

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a progressive disease with high mortality. Patient characteristics associated with diagnostic delays are not well described.
Methods
Subjects who had not been diagnosed with IPF prior to referral and received a new diagnosis of IPF at an enrolling centre for the IPF-PRO (Idiopathic Pulmonary Fibrosis Prospective Outcomes) Registry were characterised as having a longer (>1 year) or shorter (≤1 year) time from symptom onset to diagnosis and from first imaging evidence of fibrosis to diagnosis. Patient characteristics, evaluations and time to death or lung transplant were compared between these cohorts.
Results
Among 347 patients with a symptom onset date, 49% were diagnosed with IPF >1 year after symptom onset. These patients were slightly younger and had more cardiac comorbidities than patients diagnosed ≤1 year after symptom onset. Among 454 patients with a date for imaging evidence of fibrosis, 78% were diagnosed with IPF ≤1 year later. A greater proportion of patients with >1 year versus ≤1 year from imaging evidence of fibrosis to diagnosis had cardiac comorbidities and gastro-oesophageal reflux. There was no significant difference in time to death or lung transplant between groups by time to diagnosis.
Conclusions
The time from symptom onset to diagnosis remains over 1 year in approximately half of the patients with IPF, but once imaging evidence is obtained, most of the patients are diagnosed within a year. Cardiac conditions and gastro-oesophageal disorders were more commonly reported in patients with a longer time to diagnosis.
Keywords: interstitial fibrosis
Key messages.
How long does it take from onset of symptoms and from first imaging evidence of pulmonary fibrosis to diagnosis of idiopathic pulmonary fibrosis (IPF)?
Among patients who received their first diagnosis of IPF at an enrolling centre in the IPF-PRO (Idiopathic Pulmonary Fibrosis Prospective Outcomes) Registry, approximately 50% were diagnosed more than 1 year after symptom onset, while approximately 80% were diagnosed within 1 year of imaging evidence of pulmonary fibrosis.
Our results show that despite improved awareness of IPF, there remains a long period from symptom onset to diagnosis in a large proportion of patients and certain comorbidities are associated with a longer time to diagnosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrosing interstitial lung disease (ILD) with poor prognosis.1 Median survival from diagnosis of IPF is 2 to 5 years,2–4 but the course of the disease is highly variable.5–7 In its early stages, IPF may be asymptomatic. The most common symptoms are exertional dyspnoea and dry cough.1 7 8
IPF shares similar presenting symptoms with asthma and chronic obstructive pulmonary disease (COPD), but is much rarer and is challenging to diagnose, leading to it being under-recognised.8–10 Diagnosis is typically made by a specialist after exclusion of known causes of ILD (eg, connective tissue disease, environmental exposures). Within the appropriate clinical context, a usual interstitial pneumonia pattern on high-resolution CT (HRCT) is sufficient to diagnose IPF without the need for lung biopsy; however, for patients with indeterminate patterns on HRCT, lung biopsy and/or bronchoalveolar lavage can be considered.1 Given its complexity, a multidisciplinary discussion is recommended, but not required, in the guidelines to inform diagnostic decision-making.1 11
Extended time to the diagnosis of IPF has been reported in multiple studies.6 9 10 12–17 The reasons for this are multifactorial, including patient-specific factors, healthcare-related factors and the limitations of diagnostic modalities. The length of time to diagnosis is difficult to characterise due to recall bias and the insidious onset of symptoms. A recent prospective study of all patients with incident IPF seen at two ILD centres in Denmark (n=204) found that the time from onset of symptoms to diagnosis of IPF was a median of 2.1 years, and was more than 5 years in 25% of patients.10 Airway obstruction and use of inhaled therapy were associated with longer time to diagnosis. Misdiagnosis was reported by 41% of patients, while 49% of patients had received antibiotics for pneumonia in the 2 years before IPF was diagnosed.10 In a European survey of patients with IPF, median time from onset of symptoms to diagnosis of IPF was 1.5 years; 58% of patients were diagnosed more than 1 year after onset of symptoms and 55% of patients reported seeing at least three physicians prior to the diagnosis.9 Similarly, in a US survey of 1448 patients and caregivers in 2003 to 2004, 55% reported at least 1 year from symptoms to diagnosis and 38% had seen at least three healthcare providers prior to their diagnosis.18
Misdiagnoses and pursuit of alternative diagnoses can also lead to diagnostic delay. In a large case-control study of a UK primary health database, patients ultimately diagnosed with IPF were more likely than matched controls to have been diagnosed with COPD and heart failure in the year prior to diagnosis.8 Even after referral to a specialist, diagnosis of IPF can be delayed. Claims data from Medicare beneficiaries with IPF (n=7306) showed that extended time to diagnosis may occur even after chest imaging, pulmonary function tests and evaluation by a pulmonologist; almost 33% of patients had their first CT scan >3 years prior to diagnosis of IPF, and 35% had seen a pulmonologist >3 years prior to diagnosis.16 The importance of early and accurate diagnosis was highlighted in the European IPF Patient Charter.19 Earlier diagnosis of IPF enables earlier initiation of medications that slow disease progression, provision of supportive care like referral to pulmonary rehabilitation, treatment of gastro-esophageal reflux disease (GERD) and other comorbidities, and referral for lung transplant evaluation.
Registries can provide important insights into diseases and, in particular, less common diseases like IPF.20 The Idiopathic Pulmonary Fibrosis Prospective Outcomes (IPF-PRO) Registry (NCT01915511) is an ongoing observational US registry of patients with IPF that aims to improve understanding of the clinical course of IPF, its impact on patients and practices in diagnosis and care.21 Clinical data are collected retrospectively for the period prior to enrolment, at enrolment and then prospectively at regular intervals during follow-up. The objectives of these analyses were to: (1) describe the time from symptom onset and from first imaging evidence of pulmonary fibrosis to diagnosis in patients newly diagnosed with IPF at an enrolling centre, (2) describe the evaluations performed in the 12 months prior to patients receiving a new diagnosis of IPF and the evaluations and diagnostic tests performed at the time of diagnosis at the enrolling centre, (3) investigate the relationships between patient characteristics and disease evaluations and time to diagnosis and (4) determine whether a longer time to diagnosis was associated with an increased risk of death or lung transplant.
Materials and methods
Patients
The IPF-PRO Registry comprises 1002 IPF patients enrolled from a network of academic and community practice ILD centres across the USA from 5 June 2014 through 11 March 2019. Our analysis excluded patients who had a diagnosis of IPF prior to evaluation at the enrolling centre (n=444) or whose diagnostic status at the time of enrolment was unknown (n=4). As the analysis required a date for either onset of symptoms or first imaging evidence of pulmonary fibrosis, patients were excluded if the diagnosis date was recorded as year only (n=2) or if they did not have a usable date for either symptom onset or imaging evidence of pulmonary fibrosis prior to the diagnosis date (n=54).
Descriptive statistics were used to characterise patients with a new diagnosis of IPF. Continuous variables are presented as median (Q1, Q3); categorical variables are presented as frequency (%). The number of missing values are reported for each variable.
Patients and public involvement
Patients were not involved in the design, recruitment to or conduct of the study included in this analysis.
Derivation of time from symptom onset or imaging evidence of pulmonary fibrosis to diagnosis
The date of symptom onset was the patient-reported date of symptom onset as recorded in the medical record. The date of first imaging evidence of pulmonary fibrosis was the date of the first available HRCT scan documenting fibrosis in the medical record. The date of IPF diagnosis was the date confirmed in the medical record by the healthcare provider at the enrolling centre.
For most analyses, patients were divided into two groups based on time from symptom onset to diagnosis (≤1 year vs >1 year) and into two groups based on time from first imaging evidence of pulmonary fibrosis to diagnosis (≤1 year vs >1 year). Where month, date or year was missing, the available data were reviewed and patients categorised, if possible, as described in the supplemental material. For the purpose of summarising the distributions, the time to diagnosis from symptom onset and from first imaging evidence of pulmonary fibrosis were calculated, in months, for each patient. This was carried out only in patients for whom data completeness allowed and, specifically, where month-day-year or month-year were present (for the latter, day set to 15th) or where only year was present and was ≥1 year prior to the diagnosis year (month/day set to June 30th). For the majority of the latter patients, the year was ≥3 years prior to diagnosis.
Variables of interest
All data were collected at the time of enrolment and included variables at the time of symptom onset or first imaging evidence of pulmonary fibrosis, variables within 12 months prior to diagnosis, variables >12 months prior to diagnosis and variables at the time of referral to the enrolling centre. Variables collected at enrolment that were assumed to apply to the time of symptom onset or imaging evidence of pulmonary fibrosis included: age (corrected from age at enrolment), sex, smoking history and earliest symptoms of IPF (dyspnoea, cough, weight loss or fatigue, as noted in the medical record). Variables collected ≤12 months and >12 months prior to referral included surgical lung biopsy, HRCT and cardiac catheterisation. Additional variables collected in the 12 months prior to referral included referral to a pulmonary specialist other than the enrolling centre, cardiac stress test, echocardiogram, bronchoscopy, serologies for connective tissue disease, evaluation of exposure to drugs with known pulmonary toxicity, evaluation for environmental or occupational exposures associated with ILD and referral for lung transplant evaluation. Variables collected after referral and prior to enrolment included HRCT, echocardiogram, cardiac catheterisation, bronchoscopy, serologies for connective tissue disease, family history of ILD, pulmonary function tests, multidisciplinary discussion and IPF diagnosis category (definite, probable or possible IPF based on the ATS/ERS/JRS/ALAT 2011 guidelines).22
Analysis of association between time to diagnosis and risk of death or lung transplant
Using Cox proportional hazards models, we assessed the association between a shorter (≤1 year) or longer (>1 year) time from symptom onset or from first imaging evidence of fibrosis to diagnosis and a combined endpoint of death or lung transplant. Each model was stratified by the use of antifibrotic medication at enrolment; this accounted for differences between the treatment groups without directly estimating the treatment effect. Clinical variables previously identified as being associated with death or lung transplant in this registry (use of supplemental oxygen at rest or with activity, forced vital capacity (FVC) % predicted and diffusing capacity for carbon monoxide (DLCO) % predicted)23 and age were included in each model.
Results
Patients
Our analysis cohort included 498 patients with a new diagnosis of IPF and a useable date for either symptom onset or first imaging evidence of pulmonary fibrosis. Among these patients, 347 had a useable date for symptom onset, 454 had a useable date for first imaging evidence of pulmonary fibrosis and 303 patients had useable dates for both (figure 1).
Figure 1.
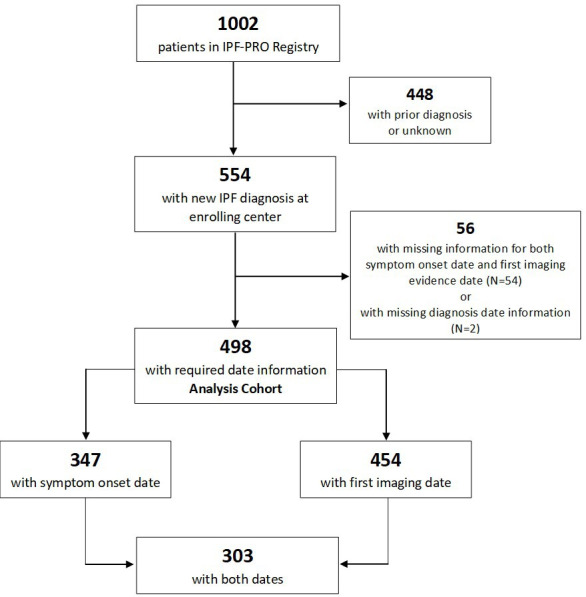
CONsolidated Standards of Reporting Trials diagram. IPF, idiopathic pulmonary fibrosis, IPF-PRO, Idiopathic Pulmonary Fibrosis Prospective Outcomes.
Table 1 summarises key data from the overall analysis cohort, the subset with a symptom onset date, the subset with an imaging date and those excluded from the analysis due to both dates being missing. In general, patient characteristics were similar across these groups. In the overall analysis cohort, median (Q1, Q3) age at enrolment was 70 (65 to 75) years and patients were predominantly male (76%), white (95%) and former smokers; common comorbidities included GERD (53%), coronary artery disease (31%), obstructive sleep apnoea (29%) and diabetes (21%). Some comorbidities were present at higher frequencies in the excluded patients. The proportion of patients with a family history of ILD in the analysis cohort was 16% and in the excluded patients was 22%. Across all the subsets, about one-third of the patients used oxygen at rest and one-fifth of the patients used oxygen with activity. Medication use at enrolment was similar across the cohorts, with 20% to 30% of patients receiving an antifibrotic medication. Notably, about 20% of the patients had been hospitalised for a respiratory indication in the prior year. Most of the patients had private insurance and/or Medicare. The majority of the patients lived in a non-rural area based on their zip code. Online supplementary table 1 presents all variables.
Table 1.
Patient characteristics at the time of enrolment
| N | Overall analysis cohort | Subset of patients with known date for symptom onset | Subset of patients with known date for first imaging evidence of pulmonary fibrosis | Patients excluded from analysis cohort |
| 498 | 347 | 454 | 56 | |
| Demographics | ||||
| Age, years | 70 (65 to 75) | 70 (64 to 75) | 71 (64 to 75) | 69 (64 to 76) |
| Male | 376 (75.5%) | 264 (76.1%) | 339 (74.7%) | 40 (71.4%) |
| White | 463 (95.1%) | 323 (94.7%) | 420 (94.6%) | 52 (96.3%) |
| Smoking history | ||||
| Non-smoker | 167 (33.7%) | 118 (34.2%) | 150 (33.2%) | 17 (30.4%) |
| Smoker | 329 (66.3%) | 227 (65.8%) | 302 (66.8%) | 39 (69.6%) |
| Current | 12 (2.4%) | 9 (2.6%) | 12 (2.7%) | 0 |
| Past | 317 (63.9%) | 218 (63.2%) | 290 (64.2%) | 39 (69.6%) |
| Comorbidities | ||||
| Gastro-oesophageal reflux disease | 265 (53.4%) | 194 (56.1%) | 239 (52.9%) | 31 (55.4%) |
| Coronary artery disease | 152 (30.7%) | 97 (28.0%) | 141 (31.3%) | 22 (39.3%) |
| Obstructive sleep apnoea | 143 (28.9%) | 102 (29.7%) | 128 (28.4%) | 21 (37.5%) |
| Diabetes | 105 (21.1%) | 77 (22.3%) | 93 (20.5%) | 14 (25.0%) |
| Hiatal hernia | 81 (16.4%) | 61 (17.7%) | 73 (16.2%) | 10 (18.2%) |
| Atrial fibrillation or flutter | 47 (9.5%) | 30 (8.7%) | 42 (9.3%) | 10 (17.9%) |
| Congestive heart failure | 36 (7.3%) | 19 (5.5%) | 34 (7.6%) | 5 (8.9%) |
| Family history | ||||
| Family history of ILD (grandparents, parents, siblings) | 79 (16.5%) | 54 (16.1%) | 69 (15.8%) | 11 (21.6%) |
| Supplemental oxygen | ||||
| With activity | 156 (31.8%) | 121 (35.5%) | 133 (29.7%) | 19 (33.9%) |
| At rest | 101 (20.5%) | 82 (24.0%) | 85 (19.0%) | 11 (19.6%) |
| Medications | ||||
| Proton pump inhibitor | 248 (55.4%) | 186 (58.7%) | 222 (54.7%) | 27 (52.9%) |
| Pirfenidone | 136 (27.3%) | 103 (29.7%) | 121 (26.7%) | 12 (21.4%) |
| Anticoagulant | 107 (23.9%) | 69 (21.8%) | 96 (23.7%) | 9 (17.6%) |
| Nintedanib | 107 (21.5%) | 74 (21.3%) | 100 (22.0%) | 19 (33.9%) |
| Oral steroid | 55 (12.3%) | 46 (14.5%) | 46 (11.3%) | 8 (15.7%) |
| Disease severity | ||||
| CPI | 52.7 (44.7 to 59.7) | 53.8 (45.5 to 60.5) | 52.5 (44.4 to 59.3) | 52.1 (46.0 to 57.5) |
| GAP score | 4.0 (3.0 to 5.0) | 4.0 (3.0 to 5.0) | 4.0 (3.0 to 5.0) | 4.0 (3.0 to 5.0) |
| GAP stage | ||||
| I | 131 (31.2%) | 88 (29.3%) | 125 (33.0%) | 13 (28.3%) |
| II | 221 (52.6%) | 156 (52.0%) | 197 (52.0%) | 26 (56.5%) |
| III | 68 (16.2%) | 56 (18.7%) | 57 (15.0%) | 7 (15.2%) |
Data are median (Q1, Q3) or n (%). Additional patient characteristics are shown in online supplementary table 1.
CPI, composite physiological index; GAP, gender, age, lung physiology; ILD, interstitial lung disease.
bmjresp-2020-000567supp001.pdf (131.1KB, pdf)
Distribution of time from symptom onset to diagnosis
Among the 347 patients with a symptom onset date, the median (Q1, Q3) time from symptom onset to diagnosis was 13.6 (5.9 to 39.5) months. The maximum was 274.3 months. About half (49%) of the patients had a time from symptom onset to diagnosis of >1 year. Of these patients, 16% had a time from symptom onset to diagnosis >1 to 2 years, 12.5% >2 to 3 years and 23.7% >3 years. Patients diagnosed within 1 year of symptom onset were evenly split between <6 months and 6 to 12 months. Patients in whom the time from symptom onset to diagnosis was ≤1 year were slightly older than those with a longer time to diagnosis (median 69 vs 67 years), but the two groups did not differ in sex, family history of ILD or smoking history (online supplementary table 1a).
Distribution of time from imaging evidence of pulmonary fibrosis to diagnosis
Among the 454 patients with an imaging date, the median (Q1, Q3) time from first imaging evidence of pulmonary fibrosis to diagnosis was 3.5 months (1.1 to 9.6). The maximum was 200.9 months. Most patients (78%) had imaging evidence of pulmonary fibrosis ≤1 year prior to diagnosis (≤6 months in 65% of the patients). The time from imaging evidence of pulmonary fibrosis to diagnosis was >1 to 2 years in 8.7% of patients, >2 to 3 years in 3.6% of patients and >3 years in 9.8% of patients.
Relationships among symptom onset, imaging evidence of pulmonary fibrosis and diagnosis
In the subset of 303 patients who had dates for both symptom onset and imaging, 46.5% had both a time from symptom onset to diagnosis and a time from imaging evidence of pulmonary fibrosis to diagnosis ≤1 year, while 28.7% had a time from symptom onset to diagnosis >1 year and a time from imaging evidence of pulmonary fibrosis to diagnosis ≤1 year. A further 19.5% of the patients had both a time from symptom onset to diagnosis and a time from imaging evidence of pulmonary fibrosis to diagnosis >1 year. It was uncommon for patients to have time from imaging evidence to diagnosis >1 year and time from symptom onset to diagnosis ≤1 year (5.3% of patients).
Among the 292 patients for whom months for symptom onset and first imaging evidence of pulmonary fibrosis were available, 72.9% of patients had symptoms documented prior to first imaging evidence of pulmonary fibrosis and 12.7% had imaging evidence of pulmonary fibrosis documented before symptom onset.
Initial IPF symptoms and patient characteristics at the time of symptom onset and at enrolment among patients with longer (>1 year) versus shorter (≤1 year) time to diagnosis
At the time of symptom onset, patients with a longer time from symptom onset to diagnosis were slightly younger (median (Q1, Q3) age 67 (59 to 72) vs 69 (63 to 73) years, p=0.02). There were no significant differences in sex or smoking status between the longer and shorter time to diagnosis groups (online supplementary table 2a). The most common symptoms reported at onset were dyspnoea (82%) and cough (63%), followed by fatigue (27.4%) and weight loss (3.7%). There were no differences in the proportions of patients with these symptoms, or in the number of symptoms, between the longer versus shorter time to diagnosis groups.
At the time of enrolment, the frequencies of several comorbidities were different between the longer and shorter time to diagnosis groups. A numerically lower proportion of patients with a longer versus shorter time to diagnosis had chronic kidney disease, while a numerically higher proportion had GERD, obstructive sleep apnoea, coronary artery disease, atrial fibrillation/atrial flutter and/or deep venous thrombosis/pulmonary embolism (online supplementary table 2b). Numerically higher proportions of patients in the longer than shorter time to diagnosis group were receiving oxygen, oral steroids, bronchodilators and anticoagulants at enrolment. Patients with a longer time to diagnosis had slightly higher FVC % predicted than patients with a shorter time to diagnosis, but the difference was not clinically significant (online supplementary table 2b).
Patient characteristics at the time of first imaging evidence of pulmonary fibrosis and at enrolment among patients with longer (>1 year) versus shorter (≤1 year) time between first imaging evidence of pulmonary fibrosis and diagnosis
In the majority (78%) of patients, the time between first imaging evidence of pulmonary fibrosis and diagnosis was ≤1 year. At the time of first imaging evidence of pulmonary fibrosis, there were no significant differences in age, sex or smoking status between the patients with a time to diagnosis of >1 and ≤1 year (online supplementary table 3a). At the time of enrolment into the registry with a new diagnosis of IPF, patients in whom the time from imaging evidence of pulmonary fibrosis to diagnosis was ≤1 year had numerically higher percentages of atrial fibrillation/flutter, hiatal hernia and diabetes than those with >1 year to diagnosis (online supplementary table 3b). Patients with a longer versus shorter time to diagnosis had slightly higher FVC and forced expiratory volume during 1 second (FEV1) % predicted at enrolment, but the differences were not clinically significant. Similar proportions of patients in the two groups were receiving oxygen at rest and during activity.
Evaluations within 12 months prior to referral to the enrolling centre
Table 2 details the evaluations performed prior to referral and at the enrolling centre. There was no difference in the proportion of patients who had surgical lung biopsy in the 12 months prior to enrolment across the groups by time from symptom onset to diagnosis or time from first imaging evidence to diagnosis (about 10% across groups). HRCT scan usage in the 12 months prior to enrolment was numerically higher in patients with a shorter versus longer time from imaging evidence of pulmonary fibrosis to diagnosis, but similar between patients by time from symptom onset to diagnosis. About one-third of the patients had seen a pulmonary specialist prior to referral to the enrolling centre. Notably, about 20% of the patients had a prior cardiac catheterisation. Approximately 20% of the patients had undergone a cardiac stress test and approximately 34% had an echocardiogram in the prior year. Cardiac testing was numerically higher in patients with cardiac disease (coronary artery disease, atrial fibrillation/flutter and/or congestive heart failure) at enrolment. However, there was no difference in cardiac testing in the longer versus shorter time to diagnosis cohorts within those who had a cardiac comorbidity. Bronchoscopy was relatively infrequent (<10% of patients). A minority of patients had undergone evaluation for drug, environmental or occupational exposures.
Table 2.
Evaluations performed before and after referral to enrolling site, by time to diagnosis
| N | Overall analysis cohort | Time from symptom onset to diagnosis | Time from first imaging evidence of pulmonary fibrosis to diagnosis | ||
| ≤1 year | >1 year | ≤1 year | >1 year | ||
| 498 | 174 | 173 | 356 | 98 | |
| Evaluations prior to referral* | |||||
| Surgical lung biopsy | 66 (13.9%) | 21 (12.4%) | 26 (15.8%) | 39 (11.4%) | 15 (16.5%) |
| Where time of evaluation known: | |||||
| Within 12 months prior to referral | 46 (9.7%) | 17 (10.1%) | 15 (9.1%) | 31 (9.1%) | 9 (9.9%) |
| HRCT | 339 (71.1%) | 115 (67.6%) | 122 (73.5%) | 238 (69.4%) | 75 (82.4%) |
| Where time of evaluation known: | |||||
| Within 12 months prior to referral | 261 (56.4%) | 94 (56.0%) | 86 (54.1%) | 210 (62.1%) | 36 (40.9%) |
| Seen by a pulmonary specialist | 224 (45.1%) | 79 (45.4%) | 97 (56.4%) | 142 (39.9%) | 57 (58.8%) |
| Where time of evaluation known: | |||||
| Within 12 months prior to referral | 164 (34.2%) | 73 (42.4%) | 60 (35.9%) | 119 (34.5%) | 27 (29.3%) |
| Cardiac catheterisation | 87 (18.3%) | 28 (16.5%) | 34 (20.9%) | 61 (17.8%) | 16 (17.8%) |
| Where time of evaluation known: | |||||
| Within 12 months prior to referral | 25 (5.3%) | 6 (3.6%) | 7 (4.3%) | 21 (6.2%) | 3 (3.4%) |
| Evaluations in 12 months prior to referral | |||||
| Echocardiogram | 165 (34.8%) | 56 (32.9%) | 57 (34.8%) | 117 (34.3%) | 33 (36.7%) |
| Cardiac stress test | 105 (22.0%) | 36 (21.2%) | 38 (22.9%) | 73 (21.3%) | 20 (22.0%) |
| Serologies for connective tissue disease | 114 (23.9%) | 37 (21.8%) | 47 (28.5%) | 87 (25.4%) | 19 (20.9%) |
| Environmental or occupational exposures | 115 (24.2%) | 35 (20.8%) | 42 (25.5%) | 83 (24.3%) | 23 (25.6%) |
| Exposure to drugs with known pulmonary toxicity | 89 (18.8%) | 25 (14.8%) | 33 (20.0%) | 62 (18.2%) | 19 (21.1%) |
| Bronchoscopy | 35 (7.4%) | 17 (10.0%) | 15 (9.0%) | 23 (6.7%) | 7 (7.7%) |
| Referral for lung transplant evaluation | 9 (1.9%) | 3 (1.8%) | 5 (3.0%) | 8 (2.3%) | 0 |
| Evaluations at the enrolling centre | |||||
| Diagnostic criteria22 | |||||
| Definite IPF | 321 (64.5%) | 111 (63.8%) | 109 (63.0%) | 233 (65.4%) | 66 (67.3%) |
| Probable IPF | 137 (27.5%) | 48 (27.6%) | 52 (30.1%) | 95 (26.7%) | 25 (25.5%) |
| Possible IPF | 40 (8.0%) | 15 (8.6%) | 12 (6.9%) | 28 (7.9%) | 7 (7.1%) |
| HRCT | 352 (71.3%) | 119 (68.8%) | 133 (77.3%) | 237 (67.1%) | 78 (80.4%) |
| Clinically significant emphysema on HRCT | 62 (12.5%) | 17 (9.8%) | 23 (13.3%) | 41 (11.6%) | 17 (17.5%) |
| Pulmonary function tests | 482 (96.8%) | 170 (97.7%) | 168 (97.1%) | 343 (96.3%) | 96 (98.0%) |
| Serologies for connective tissue disease | 291 (61.3%) | 100 (59.2%) | 112 (68.3%) | 196 (57.3%) | 64 (70.3%) |
| Echocardiogram | 129 (27.0%) | 37 (21.8%) | 57 (34.5%) | 77 (22.4%) | 37 (40.7%) |
| Cardiac catheterisation | 30 (6.3%) | 10 (5.9%) | 10 (6.1%) | 21 (6.1%) | 7 (7.7%) |
| Multidisciplinary discussion | 218 (43.8%) | 75 (43.1%) | 84 (48.6%) | 142 (39.9%) | 52 (53.1%) |
*Time period of evaluation (prior) was unknown for 3% of surgical lung biopsies, 4% of HCRT, 8% of pulmonary specialist referrals and 5% of cardiac catheterisations.
HRCT, high-resolution CT; IPF, idiopathic pulmonary fibrosis.
When the diagnosis of IPF was made at the enrolling centre, >60% of patients met criteria for definite IPF (as per22) according to the investigator’s assessment. In patients with a longer (>1 year) time from symptom onset or from first imaging evidence of pulmonary fibrosis to diagnosis, HRCT scan, echocardiogram and serologies for connective tissue diseases were more commonly part of the diagnostic evaluation. Multidisciplinary discussion was used to inform diagnosis in fewer than half of the patients prior to enrolment in the registry (table 2).
Association between time to diagnosis and risk of death or lung transplant
Based on univariable (unadjusted) analyses, the HR for the combined endpoint of death or lung transplant in patients with a time from symptom onset to diagnosis >1 versus ≤1 year was not significant (HR 0.83 (CI 0.53 to 1.30), p=0.41). Similarly, the HR for death or lung transplant in patients with a time from imaging evidence of pulmonary fibrosis to diagnosis >1 versus ≤1 year was not significant (HR 0.85 (CI 0.51 to 1.43); p=0.55) (figure 2).
Figure 2.

Kaplan-Meier plots of time to death or lung transplant by (A) time from symptom onset to diagnosis and (B) time from first imaging evidence of idiopathic pulmonary fibrosis to diagnosis.
In an adjusted model that included clinical variables associated with death or lung transplant in this registry (use of supplemental oxygen at rest or with activity, FVC % predicted and DLCO % predicted) and age, the HRs were <1, but these were not statistically significant. Notably, the HRs in the adjusted model were lower than in the unadjusted model (time from symptom to diagnosis HR 0.76 (CI 0.48 to 1.21); p=0.25]; time from imaging evidence of pulmonary fibrosis to diagnosis HR 0.72 (CI 0.42 to 1.24); p=0.24]), suggesting that once markers of disease severity are accounted for, patients with >1 year from symptom onset or first imaging evidence of pulmonary fibrosis to diagnosis may have a different trajectory to those with more recent onset of symptoms or imaging evidence of pulmonary fibrosis.
Discussion
In the last 20 years, three widely referenced guidelines for the diagnosis of IPF have been published (in 2000, 2011 and 2018),1 22 24 raising awareness of IPF. In this analysis of patients who received a new diagnosis of IPF at enrolment into the IPF-PRO Registry, approximately half of the patients had documented symptoms >1 year prior to diagnosis, consistent with prior studies.6 9 10 13 15 17 18 We regarded a time frame of 12 months after symptom onset as ‘reasonable’ for patients with IPF to be diagnosed and were able to analyse detailed data collected in the registry on evaluations conducted in the 12 months prior to enrolment. We empirically chose to look at groups based on time to diagnosis since first imaging evidence of fibrosis over the same time frame. The majority of patients in our study were diagnosed within a year of the first imaging evidence of pulmonary fibrosis. While several studies have examined the time from onset of symptoms to diagnosis of IPF, we believe that this is the first study to analyse the time from first imaging evidence of pulmonary fibrosis to diagnosis of IPF. The time from first imaging evidence to diagnosis was much shorter than the time from symptom onset to diagnosis, suggesting that the critical step is getting an HRCT scan performed. A minority of patients had imaging evidence first and symptoms later, indicating that radiographic changes can be seen before patients report symptoms. In these cases, it is possible that the imaging was done for another reason and the pulmonary fibrosis noted incidentally, thus prompting referral to a specialist. A recent analysis of data from 146 ILD patients at two centres found that diagnostic testing, including CT scan, that noted changes consistent with ILD had a shorter time to pulmonary referral compared with testing that did not note ILD features.17 This highlights the critical step of diagnostic testing, including CT imaging.
Comorbidities may potentially play a role in extending the time to diagnosis of IPF. Patients who had a longer (>1 year) time from symptom onset or imaging evidence of fibrosis to diagnosis of IPF had higher rates of cardiac conditions, particularly atrial fibrillation/atrial flutter, as well as gastro-oesophageal conditions, including GERD (cohort based on symptom onset) and hiatal hernia (cohort based on imaging evidence of pulmonary fibrosis) at enrolment. A limitation of the registry data is that we cannot confirm whether these patients were diagnosed with these comorbidities before or after symptoms or imaging evidence of pulmonary fibrosis developed, or if they were misdiagnoses prior to the diagnosis of IPF. However, given prior reports of misdiagnoses leading to delays in diagnosis of IPF, it is possible that these cardiac comorbidities, with potentially similar presentations of dyspnoea, are more frequent in patients with a longer time to diagnosis. The finding of a slightly higher frequency of gastro-oesophageal disorders at enrolment in patients with a longer time to diagnosis corresponds to a prior study in which GERD was associated with delayed diagnosis.25
Our analysis considered medical tests done within a year prior to diagnosis as well as testing done at the enrolling centres. Compared with data from a Medicare cohort,16 our analysis indicated a higher percentage of patients with imaging performed within a year of diagnosis (79% vs 42.5%). Only 40% of the Medicare cohort had autoimmune serologies billed in the 5 years prior to diagnosis, whereas in our registry, such serologies were performed in 24% of patients prior to referral and in 61% of patients at the enrolling centre. These differences may reflect our broader insurance base, more detailed medical chart review (rather than analysis of billing records), specialist involvement at the enrolling centre or more recent cohort. Rates of surgical lung biopsy were similar in both studies (about 10%), reflecting the practice of limiting this surgical procedure in the diagnosis of IPF. Despite diagnostic guidelines encouraging the use of multidisciplinary discussion only about half of the patients had such a discussion reported in the medical chart. This may reflect the limitations of a retrospective chart review (as the discussion may not be noted in the medical record) or the high rate of ‘definite IPF’ in this cohort.
For both time from symptom onset and time from first imaging evidence of pulmonary fibrosis, we found no significant difference between a longer or shorter time to diagnosis and risk of death or lung transplant. This finding is in contrast to a single-centre study suggesting that a longer time to diagnosis of IPF is associated with worse survival.26 It is possible that our finding reflects the limited follow-up beyond 24 months, the size of the cohort, use of antifibrotic medications or the similarities in demographics and lung function between patients with longer and shorter times to diagnosis at the time of enrolment.
Our analyses should be evaluated in the context of several limitations. An inherent bias of the registry is that only patients diagnosed at enrolling sites were included. The IPF-PRO Registry centres represent a range of ILD practices, including academic and community centres across the USA, strengthening the generalisability of the sample. However, patients enrolled at these specialised ILD centres may not reflect patients seen in the wider community or those who chose not to participate in the registry. While the registry collected the date of the HRCT scan showing fibrosis, it did not collect the actual images and it is possible that at that time the HRCT image did not warrant a diagnosis of IPF, even if the patient had been seen by a specialist. This is difficult to classify as a ‘delay’ since the disease may have been at an inconclusive stage. It is important to note that the date used was the date of first imaging evidence of pulmonary fibrosis rather than conclusive for IPF. The registry does not collect the reason for the HRCT scan, thus we cannot assess if it was performed for respiratory concerns, prior chest radiograph abnormalities or a non-respiratory concern. In addition, we do not know whether the symptoms noted in the medical record were attributed to conditions other than IPF. Data collection in the registry is dependent on medical records available to the site. While the registry placed emphasis on imaging, symptoms and evaluation in the 12 months prior to enrolment, it is possible that medical testing not known to the enrolling site was conducted. In addition, there was overlap between the symptom and imaging cohorts (as 303/498 subjects had dates for both symptoms and imaging recorded) and this may have introduced a bias into the results.
Conclusions
In patients with IPF, the time from symptom onset to diagnosis remains over 1 year in about half of the patients, but once imaging evidence of pulmonary fibrosis is obtained, most patients receive a diagnosis within 1 year. Cardiac conditions and gastro-oesophageal disorders were reported more frequently in patients with a longer (>1 year) versus shorter time to diagnosis of IPF. There was no significant difference between the longer (>1 year) versus shorter time to diagnosis groups in a combined endpoint of risk of death or lung transplant.
Acknowledgments
The authors acknowledge the IPF-PRO Registry principal investigators: Albert Baker, Lynchburg Pulmonary Associates, Lynchburg, Virginia; Scott Beegle, Albany Medical Center, Albany, New York; John Belperio, University of California Los Angeles, Los Angeles, California; Rany Condos, NYU Medical Center, New York, New York; Francis Cordova, Temple University, Philadelphia, Pennsylvania; Daniel A Culver, Cleveland Clinic, Cleveland, Ohio; Daniel Dilling, Loyola University Health System, Maywood, Illinois; John Fitzgerald, UT Southwestern Medical Center, Dallas, Texas; Kevin R Flaherty, University of Michigan, Ann Arbor, Michigan; Kevin Gibson, University of Pittsburgh, Pittsburgh, Pennsylvania; Mridu Gulati, Yale School of Medicine, New Haven, Connecticut; Kalpalatha Guntupalli, Baylor College of Medicine, Houston, Texas; Nishant Gupta, University of Cincinnati Medical Center, Cincinnati, Ohio; Amy Hajari Case, Piedmont Healthcare, Austell, Georgia; David Hotchkin, The Oregon Clinic, Portland, Oregon; Tristan Huie, National Jewish Health, Denver, Colorado; Robert Kaner, Weill Cornell Medical College, New York, New York; Hyun Kim, University of Minnesota, Minneapolis, Minnesota; Lisa Lancaster, Vanderbilt University Medical Center, Nashville, Tennessee; Joseph A Lasky, Tulane University, New Orleans, Louisiana; Doug Lee, Wilmington Health and PMG Research, Wilmington, North Carolina; Timothy Liesching, Lahey Clinic, Burlington, Massachusetts; Randolph Lipchik, Froedtert & The Medical College of Wisconsin Community Physicians, Milwaukee, Wisconsin; Jason Lobo, UNC Chapel Hill, Chapel Hill, North Carolina; Tracy Luckhardt (formerly Joao de Andrade), University of Alabama at Birmingham, Birmingham, Alabama; Yolanda Mageto, Baylor University Medical Center at Dallas, Dallas, Texas; Numaan Malik, University of Virginia, Charlottesville, Virginia; Prema Menon, Vermont Lung Center, Colchester, Vermont; Lake Morrison, Duke University Medical Center, Durham, North Carolina; Andrew Namen, Wake Forest University, Winston-Salem, North Carolina; Justin Oldham, University of California, Davis, Sacramento, California; Tessy Paul, University of Virginia, Charlottesville, Virginia; Anna Podolanczuk, Columbia University Medical Center/New York Presbyterian Hospital, New York, New York; Mary Porteous, University of Pennsylvania, Philadelphia, Pennsylvania; Rishi Raj, Stanford University, Stanford, California; Murali Ramaswamy, PulmonIx LLC, Greensboro, North Carolina; Tonya Russell, Washington University, St. Louis, Missouri; Paul Sachs, Pulmonary Associates of Stamford, Stamford, Connecticut; Zeenat Safdar, Houston Methodist Lung Center, Houston, Texas; Shirin Shafazand, University of Miami, Miami, Florida; Ather Siddiqi, Renovatio Clinical, The Woodlands, Texas; Barry Sigal, Salem Chest and Southeastern Clinical Research Center, Winston-Salem, North Carolina; Mary Strek, University of Chicago, Chicago, Illinois; Sally Suliman, University of Louisville, Louisville, Kentucky; Jeremy Tabak, South Miami Hospital, South Miami, Florida; Rajat Walia, St. Joseph’s Hospital, Phoenix, Arizona; Timothy Whelan, Medical University of South Carolina, Charleston, South Carolina. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors received no direct compensation related to the development of this manuscript. Editorial support was provided by Julie Fleming, BSc, and Wendy Morris, MSc, of FleishmanHillard Fishburn, London, UK, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. A podcast of the lead author discussing these data can be downloaded from: https://www.usscicomms.com/respiratory/snyder/IPF-PROtimetodiagnosis.
Footnotes
Collaborators: On behalf of the IPF-PRO Registry investigators.
Contributors: All authors were involved in data acquisition or analysis, in the interpretation of the data and in drafting the manuscript.
Funding: The IPF-PRO Registry is funded by Boehringer Ingelheim Pharmaceuticals, Inc, and coordinated by the Duke Clinical Research Institute. Boehringer Ingelheim Pharmaceuticals, Inc, was involved in the design of this study and the writing of this manuscript.
Competing interests: LDS, CM, MLN and ASH are employees of the Duke Clinical Research Institute, which receives funding support from Boehringer Ingelheim Pharmaceuticals, Inc, to coordinate the IPF-PRO Registry. CM receives research support from the National Institutes of Health (5T32HL007538-35) and the CHEST Foundation. CHH reports non-financial support from Boehringer Ingelheim in the form of statistical support and writing assistance for other projects. LL reports grants and other from Boehringer Ingelheim and Genentech, and grants from Bellerophon, Celgene and Novartis. KRF reports grants and personal fees from Boehringer Ingelheim and Roche/Genentech, and personal fees from Bellerophon, Blade Therapeutics, Celgene, FibroGen, Respivant, Sanofi Genzyme and Veracyte. IN reports personal fees from Boehringer Ingelheim, Genentech and ImmuneWorks. SB and CSC are employees of Boehringer Ingelheim Pharmaceuticals, Inc. JAdA reports personal fees from Boehringer Ingelheim. TPMW reports grants and personal fees from Boehringer Ingelheim and Genentech, and grants from Biogen, Celgene, Galapagos, Gilead Sciences, Global Blood Therapeutics, Kadmon, Nitto and the Pulmonary Fibrosis Foundation.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication: Not required.
Ethics approval: The study was approved by the Duke University Institutional Review Board (Pro00046131). The protocol was also approved by the relevant Institutional Review Boards and/or local Independent Ethics Committees prior to patient enrolment at each site listed in the Acknowledgements.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. The data sets analysed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
References
- 1.Raghu G, Remy-Jardin M, Myers JL, et al. . Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198:e44–68. 10.1164/rccm.201807-1255ST [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, Chen S-Y, Yeh W-S, et al. . Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014;2:566–72. 10.1016/S2213-2600(14)70101-8 [DOI] [PubMed] [Google Scholar]
- 3.Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther 2018;35:724–36. 10.1007/s12325-018-0693-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaunisto J, Salomaa E-R, Hodgson U, et al. . Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res 2019;5: :00170-2018–2018. 10.1183/23120541.00170-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40. 10.1164/rccm.201006-0894CI [DOI] [PubMed] [Google Scholar]
- 6.Doubková M, Švancara J, Svoboda M, et al. . Empire registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 2018;12:1526–35. 10.1111/crj.12700 [DOI] [PubMed] [Google Scholar]
- 7.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med Overseas Ed 2018;378:1811–23. 10.1056/NEJMra1705751 [DOI] [PubMed] [Google Scholar]
- 8.Hewson T, McKeever TM, Gibson JE, et al. . Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax 2017. doi: 10.1136/thoraxjnl-2017-210177. [Epub ahead of print: 11 Oct 2017]. [DOI] [PubMed] [Google Scholar]
- 9.Schoenheit G, Becattelli I, Cohen AH. Living with idiopathic pulmonary fibrosis: an in-depth qualitative survey of European patients. Chron Respir Dis 2011;8:225–31. 10.1177/1479972311416382 [DOI] [PubMed] [Google Scholar]
- 10.Hoyer N, Prior TS, Bendstrup E, et al. . Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir Res 2019;20:103. 10.1186/s12931-019-1076-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch DA, Sverzellati N, Travis WD, et al. . Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med 2018;6:138–53. 10.1016/S2213-2600(17)30433-2 [DOI] [PubMed] [Google Scholar]
- 12.Cottin V. Current approaches to the diagnosis and treatment of idiopathic pulmonary fibrosis in Europe: the air survey. Eur Respir Rev 2014;23:225–30. 10.1183/09059180.00001914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behr J, Kreuter M, Hoeper MM, et al. . Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J 2015;46:186–96. 10.1183/09031936.00217614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell A-M, Ripamonti E, Vancheri C. Qualitative European survey of patients with idiopathic pulmonary fibrosis: patients' perspectives of the disease and treatment. BMC Pulm Med 2016;16:10. 10.1186/s12890-016-0171-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guenther A, Krauss E, Tello S, et al. . The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 2018;19:141. 10.1186/s12931-018-0845-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mooney J, Chang E, Lalla D, et al. . Potential delays in diagnosis of idiopathic pulmonary fibrosis in Medicare beneficiaries. Ann Am Thorac Soc 2019;16:393–6. 10.1513/AnnalsATS.201806-376RL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pritchard D, Adegunsoye A, Lafond E, et al. . Diagnostic test interpretation and referral delay in patients with interstitial lung disease. Respir Res 2019;20:253. 10.1186/s12931-019-1228-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collard HR, Tino G, Noble PW, et al. . Patient experiences with pulmonary fibrosis. Respir Med 2007;101:1350–4. 10.1016/j.rmed.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 19.Bonella F, Wijsenbeek M, Molina-Molina M, et al. . European IPF patient charter: unmet needs and a call to action for healthcare policymakers. Eur Respir J 2016;47:597–606. 10.1183/13993003.01204-2015 [DOI] [PubMed] [Google Scholar]
- 20.Culver DA, Behr J, Belperio JA, et al. . Patient registries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2019;200:160–7. 10.1164/rccm.201902-0431CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Brien EC, Durheim MT, Gamerman V, et al. . Rationale for and design of the idiopathic pulmonary Fibrosis-PRospective outcomes (IPF-PRO) registry. BMJ Open Respir Res 2016;3:e000108. 10.1136/bmjresp-2015-000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raghu G, Collard HR, Egan JJ, et al. . An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snyder L, Neely ML, Hellkamp AS, et al. . Predictors of death or lung transplant after a diagnosis of idiopathic pulmonary fibrosis: insights from the IPF-PRO registry. Respir Res 2019;20:105. 10.1186/s12931-019-1043-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.American Thoracic Society American thoracic Society. idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American thoracic Society (ats), and the European respiratory Society (ERS). Am J Respir Crit Care Med 2000;161:646–64. 10.1164/ajrccm.161.2.ats3-00 [DOI] [PubMed] [Google Scholar]
- 25.Cosgrove GP, Bianchi P, Danese S, et al. . Barriers to timely diagnosis of interstitial lung disease in the real world: the intensity survey. BMC Pulm Med 2018;18:9. 10.1186/s12890-017-0560-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamas DJ, Kawut SM, Bagiella E, et al. . Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011;184:842–7. 10.1164/rccm.201104-0668OC [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjresp-2020-000567supp001.pdf (131.1KB, pdf)