


Abstract
RNA-seq experiments previously performed by our laboratories showed enrichment in intronic sequences and alterations in alternative splicing in dengue-infected human cells. The transcript of the SAT1 gene, of well-known antiviral action, displayed higher inclusion of exon 4 in infected cells, leading to an mRNA isoform that is degraded by non-sense mediated decay. SAT1 is a spermidine/spermine acetyl-transferase enzyme that decreases the reservoir of cellular polyamines, limiting viral replication. Delving into the molecular mechanism underlying SAT1 pre-mRNA splicing changes upon viral infection, we observed lower protein levels of RBM10, a splicing factor responsible for SAT1 exon 4 skipping. We found that the dengue polymerase NS5 interacts with RBM10 and its sole expression triggers RBM10 proteasome-mediated degradation. RBM10 over-expression in infected cells prevents SAT1 splicing changes and limits viral replication, while its knock-down enhances the splicing switch and also benefits viral replication, revealing an anti-viral role for RBM10. Consistently, RBM10 depletion attenuates expression of interferon and pro-inflammatory cytokines. In particular, we found that RBM10 interacts with viral RNA and RIG-I, and even promotes the ubiquitination of the latter, a crucial step for its activation. We propose RBM10 fulfills diverse pro-inflammatory, anti-viral tasks, besides its well-documented role in splicing regulation of apoptotic genes.
INTRODUCTION
Dengue virus (DENV) is the most prevalent arbovirus worldwide, found in over 100 tropical and sub-tropical countries (1). It is transmitted mainly by Aedes aegypti and Aedes albopictus mosquitoes. Over half of the global population is at risk for dengue infection, with 100 million symptomatic cases being reported every year (2). DENV is part of the Flaviviridae family, which also includes Zika (ZIKV), yellow fever, Japanese encephalitis, and West Nile viruses. They are enveloped, icosahedral viruses, with a positive-sense and single-stranded RNA (ssRNA) genome that encodes one open reading frame that, upon proteolytic processing, gives rise to three structural (capsid, precursor membrane (prM), and envelope) and seven nonstructural (NS) proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5). The genome is approximately 11 kb in length, containing a type I cap at the 5′ end and lacking a 3′ poly(A) tail (3,4). Like other RNA viruses, the DENV genome encodes a limited set of proteins, relying on the host machinery for productive replication (5).
For DENV, as for many RNA viruses, an initial step in the establishment of the cellular innate antiviral response is the sensing of cytosolic RNA by retinoic acid-inducible gene I (RIG-I) like receptors (RLRs). Expressed by most cells of the human organism, RLRs belong to the family of DExD/H-box helicases and include three members of relevance: RIG-I, melanoma differentiation-associated antigen 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) (6). Studies of gene-deficient mice indicate that RLRs are critical sensors of viral infection. RLRs are expressed at low concentrations in the resting cell and, upon stimulation by viral infection, they trigger signaling pathways that promote the IRF3-, IRF7-dependent expression of type I and type III interferons (IFNs), as well as the NF-kappa B-dependent expression of pro-inflammatory cytokines. In the case of DENV, it was reported that RIG-I recognizes the 5′ region of the viral genome, in particular a double stranded RNA structure bearing 5′-triphosphates that is only present during viral genome replication (7). This recognition is followed by a multi-step activation process including conformational changes and several post-translational modifications of RIG-I that are crucial to unleash its full activity and consequently its downstream signaling pathway. One of the first RIG-I post-translational modifications is the non-degradative ubiquitination at multiple sites, involving the activity of different ubiquitin E3 ligases (8,9).
Several transcriptomic studies identified spermidine/spermine-N1-acetyltransferase (SAT1) as a downstream target of type I IFN stimulation in cell culture and in vivo (10). SAT1 regulates the cellular content of polyamines, which are small, abundant, aliphatic molecules present in all mammalian cells. Within the context of the cell, they play a myriad of roles, from modulating nucleic acid conformation to promoting cellular proliferation and signaling (11). In addition, polyamines have emerged as important molecules in virus-host interactions. Many viruses have been shown to require polyamines for one or more aspects of their replication cycle, including DNA and RNA polymerization, nucleic acid packaging, and protein synthesis (12,13). When SAT1 acetylates polyamines, they are either secreted from the cell or oxidized by acetylpolyamine oxidase. The fact that SAT1 reduces polyamine availability and is upregulated by IFN assigns it an anti-viral role (14).
Alternative splicing provides an important regulatory step for the control of SAT1 mRNA production. SAT1 pre-mRNA can undergo alternative splicing giving rise to ‘SAT1-IN’ mRNA isoform, which includes an additional 110-nt exon containing multiple premature termination codons downstream of constitutive exon 3 and upstream of constitutive exons 5, 6 and 7. This renders the SAT1-IN splice variant a target for nonsense-mediated mRNA decay (NMD) (Figure 1A). Polyamines or analogs inhibit inclusion of this ‘poison’ exon, leading to a higher proportion of stable and protein-coding SAT1 mRNA, the isoform that lacks exon 4 ‘SAT1-EX’ (15,16). The rapid loss of SAT1-IN mRNA via NMD may limit any accumulation in cells, although SAT1-IN mRNA has been found to accumulate in response to X-rays (17), viruses (18) and hypoxia (19). In our previous work, in which we proposed that DENV NS5 protein hijacks the cellular spliceosome and modulates splicing, RNA-seq experiments revealed an accumulation of SAT1-IN mRNA upon DENV infection (20).
Figure 1.

Dengue virus infection modulates SAT1 alternative splicing. (A) Diagram of SAT1 pre-mRNA splicing giving rise to two different splicing isoforms, a protein coding (EX isoform) and a non-coding one (IN isoform). Arrows indicate the position of the primer pairs used for end-point and qPCR. (B)SAT1 RNA-seq reads indicating the position of alternative 110 nt-exon 4 from mock and dengue infected A549 cells (20) at 24 h post-infection (hpi). (C) Reverse transcription followed by end-point PCR identifying SAT1 splicing isoforms by electrophoresis in agarose gel, from total RNA of A549 cells at 24 hpi. (D) Quantified SAT1 inclusion/exclusion ratio with specific primers for each splicing isoform by RT-qPCR, from total RNA of A549 cells at 24 hpi with DENV or ZIKV. (E–H) RT-qPCRs of viral RNA (E), Interferon Stimulated Gene 15 (ISG15) (F), SAT1-IN and EX isoforms (G) and SAT1-IN and EX isoforms relative to SAT1 pre-mRNA (H) from A549 cells at the indicated time points post-infection. Viral infection was also monitored by plaque formation assay (PFU: Plaque Forming Units) (E). Average values from triplicates are shown with standard deviation and p-values, determined using a paired two-tailed t-test. Significant p-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05).
SAT1 pre-mRNA alternative splicing is known to be regulated by the RNA-binding protein RBM10 (21). This protein contains two RNA recognition motifs (RRM), two zinc fingers and one G patch motif, features that are shared with other splicing-related factors, including protein components of the small nuclear ribonucleoprotein particles (snRNPs) that constitute the spliceosome (22,23). Proteomic studies carried out by several laboratories detected the association of RBM10 with purified splicing complexes (24–29), further supporting its role as a regulator of pre-mRNA splicing. Moreover, RBM10 and its paralog RBM5 have been shown to regulate alternative splicing of apoptosis related genes, assigning both factors a tumor suppressor role (30–33). RNA-seq experiments identified significant splicing changes following RBM10 depletion or over-expression in HEK 293T cells. A deeper analysis of the affected alternative splicing events identified a predominance of enhanced exon skipping, correlating with a robust binding of RBM10 to upstream and downstream introns flanking the corresponding alternative exons (34). In addition, RBM10 has been reported to regulate its own alternative splicing (35), by promoting exon 6 and 12 skipping. This gives rise to NMD-targeted mRNA isoforms regulating its own protein levels. Importantly, lung adenocarcinoma-associated mutations affecting splice sites of RBM10 exons 6 or 12 abolish exon inclusion and correlate with reduced RBM10 mRNA levels. Of note, recent mass spectrometry-based interactome studies revealed that RBM10 interacts with ZIKV NS5 protein, the viral polymerase that was also reported to bind several core splicing components, both in Zika and DENV viruses (20,36).
Previous work from our laboratories has revealed that DENV infection as well as DENV NS5 per se trigger changes in alternative splicing patterns of human host cell lines (20). Here, we report that among several affected pre-mRNAs, SAT1 splicing patterns are modulated upon DENV infection. Consistent with this, we observed a decrease in RBM10 protein levels, a splicing factor known to regulate SAT1 splicing (21). We found that DENV NS5 binds RBM10 and mediates its proteasomal degradation. Additionally, RBM10 over-expression not only restores SAT1 splicing to the levels observed in mock conditions but also attenuates DENV replication, while RBM10 depletion enhances viral proliferation. These observations led us to assign an anti-viral role to RBM10, supported also by the evidence that transcriptional induction downstream of RLR activation is attenuated when cells are infected with DENV in the context of RBM10 knock-down. We propose that RBM10 exerts this pro-inflammatory function, at least in part, by interacting with RIG-I receptor, which could be facilitated by cytoplasmic DENV RNA, independently of the type I IFN receptor (IFNR) signaling pathway. In line with this idea, we show that RBM10 over-expression enhances RIG-I ubiquitination, a crucial step along the activation of this viral sensor required for the establishment of a robust innate immune response. In this context, it is possible that DENV protein NS5 restricts RBM10 from playing an anti-viral role, not only by physically interacting with it, but also by triggering its proteasomal degradation.
MATERIALS AND METHODS
Cell lines
A549 cells (human lung adenocarcinoma epithelial cell line, ATCC, CCL-185) were maintained in Dulbecco's modified Eagle's medium/F-12 (Ham's) and HEK 293T cells (human embryonic kidney cell lines, ATCC CRL-1573) were maintained in Dulbecco's modified Eagle's medium. The A549 media was supplemented with 5% fetal bovine serum and 50 μg/ml gentamicin while the HEK 293T media with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin.
DENV infections, IFN treatment and DNA/RNA transfections
DENV-2 infections, strain 16681, were performed with a multiplicity of infection (MOI) ∼1. The viral adsorption was carried out at 4°C with agitation and, after one hour of incubation, replaced with fresh media. For IFN treatments, 50 000 U/ml of interferon α 2b (Biosidus) were added to cell media. Plasmid DNA and siRNAs (Dharmacon) were transfected with 293-Free™ Transfection Reagent (Merck) and Lipofectamine 2000 (Thermo Fisher), respectively, according to manufacturer's instructions. Poly I:C intracellular administration was performed by transfecting polyinosinic-polycytidilic acid [poly(I:C)] HMW (InvivoGen) with Lipofectamine 2000, at a ratio of 10:1 and a final concentration of Poly I:C of 10 μg/ml. Treatment with JAK inhibitor I (SC-204021) was performed at a final concentration of 1 μM for 24 h and the corresponding control was performed with the appropriate volume of DMSO.
T7-RBM10 plasmid was kindly provided by Dr Cáceres’ laboratory (MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, UK) (37), Streptag-NS5 has been previously described (20), and Flag-RIG I plasmid was kindly provided by Dr Fujita's laboratory (Institute for Frontier Life and Medical Sciences, Kyoto University, Japan).
End point and quantitative PCR
Total cellular RNA was isolated by using Trizol (Invitrogen) and reverse transcribed with random deca-oligonucleotide primer mix. End-point PCR products were electrophoresed in 1–1.5% ethidium bromide-containing agarose gels. Quantitative PCRs (qPCRs) were performed by using SYBR Green dye and specific primers for different pre-mRNAs, mRNAs and DENV RNA genome. GAPDH mRNA was used as a control housekeeping gene. The specific primers used for PCR and qPCR are listed in Supplementary Table S1.
When indicated, RNA samples were prepared from different subcellular fractions: chromatin; nucleoplasm or cytoplasm obtained as described in Supplementary Material.
DENV replication, Poly I:C intracellular administration and IFN treatment in the context of RNA interference
RNA interference experiments were carried out using siGENOME ON-TARGET plus SMART pool siRNA oligonucleotides (Dharmacon RNA Technologies). The control non-related siRNA used was directed against firefly luciferase. Twenty-four hours after seeding in 24-well plates, A549 cells were transfected with the corresponding siRNA. Briefly, 20 pmol of siRNA in 50 μl of Opti-MEM (Invitrogen) were mixed with 2 μl of Lipofectamine in 50 μl of Opti-MEM and incubated for 20 min. The mix was added to a 50% confluent A549 cells monolayer and incubated overnight. After 24 h of transfection, cells were mock infected or infected, as indicated above. Alternatively, cells were transfected with Poly I:C or treated with IFN. RNA was purified using Trizol at 48 h post-infection (hpi), at 9 h after Poly I:C transfection or 24 h after IFN addition. RT-PCR and qPCR were performed as previously described using primers listed on Supplementary Table S1.
Antibodies
Rabbit polyclonal antibodies against RBM10 were purchased from Abcam (ab72423) and Sigma-Aldrich (HPA034972), anti-RIG-I from Santa Cruz (D-12, SCBT), anti-tubulin (mouse monoclonal) from Sigma-Aldrich (T9026), anti-T7 tag (mouse monoclonal) from Merck (69522), anti-H3 (rabbit polyclonal) from Abcam (ab1791), and anti-dsRNA antibody from SCICONS (mAb J2). Anti-NS5 antibodies (rabbit polyclonal and mouse polyclonal) were obtained in our laboratory and anti-Streptag antibody (mouse monoclonal) was purchased from EMD Millipore (71590-3).
Western blots
Protein samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare Amersham). Membranes were blocked and then incubated with primary antibodies. After washing, membranes were incubated with IRDye® 800CW or 680RD (LI-COR Biosciences) secondary antibodies. Bound antibody was detected using an Odyssey imaging system (LI-COR Biosciences). Western blots were performed at least three times from independent experiments and representative images are shown in each case.
Immunofluorescence assays
Immunofluorescence assays were performed as previously described (38). Briefly, A549 cells were seeded into 24-well plates containing glass coverslips. Twenty-four hours later, cells were mock infected or infected with DENV. After 24 h, coverslips were collected and the cells were fixed with paraformaldehyde 4% in PBS pH 7.4 at room temperature for 10 min. PFA-fixed cells were then permeabilized with 1% Triton X-100 for 5 min at room temperature. Coverslips were blocked with BSA 3% in PBS and incubated with primary antibody (appropriate dilution in blocking solution) for 1 hour at RT. After three washes with PBS, incubation with secondary antibody (Alexa Fluor® 488 and 647, 1:500 in blocking solution) was performed for 1 h and washed 4 times with PBS. Coverslips were mounted with a drop of mounting medium (Vectashield) and images were obtained with an Olympus FV300 confocal microscope. Fluorescence intensity analysis of images was performed with Cell Profiler software (v 3.1.5) after generating a suitable pipeline for this purpose.
Subcellular fractionation
Preparation of nuclear and cytoplasmic proteins from A549 cells was conducted following a small-scale fractionation protocol described by (39). Briefly, ∼6 × 106 A549 cells were mock infected or infected with DENV. After 48 h, cells were harvested using a cell scraper and washed with 0.5 ml of PBS. A 0.1 ml aliquot was separated as whole cell extract and the rest pelleted at 100 rcf for 2 min. After centrifugation, cell pellets were resuspended in HLB buffer (10 mM Tris–HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.3% Igepal CA630, 10% glycerol and 1× protease inhibitors, Roche). Incubation was carried out on ice for 10 min. Samples were centrifuged at 4°C and 800 g for 8 min. The pellet corresponds to intact nuclei and the supernatant to cytoplasmic fraction. 2× Laemmli sample buffer was added to all fractions and they were sonicated twice for 5 s each, heated for 5 min at 95°C and analyzed by SDS PAGE/western blot.
Immunoprecipitation of RNP complexes
Cells were harvested and lysed in RIPA buffer (50 mM Tris–HCl (pH 7.5), 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.05% (w/v) SDS, 1 mM EDTA, 150 mM NaCl) containing complete protease inhibitor and RNasin (Promega). Extracts were sonicated at high amplitude with three 10-s bursts, and insoluble material was pelleted. Anti-RBM10 or control IgG were added to the supernatant and incubated overnight. When indicated, RNAse A (0,1 mg/ml) was previously added to lysates. Complexes were incubated with GammaBind G sepharose beads (Thermo Fisher) for 1 h and washed three times in wash buffer (50 mM Tris–HCl (pH 7.5), 0.1% (v/v) NP-40, 1 mM EDTA, 125 mM NaCl). For western blot analysis, beads were resuspended in 2× Laemmli sample buffer. For RNA immunoprecipitation analysis, beads were resuspended in Trizol and subjected to RNA extraction followed by RT-qPCR. cDNA was prepared from one-half of the RNA using 10-mer random primers. The cDNA and no-reverse transcription control were analyzed by qPCR with primer pairs spanning DENV RNA and SAT1 pre-mRNA as detailed in the primer list (Supplementary Table S1) and visualized as the average ratio of immunoprecipitated RNA divided by the input.
Purification of 6xHis-ubiquitin-conjugated proteins
HEK 293T cells were transfected in 35-mm culture wells with the indicated plasmids. After 48 h, His-ubiquitin conjugates were purified under denaturing conditions using Ni-NTA-agarose beads according to the manufacturer's instructions (Qiagen). Briefly, transfected cells were harvested in ice-cold PBS. An aliquot was taken as input and the remaining cells were lysed in 6 M guanidinium–HCl containing 100 mM Na2HPO4/NaH2PO4, 10 mM Tris–HCl pH 8.0, 5 mM imidazole. Samples were sonicated to reduce the viscosity and proteins were purified using Ni-NTA beads (Qiagen) according to (40). Samples were subsequently washed with wash buffer I (8 M urea, 10 mM Tris–HCl pH 8.0, 100 mM Na2HPO4/NaH2PO4, 5 mM imidazole), wash buffer II (8 M urea, 10 mM Tris–HCl, 100 mM Na2HPO4/NaH2PO4, 0.2% Triton X-100, 5 mM imidazole, pH 6.3), and wash buffer III (8 M urea, 10 mM Tris–HCl pH 6.3, 100 mM Na2HPO4/NaH2PO4, 0.1% Triton X-100, 5 mM imidazole. Samples were eluted in 2× Laemmli sample buffer containing 200 mM imidazole for 5 min at 95°C.
Statistics
Typically, three independent experiments in triplicate repeats were conducted for each condition examined. Average values are shown with standard deviation and P-values, measured with a paired two-tailed t-test. Significant P-values are indicated by the asterisks above the graphs (***P < 0.001; **P < 0.01; *P < 0.05).
RESULTS
Dengue virus infection modulates SAT1 alternative splicing
As mentioned earlier, SAT1 has received increasing attention for its anti-viral role through depletion of the cellular polyamines required for viral replication. It caught our attention that RNA-seq experiments previously performed by our laboratories (20) evidenced changes in the splicing pattern of SAT1, among several other splicing events that were altered upon DENV infection (Figure 1A and B). In particular, we observed an increase in reads corresponding to alternative exon 4 at 24 and 36 hours post-infection (hpi) compared to uninfected (Mock) cells. SAT1 mRNA isoforms either containing or lacking exon 4 were detected by RT-PCR (Figure 1A and C). We proceeded to quantify them by real time PCR (qPCR) and, confirming our previous RNA-seq results, a significant increase in exon 4 inclusion at 24 hpi was clearly observed. This effect was also observed after infecting A549 cells with ZIKV, another member of the Flaviviridae family (Figure 1A and D).
SAT1 splicing changes were analyzed by RT-qPCR throughout a wider time frame (0–36 hpi). To monitor infection over time, we quantified viral genome within cells and induction of Interferon Stimulated Gene 15 (ISG15) by RT-qPCR (Figure 1E and F), which is slightly delayed from viral replication detection, in agreement with previous reports (41,42). The progress of the infection was also verified by quantification of virus titers by plaque assay (Figure 1E, PFU/ml). We actually found that exon inclusion (IN) over exon exclusion (EX) ratio decreases at 6 hpi due to SAT1-EX enhancement (Figure 1G and H), which is consistent with the well-known SAT1 early induction as an anti-viral protein (10). However, IN/EX ratio is reversed already at 24 hpi and keeps increasing until 36 hpi (Figure 1G). This reversion in splicing IN/EX ratio is explained by SAT1-IN isoform accumulation and seems to mitigate the early induction of SAT1-coding isoform (EX) (Figure 1H). Taking into account that the NMD pathway takes place in the cytoplasm, the detection of SAT1 splicing changes at the level of chromatin-associated RNA, as well as nucleoplasmic RNA, rules out a possible accumulation of the SAT1-IN isoform as a mere consequence of NMD attenuation due to the translation blockade that has been reported to take place upon viral infection (43) (Supplementary Figure S1). Altogether these results corroborate a change in the splicing pattern of SAT1 pre-mRNA, increasing the proportion of the non-coding isoform, as a result of DENV infection.
RBM10 regulates SAT1 alternative splicing and modulates DENV infection
As reported in earlier studies, RBM10 role in alternative splicing is mainly fostering exon skipping (34) and SAT1 exon 4 has been identified as one of its targets (21). We assessed RBM10 activity either by its depletion or over-expression in A549 cells. We found a significant increase of SAT1 exon 4 inclusion when RBM10 was silenced by siRNA, and, consistently, the opposite effect when T7-tagged RBM10 was over-expressed in these cells (Figure 2A and B). siRNA specificity as well as RBM10 action on SAT1 splicing were validated by rescue experiments in which the effect of RBM10 depletion on SAT1 alternative splicing was reversed by over-expression of T7-RBM10 using an expression vector that lacks full complementarity with the pool of RBM10 siRNAs (Supplementary Figure S2A and B).
Figure 2.

RBM10 regulates SAT1 alternative splicing and modulates DENV infection (A, B) Quantified inclusion/exclusion ratio in A549 cells either transfected with siRNA against RBM10 (A) or an expression vector for T7-RBM10 (B). (C) RNA from A549 cells transfected with the indicated siRNAs or DNA plasmids for 24 h and infected with DENV for additional 48 h was quantified by RT-qPCR for SAT1 inclusion/exclusion ratio. (D) RBM10 knock-down and over-expression in A549 infected cells were monitored by western blot (M: Mock; DV: DENV). (E) Viral infection was monitored by RT-qPCR of viral RNA and by plaque formation assay (PFU: Plaque Forming Units). Average values from triplicates are shown with standard deviation and P-values, determined using a paired two-tailed t-test. Significant p-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05). (F) Model of SAT1 pre-mRNA splicing regulation upon RBM10 protein level alterations or DENV infection.
To further explore this result, we used the RBM10 depletion/over-expression background to evaluate SAT1 alternative splicing upon DENV infection. We transfected cells with siRBM10 or T7-RBM10 expression vector and 24 h later, infected them with DENV. Remarkably, RBM10 depletion enhanced the effect of viral infection on IN/EX ratio even further, whereas T7-RBM10 over-expression was able to prevent the infection-triggered increase of the IN/EX ratio at 48 hpi, as assayed by RT-PCR (Supplementary Figure S2C) and qPCR (Figure 2C). Moreover, we quantified viral genome within cells by qPCR and found higher levels of viral replication in RBM10-depleted cells, compared to siControl-transfected cells (Figure 2E, left panel). In contrast, viral replication was severely attenuated when RBM10 was over-expressed, compared to transfection with an empty vector (Figure 2E, right panel). The latter result was further confirmed by plaque assay (PFU/ml), a less sensitive but widely used measurement for viral titering. In all conditions, RBM10 protein levels were monitored by western blot (Figure 2D). These results lead us to propose an anti-viral role for RBM10, which is consistent with its already characterized pro-apoptotic function. Overall, advanced DENV infection seems to have the same effect on SAT1 pre-mRNA splicing as RBM10 knock-down (Figure 2F), which actually promotes viral replication and has an additive effect on SAT1 splicing when combined with infection (Figure 2C left panel and Supplementary Figure S2C).
To examine the global impact of DENV infection on RBM10 regulated splicing, we analyzed our RNA-seq data (20) together with the data set already published by Wang et al. (34), which evaluated differentially spliced exons upon RBM10 knock-down or over-expression in HEK 293T. The fact that we found several differentially spliced alternative exons that are shared by both data sets (Supplementary Table S2) suggests that RBM10 might account for DENV-triggered host cell splicing modulation beyond the particular case of SAT1 transcript. In this context, it is tempting to speculate that RBM10 action on a given combination of pre-mRNA targets could provide a more restrictive cellular milieu for viral replication, while viral tampering with the splicing pattern of several RBM10-dependent events upon infection could revert this situation, thus promoting a more permissive cellular environment for viral proliferation. It is likely that higher sequencing depth from RBM10-depleted infected cells would shed more light on this changing alternative splicing scenario.
DENV NS5 targets RBM10 for proteasomal degradation
Since RBM10 knock-down seemed to promote viral replication and RBM10 also accounted for many splicing changes observed upon DENV infection, we wondered whether DENV had any impact on RBM10. Therefore, protein levels were analyzed by western blot assay showing a decrease upon DENV infection (Figure 3A). This observation does not seem to be cell-type dependent, since it was observed both in A549 and HEK 293T cells (Supplementary Figure S3A). Additionally, and as previously reported, A549 cells have less RBM10 mRNA than HEK 293T cells, which inversely correlates with dengue viral replication ability within these two cell lines (Supplementary Figure S3B and C).
Figure 3.

DENV NS5 interacts with RBM10 and targets it for proteasomal degradation. (A) A549 cells were infected with DENV for 48 h and whole cell lysates were analyzed by western blot of RBM10, tubulin and NS5. (B) A549 cells were infected with DENV for 48 h and subjected to subcellular fractionation. Western blot of RBM10, tubulin, histone H3 and NS5 was performed from WCE: whole cell extract; C: cytoplasm and N: nucleus. (C) A549 cells were infected with DENV for 48 h and subjected to indirect immunofluorescence of RBM10 and NS5. (D) Western blot of T7-immunoprepicitated RBM10 and co-precipitated Streptag-NS5 in the absence or presence of RNase A. (E) A549 cells were transfected with empty vector or Streptag-NS5 expression vector (0.1, 0.5 and 1 μg) and treated with 2 μM MG132 for 3 h. RBM10, NS5 and tubulin levels were assessed by western blot. (F) A549 cells were transfected with empty vector or NS5 expression vector and after 48 h SAT1 inclusion/exclusion ratio was quantified by RT-qPCR. Average values from triplicates are shown with standard deviation and p-values, determined using a paired two-tailed t-test. Significant P-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05).
Subcellular fractionation evidenced that RBM10 co-fractionates with the DENV NS5 protein, which was also observed by indirect immunofluorescence, being both mainly detected within cell nucleus (44,45) (Figure 3B and C). RBM10 has already been reported to interact with ZIKV NS5 (36) and, in agreement with all these findings, RBM10 co-immunoprecipitated with DENV NS5, even after treatment with RNase A, suggesting that this interaction is RNA-independent (Figure 3D). In our previous report, mass spectrometry experiments failed to identify RBM10 as an NS5 partner (20). Nevertheless, we found that several spliceosomal proteins, in particular those belonging to U5 snRNP, were pulled down together with NS5. Therefore, we suggest that RBM10 might interact with NS5, not necessarily as a direct partner but in the context of active spliceosomes. However, and in contrast to RBM10, none of the spliceosomal proteins that were found to co-precipitate with NS5 in our previous study showed changes in protein levels upon DENV infection (20). We then wondered what could be the mechanism responsible for RBM10 decrease, and whether NS5 alone could be the main effector. To address this, we transfected increasing amounts of NS5 in A549 cells and after 48 h assessed RBM10 protein levels by western blot. It was indeed of note that RBM10 levels decreased with NS5 over-expression, and that this effect was no longer observed when proteasome was inhibited by MG132 (Figure 3E and Supplementary Figure S4A), neither when cells were treated with IFN nor transfected with Poly I:C, in the absence of infection or NS5 expression (Supplementary Figure S4B). These results led us to conclude that DENV NS5 protein was targeting RBM10 for proteasomal degradation. We then analyzed SAT1 splicing patterns after sole NS5 over-expression. In agreement with previous findings, the inclusion over exclusion ratio increased similarly to an RBM10 knock-down scenario, and concomitantly with a decrease in RBM10 levels (Figure 3F and Supplementary Figure S4A). This observation can be explained by NS5-targeted RBM10 down-regulation but also by the already mentioned evidence that supports NS5 intrusion into cellular spliceosomes (20).
RBM10 is involved in the RLR signaling pathway
Considering that DENV triggered a decrease in RBM10 protein levels, a condition that according to the aforementioned results favors viral replication, we used the RBM10 depletion/over-expression background to analyze the cellular innate immune response upon DENV infection (Figure 4). We quantified IFNα, RIG-I, IL-8 and ISG15 mRNAs by RT-qPCR and detected that DENV-triggered induction of these transcripts was abolished when RBM10 was silenced (Figure 4A). In contrast, over-expression of T7-RBM10 induced all of these transcripts even in the absence of infection (Figure 4B). In every case, RBM10 protein levels were monitored by western blot (Figure 4C). This led us to propose a possible involvement of RBM10 in the RLR signaling pathway, which senses viral cytosolic RNA and transiently induces the expression of type I IFN and NF-kappa B-dependent pro-inflammatory cytokines.
Figure 4.
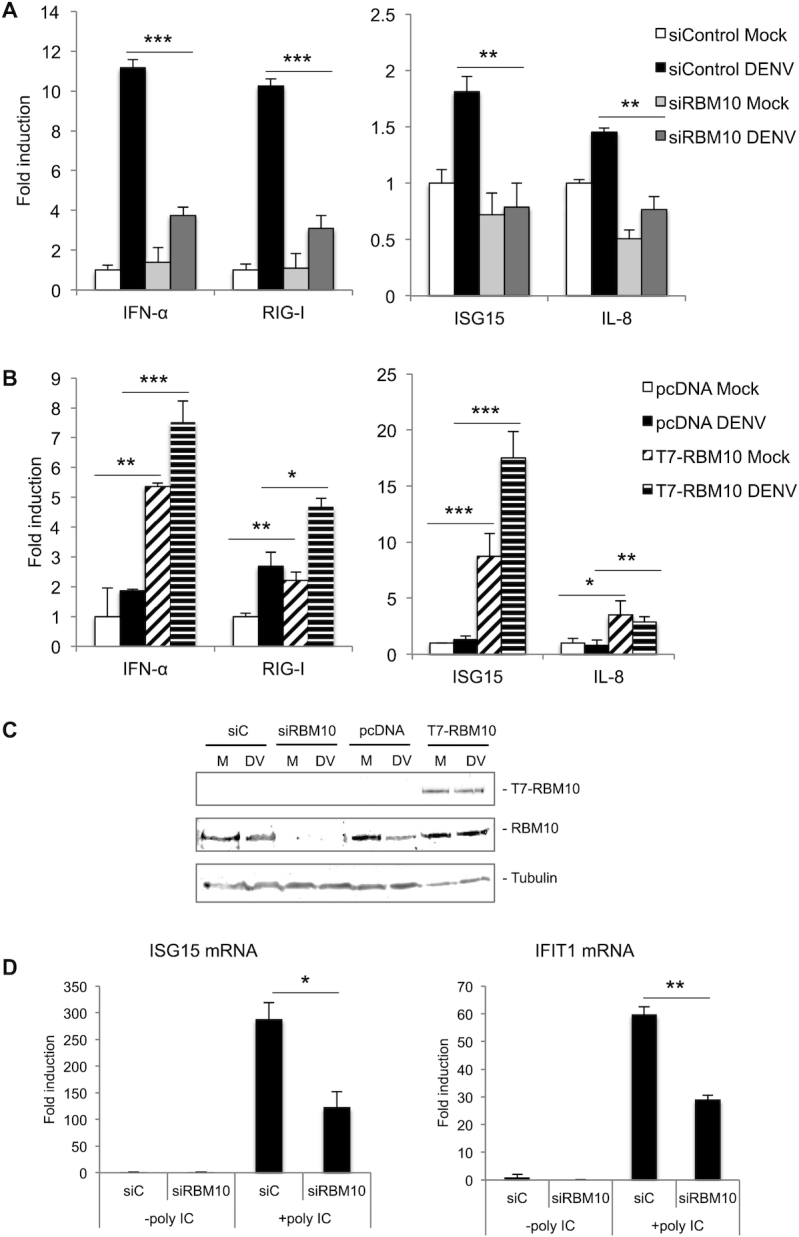
RBM10 is involved in the anti-viral innate response. (A–C) A549 cells were transfected with the indicated siRNAs (40 nM) or expression vectors (200 ng) for 24 h and infected with DENV for additional 48 h. RIG-I, IFN α, IL-8 and ISG15 mRNA levels were analyzed by RT-qPCR (A, B) and RBM10 and tubulin protein levels by western blot (M: Mock; DV: DENV) (C). (D) A549 cells were transfected with the indicated siRNAs (40 nM) for 72 h during which, the last 9 h included Poly I:C (10 μg /ml) intracellular administration. Upon harvesting, RNA was prepared and analyzed by RT-qPCR for ISG15 and IFIT1 mRNAs. Average values from triplicates are shown with standard deviation and P-values, determined using a paired two-tailed t-test. Significant P-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05).
Supporting a role for RBM10 in the innate immune response, we found that RBM10 depletion also interferes with the induction of Type I IFN (IFNβ) and ISGs (ISG15 and IFIT1) mRNAs upon cellular treatment with Poly I:C, a synthetic analogue of viral RNA (Figures 4D and 5A). Remarkably, these results point to a more general and widespread role of RBM10 in modulating the innate immune response, as part of a defense mechanism that could be even extrapolated to other RNA viruses.
Figure 5.
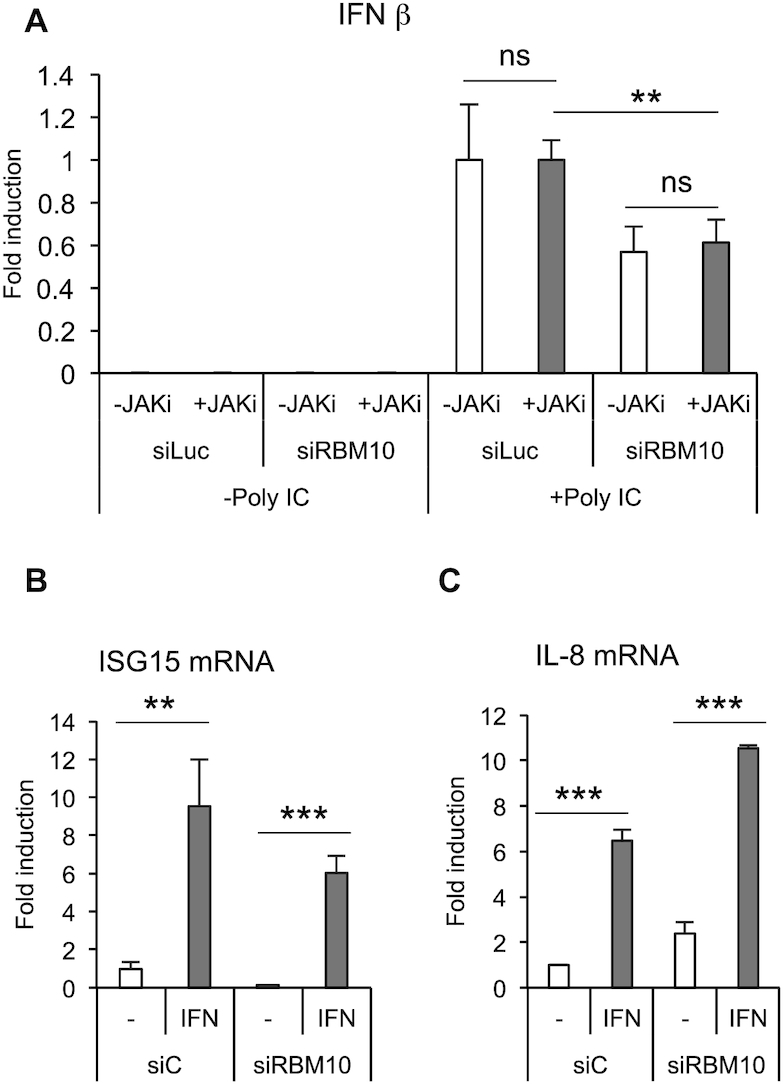
RBM10 is involved in the RLR signaling pathway. (A) A549 cells were transfected with the indicated siRNAs (40 nM) for 48 h, then mock treated (DMSO) or treated with 1 μM JAK inhibitor for additional 24 h during which, the last 9 h included Poly I:C intracellular administration. RNA was prepared and analyzed by RT-qPCR for IFN β. (B and C) A549 cells were transfected with the indicated siRNAs (40 nM) for 24 h and then treated with IFN α (50 000 U/ml) for additional 24 h. ISG15 (B) and IL-8 (C) mRNA levels were analyzed by RT-qPCR. Average values from triplicates are shown with standard deviation and P-values, measured with a paired two-tailed t-test. Significant P-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05).
To further dissect RBM10 action, we took advantage of the Poly I:C treatment as a way of inducing the innate immune response in the absence of viral infection and therefore in the absence of any viral counteracting activity. We thus combined RBM10 depletion, intracellular administration of Poly I:C and treatment of A549 cells with a kinase inhibitor that specifically blocks the activity of the Janus kinase (JAK), a protein required for the activation of IFNR signaling pathway. Results from these experiments showed that the effect of RBM10 silencing on Poly I:C-triggered INFβ induction is not significantly altered by inhibition of the IFNR downstream pathway (Figure 5A). The efficacy of the JAK inhibitor was monitored by measuring ISG15 mRNA levels, as ISG15 promoter is one of the targets of the JAK-STAT signaling cascade downstream of IFNR (Supplementary Figure S5A). The above results are in agreement with the fact that knocking down RBM10 does not impair neither ISG15 nor IL-8 induction upon IFN treatment (Figure 5B and C), while it does so upon DENV infection, as already shown in Figure 4A. In summary, these data point to a possible action of RBM10 in the RLRs signaling cascade and independently of the IFNR signaling pathway.
Since RLR signaling begins in the cytoplasm, we also analyzed any possible changes in the sub-cellular distribution of RBM10 upon infection. Detection of endogenous RBM10 as well as viral RNA by immunofluorescence suggests a decrease in the ratio of RBM10 nuclear to cytoplasmic localization upon viral infection (Figure 6A). Whether this partial re-distribution can account for the observed changes in RBM10-mediated alternative splicing awaits further investigation. We then tested the interaction between RBM10 and RIG-I, the receptor involved in viral RNA recognition, by co-immunoprecipitation and found that not only do they interact (Figure 6B), but also that RBM10 co-precipitates with DENV RNA in A549 infected cells (Figure 6C, gray bars), suggesting these two proteins could be part of cytoplasmic ribonucleoprotein complexes. However, the interaction between RIG-I and RBM10 was also detected upon IFN treatment and even maintained in the presence of RNase (Figure 6B), which suggests that the viral RNA is not strictly necessary but might contribute to the association between these two proteins. Remarkably, in infected cells we also detected a smaller fraction of RBM10-bound SAT1 pre-mRNA compared to mock conditions (Figure 6C, black bars). In this context, a large pool of RBM10-free SAT1 pre-mRNA is processed towards the exon 4-containing isoform, ultimately favoring viral replication. These observations could be interpreted as two sides of the same coin. On one side, RBM10 may exert its antiviral role in part by stimulating the innate immune response, possibly through its interaction with RIG-I and the viral RNA and/or through its splicing regulatory function. On the other side, viral counteracting activities tend to diminish RBM10 levels and therefore RBM10 action on SAT1 and/or other pre-mRNA targets.
Figure 6.

RBM10 binds RIG-I and DENV genome and promotes RIG-I ubiquitination. (A) RBM10 and dsRNA (a marker of viral RNA) localization was visualized by indirect immunofluorescence and nuclei-cytoplasmic ratio was quantified. (B) A549 cells were either treated with IFN α or DENV infected and subjected to co-immunoprecipitation assays with anti-RBM10. RNase A (0.1 mg/ml) was added to lysates when indicated. Input and immunoprecipitated fractions were analyzed by western blot with the corresponding antibodies. (C) RNA-immunoprecipitation (RIP) assays with anti-RBM10 of lysates from A549 infected cells at 48 hpi. RT-qPCRs were performed with primers for SAT1 pre-mRNA and viral RNA and represented as RIP/input RNA. Average values from triplicates are shown with standard deviation and p-values, determined using a paired two-tailed t-test. Significant P-values are indicated by the asterisks above the graphs (***P< 0.001; **P< 0.01; *P< 0.05). (D) HEK 293T were transfected with the indicated expression vectors and after 48 h subjected to Ni-NTA affinity purification. Eluates and input fractions were analyzed by western blot with the corresponding antibodies.
Considering the above results pointing to RBM10 as a modulator of the RLR signaling cascade, we evaluated whether it could exert any effect on RIG-I ubiquitination, one of the steps required for the activation of this cytoplasmic receptor upon viral RNA sensing. For this purpose, cells were co-transfected with an expression vector for 6xHis-Ubiquitin, one for Flag-tagged RIG-I and increasing amounts of the T7-RBM10 coding plasmid. Purification of newly ubiquitinated proteins was achieved by nickel affinity chromatography followed by detection of ubiquitin-conjugated RIG-I by western blot. The results from these experiments showed an increase in ubiquitinated RIG-I triggered by RBM10 (Figure 6D) in a concentration-dependent manner (Supplementary Figure S5B), without any effect on Flag-RIG-I protein levels. These observations are consistent with an RBM10-stimulated RIG-I activation. Further studies will be required to conclude whether this effect is dependent on RBM10 activity as a splicing regulator or could represent a novel, splicing-independent and cytoplasmic role for this protein.
DISCUSSION
Here, we report that SAT1 exon 4 alternative splicing is affected by DENV infection of cultured human cells. It is of note that the protein levels of the splicing factor already characterized to regulate this splicing event, RBM10, are also affected. We propose that the DENV protein NS5 is responsible for RBM10 proteasomal degradation since these two proteins interact and, more importantly, NS5 over-expression per se decreases RBM10 protein levels, in a proteasome-dependent manner. Moreover, RBM10 over-expression in infected cells not only restores SAT1 splicing patterns to mock conditions but also severely diminishes viral replication. In contrast, RBM10 knock-down enhances SAT1 splicing change and viral replication. Altogether, these results led us to assign RBM10 an anti-viral role. Indeed, induction of IFN and pro-inflammatory cytokines downstream of RLR activation is attenuated upon RMB10 depletion. Co-immunoprecipitation experiments suggest an RBM10 interaction with the RIG-I/DENV RNA ribonucleoprotein complex, adding a new layer of complexity to RBM10 pro-inflammatory anti-viral function besides its already described nuclear role in splicing regulation (23,30–33,58). Furthermore, RBM10 enhances RIG-I ubiquitination, a crucial step required for activation of the innate response.
We started this study by analyzing splicing changes upon DENV infection from our previously reported RNA-seq experiments (20). Host cell splicing changes upon viral infections as well as interference of the splicing machinery by viral components have also been reported by other laboratories (46–48). In our case, we had found a significant increase in intron retention and, to a lesser extent, changes in alternative splicing patterns (20). Taking into account the previously reported anti-viral function of SAT1 (10,12) and the changes in the splicing pattern of its pre-mRNA caused by other viruses (18), this transcript appeared as an interesting candidate to begin elucidating the molecular mechanism underlying DENV-triggered splicing regulation within the complex and dynamic scenario of virus-host cell interaction. In fact, in our RNA-seq dataset, in addition to increased reads corresponding to alternative exon 4 upon DENV infection, we also detected the retention of a big intron encompassing exon 4 and flanking introns. This could indicate an altered function of RBM10, which was reported in several occasions to facilitate SAT1 exon 4 skipping. The model proposed is that RBM10 binding in the vicinity of splicing sites might repress the splicing of introns and delay the splicing choice, thereby facilitating the skipping of cassette exons (34). The fact that DENV infection affected RBM10 protein levels as well as SAT1 alternative splicing favoring exon 4 inclusion is consistent with this proposed model.
We were able to detect changes in SAT1 splicing and RBM10 protein levels both in A549 and HEK 293T cells. This represented a validation of our previous RNA-seq results, performed in A549 cells. More importantly, splicing-impairing mutations within RBM10 have been reported in A549 cells. Indeed, RBM10 pro-apoptotic functions have been attributed, at least in part, to its ability to regulate NUMB splicing, which is diminished in these cells (21,32). In this respect, it is important to mention that we used wild type RBM10 for over-expression assays in our study, which may account for induction of inflammatory genes even under mock conditions (Figure 4B), particularly considering the widely reported RBM10 lower expression in A549 cells due to its self-splicing deregulation (35). In any case, this highlights RBM10 relevance in inflammation and protection from infection. All things considered, we propose that there is a conserved RBM10-mediated splicing regulation upon DENV infection of the two cell lines tested, which is supported by changes in SAT1 splicing and RBM10 protein levels as well as a significant overlap of other splicing events regulated both in DENV-infected A549 and HEK 293T RBM10-KD cells. Moreover, A549 cells represent a suitable cell culture model for analyzing RLR signaling pathway and inflammation upon DENV infection (49), which might be attributed in part to their RBM10 lower levels leading to delayed apoptosis as well as higher DENV infectivity and higher impact on RBM10-dependent functions upon its depletion.
RBM10 down-regulation upon DENV infection can be explained by NS5-targeted proteasomal degradation. We were able to detect a decrease in RBM10 protein levels not only in infected cells but also in the context of NS5 over-expression. However, IFN treatment or Poly I:C administration does not seem to exert this effect. Mapping the exact region within NS5 responsible for triggering RBM10 degradation, as well as understanding whether the physical interaction of these two proteins is required for this regulatory mechanism warrants further investigation. Several studies already reported the ability of NS5, or other viral protein, to target host proteins for degradation (50–54). Consistently, NS5 over-expression also affected SAT1 splicing patterns, which could be due to diminished RBM10 and/or to spliceosome misassembly, what was proved to be triggered by NS5 (20). RBM10 has been shown to be involved in spliceosome assembly (24,29) and we propose that it could be within the context of the spliceosome that NS5 and RBM10 interact. The interaction of these two proteins is mainly supported by co-immunoprecipitation assays and is consistent with their subcellular co-fractionation and nuclear co-localization. However, our previous mass spectrometry analysis failed to detect RBM10 as an NS5 partner, even though it was significantly enriched in ZIKV NS5 interactome (36). This discrepancy might be due to DENV NS5-targeted RBM10 proteasomal degradation. In any case, involvement of NS5 and RBM10 in spliceosome assembly is strongly supported by previous evidence (20,24,26,29). The novelty for RBM10, revealed by the current study, is that the cellular levels of this protein are reduced by DENV infection or NS5 expression, whereas the levels of several analyzed spliceosomal proteins that were identified as NS5-binders are not affected under similar conditions (20).
RBM10 involvement in splicing is further supported by its modular domain organization, which displays two RNA binding motifs and three nuclear localization signals (23). It is presumably due to these that its subcellular partitioning is mainly nuclear, even though we detected a decrease in nuclei-to-cytoplasmic localization ratio upon infection. Whether RBM10 interaction with DENV RNA and RIG-I receptor, which is known to detect viral cytosolic RNA and to trigger anti-viral signaling towards nuclear transcription, can account for this partial re-localization of RBM10 warrants further investigation. Our results suggest that RBM10 and RIG-I interact also in the absence of DENV RNA (i.e. IFN alone or DENV with RNase A treatment, Figure 6B). However, DENV RNA could still function as a scaffold and facilitate the encounter. Several splicing and RNA processing factors have been found to interact with DENV genome by mass spec analysis, particularly with the 3′UTR region (55–57). Since several sequences have been spotted as potential RBM10 binding sites (23), further investigation is required to identify them within the DENV genome as well as to decipher the functional consequences of this RBM10–viral RNA interaction. In any case, we report that RBM10 associates with RIG-I, which could provide a novel cytoplasmic role for RBM10 in regulating inflammation, although we do not rule out that RBM10 also plays pro-inflammatory tasks in the nucleus, where it is mainly localized. In fact, it has been reported that RBM10 promotes inflammation by modulating alternative splicing of Dnmt3b, which codes for a DNA methyl transferase known to regulate the activity of NF-κB-responsive promoters and consequently inflammation development (58).
It is known that eukaryotic cells rely on the innate immune response to limit viral replication and a great amount of proteins has been identified to participate and/or cooperate with this cellular program (59–62). Our results point to RBM10 as fitting into this group and exerting an anti-viral function at least in part through its ability to regulate splicing patterns towards the production of anti-viral and pro-inflammatory mRNA isoforms, in particular with respect to SAT1 pre-mRNA. However, we cannot rule out other non-splicing related functions of RBM10, especially after observing its co-purification with DENV RNA as well as with the viral sensor RIG-I. Whether these interactions, which are supposed to take place in the cytoplasm, have anything to do with the regulation of RBM10 activity as a pre-mRNA processing factor (21,32,34,37,63) and/or are related to a novel cytoplasmic role for RBM10 remains to be further studied. Also, we have detected that RBM10 stimulates at least one step along the RIG-I activation process that is its ubiquitination. Again, whether this effect depends on RBM10–RIG-I interaction or is mediated by RBM10-dependent splicing regulation is still an intriguing question (Figure 7, left side). On the other hand, viral pathogens have evolved sophisticated mechanisms to counteract the cellular response (62). Considering our previous and current results, we propose that several events could take place in the context of DENV infection and contribute to alter host cell splicing patterns and to attenuate the innate response leading to a more permissive environment for viral replication. So far we can mention the already reported intrusion of NS5 into the spliceosome (20) as well as NS5–RBM10 interaction, NS5-triggered RBM10 degradation, and potentially RBM10 partial re-localization, as putative levels of viral action (Figure 7, right side).
Figure 7.

Impact of DENV on RBM10 and its consequences on splicing, inflammation and viral replication. Nuclear RBM10 regulates alternative splicing favoring anti-viral mRNA isoforms, among which SAT1 isoforms were investigated in this report. Upon DENV infection, NS5 intrudes into the spliceosome, as previously reported by our laboratories, and also targets RBM10 for proteasomal degradation, favoring pro-viral splicing isoforms. On the other hand, RMB10 also interacts with viral genome and RIG-I and promotes the ubiquitination of the latter, which is known to be required for its activation and the induction of the innate immune response. Overall, RBM10 is required for the induction of interferon and pro-inflammatory cytokines. Whether this is a result of RBM10 involvement in splicing regulation, or in RIG-I/DENV genome ribonucleoprotein complex, or both, remains an intriguing question. The fact is that higher protein levels of RBM10 correlate with an anti-viral, pro-inflammatory scenario while lower levels with a pro-viral, anti-inflammatory one.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Maria Mora González Lopez Ledesma and Valeria Buggiano for technical help, Luciana Giono for text revising and editing, and Federico Giovannoni, Kristin Patrick, as well as members of the Petrillo, de la Mata, Muñoz, Schor and Kornblihtt laboratories for helpful and encouraging discussion.
Contributor Information
Berta Pozzi, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
Laureano Bragado, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
Pablo Mammi, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
María Florencia Torti, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Biológica, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, IQUIBICEN, Buenos Aires, Argentina.
Nicolás Gaioli, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
Leopoldo G Gebhard, CONICET-Universidad Nacional de Quilmes, Laboratorio de Virus Emergentes, Departamento de CyT, Buenos Aires, Argentina.
Martín E García Solá, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
Rita Vaz-Drago, Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Portugal.
Néstor G Iglesias, CONICET-Universidad Nacional de Quilmes, Laboratorio de Virus Emergentes, Departamento de CyT, Buenos Aires, Argentina.
Cybele C García, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Biológica, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, IQUIBICEN, Buenos Aires, Argentina.
Andrea V Gamarnik, CONICET-Fundación Instituto Leloir (FIL), Buenos Aires, Argentina.
Anabella Srebrow, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Fisiología, Biología Molecular y Celular, Buenos Aires, Argentina; CONICET-Universidad de Buenos Aires, Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), Buenos Aires, Argentina.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agencia Nacional de Promoción Científica y Tecnológica de Argentina (ANPCyT) [2014-2888, 2015-1731, 2017-0111 to A.S. and 2015-2555, 2017-1717 to A.V.G.]; Universidad de Buenos Aires, Argentina (UBACyT) [20020170100045BA to A.S.]; NIH (NIAID) [R01.AI095175 to A.V.G.]; Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina (CONICET) [PIP 11220170100171CO to C.C.G]; B.P. has been a postdoctoral fellow from CONICET from 2017 to 2019 and is currently a postdoctoral fellow at the Institute of Cell Biology in the University of Bern, Switzerland; L.B. and M.E.G.S. are recipients of doctoral fellowships from CONICET; M.F.T. is a doctoral fellowship recipient from ANPCyT; N.G. has been an undergraduate fellowship recipient from the University of Buenos Aires (2018–2020); P.M. has been a doctoral fellow from CONICET (2015–2019) and is currently a postdoctoral fellow supported by H2020-Marie Sklodowska-Curie Research and Innovation Staff Exchanges [734825-LysoMod]; R.V.D. has been a visiting post-doctoral fellow at the Srebrow lab from IMM (Lisbon, Portugal) supported by the same program. A.S., A.V.G., C.C.G., N.G.I. and L.G.G. are career investigators from CONICET.
Conflict of interest statement. None declared.
REFERENCES
- 1. Brady O.J., Gething P.W., Bhatt S., Messina J.P., Brownstein J.S., Hoen A.G., Moyes C.L., Farlow A.W., Scott T.W., Hay S.I.. Refining the global spatial limits of dengue virus transmission by Evidence-Based consensus. PLoS Negl. Trop. Dis. 2012; 6:e1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Messina J.P., Brady O.J., Scott T.W., Zou C., Pigott D.M., Duda K.A., Bhatt S., Katzelnick L., Howes R.E., Battle K.E. et al.. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014; 22:138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sugrue R.J., Fu J., Howe J., Chan Y.-C.C.. Expression of the dengue virus structural proteins in Pichia pastoris leads to the generation of virus-like particles. J. Gen. Virol. 1997; 78:1861–1866. [DOI] [PubMed] [Google Scholar]
- 4. Gebhard L.G., Filomatori C. V., Gamarnik A. V.. Functional RNA elements in the dengue virus genome. Viruses. 2011; 3:1739–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischl W., Bartenschlager R.. Exploitation of cellular pathways by dengue virus. Curr. Opin. Microbiol. 2011; 14:470–475. [DOI] [PubMed] [Google Scholar]
- 6. Jensen S., Thomsen A.R.. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012; 86:2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chazal M., Beauclair G., Gracias S., Najburg V., Simon-Lorière E., Tangy F., Komarova A. V., Jouvenet N.. RIG-I recognizes the 5′ region of dengue and zika virus genomes. Cell Rep. 2018; 24:320–328. [DOI] [PubMed] [Google Scholar]
- 8. Liu Y., Olagnier D., Lin R.. Host and viral modulation of RIG-I-Mediated antiviral immunity. Front. Immunol. 2016; 7:662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xian H., Xie W., Yang S., Liu Q., Xia X., Jin S., Sun T., Cui J.. Stratified ubiquitination of RIG-I creates robust immune response and induces selective gene expression. Sci. Adv. 2017; 3:e1701764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mounce B.C., Poirier E.Z., Passoni G., Simon-Loriere E., Cesaro T., Prot M., Stapleford K.A., Moratorio G., Sakuntabhai A., Levraud J.P. et al.. Interferon-Induced Spermidine-Spermine acetyltransferase and polyamine depletion restrict zika and chikungunya viruses. Cell Host Microbe. 2016; 20:167–177. [DOI] [PubMed] [Google Scholar]
- 11. Pegg A.E. Spermidine/spermine-N1-acetyltransferase: a key metabolic regulator. Am. J. Physiol. - Endocrinol. Metab. 2008; 294:E995–E1010. [DOI] [PubMed] [Google Scholar]
- 12. Smirnova O.A., Keinanen T.A., Ivanova O.N., Hyvonen M.T., Khomutov A.R., Kochetkov S.N., Bartosch B., Ivanov A. V.. Hepatitis C virus alters metabolism of biogenic polyamines by affecting expression of key enzymes of their metabolism. Biochem. Biophys. Res. Commun. 2017; 483:904–909. [DOI] [PubMed] [Google Scholar]
- 13. Smirnova O.A., Isaguliants M.G., Hyvonen M.T., Keinanen T.A., Tunitskaya V.L., Vepsalainen J., Alhonen L., Kochetkov S.N., Ivanov A. V.. Chemically induced oxidative stress increases polyamine levels by activating the transcription of ornithine decarboxylase and spermidine/spermine- N1-acetyltransferase in human hepatoma HUH7 cells. Biochimie. 2012; 94:1876–1883. [DOI] [PubMed] [Google Scholar]
- 14. Li M.M.H., MacDonald M.R.. Polyamines: Small molecules with a big role in promoting virus infection. Cell Host Microbe. 2016; 20:123–124. [DOI] [PubMed] [Google Scholar]
- 15. Hyvönen M.T., Uimari A., Keinänen T.A., Heikkinen S., Pellinen R., Wahlfors T., Korhonen A., Närvänen A., Wahlfors J., Alhonen L. et al.. Polyamine-regulated unproductive splicing and translation of spermidine/spermine N1-acetyltransferase. RNA. 2006; 12:1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hyvönen M.T., Uimari A., Vepsäläinen J., Khomutov A.R., Keinänen T.A., Alhonen L.. Tissue-specific alternative splicing of spermidine/spermine N 1-acetyltransferase. Amino Acids. 2012; 42:485–493. [DOI] [PubMed] [Google Scholar]
- 17. Ichimura S., Nenoi M., Mita K., Fukuchi K., Hamana K.. Accumulation of spermidine/spermine N1-acetyltransferase and alternatively spliced mRNAs as a delayed response of HeLa S3 cells following X-ray irradiation. Int. J. Radiat. Biol. 2004; 80:369–375. [DOI] [PubMed] [Google Scholar]
- 18. Nikiforova N.N., Velikodvorskaja T. V., Kachko A. V., Nikolaev L.G., Monastyrskaya G.S., Lukyanov S.A., Konovalova S.N., Protopopova E. V., Svyatchenko V.A., Kiselev N.N. et al.. Induction of alternatively spliced spermidine/spermine N1-acetyltransferase mRNA in the human kidney cells infected by Venezuelan equine encephalitis and tick-borne encephalitis viruses. Virology. 2002; 297:163–171. [DOI] [PubMed] [Google Scholar]
- 19. Kim K., Ryu J.H., Park J.W., Kim M.S., Chun Y.S.. Induction of a SSAT isoform in response to hypoxia or iron deficiency and its protective effects on cell death. Biochem. Biophys. Res. Commun. 2005; 331:78–85. [DOI] [PubMed] [Google Scholar]
- 20. De Maio F.A., Risso G., Iglesias N.G., Shah P., Pozzi B., Gebhard L.G., Mammi P., Mancini E., Yanovsky M.J., Andino R. et al.. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog. 2016; 12:e1005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J., Sun Y., Huang Y., Song F., Huang Z., Bao Y., Zuo J., Saffen D., Shao Z., Liu W. et al.. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci. Rep. 2017; 7:40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loiselle J.J., Sutherland L.C.. RBM10: Harmful or helpful-many factors to consider. J. Cell. Biochem. 2018; 119:3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rappsilber J., Ryder U., Lamond A.I., Mann M.. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2003; 13:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makarov E.M., Owen N., Bottrill A., Makarova O. V.. Functional mammalian spliceosomal complex e contains SMN complex proteins in addition to U1 and U2 snRNPs. Nucleic Acids Res. 2012; 40:2639–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agafonov D.E., Deckert J., Wolf E., Odenwalder P., Bessonov S., Will C.L., Urlaub H., Luhrmann R.. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Biol. 2011; 31:2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Behzadnia N., Golas M.M., Hartmuth K., Sander B., Kastner B., Deckert J., Dube P., Will C.L., Urlaub H., Stark H. et al.. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 2007; 26:1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bessonov S., Anokhina M., Will C.L., Urlaub H., Lührmann R.. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008; 452:846–850. [DOI] [PubMed] [Google Scholar]
- 29. Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C.L., Pena V., Lührmann R., Stelzl U.. Dynamic Protein-Protein interaction wiring of the human spliceosome. Mol. Cell. 2012; 45:567–580. [DOI] [PubMed] [Google Scholar]
- 30. Sutherland L.C., Wang K., Robinson A.G.. RBM5 as a putative tumor suppressor gene for lung cancer. J. Thorac. Oncol. 2010; 5:294–298. [DOI] [PubMed] [Google Scholar]
- 31. Hernández J., Bechara E., Schlesinger D., Delgado J., Serrano L., Valcárcel J.. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016; 13:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bechara E.G., Sebestyén E., Bernardis I., Eyras E., Valcárcel J.. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell. 2013; 52:720–733. [DOI] [PubMed] [Google Scholar]
- 33. Wang K., Bacon M.L., Tessier J.J., Rintala-Maki N.D., Tang V., Sutherland L.C.. RBM10 modulates apoptosis and influences TNF-α gene expression. J. Cell Death. 2012; 5:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y., Gogol-Döring A., Hu H., Fröhler S., Ma Y., Jens M., Maaskola J., Murakawa Y., Quedenau C., Landthaler M. et al.. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol. Med. 2013; 5:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun Y., Bao Y., Han W., Song F., Shen X., Zhao J., Zuo J., Saffen D., Chen W., Wang Z. et al.. Autoregulation of RBM10 and cross-regulation of RBM10/RBM5 via alternative splicing-coupled nonsense-mediated decay. Nucleic Acids Res. 2017; 45:8524–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shah P.S., Link N., Jang G.M., Sharp P.P., Zhu T., Swaney D.L., Johnson J.R., Von Dollen J., Ramage H.R., Satkamp L. et al.. Comparative Flavivirus-Host protein interaction mapping reveals mechanisms of dengue and zika virus pathogenesis. Cell. 2018; 175:1931–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodor J., FitzPatrick D.R., Eyras E., Cáceres J.F.. The RNA-binding landscape of RBM10 and its role in alternative splicing regulation in models of mouse early development. RNA Biol. 2017; 14:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Samsa M.M., Mondotte J.A., Iglesias N.G., Assunção-Miranda I., Barbosa-Lima G., Da Poian A.T., Bozza P.T., Gamarnik A.V., Samsa M.M., Mondotte J.A. et al.. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009; 5:e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gagnon K.T., Li L., Janowski B.A., Corey D.R.. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc. 2014; 9:2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tatham M.H., Rodriguez M.S., Xirodimas D.P., Hay R.T.. Detection of protein SUMOylation in vivo. Nat. Protoc. 2009; 4:1363–1371. [DOI] [PubMed] [Google Scholar]
- 41. Miorin L., Albornoz A., Baba M.M., D’Agaro P., Marcello A.. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus Res. 2012; 163:660–666. [DOI] [PubMed] [Google Scholar]
- 42. Overby A.K., Popov V.L., Niedrig M., Weber F.. Tick-Borne encephalitis virus delays interferon induction and hides its Double-Stranded RNA in intracellular membrane vesicles. J. Virol. 2010; 84:8470–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roth H., Magg V., Uch F., Mutz P., Klein P., Haneke K., Lohmann V., Bartenschlager R., Fackler O.T., Locker N. et al.. Flavivirus infection uncouples translation suppression from cellular stress responses. MBio. 2017; 8:e02150-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tay M.Y.F., Fraser J.E., Chan W.K.K., Moreland N.J., Rathore A.P., Wang C., Vasudevan S.G., Jans D.A.. Nuclear localization of dengue virus (DENV) 1–4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antiviral Res. 2013; 99:301–306. [DOI] [PubMed] [Google Scholar]
- 45. Kumar A., Buhler S., Selisko B., Davidson A., Mulder K., Canard B., Miller S., Bartenschlager R.. Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J. Virol. 2013; 87:4545–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ku C.C., Che X.B., Reichelt M., Rajamani J., Schaap-Nutt A., Huang K.J., Sommer M.H., Chen Y.S., Chen Y.Y., Arvin A.M.. Herpes simplex virus-1 induces expression of a novel MxA isoform that enhances viral replication. Immunol. Cell Biol. 2011; 89:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Álvarez E., Castelló A., Carrasco L., Izquierdo J.M.. Poliovirus 2a protease triggers a selective nucleo-cytoplasmic redistribution of splicing factors to regulate alternative pre-mRNA splicing. PLoS One. 2013; 8:e73723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Almstead L.L., Sarnow P.. Inhibition of U snRNP assembly by a virus-encoded proteinase. Genes Dev. 2007; 21:1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reynolds A., Anderson E.M., Vermeulen A., Fedorov Y., Robinson K., Leake D., Karpilow J., Marshall W.S., Khvorova A.. Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA. 2006; 12:988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Porritt R.A., Hertzog P.J.. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015; 36:150–160. [DOI] [PubMed] [Google Scholar]
- 51. Aslam B., Ahmad J., Ali A., Paracha R.Z., Tareen S., Khusro S., Ahmad T., Muhammad S.A., Niazi U., Azevedo V.. Structural modeling and analysis of dengue-mediated inhibition of interferon signaling pathway. Genet. Mol. Res. 2015; 14:4215–4237. [DOI] [PubMed] [Google Scholar]
- 52. Ashour J., Laurent-Rolle M., Shi P.-Y., Garcia-Sastre A.. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009; 83:5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schwartz A.L., Ciechanover A.. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009; 49:73–96. [DOI] [PubMed] [Google Scholar]
- 54. Çevik R.E., Cesarec M., Filipe Da Silva, Licastro A., McLauchlan D., Marcello A.. Hepatitis C virus NS5A targets nucleosome assembly protein NAP1L1 To control the innate cellular response. J. Virol. 2017; 91:e00880-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Viktorovskaya O. V., Greco T.M., Cristea I.M., Thompson S.R.. Identification of RNA binding proteins associated with dengue virus RNA in infected cells reveals temporally distinct host factor requirements. PLoS Negl. Trop. Dis. 2016; 10:e0004921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Phillips S.L., Soderblom E.J., Bradrick S.S., Garcia-Blanco M.A.. Identification of proteins bound to dengue viral RNA In Vivo reveals new host proteins important for virus replication. MBio. 2016; 7:e01865-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ward A.M., Bidet K., Yinglin A., Ler S.G., Hogue K., Blackstock W., Gunaratne J., Garcia-Blanco M.A.. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011; 8:1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Atsumi T., Suzuki H., Jiang J.J., Okuyama Y., Nakagawa I., Ota M., Tanaka Y., Ohki T., Katsunuma K., Nakajima K. et al.. Rbm10 regulates inflammation development via alternative splicing of Dnmt3b. Int. Immunol. 2017; 29:581–591. [DOI] [PubMed] [Google Scholar]
- 59. Gack M.U. TRIMming flavivirus infection. Cell Host Microbe. 2011; 10:175–177. [DOI] [PubMed] [Google Scholar]
- 60. Giovannoni F., Damonte E.B., García C.C.. Cellular promyelocytic leukemia protein is an important dengue virus restriction factor. PLoS One. 2015; 10:e0125690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Giovannoni F., Ladelfa M.F., Monte M., Jans D.A., Hemmerich P., García C.. Dengue non-structural protein 5 polymerase complexes with promyelocytic leukemia protein (PML) Isoforms III and IV to disrupt PML-nuclear bodies in infected cells. Front. Cell. Infect. Microbiol. 2019; 9:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tremblay N., Freppel W., Sow A.A., Chatel-Chaix L.. The interplay between dengue virus and the human innate immune system: A game of hide and seek. Vaccines. 2019; 7:E145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mohan N., Kumar V., Kandala D.T., Kartha C.C., Laishram R.S.. A splicing-independent function of RBM10 controls specific 3′ UTR processing to regulate cardiac hypertrophy. Cell Rep. 2018; 24:3539–3553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.