

Recently, in the European Heart Journal, we described how the European Union (EU) Regulation on Medical Devices (MDR)1 will affect our daily clinical practice.2 That law—which increases the requirements for clinical evidence before new high-risk medical devices are approved—was due to be applied from 26 May 2020 but, in common with many aspects of our lives, it has been disrupted by the COVID-19 pandemic. Manufacturers, notified bodies, national regulatory agencies, and the European Commission (EC) have all been preoccupied with responding to the crisis, so a 1-year postponement has been agreed.
In the meantime, it is possible to draw some tentative conclusions from recent events. What are we learning about the conduct of clinical trials and about EU systems for evaluating new drugs and devices? Are they ‘fit for purpose’ in responding to a major public health challenge? How should clinicians and scientists be contributing to regulatory governance?
The infodemic
The new SARS-CoV-2 virus was first isolated from a patient admitted to hospital in Wuhan China on 26 December 2019. Its full genome—only 29 903 nucleotides in length, >100 000 times shorter than the 3.2 × 109 bp of code in the human genome—was reported on 7 January 2020.3,4 The Director General of the World Health Organization (WHO), Tedros Adhanom Ghebreyesus, declared a Public Health Emergency of International Concern (PHEIC) on 30 January5 and a pandemic on 12 March 2020. At the Munich Security Conference, he stated memorably that ‘We’re not just fighting an epidemic; we’re fighting an infodemic’.6
The scientific community has mobilized to an unprecedented degree to investigate the SARS-CoV-2 virus, and that effort has resulted in an explosion of publications. By 12.00 h Central European Time on 31 May 2020, 17 533 papers relating to COVID-19 had been listed on the US National Library of Medicine PubMed database. That outstrips the reading capacity of an individual7 so it is unsurprising that the total included 139 papers listed as ‘Systematic Reviews’. Yet checking the small number coded as ‘Clinical Trials’ reveals only four drug studies, which randomized 22–150 patients to receive treatment for 4–21 days.8–11 Nonetheless, a tsunami of research is coming to engulf us: by the same date, 1833 clinical studies for COVID-19 had been registered on ClinicalTrials.gov. Many are suboptimally designed however, and some early results released as pre-prints have been poorly reported and inappropriately cited.12 Within all this noise, important signals may be submerged.
Good clinical research
What has been encouraging has been the capacity of the system for approving clinical trials to cut through what has been perceived to be excessive bureaucracy.13,14 The largest double-blind controlled trial of hospitalized patients with COVID-19, the RECOVERY study in the UK, went from first protocol to enrolling its first patient in 9 days. By 31 May, 11 004 patients in 176 hospitals had been randomized to usual care or usual care with lopinavir–ritonavir, dexamethasone, hydroxychloroquine, or azithromycin.15 RECOVERY has an adaptive design, so protocol amendments have added a second randomization to tocilizumab for patients who deteriorate, and another arm to give convalescent plasma containing antibodies against SARS-CoV-2. A similar study, the Solidarity trial, that is being coordinated by the WHO, has enrolled >3500 patients in 17 countries.16
Such experience should prove invaluable to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) whose working group E6 is ‘renovating’ its guidance on Good Clinical Practice (GCP).17,18 A simultaneous initiative funded by the Wellcome Trust and the Gates Foundation with the African Academy of Sciences, called the Good Clinical Trials Collaborative (GCTC), is developing new clinical practice guidelines for trials.19 Before reaching any conclusions, the GCTC is now consulting widely to learn from the experience of trialists during the COVID-19 pandemic; the ICH working group should do the same, so that it can consider how essential ethical and scientific principles can be respected without compromise, while simplifying and streamlining approval processes and monitoring.
Unfortunately, the absence of good evidence for effective therapy of COVID-19 has not prevented speculation or a plethora of premature ‘guidelines’ (mentioned in 804 results from the PubMed search). As shown in a review prepared for the Polish health technology assessment agency,20 there is no consensus (Table 1)—because there is insufficient evidence. The need for open sharing of data and reflection before rushing to judgement is obvious.
Table 1.
Summary of therapeutic ‘guidelines’ in COVID-19
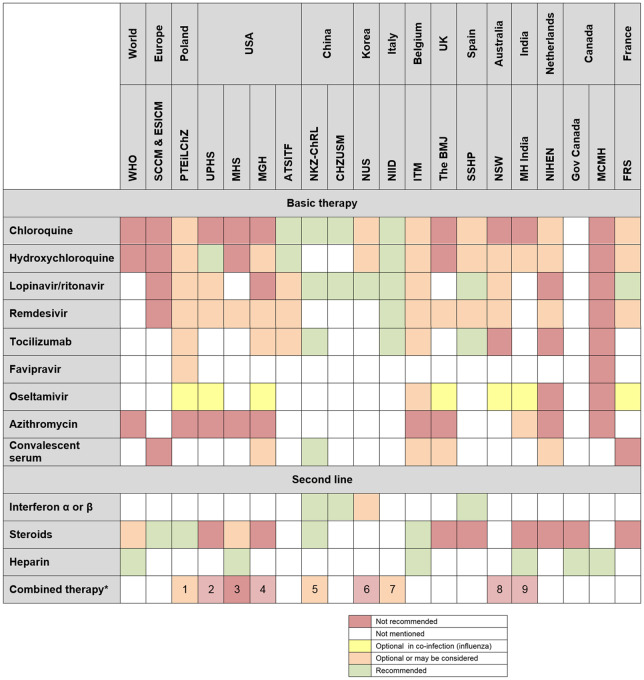
|
All types of combined therapies referenced in the guidelines.
1, lopinavir/ritonavir + chloroquine or hydroxychloroquine, lopinavir/ritonavir + chloroquine or hydroxychloroquine + tocilizumab; 2, lopinavir/ritonavir + ribavirin; 3, lopinavir/ritonavir + amiodarone, quetiapine or simvastatin; 4. hydroxychloroquine + remdesivir; 5, ribavirin + interferon or lopinavir/ritonavir; 6, lopinavir/ritonavir + chloroquine or hydroxychloroquine; 7, chloroquine/hydroxychloroquine + lopinavir/ritonavir or darunavir + ritonavir, lopinavir/ritonavir + chloroquine or hydroxychloroquine + tocilizumab; 8, lopinavir/ritonavir ± hydroxychloroquine; 9, hydroxychloroquine + azithromycin.
WHO, World Health Organization; SCCM & ESICM, Society of Critical Care Medicine and the European Society of Intensive Care Medicine; PTEiLChZ, Polish Society of Epidemiology and Infectious Diseases; UPHS, University of Pennsylvania Health System; MHS, Military Health System; MGH, Massachusetts General Hospital; ATSITF, American Thoracic Society‐led International Task Force; NKZ-ChRL, National Health Commission of Peoples Republic of China; CHZUSM, Children’s Hospital, Zhejiang University School of Medicine; NUS, NUS Saw Swee Hock School of Public Health; NIID, National Institute for the Infectious Diseases ‘L. Spallanzani’ IRCCS; ITM, Institute of Tropical Medicine w Antwerpii; BMJ, The British Medical Journal; SSHP, Spanish Society of Hospital Pharmacy; NSW, New South Wales Government, Department of Health; MH India, Government of India, Ministry of Health & Family Welfare Directorate General of Health Services; NIHEN, The National Institute for Health and the Environment of the Netherlands; Gov Canada, Government of Canada; MCMH, British Columbia Ministry of Health; FRS, French Resuscitation Society.
Adapted with permission from Nizankowski et al.20
If we had comprehensive clinical registries, with the ability to randomize patients within them to different therapeutic strategies and then extract and aggregate data, questions such as whether taking hydroxychloroquine is safe and effective for COVID-19 could be addressed reliably, whereas large-scale observational data with multiple confounding variables give potentially misleading and clinically uninformative results.21 To facilitate such studies, clarity on the implications of the EU General Data Protection Regulation22 for medical research is becoming urgent.23 US and French authorities authorized the use of antimalarial drugs24 but the emergency authorization of hydroxychloroquine in the USA was based on ‘limited in-vitro and anecdotal clinical data in case series’ and it has been criticized for lack of transparency.25 A rapidly conducted registry analysis21 led to the temporary suspension of its use in the WHO Solidarity trial , but there is no substitute for proper clinical trials.26 After serious questions were raised, the registry publication has already been retracted.
Approving medical devices
It is a mistake to consider the evaluation of medical devices any less important than is required for new drugs. Less stringent requirements for regulatory approval of high-risk medical devices in Europe, compared with the USA, led in the past to an almost three-fold excess of device alerts or recalls.27 In the USA, high-risk devices approved by a new accelerated process had higher recall rates and shorter times on the market before serious recalls than devices that underwent standard review.28 The MDR has been criticized by some for potentially impeding innovation, but there are provisions for special approval when there are unmet needs.29 We are learning now how challenging it can be to implement those systems across Europe when there is no central coordinating agency for medical devices.
Personal protective equipment (PPE)
Although protective garments for PPE qualify as low-risk (class I) medical devices, if they need to be sterile then they have to be approved by a notified body before CE marking. Since April 2019, newly manufactured PPE products have also had to meet the requirements of regulation (EU) 2016/425 which include type examination and some form of surveillance.30 In a public health crisis, EU member states can bypass those procedures and allow temporary access for needed devices31 but each national agency has to do that individually, so there have been variations and delays between countries in making the same device available.
The European Standardization Organisation (CEN) has made relevant standards freely available, and new manufacturers have entered the market. Nonetheless, hospitals and governments have been competing to secure adequate supplies, which has led to an increase in prices. Europol issued a warning about counterfeit goods including surgical masks.32
Ventilators
Ventilators are complex class IIb devices that incorporate sophisticated medical software, for which there are well established international standards. Considering how difficult it would be to satisfy those rapidly, by developing a new ventilator from scratch, the EC issued guidance on 23 April to encourage manufacturers of CE-marked ventilators to scale up their production.33 More recently, the Working Group on New Technologies proposed that a ‘COVID-19 ventilator’ made by ‘non-traditional actors including consortia of university hospitals, doctors, engineers and regulators’ could be used without CE marking if it was given a derogation by a national regulatory agency. Its specifications did not include pressure monitoring or alarms, although patients with COVID-19 and severe respiratory failure may need more, not less, sophisticated ventilators.
The rush to increase numbers of available PPE and ventilators has exposed the lack of a pre-conceived and coordinated plan to cope with emergencies. New entries to the market of unapproved devices such as the ‘Shangrila 510’ ventilator have had to be scrapped.34
As with drugs for COVID-19, there have been varying guidelines concerning the use of non-invasive ventilation with continuous positive airways pressure—which was discouraged in initial guidance from Australia and New Zealand35 then later encouraged in the UK.36 Recently the National Institute of Health in the USA has taken an equivocal stance.37
In vitro diagnostic tests
According to the EU Regulation on In Vitro Diagnostic Medical Devices (IVDR),38 tests to detect SARS-CoV-2 qualify as high risk or Group D because the infective agent is associated with high individual risk and high public health risk. The IVDR will not be applied until 2022 at the earliest, so currently SARS-CoV-2 detection kits can be validated according to the IVD Directives (which are open to national interpretation) or the IVDR. Each new test can be approved rapidly but only within each country and by derogation from relevant EU laws, so each regulatory agency has been setting its own criteria. The EC has not used its authority under IVDR Article 54 to apply EU-wide derogations. Such regulatory complexity may discourage IVD manufacturers from prioritizing distribution to European countries.
Laboratories responded promptly by producing many ‘laboratory developed tests’.39 The first commercial tests were available after several weeks. A review of all tests published up to 4 May 2020 identified 39 individual studies to detect the SARS-CoV-2 virus.40 Ideally, these would detect viral RNA with high sensitivity so that all infected individuals can be identified (as well as high specificity so that only infected individuals give positive results). The pooled sensitivity of an initial RT–PCR (reverse transcription–polymerase chain reaction) test for the virus was 89%. Recently the Food and Drug Administration (FDA) warned that a point-of-care test to diagnose COVID-19 may return false-negative results.41
The optimal test for antibodies would have high specificity, as a false-positive result may wrongly reassure someone that they have already had the infection and therefore have some immunity. The specificities of 25 tests to identify antibodies to SARS-CoV-2 were 88.9–100%.40 Their sensitivities were 18.4–96.1%, so false-negative results may be common. The method used to detect convalescent antibodies is important since sensitivities were 85% (95% confidence interval 70–94%) for tests using enzyme-linked immunosorbent assays (ELISAs) but 55–70% for lateral flow immunoassays.42
Variable diagnostic performance is a basic consideration that some politicians have been slow to understand, and it is not yet known if and for how long antibodies will provide protection. The EC reported in April 2020 that 192 COVID-19 devices had already been CE marked under Directive 98/79/EC7, specifically 78 RT–PCR tests, 13 rapid antigen tests, and 101 antibody tests.43 Their reliability must be fully reported. The EU will establish a network of COVID-19 reference laboratories, together with a supporting platform.44
A learning healthcare system includes good regulatory science
Some initial messages are emerging. Politicians and regulators—and the public and patients—depend on good scientific advice. The questions raised can be answered only by experts; certainty is impossible so transparency is essential.45 Manufacturers, laboratory scientists, and clinical trialists have demonstrated that rapid innovation is possible, but what we need is a science-based regulatory system with more capacity and flexibility and a well-prepared strategy for responding rapidly to a crisis in the interests of patients.46 That means sharing and rationalizing processes as widely as possible, and involving the academic community.
There may be other occasions when new drugs, devices, and tests need to be used on compassionate grounds before there has been time to evaluate them properly. There should be EU-wide systems for making derogations. Regulators should be able to grant conditional approvals, with strict requirements to monitor performance in a systematic way and with agreements to share risks.47 The EC has issued guidance on public procurement and antitrust issues.44 Joint health technology assessments and pricing initiatives could also contribute.48
The EU has offered substantial research funding. A major medical publisher (Elsevier) has made its publications relating to COVID-19 open access and publicly available ‘for as long as the COVID-19 resource centre remains active’. More investment in research and sustainable methods of promoting open access to all the evidence are important. European medical associations can contribute to the rapid dissemination of scientifically validated results if they are seen as partners and not just validatory stakeholders.
In our paper on the MDR,2 we describe the opportunities that it will provide for applying evidence to guide our clinical practice when prescribing high-risk devices. The COVID-19 pandemic has clearly reinforced the need for scientists and physicians collectively to engage with regulators to develop appropriate systems for evaluating and approving both laboratory tests and new medical devices, as well as with the European Medicines Agency for drugs. The Regulatory Affairs committees of the ESC and the Biomedical Alliance in Europe (representing 33 medical specialist associations) nominate colleagues to be stakeholder members of European regulators’ committees. Now their agendas should be everybody’s business.
Alan Fraser chairs the Regulatory Affairs Committees of the ESC and the Biomedical Alliance in Europe. Piotr Szymański is chairman elect of the ESC committee. Elizabeth Macintyre chairs the Working Group on In Vitro Diagnostic Medical Devices of the Biomedical Alliance in Europe. Martin Landray leads the Good Clinical Trials Collaborative and is a member of the ESC Regulatory Affairs Committee.
References
References are available as supplementary material at European Heart Journal online.

Supplementary Material
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.