Franker M., Rannikko E.H., Cai D., Prus P., Mågård M., Westerlund N., Verhey K.J., et al. 2016. KIF5C S176 Phosphorylation Regulates Microtubule Binding and Transport Efficiency in Mammalian Neurons. Front. Cell. Neurosci. 10:57 10.3389/fncel.2016.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez F., Diamantopoulos G.S., Stalder R., and Kreis T.E.. 1999. CLIP-170 highlights growing microtubule ends in vivo. Cell. 96:517–527. 10.1016/S0092-8674(00)80656-X [DOI] [PubMed] [Google Scholar]
- Peris L., Thery M., Fauré J., Saoudi Y., Lafanechère L., Chilton J.K., Gordon-Weeks P., Galjart N., Bornens M., Wordeman L., et al. 2006. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 174:839–849. 10.1083/jcb.200512058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre P., Scheel J., Rickard J.E., and Kreis T.E.. 1992. CLIP-170 links endocytic vesicles to microtubules. Cell. 70:887–900. 10.1016/0092-8674(92)90240-D [DOI] [PubMed] [Google Scholar]
- Pierre P., Pepperkok R., and Kreis T.E.. 1994. Molecular characterization of two functional domains of CLIP-170 in vivo. J. Cell Sci. 107:1909–1920. [DOI] [PubMed] [Google Scholar]
- Rickard J.E., and Kreis T.E.. 1991. Binding of pp170 to microtubules is regulated by phosphorylation. J. Biol. Chem. 266:17597–17605. [PubMed] [Google Scholar]
- Tanenbaum M.E., Galjart N., van Vugt M.A.T.M., and Medema R.H.. 2006. CLIP-170 facilitates the formation of kinetochore-microtubule attachments. EMBO J. 25:45–57. 10.1038/sj.emboj.7600916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma C.R., Matov A., Gutbrodt K.L., Hoerner C.R., Smole Z., Krek W., and Danuser G.. 2010. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J. Cell Biol. 190:991–1003. 10.1083/jcb.201006059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropini C., Roth E.A., Zanic M., Gardner M.K., and Howard J.. 2012. Islands containing slowly hydrolyzable GTP analogs promote microtubule rescues. PLoS One. 7 e30103 10.1371/journal.pone.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haren J., Charafeddine R.A., Ettinger A., Wang H., Hahn K.M., and Wittmann T.. 2018. Local control of intracellular microtubule dynamics by EB1 photodissociation. Nat. Cell Biol. 20:252–261. 10.1038/s41556-017-0028-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan P.S., Miura P., Henderson M., Byrne B., and Vaughan K.T.. 2002. A role for regulated binding of p150(Glued) to microtubule plus ends in organelle transport. J. Cell Biol. 158:305–319. 10.1083/jcb.200201029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemu A., Szczesna E., Zehr E.A., Spector J.O., Grigorieff N., Deaconescu A.M., and Roll-Mecak A.. 2018. Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science. 361:eaau1504. doi:. 10.1126/science.aau1504 [DOI] [PMC free article] [PubMed]
- Verhey K.J., Meyer D., Deehan R., Blenis J., Schnapp B.J., Rapoport T.A., and Margolis B.. 2001. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152:959–970. 10.1083/jcb.152.5.959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R.A., O’Brien E.T., Pryer N.K., Soboeiro M.F., Voter W.A., Erickson H.P., and Salmon E.D.. 1988. Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J. Cell Biol. 107:1437–1448. 10.1083/jcb.107.4.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrich A., Honnappa S., Jaussi R., Okhrimenko O., Frey D., Jelesarov I., Akhmanova A., and Steinmetz M.O.. 2007. Structure-function relationship of CAP-Gly domains. Nat. Struct. Mol. Biol. 14:959–967. 10.1038/nsmb1291 [DOI] [PubMed] [Google Scholar]
- Wittmann T., and Waterman-Storer C.M.. 2005. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J. Cell Biol. 169:929–939. 10.1083/jcb.200412114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Li H., Liu X.S., Deng A., and Liu X.. 2009. Cdc2-mediated phosphorylation of CLIP-170 is essential for its inhibition of centrosome reduplication. J. Biol. Chem. 284:28775–28782. 10.1074/jbc.M109.017681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J., Whitmarsh A.J., Cavanagh J., Sharma M., and Davis R.J.. 1999. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 19:7245–7254. 10.1128/MCB.19.10.7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeke A., Misheva M., Reményi A., and Bogoyevitch M.A.. 2016. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 80:793–835. 10.1128/MMBR.00043-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
presents parameters of MT dynamic instability related to Fig. S3.
shows statistical values related to Table S1.
presents parameters of MT dynamic instability related to Fig. 4.
shows statistical values related to Table S3.
presents parameters of MT dynamic instability related to Fig. 5.
shows statistical values related to Table S5.
presents parameters of MT dynamic instability related to Fig. 6.
shows statistical values related to Table S7.
presents CLIP-170 comet and remnant parameters related to Fig. 7 B.
shows statistical values related to Table S9.
presents CLIP-170 comet and remnant parameters related to Fig. S5.
shows statistical values related to Table S11.