


Abstract
First triplets of mRNA coding region affect the yield of translation. We have applied the flowseq method to analyze >30 000 variants of the codons 2–11 of the fluorescent protein reporter to identify factors affecting the protein synthesis. While the negative influence of mRNA secondary structure on translation has been confirmed, a positive role of rare codons at the beginning of a coding sequence for gene expression has not been observed. The identity of triplets proximal to the start codon contributes more to the protein yield then more distant ones. Additional in-frame start codons enhance translation, while Shine–Dalgarno-like motifs downstream the initiation codon are inhibitory. The metabolic cost of amino acids affects the yield of protein in the poor medium. The most efficient translation was observed for variants with features resembling those of native Escherichia coli genes.
INTRODUCTION
The mechanisms that determine the efficiency of translation are of fundamental value for our understanding of gene expression and allocation of intracellular resources. No less important is this knowledge for the optimization of exogenous gene expression in biotechnology. Despite decades of research, the rules that govern the translation efficiency are still controversial. Several features of a coding sequence have been suggested to contribute to the protein yield. Folding of the mRNA coding region adjacent to the start codon might preclude ribosome binding and is frequently viewed as a primary determinant for the efficiency of translation (1–5). A species-specific (6,7) feature is synonymous codon usage, numerically expressed as codon or tRNA adaptation indices (CAI (7) and tAI (8), respectively). Moreover, codon usage varies significantly between mRNAs encoded in the same genome (9,10) and even along a particular mRNA (11,12). Enrichment for frequently used codons along mRNA is generally believed to enhance translation (13–15) and mRNA stability (13,16) which are essential for natural gene expression as well as for the biotechnological applications (17,18). However, a genome-wide computational analysis of a number of species has revealed that the 5′-most part of a coding region, called ramp, is characterized on average by lower tAI (11,19), a more frequent occurrence of codons for positively charged amino acids (19), and an uneven distribution of secondary structure elements (20). Slow translation of the ramp region has been hypothesized to reduce ribosome collisions downstream (11,20).
An analysis of ribosome-protected mRNA fragments (riboseq) (21) revealed an increased ribosome density at the beginning of coding regions, initially interpreted as a proof of a slowly translated ramp (11), although later it was explained by elevated initiation rates for short ORFs (3). A bias towards positively charged amino acids at the start of coding regions was explained by specific sequence requirements for transmembrane proteins (22), while lower CAI of the ramp region was ascribed to selection against stable secondary structures sequestering ribosome binding sites (1) in species where rare triplets are AU-rich (23,24).
Numerous controversies are associated with general understanding of factors that slow the ribosome progression along mRNA, such as codons recognized by rare tRNA species (11,25), triplets that require wobble decoding (26,27), Shine–Dalgarno (SD)-like sites (28,29) and codons for particular amino acids (27–29). Several difficulties hamper identification of coding sequence features that affect the protein production. Computational analysis of mRNA sequences does not allow one to distinguish the cause of particular sequence bias, be it selection towards translation efficiency of particular mRNA, saving resources for the translation of other mRNAs, or other reasons. A direct estimation of natural mRNAs translation efficiencies by riboseq has produced a wealth of data, but restriction of the sampling space by a limited number of genes encoded in a genome makes it difficult to assess the contribution of each factor on the protein yield per mRNA.
The flowseq method proved to be an effective tool to evaluate an influence of different mRNA features on protein biosynthesis (1,2,30–32) in a single massively parallel experiment. Previous applications of this method involved a variety of experimental designs. After the pioneering work of Kudla et al. (2), the significance of coding region determinants on the translation yield was addressed with a library of 137 variants of the first 10 codons derived from natural Escherichia coli genes (1), a set of 244 000 reporters containing computationally designed large 96 nt mRNA regions aimed to evenly represent possible combinations of features known to influence translation (32), and a set of 259 134 reporters created to exhaustively sample the sequence space of codons 3–5 (31). Our previous studies relied on a dual fluorescent protein reporter (33) to assess the influence of the 5′-UTR mRNA region on the translation efficiency for rationally designed (34) and randomized (30) plasmid libraries. Here, we apply the flowseq pipeline to dissect the influence of codons 2–11 on the translation yield, aiming to complement previously published efforts (1,31,32).
MATERIALS AND METHODS
Strains
Escherichia coli JM109 strain was used for cloning procedures. BW25113 and ΔArg (derivative of BW25113, created by replacing of argY, argZ and argQ by kanR, the kanamycin resistance cassette, according to the Datsenko–Wanner protocol (35) were used for cell sorting experiments.
Construction of reporter plasmids and randomized library of reporter constructs
The pRFPCER plasmid (33) was cut by BsmBI and ligated with DNA duplex, obtained by annealing of oligonucleotides 5′–TGAAAGAGACGGACGAGAGCGGATCCC–3′ and 5′–TTCAGGGATCCGCTCTCGTCCGTCTCT–3′. As a result, a BamHI site was inserted downstream of the ATG start codon to produce pRFPCER2. Library preparation was done as described previously (30), pRFPCER2 was digested with SacII and ligated with the DNA duplex obtained by annealing of oligonucleotides 5′–CACACAACACCGGAGCAACTATG–3′ and 5′–AGCTTACGGATCCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCATAGTTGCTCCGTGTTGTGTGGC–3′. The Klenow fragment was used to synthesize the complementary strand of the randomized part. The linearized plasmid with two randomized duplexes at the ends was digested with BamHI and circularized by self-ligation. As a result, the 30N randomized part was inserted immediately downstream the ATG start codon.
Escherichia coli JM109 strain was electroporated by the obtained library with effectivity of 106–107 CFU/ μg of DNA and cells were grown overnight in the LB medium with ampicillin. The plasmid library was purified by PureYield™ Plasmid Miniprep System (Promega). BW25113 or ΔArg strains were electroporated by the library and grown overnight in the LB or M9 medium supplemented with ampicillin prior to cell sorting.
The pRFPCER2 plasmid was subjected to PCR with the following oligonucleotides: LguI rev TCTCTTTCAGGCGCTCTTCTCATAGTTGCTCCGGTGTTG
BamHI F CGGACGAGAGGCGGATCCCTGAAAGAGACGGACGAGAGC and circularized by self-ligation. Obtained pRFPCER3 plasmid was digested by BamHI and LguI enzymes. Then digested plasmid was mixed with 160 preliminary annealed oligonucleotides (see Supplementary Table S2 for sequences of the variable part of resulted coding regions) and ligated. Cells of E. coli strain JM109 were transformed by the products of ligation, obtained plasmids were confirmed by sequencing.
Cell sorting
Overnight cell cultures were washed twice with sterile PBS and sorted with cell sorter BD FACSAriaTMIII, ∼2.5 million of cells were analyzed and sorted into 12 bins according to the ratio of CER and RFP. Sorted cells were grown overnight in the LB medium with ampicillin and used for plasmid purification and fluorescence measurement as described previously (30).
Preparation of amplicon libraries and sequencing
1 ng of plasmids from each fraction was used for PCR amplification with barcoded oligonucleotides complementary to the constant part of the plasmids surrounding the randomized part. The quality of PCRs was estimated by agarose gel. The paired-end library was prepared from pooled barcoded amplicons using a NEBNext Ultra DNA library prep kit for Illumina (NEB). Amplicon deep sequencing was conducted on a MiSeq genome sequencer (2 × 300 cycles, Illumina) in SB RAS Genomics Core Facility (ICBFM SB RAS, Novosibirsk, Russia). Constant regions in reads were removed by CUTADAPT (36). The read data reported in this study were submitted to the GenBank under the study accession PRJNA476703 and the sample accession SRS3434030.
Variant calling
All raw reads shorter than 140 and longer than 155 nucleotides were removed. We selected reads that contained constant parts of the amplicons from ATG to AGA for forward reads and TCT to CAT for reverse reads that were converted to complementary forward reads for further analysis. All reads without detectable or with more than one occurrence of the constant part as well as sequences with redundant nucleotides were removed. All remaining sequences containing the expected constant parts flanking specific sequences of the randomized region were grouped by sample, replica, and fraction according to barcodes.
Error correction
After variant calling, occurrences of each variant across 12 bins were counted. We applied the error correction procedure from our previous work (30). Briefly, total counts of all sequence variants were calculated, and variants were separated into rare (four or less occurrence) and common (five or mores occurrence) according to an empirical frequency threshold of 4. The distances between common variants were calculated, so that any mismatch, insertion or deletion that is needed to transform one sequence to another increased the distance by 1. It was noted that the distances between sequences fell into two groups, being less or equal to 6 and 20 or more (Supplementary Figure S1E). Our interpretation of that was that a closely related group of sequences separated by the distances six or less were actually one sequence variant blurred by PCR and sequencing mistakes. Thus, for each group of variants separated by distances six or less we selected the most abundant variant and reassigned occurrences of all other closely related common and rare reads to this variant. Finally, if a rare variant was not similar, at the given threshold, to any common one, it was included into the dataset without merging. After correction for the PCR and sequencing errors, the resulting distribution of the similarity was statistically indistinguishable (P-value = 0.22) from ideal randomization (Supplementary Figure S1F).
Fraction calculation and filtering
The translation efficiency fraction (TEF) was assigned to variants following ref. (30). Briefly, if the same sequence variant could be found in several bins sorted by the CER/RFP ratio, we calculated the mean bin in the Gaussian approximation. We calculated the proportion of variant occurrences in the mean bin and adjacent bins and discarded sequence variants with over 20% occurrences outside this peak, assuming that these sequence variants were distributed too broadly. We also discarded sequence variants whose mean bins in two replicates differed by two or more. As the number of variants in bins differed dramatically and in some of them were insufficient to overcome statistical noise, the four highest-efficiency bins were merged, and the remaining bins were merged in groups of two, hence forming the final five translation efficiency fractions (TEFs).
All nucleotide sequences were translated according to the standard genetic code (one frame only), all variants with stop codons were filtered out. Protein-coding genes were selected using genome annotation NC_000913.v2. The same region covering nucleotide positions 4–33 was used for comparison with TEFs.
Statistical analysis
Calculation of the RNA secondary structure
To calculate the RNA folding energy, we assembled each variant as UTR + AUG + 30NT + constant part, where the UTR sequence is AGAAGGAGAUAUCAU, AUG is the start codon, 30NT is 30 nucleotides of the randomized part variant, and the constant part is GGAUCCCUGAAAGAGACGGACGAGAGCGGCCUGGUGAGCAAGGGCGAGGA. Then we run RNAfold ver. 2.1.7 from the Vienna RNA package to calculate the folding energy dG with default parameters (37).
Calculation of the SD-likeness
For each hexamer of a variant its SD-likeness was calculated as the free energy of hybridization with the anti-SD sequence CACCUCCU in the 16S rRNA using RNAfold from the Vienna RNA package (37). Each variant was assigned with the minimum hybridization energy of the constituent hexamers.
Calculation of the metabolic cost
For each codon position 2–11 in each fraction we calculated the average metabolic cost of the encoded amino acid. The metabolic costs of amino acid biosynthesis were taken from ref. (38).
Random shuffling
To account for confounding effects of uneven nucleotide frequencies in the TEFs, we constructed 10 000 sets of shuffled variants. To do that, we randomly permuted nucleotides in each position, separately for each fraction. Hence, we retained the original positional nucleotide frequencies in each class, disrupting other features such as codons, RNA structures etc. Thus we obtained a set of shuffled datasets of mRNAs that retained nucleotide frequencies at each coding region position for each TEF, while the sequences were randomized. We repeated all analyses on these shuffled datasets and used the results to estimate statistical significance of our observations. This comparison allowed us to assess whether a sequence-depended feature was enriched in a particular TEF solely due to the TEF’s nucleotide composition.
Comparison of distributions
For each TEF, the distributions of RNA folding energy and SD-likeness were constructed, as well as the distributions for all classes combined. The lower quartile of the combined distribution was used as a threshold. Hence, the distribution in each fraction was divided into the head (above the threshold) and tail (below the threshold) parts. We then calculated the value of χ2 for the 2 × 5 contingency table (head / tail size vs. fraction). The same procedure was performed for the shuffled datasets. Statistical significance was calculated as the fraction of shuffled sequences having χ2 larger than the observed value, or 10−4 if none of the shuffled sequences had a higher χ2. Significance of the difference with the set of E. coli genes was estimated in a similar way with the 2 × 2 contingency table (head/tail size versus given fraction/E. coli).
Influence of codons and amino acids
For each codon at each position 2–11, we calculated the positional codon frequency in each TEF. We retained only those pairs codon+position, for which the frequency monotonically depended on the fraction (the absolute value of the Spearman correlation coefficient being greater than 0.8; monotonic codons or pairs). The effect of a codon at a given position was calculated as the coefficient of linear regression of the codon positional frequency on the fraction number; for non-monotonic pairs the effect was assumed to be zero. We then calculated the effects in the shuffled datasets and calculated the P-value and Z-score of the observed value relative to the constructed distribution, as described in two preceding paragraphs, separately for each codon+position pair.
The effect of amino acids was calculated as the weighted sum of effects of codons encoding this amino acid with weights equal to the frequencies of these codons in E. coli genes. The effect of a position was calculated as the average absolute value of effects of all monotonic codons at this position.
The code used for the analysis is available at https://github.com/homo-sapiens34/leading-codons.
RNA/DNA ratio determination for rationally designed reporter plasmids
Prepared plasmids were mixed in equimolar ratio and used for transformation of BW25113 strain cells. As a result, more when 105 individual clones were obtained. A mixture of the obtained clones was grown to middle log-phase (A600 = 0.5) and used for plasmid DNA and total RNA preparation. cDNA was made by reverse transcription of total RNA with oligonucleotide CGGACACGCTGAACTTGT. Amplicons for sequencing were prepared by PCR from plasmid DNA or cDNA with oligonucleotides CGGACACGCTGAACTTGT and CACACAACACCGGAGCAAC and subjected to sequencing as described above.
Rational design of target sequences
To confirm the effects revealed by the analysis of the flow-seq data, we designed a set of target sequences placed as codons 2–11 into the reporter construct for experimental validation. For each tested feature, we designed a set of sequences varying this feature while keeping constant the remaining features.
To test the positional effects of particular codons, we used a set of sequences containing one, two, or three codons predicted to have a negative impact on translation if present at a particular spacer position. The sequences had no additional start codons, no SD-like regions, and all had the predicted RNA folding energy 15.8 ± 1.7 kcal/mol. Similarly, two more sets were used to test the influence of the position of SD-like sequences and additional start codons. Finally, to examine the impact of the amino acid metabolic cost, we tested two pairs of sequences having almost equivalent codon content and folding energy while varying in the cost of the encoded amino acids.
The complete list of designed target sequences is provided in Supplementary Table S2.
RESULTS AND DISCUSSION
Creation and flowseq analysis of the reporter library
To dissect the influence of the ramp region on the efficiency of reporter protein synthesis we inserted a 30-nucleotide randomized region (codons 2–11) immediately downstream of the ATG start codon of the Cerullian (CER) fluorescent protein gene in a reporter construct (30,33,34) that additionally contained a red fluorescent protein (RFP) gene used as an internal control (Figure 1A). While the theoretically possible number of randomized 30 nt sequence variants, 430, by far exceeds possibilities of experimental analysis, transformation of the E. coli host limited the complexity of the library to 5 × 106 independent clones. The resulting library in duplicate was sorted into five fractions by the CER translation efficiency related to that of RFP (translation efficiency fractions, TEFs; Figure 1A, Supplementary Figure S1A) and subjected to next generation sequencing (NGS) to deduce the sequences of randomized region. Overall, 32 376 variants of the ramp sequence (Supplementary Table S1) were found in both replicas of the library (Supplementary Figure S1B) and considered for further analysis, thereby representing a diversity of ramp regions sharply exceeding that of natural E. coli genes and comparable with earlier datasets examined by flowseq (1,31,32). Replicas demonstrated a relatively good reproducibility, so that similar mRNA variants are usually found within the same or immediately adjacent bins (Supplementary Figure S1B). Measurement of CER and RFP fluorescence in the bulk sorted fractions (Supplementary Figure S1C) demonstrated a good correspondence with the results of FACS analysis. The majority of randomized sequences demonstrated medium translation yield, while efficiently translated and completely inactive mRNA variants were an order of magnitude less abundant (Supplementary Figure S1D).
Figure 1.
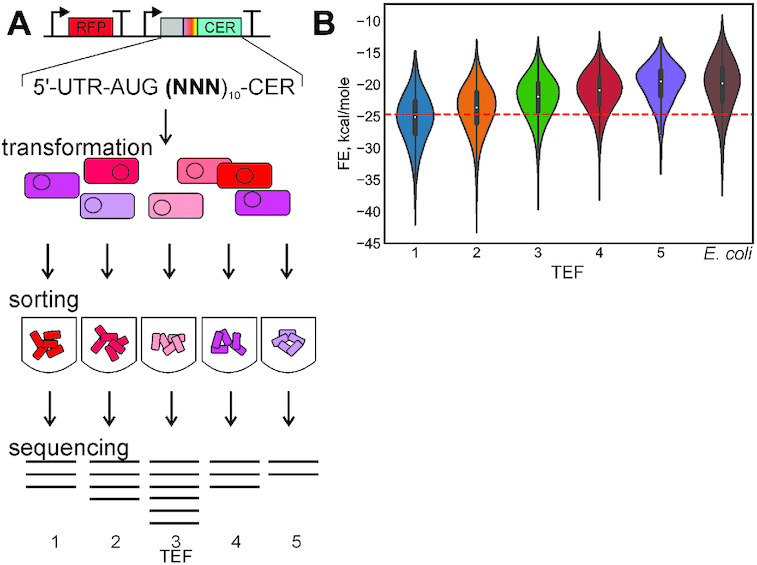
The principal scheme of the flowseq experiment. (A) Top to bottom: library construction, transformation of E. coli cells by the plasmid library, sorting by CER/RFP fluorescence into five TEF, sequencing. (B) Violin plot of the distributions of the RNA folding free energy in five TEF and in natural E. coli genes, P-value<10−4 (random shuffling, see Methods, Statistical Analysis).
Sequences with in-frame stop codons were found predominantly in TEFs 1–2 (Supplementary Figure S2A), which are characterized by the lowest efficiency of translation. Prior to further analysis such sequences were filtered out.
The analysis of the folding energy of CER mRNA variants (Figure 1B) revealed, in agreement with earlier studies (1,2,4,5,23,25), a decreased stability of secondary structures in efficiently translated mRNAs, close to that in E. coli genes. In line with this observation, a significant bias in the nucleotide composition of the ramp region of CER mRNA pools differing by the CER/RFP expression ratio was observed (Supplementary Figure S2B). Efficiently translated mRNAs were enriched in adenines and to lesser extent uridines in the randomized region. The observed distribution is close to the nucleotide composition bias of the codon 2–11 region of natural E. coli ORFs (Supplementary Figure S2B, rightmost bar). Enrichment for A and U in the 5′-end of efficiently translated ORFs was detected in other flowseq experiments as well (1,31,32).
The compositional bias of the ramp region of efficiently translated mRNAs likely reflects the requirement for a weak secondary structure. Since other coding region features, such as codon composition, CAI/tAI, and the likelihood to pair with the 3′-end region of the 16S rRNA are expected to depend on the nucleotide composition, to offset the influence of the latter we have generated ten thousand randomized datasets of the same size as the flowseq dataset and preserving position-specific nucleotide frequencies observed in each TEF (Materials and Methods, Statistical Analysis, Random Shuffling). All mRNA features analyzed further were compared with this randomized set to assess their statistical significance.
Another possible source of error in the interpretation of the data could be different stability or hypothetically variable transcription efficiency of mRNA variants possessing different codons 2–11. It is well known that, in general, features of the entire mRNA sequence, such as codon usage influence mRNA abundance in E. coli (13), yeast (39) zebrafish (40) and human (16). To check whether the mRNA amount varies at a scale comparable to the CER/RFP ratio in the reporter system we use, we created a representative set of ca. 150 reporter plasmids with combinations of CER mRNA features in codons 2–11 both favorable and inhibitory for the translation (Supplementary Table S2) and used this set to transform the E. coli host. The measurement of the CER/RFP ratio in these cultures was accompanied by determination of the mRNA abundance normalized to the corresponding DNA abundance (Supplementary Figure S2C). We observed only minor, one order of magnitude, variations in the mRNA level, while the CER/RFP levels varied >3 orders of magnitude on the same set of reporters. Thus, we conclude that the translation efficiency, but not mRNA abundance is the major contributor to the observed difference in the fluorescent protein yield in the experimental setup used. With this result in mind we consider the CER/RFP ratios to be a reasonable approximation of the translation efficiency.
Influence of SD-like sequences in the coding region on translation efficiency
Ribosome pausing at SD-like sites in E. coli, initially detected by the riboseq approach (28), was later suspected to be an artifact of the sample preparation procedure (29). To check whether such sequences in the ramp region of CER mRNA would lead to a reduction of the translation efficiency, we analyzed the presence of subsequences complementary to the 16S rRNA 3′-end region for all mRNAs in the dataset (Figure 2A). The reduced occurrence of SD-like patches in the ramp region of efficiently translated mRNAs demonstrates their negative influence on the protein biosynthesis, supporting earlier observations obtained with a limited set of model mRNAs (41). This observation is robust after accounting for the nucleotide composition (P-value < 10−4). Remarkably, the distribution of anti-SD binding energies in high-efficiency TEFs is similar to that of E. coli genes (Figure 2A, rightmost plot).
Figure 2.

Influence of SD-like sequences in the beginning of the coding region on translation efficiency. (A) Violin plot of distributions of the energy of potential base-pairing with the 16S rRNA 3′-terminal region in five TEF and in natural E. coli genes, P-value <10−4 (random shuffling, see Methods, Statistical Analysis). Dashed line: lower quartile of the combined distribution for all TEFs. (B) Translation efficiency of the designed constructs. Schematic representation (left side of the panel) and translation efficiencies (right side of the panel) of the constructs. All constructs designations are indicated next to the schematic representations. SD-like sequences are shown in red boxes. Translation efficiencies of the CER reporter were normalized to the reference RFP construct and indicated as a diagram. Exact values are shown next to the corresponding bars. The sequences are listed in Supplementary Table S2.
To validate the predicted influence of SD-like sequences in the 5′-end of the coding region on the CER protein production, we designed and created a set of reporter constructs with AGGAGG SD-like sequence placed at nucleotide positions 4–28 of the coding region (Figure 2B, left part; see Materials and Methods, Rational Design of Target Sequences and Supplementary Table S2 for exact sequences and more detailed data). To offset the influence of the secondary structure on the translation efficiency, all reporter mRNA sequences were designed to have a uniform folding energy of –13 ± 0.2 kcal/mol, corresponding to the least structured mRNAs in the randomized pool (Supplementary Figure S3A). The efficiency of the CER/RFP production driven by this set of reporters (Figure 2B, right panel) corroborated the observations based on the flowseq analysis (Figure 2A): SD-like motifs in the beginning of the ORF downregulated translation. This effect could not be attributed to the placement of specific codons into the CER ORF, as it did not depend on the position of the SD-like patch relative to the reading frame of CER. However, the inhibitory properties of SD-like sequence in the coding region depended on the distance to the start codon (Figure 2B, compare CER/RFP ratios across reporters with SD positions +4 to +28). This effect may be explained by the sequestration of the ribosome binding site and start codon by ribosomes stalling at the downstream SD-like sequence.
Influence of specific codons on translation efficiency
To assess the contribution of codons in the ten triplets following the initiator AUG, we calculated position-specific codon frequencies for individual TEFs and compared them with the randomized set of mRNAs. In each position, we calculated the number of codons whose frequencies monotonically depended (increased or decreased) on the translation efficiency. The number of such codons was significantly higher in the observed data compared to shuffled control datasets (Materials and Methods, Statistical Analysis, Random Shuffling; Supplementary Figure S3B), demonstrating that codon selection indeed is not random. We then calculated the effect of such monotonic codons on the translation efficiency as the coefficient of the linear regression of the frequency on the TEF number (Figure 3A, left panel; here and below see Materials and Methods, Statistical Analysis, Influence of Codons and Amino Acids for computational details). Most of individual codon preferences in highly expressed reporters could be explained by positional nucleotide composition, as shown by the comparison with shuffled control datasets (Figure 3A, right panel). However, some triplets (Figure 3A, right panel), as well as encoded amino acids (Supplementary Figure S4A) are apparently either beneficial or inhibitory for translation even after taking into account the nucleotide composition bias. The average effect of the codon identity on the protein yield was higher in AUG-proximal positions and decreased farther from the start codon (Figure 3B), in line with positional dependence of the codon usage bias in natural mRNAs (11,12).
Figure 3.

Influence of codons 2–11 on translation efficiency. (A) Differences of triplet frequencies at codon positions 2–11 (columns) as a function of TEF. The color of a cell represents the linear regression coefficient of the triplet frequency dependence on the TEF number (left) or Z-score of the given codon at a given position (right). The color is green for positive values, red for negative values (the color code is shown below the panel). Zero (yellow) denotes codons with non-monotonic dependence of frequency on fraction. Cells with P-values <0.01 are boxed (six such cells in each heatmap are expected for random sequences). (B) Position specificity of the codon influence on translation efficiency. Shown is the average Z-score of effects of codons in a particular position (corrected for the nucleotide content, see Methods). Codons with frequencies with non-monotonic dependence on the translation efficiency are excluded. Pearson's r = −0.8, P-value = 0.005. (C, D) Translation efficiency of the designed constructs. Schematic representation (left side of the panel) and translation efficiencies (right side of the panel) of the constructs. All constructs designations are indicated next to the schematic representations. Codons inhibitory for expression in the particular position according to the analysis of flowseq data are shown in red boxes. Translation efficiencies of the CER reporter were normalized to the reference RFP construct and indicated as a diagram. Exact values are shown next to the corresponding bars. The sequences are listed in Supplementary Table S2. (E) Correlation of the codon influence on translation for each of the sense codons at each of mRNA positions examined (2–11) and codon usage in E. coli. Shown is the correlation plot of codon effect that is the linear regression coefficient × 103 of the triplet frequency dependence on the TEF number and codon usage preference over synonymous codons in E. coli. Pearson's r = 0.33, P-value = 9.6 × 10−9. (F) Same as in (E), but Z-scores are shown instead of correlation coefficients, so that the data are corrected for nucleotide frequencies. Pearson's r = −0.12, P-value = 0.004.
To validate the predicted positive or negative effect of specific codons at positions 2–11 on the protein yield, we designed a set of specific reporter plasmids (Materials and Methods, Rational Design of Target Sequences and Supplementary Table S2). To avoid confounding effects, in all designed reporters the affinity of nucleotides +4 to +33 of the CER coding region to the 3′-end region of the 16S rRNA was minimized, while the folding energy of the secondary structure in reporter mRNA was within –15.8±1.7 kcal/mol. In these designed reporters, the majority of codons 2–11 were positionally optimal according to the flowseq data analysis, while introducing one, two, or three codons predicted to be inhibitory for protein yield into the respective positions of the ramp region. Substitution of an optimal codon by an inhibitory one at positions 2, 3, 4, 6, 7, 8, 9, and 10 of the CER coding region (Figure 3C, left scheme) resulted in a decrease of CER/RFP production for a number of constructs (Figure 3C, right plot). Placement of inhibitory codons to the sites that are on average closer to the start of the ORF (+2, +3, +4 and +7) were found to decrease protein synthesis, while this effect was negligible for more distal sites (+6, +8, +9 and +10). This result confirmed the tendency observed with flowseq (Figure 3A, B). Since the influence of a single inhibitory codon on reporter gene expression was moderate (Figure 3C), we tested whether two or three such codons would additively suppress expression (Figure 3D, left scheme) and found it to be exactly the case (Figure 3D, right plot). Similar to a single unfavorable codon, pairs (Supplementary Figure S4B) and triples (Supplementary Figure S4C) of such codons inhibited expression on average more efficiently when placed closer to the beginning of start codon.
Earlier, the codon usage (11) and amino acid occurrence (19) in the N-terminal part of natural proteins were reported to be biased, while the origin of this bias was a matter of debate (1,22,25). The negative influence of GGA, GGG, AGG codons (Figure 3A) may be caused by their similarity with the SD sequence. The inhibitory effect of conformationally flexible (Gly) or restricted (Pro) residues (Supplementary Figure S4A) is reminiscent of data on the ribosome stalling at sites containing these residues at different positions relative to the P-site (29,42,43). An unlikely caveat to this analysis is the possibility that addition of some particular N-terminal extensions to the fluorescent protein may affect its fluorescence.
The apparently beneficial role of methionine among amino acids 2–11 for the translation efficiency (Figure 3A, AUG lines, Supplementary Figure S4A, M line) is likely explained by the use of extra AUG codons as auxiliary translation initiation sites. To check this assumption, we created a set of reporter constructs containing mainly codons unfavorable for the expression in positions 2–11, while maintaining similar RNA secondary structure folding energy of –24 kcal/mol. In this set of reporters, AUG triplets were placed to the coding region of CER at positions +4 to +30 with a single-nucleotide increment (Figure 4, the left scheme, in-frame AUG codons are colored green). The yield of the CER protein was indeed on average slightly higher if the AUG codon position coincided with CER reading frame (positions +4, +7, +10, +13, +16, +19, +22, +25 and +28) in comparison with out of frame location (positions +5, +6, +8, +9, +11, +12, +14, +15, +17, +18, +20,+21, +23, +24, +26, +27, +29 and +30). Overall, the in-frame dataset differed from the out-of-frame dataset with P-value = 0.018 (the Mann–Whitney test).
Figure 4.

Influence of additional AUG codons in the ramp mRNA region on the yield of translation. Schematic representation (left side of the panel) and translation efficiencies (right side of the panel) of the constructs. All constructs designations are indicated next to the schematic representations. AUG codons are shown. Red boxes correspond to the out of frame AUG codons, while that in the CER frame are shown in green boxes. Translation efficiencies of the CER reporter were normalized to the reference RFP construct and indicated as a diagram. Exact values are shown next to the corresponding bars. The sequences are listed in Supplementary Table S2.
Influence of the codon optimality on the translation efficiency
The most controversial issue is the proposed selection for rare codons in the ramp region of natural mRNAs (1,3,11,20), which might be alternatively interpreted as a consequence of selection against formation of stable secondary structure (23,24). We have observed that the position-specific codon effect on reporter expression is positively correlated with codon usage in native E. coli genes (Figure 3E), although this correlation completely disappears after correction for positional nucleotide frequencies (Figure 3F). No correlation was observed in the comparison of codon effects and codon tAI (8) (Supplementary Figure S4D).
To address directly the possibility of influence of the ramp region tAI on translation efficiency independent from other parameters, we created a ΔArg strain of E. coli where we deleted three out of four genes encoding tRNAArgACG (Figure 5A) which resulted in a ∼5-fold decrease in the tRNAArgACG abundance (Supplementary Figure S5A). Hence, we reduced tAI of efficiently translated CGU, CGC and CGA codons fivefold. We transformed the ΔArg strain by the same reporter plasmid library as the one used for the transformation of the wild type strain and sorted cells likewise (Supplementary Table S3, Figure S5B,C). The influence of each codon on the translation efficiency was calculated as for the wild type strain and used for comparison (Figure 5B; see Supplementary Figure S5D for Z-scores). Apparently, the fivefold decrease in tAI did not result in significant changes of the CGU, CGC and CGA codons influence on the protein biosynthesis.
Figure 5.

B. Influence of tRNAArgACG gene copy number on the translation of CER reporters. (A) The scheme of serV operon in the wild type and in the ΔArg strains. (B) Scatter plot of the linear regression coefficients × 103 of the triplet frequency dependences on the TEF number in the WT (x-axis) and ΔArg (y-axis) strains. Points corresponding to the arginine codons decoded by tRNAArgACG are shown in red. (C) Translation efficiency of the designed constructs. Schematic representation (lower part of the panel) and translation efficiencies (upper part of the panel) of the constructs. Translation efficiencies of the CER reporter were normalized to the reference RFP construct and indicated as a diagram. All constructs designations are indicated next to the schematic representations and below the corresponding bars. Arginine codons decoded by tRNAArgACG are shown in red, while those decoded by other tRNAs are shown in green. The sequences are listed in Supplementary Table S2. Dark green bars correspond to the wild type strain with full set of tRNAArgACG genes, while light green bars correspond to the ΔArg strain (key is provided on the graph).
To validate this conclusion, we transformed cells of the ΔArg strain and cells of the wild type parental strain with a designed set of four reporter constructs containing three varied arginine codons following the CER start codon (Figure 5C, scheme below, Supplementary Table S2). The CER protein yield in both strains demonstrated minor differences (Figure 5C, above). The reduction of the tRNAArgACG abundance resulted in a mild increase of the CER protein yield if non-cognate AGA arginine codons were located close to the start of the reading frame. At the same time, the CGA and CGU codons cognate for tRNAArgACG, when placed in the starting region of the coding sequence, supported somewhat less efficient translation of the reporter; the CGC codon performed almost identically.
Previously published analysis of the influence of three-fold reduction of the tRNAThrUGU gene expression in yeast on endogenous mRNAs translation (25) also has not revealed any dependence of the translation efficiency on the presence of ACA codons in the ramp mRNA region upon reduction of tRNA concentration. The simplest interpretation of these data contradicts the hypothesis that low tAI of natural mRNAs ramp region influences the efficiency of their translation. However, these data do not exclude an interpretation that lower tAI of the ramp region might be explained by reasons beyond the efficiency of translation of a particular mRNA, e.g. it might be beneficial for the translation of other mRNAs via reduction of ribosome idling (11,44).
Influence of nutrients availability on translation of specific reporter mRNAs
The assessment of the translation efficiency by flowseq has been performed in the rich LB medium, where the ribosome progression is unlikely to be slowed down by the amino acid deficiency. However, in natural ecological niches bacteria might encounter resource limitation. To address this, we grew wild type E. coli cells transformed with the reporter plasmid library in the poor M9 medium and repeated the flowseq experiment and data analysis (Supplementary Table S4, Figure S6A,B). The influence of specific codons residing in the ramp region on the protein yield (Supplementary Figure S6C) demonstrated an overall good match to that observed for E. coli grown in the rich medium (Figure 2A). However, we noted a small difference in the relative influence of particular codons on the yield of CER protein in the rich and poor medium dependent on the identity of the encoded amino acid. Synthesis of different amino acids is known to require different amounts of ATP molecules and reducing equivalents (38). The use of ‘expensive’ amino acids in the N-terminal region of a protein might compromise its yield (44). We hypothesized that while growing in the poor M9 medium this effect might be exacerbated. For each reporter in the library, we calculated the total synthesis costs of amino acids 2–11 encoded by its randomized part using the cost values of individual amino acids taken from ref. (36) (see Methods for details). For each TEF, we calculated the average synthesis cost of all fragments in this TEF and then compared the ratio of these average costs for TEFs obtained by sorting of the cells grown in the minimal M9 or rich LB medium (Figure 6A). The slope of the regression line of this ratio on the TEF number is negative with P-value of 0.018. This shows that while overall the influence of amino acids synthesis costs is small, it contributes more to the translation efficiency in the poor M9 than in the rich LB medium. The average cost ratio for each TEF in M9 and LB calculated separately for each codon position (Supplementary Figure S6D) shows that the amino acid cost at position +2 is the major contributor to this effect. This result is in line with the observation that properties of codons proximal to the start codon in general influence the translation efficiency more than more distant ones.
Figure 6.

Influence of the amino acids’ metabolic cost on the efficiency of translation in the rich and poor medium. (A) The influence of the amino acids metabolic cost on the efficiency of translation in the rich and poor medium. The ratio of the average metabolic costs of amino acids encoded by the codons 2–11 of the reporter constructs that fell into a particular TEF (x-axis) in the poor and rich medium (M9/LB). The shaded area shows 95% confidence intervals for the regression line. (B) Translation efficiencies of designed constructs (y-axis) in different media (x-axis) after 2 days of cultivation to compensate for different growth rates in rich and poor media. Two pairs of reporter constructs are shown. The folding energy within the same pair (same colors of the graphs) is designed to be constant. Thick lines correspond to the reporters, whose triplets 2–11 encode the more expensive set of amino acids, while thin lines correspond to the reporters encoding cheaper amino acids. The metabolic cost of aminoacids 2–11 of the reporter (38) is shown next to the graphs. The sequences are listed in Supplementary Table S2. The following media were used: LB (LB), LB diluted 1:4 with water (LB/4), M9 supplied with glucose and expensive amino acids Tyr, His, Ile, Leu (M9+AA), M9 supplied with glucose without amino acids (M9), M9 supplied with glycerol without glucose (M9/Gly).
To challenge this finding, we designed two pairs of reporter constructs. In each pair, the metabolic costs of encoded amino acids were contrasting, while the features other when amino acid metabolic costs, such as folding energy of mRNA and SD-likeness was kept equal. We also avoided AUG codons and codons with significant inhibitory influence on translation for these mRNA pairs (Figure 6B, lines of the same color correspond to mRNAs with the same folding energy, metabolic costs are indicated next to the graphs, thick lines correspond to ‘expensive’ fragments, and thin lines, to ‘cheap’ fragments). Cells transformed by the reporter constructs were grown in either rich LB medium (Figure 6B, medium indicated below the graphs) or 4-fold diluted LB medium, M9 with amino acids and glucose, M9 with glucose, and M9 with glycerol instead of the glucose. The measurement of the translation yield (Figure 6B) revealed that if the CER protein coding part was preceded by a patch of expensive amino acids, its synthesis appeared to be sensitive to the richness of medium (Figure 6B, thick lines decline from LB to the poorer mediums), while this dependence was absent or even reversed in the opposite case (Figure 6B, thin lines remain constant or increase from LB to the poorer mediums). The likely explanation for this effect might reside in faster clearance of the ribosome binding site by an elongating ribosome if it does not have to wait for aa-tRNA charged by an expensive amino acid which is presumably scarcer in the poor medium.
CONCLUSIONS
This study illustrates the complexity of the evolutionary pressure on translated sequences which have to be optimized not only to encode a functional protein and avoid ribosome binding site sequestration by formation of RNA secondary structure. The avoidance of SD-like motifs at least in the beginning of ORF and accounting for the metabolic cost of amino acids might contribute to the evolution of natural genes and should be considered upon creation of artificial expression constructs. While optimization of the translation efficiency is an obvious pathway for the coding sequences evolution, it may not account for all biases observed in the sequence of natural genes, suggesting that factors other than just a yield of protein per mRNA might be selected for.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the members of our labs for discussions.
Contributor Information
Ilya A Osterman, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia.
Zoe S Chervontseva, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; A.A.Kharkevich Institute for Information Transmission Problems, Moscow 127051, Russia.
Sergey A Evfratov, Lomonosov Moscow State University, Moscow 119992, Russia.
Alena V Sorokina, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia.
Vladimir A Rodin, Lomonosov Moscow State University, Moscow 119992, Russia.
Maria P Rubtsova, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia.
Ekaterina S Komarova, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia.
Timofei S Zatsepin, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia.
Marsel R Kabilov, Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, Novosibirsk 630090, Russia.
Alexey A Bogdanov, Lomonosov Moscow State University, Moscow 119992, Russia.
Mikhail S Gelfand, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; A.A.Kharkevich Institute for Information Transmission Problems, Moscow 127051, Russia.
Olga A Dontsova, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia; Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow 117997, Russia.
Petr V Sergiev, Skolkovo Institute of Science and Technology, Skolkovo, Moscow region 143025, Russia; Lomonosov Moscow State University, Moscow 119992, Russia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Next generation sequencing was supported by Russian Foundation for Basic Research [17-00-00369 (17-00-00366, 17-00-00367)]; plasmid library and individual plasmids construction, cell transformation and analysis of was supported by Russian Science Foundation [17-75-30027]; computational analysis was supported by Russian Science Foundation [18-14-00358]. Funding for open access charge: Skolkovo Institute of Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1. Goodman D.B., Church G.M., Kosuri S.. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013; 342:475–479. [DOI] [PubMed] [Google Scholar]
- 2. Kudla G., Murray A.W., Tollervey D., Plotkin J.B.. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009; 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shah P., Ding Y., Niemczyk M., Kudla G., Plotkin J.B.. Rate-limiting steps in yeast protein translation. Cell. 2013; 153:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Espah Borujeni A., Cetnar D., Farasat I., Smith A., Lundgren N., Salis H.M.. Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acids Res. 2017; 45:5437–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Smit M.H., van Duin J.. Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literature data. J. Mol. Biol. 1994; 244:144–150. [DOI] [PubMed] [Google Scholar]
- 6. Grantham R., Gautier C., Gouy M., Mercier R., Pavé A.. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980; 8:r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharp P.M., Li W.H.. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. dos Reis M., Wernisch L., Savva R.. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003; 31:6976–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frenkel-Morgenstern M., Danon T., Christian T., Igarashi T., Cohen L., Hou Y.-M., Jensen L.J.. Genes adopt non-optimal codon usage to generate cell cycle-dependent oscillations in protein levels. Mol. Syst. Biol. 2012; 8:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Begley U., Dyavaiah M., Patil A., Rooney J.P., DiRenzo D., Young C.M., Conklin D.S., Zitomer R.S., Begley T.J.. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007; 28:860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 12. Hockenberry A.J., Sirer M.I., Amaral L.A.N., Jewett M.C.. Quantifying position-dependent codon usage bias. Mol. Biol. Evol. 2014; 31:1880–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boël G., Letso R., Neely H., Price W.N., Wong K.-H., Su M., Luff J., Valecha M., Everett J.K., Acton T.B. et al.. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 2016; 529:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berg O.G., Kurland C.G.. Growth rate-optimised tRNA abundance and codon usage. J. Mol. Biol. 1997; 270:544–550. [DOI] [PubMed] [Google Scholar]
- 15. Gustafsson C., Govindarajan S., Minshull J.. Codon bias and heterologous protein expression. Trends Biotechnol. 2004; 22:346–353. [DOI] [PubMed] [Google Scholar]
- 16. Wu Q., Medina S.G., Kushawah G., DeVore M.L., Castellano L.A., Hand J.M., Wright M., Bazzini A.A.. Translation affects mRNA stability in a codon-dependent manner in human cells. eLife. 2019; 8:e45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gustafsson C., Minshull J., Govindarajan S., Ness J., Villalobos A., Welch M.. Engineering genes for predictable protein expression. Protein Expr. Purif. 2012; 83:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gould N., Hendy O., Papamichail D.. Computational tools and algorithms for designing customized synthetic genes. Front. Bioeng Biotechnol. 2014; 2:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tuller T., Veksler-Lublinsky I., Gazit N., Kupiec M., Ruppin E., Ziv-Ukelson M.. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011; 12:R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tuller T., Waldman Y.Y., Kupiec M., Ruppin E.. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Charneski C.A., Hurst L.D.. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol. Biol. Evol. 2014; 31:70–84. [DOI] [PubMed] [Google Scholar]
- 23. Bentele K., Saffert P., Rauscher R., Ignatova Z., Blüthgen N.. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013; 9:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allert M., Cox J.C., Hellinga H.W.. Multifactorial determinants of protein expression in prokaryotic open reading frames. J. Mol. Biol. 2010; 402:905–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pop C., Rouskin S., Ingolia N.T., Han L., Phizicky E.M., Weissman J.S., Koller D.. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol. Syst. Biol. 2014; 10:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stadler M., Fire A.. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011; 17:2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gardin J., Yeasmin R., Yurovsky A., Cai Y., Skiena S., Futcher B.. Measurement of average decoding rates of the 61 sense codons in vivo. Elife. 2014; 3:e03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li G.-W., Oh E., Weissman J.S.. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012; 484:538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mohammad F., Woolstenhulme C.J., Green R., Buskirk A.R.. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 2016; 14:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evfratov S.A., Osterman I.A., Komarova E.S., Pogorelskaya A.M., Rubtsova M.P., Zatsepin T.S., Semashko T.A., Kostryukova E.S., Mironov A.A., Burnaev E. et al.. Application of sorting and next generation sequencing to study 5′-UTR influence on translation efficiency in Escherichia coli. Nucleic Acids Res. 2017; 45:3487–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verma M., Choi J., Cottrell K.A., Lavagnino Z., Thomas E.N., Pavlovic-Djuranovic S., Szczesny P., Piston D.W., Zaher H.S., Puglisi J.D. et al.. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019; 10:5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cambray G., Guimaraes J.C., Arkin A.P.. Evaluation of 244, 000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol. 2018; 36:1005–1015. [DOI] [PubMed] [Google Scholar]
- 33. Osterman I.A., Prokhorova I.V., Sysoev V.O., Boykova Y.V., Efremenkova O.V., Svetlov M.S., Kolb V.A., Bogdanov A.A., Sergiev P.V., Dontsova O.A.. Attenuation-based dual-fluorescent-protein reporter for screening translation inhibitors. Antimicrob. Agents Chemother. 2012; 56:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Osterman I.A., Evfratov S.A., Sergiev P.V., Dontsova O.A.. Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 2013; 41:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10. [Google Scholar]
- 37. Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akashi H., Gojobori T.. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3695–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Presnyak V., Alhusaini N., Chen Y.-H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R. et al.. Codon optimality is a Major determinant of mRNA stability. Cell. 2015; 160:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bazzini A.A., Del Viso F., Moreno-Mateos M.A., Johnstone T.G., Vejnar C.E., Qin Y., Yao J., Khokha M.K., Giraldez A.J.. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 2016; 35:2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin H., Zhao Q., Gonzalez de Valdivia E.I., Ardell D.H., Stenström M., Isaksson L.A.. Influences on gene expression in vivo by a Shine-Dalgarno sequence. Mol. Microbiol. 2006; 60:480–492. [DOI] [PubMed] [Google Scholar]
- 42. Mankin A.S. Nascent peptide in the ‘birth canal’ of the ribosome. Trends Biochem. Sci. 2006; 31:11–13. [DOI] [PubMed] [Google Scholar]
- 43. Woolstenhulme C.J., Guydosh N.R., Green R., Buskirk A.R.. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015; 11:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frumkin I., Schirman D., Rotman A., Li F., Zahavi L., Mordret E., Asraf O., Wu S., Levy S.F., Pilpel Y.. Gene architectures that minimize cost of gene expression. Mol. Cell. 2017; 65:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.