


Abstract
Transposable elements (TEs) are ubiquitous DNA segments capable of moving from one site to another within host genomes. The extant distributions of TEs in eukaryotic genomes have been shaped by both bona fide TE integration preferences in eukaryotic genomes and by selection following integration. Here, we compare TE target site distribution in host genomes using multiple de novo transposon insertion datasets in both plants and animals and compare them in the context of genome-wide transcriptional landscapes. We showcase two distinct types of transcription-associated TE targeting strategies that suggest a process of convergent evolution among eukaryotic TE families. The integration of two precision-targeting elements are specifically associated with initiation of RNA Polymerase II transcription of highly expressed genes, suggesting the existence of novel mechanisms of precision TE targeting in addition to passive targeting of open chromatin. We also highlight two features that can facilitate TE survival and rapid proliferation: tissue-specific transposition and minimization of negative impacts on nearby gene function due to precision targeting.
INTRODUCTION
Transposable elements (TEs) are ubiquitous mobile DNA sequences that can make additional copies of themselves and integrate into new positions in host genomes. Because of these properties, TEs can make up the majority, sometimes the vast majority, of eukaryotic genomes (1). Indeed, the overall architecture of many genomes is determined in large measure by the quantity and distribution of TEs, which in turn is determined by both integration preferences and selection following integration.
According to their structural and biochemical features, TEs can be classified as retrotransposons (Class I TEs) or DNA transposons (Class II TEs). Both Class I and Class II TEs can be either autonomous or non-autonomous. Autonomous elements encode proteins that are capable of mobilizing both autonomous and non-autonomous elements. Non-autonomous elements can only transpose in the presence of their cognate autonomous elements. Retrotransposons duplicate via a ‘copy-and-paste’ mechanism involving reverse transcription as a step in the process of replication. DNA transposons transpose via a ‘cut-and-paste’ mechanism, in which an element is physically excised from one position and reintegrated at a second position.
TEs have a significant impact on genome evolution. Increases in TE copy numbers result in increased genome size, complexity and instability (2). TE transposition is associated with chromosomal structural variation (3) and can also affect expression of individual genes (4). TEs have served as a rich source of novel mutations on which selection can operate and have contributed to gene evolution and phenotypic diversification (5). Despite this, changes induced by TEs are primarily neutral or deleterious to their hosts (6), and TEs are kept under strict control by host immune systems. Overlapping transcriptional and post-transcriptional silencing mechanisms have evolved in plants and animals as layered defenses that have evolved to repress TE expression and amplification (7,8). Although this system is highly efficient and results in epigenetic silencing of most TEs in most genomes, it is clear that TEs can also undergo rapid increases in copy number, and currently or recently active TEs have been identified in a wide variety of organisms (9).
The consequences of TE activity depend largely on where TEs integrate. While TE integration sites in eukaryotic genomes are broadly distributed, different TEs adopt distinct integration strategies, resulting in dramatically different insertion profiles (10). There is ample evidence that both DNA transposons and retrotransposons insert non-randomly in host genomes. For instance, in maize, Activator elements preferentially target linked genic regions (11), and maize Mutator (Mu) elements target unlinked open chromatin regions near recombination hot spots, which tend to be in the 5′ ends of genes (12). P elements in Drosophila integration has been associated with replication origins, which are also in the 5′ end of genes (13). Integration of L1 retrotransposons in the human genome also appears to be influenced by DNA replication, and is not targeted to either actively transcribed regions or to open chromatin (14,15). Some other retrotransposons, such as Ty1 in yeast, target nucleosome-bound DNA near the H2A/H2B interface upstream of Pol III-transcribed genes due to physical interaction between the transposase and RNA polymerase III subunits (16–19). In contrast, gene space is a preferred target of many TEs (10), presumably because these are regions of relatively open chromatin, which may facilitate both integration and subsequent expression of autonomous elements (20,21). However, this is not always the case. Tal1, for instance, targets centromeres (22) and Ty5 targets silenced heterochromatin (23). Given that transposases in many cases are recruited to particular genomic niches via physical interaction with pre-seated proteins (16,17,23–27), a tethering model has been proposed for TE targeting (10,26,27). In addition, the timing of transposition is critical for copy number increase of cut-and-paste TE element and is therefore important for TE amplification. For instance, transposition of Activator primarily takes place after replication of the donor site but before replication of the target site, resulting in a net increase in copy number following cell replication (28).
Although there has been a great deal of analysis of de novo insertions of TEs in a variety of species, there have been few broad comparisons of TE insertion profiles in both animals and plants. Gene expression is often associated with open, accessible chromatin, which in turn is associated with insertion of a number of elements in both plants and animals (12,13). Further, RNAseq gene expression data are available from a broad range of tissues in multiple species, making direct comparisons relatively easy (29–31). With this in mind, we performed a comparison of multiple TEs in multiple species and examined TE distribution in the context of genome-wide transcriptional landscapes using multiple relatively unselected de novo transposon insertion datasets collected by many groups, including our own. We identify two distinct types of Pol II-associated TE targeting strategies, as well as those that are independent of Pol II transcription, and we provide evidence for convergent evolution among plant and animal TE families. We also provide data that suggests that TEs have evolved strategies to minimize their effects on host gene expression, even when those TEs specifically target gene space.
MATERIALS AND METHODS
Collecting de novo transposon coordinates and RNA-seq datasets
The TE families that were analyzed in this study have all been employed as efficient mutagens and each has been used for sequence-indexed mutant library construction. The UniformMu, Dissociation (Ds)-GFP, Transposon of Oryza sativa 17 (Tos17), Ds, Suppressor-mutator (Spm), P, piggyBac (Pb) and Minos (Mb) de novo transposon insertion collections in maize, rice and Drosophila are publicly available. Coordinates of TE insertions were retrieved from relevant websites and databases (32–38). Somatic Mu elements (SomaticMu elements) have been generated by performing Mu-seq with leaves collected from high-copy Mu-active maize seedlings and coordinates were called using the same pipeline as was used for the germinally inserted UniformMu collection (39). In the >320,000 SomaticMu insertions analyzed, a few hundred germinal insertions (ancient and germinally transmitted background insertions) were not removed as their impact on the overall distribution of SomaticMu insertions is neglectable. We also collected 1358 annotated Pack-MULEs in the maize genome (ftp://ftp.gramene.org/pub/gramene/release61/gff3/zea_mays/repeat_annotation/B73v4.TE.filtered.gff3.gz) and 2959 Pack-MULEs in the rice genome from the literature (40). The TE coordinates in each organism were made consistent with current genome assembly versions (AGPv4 B73 for maize, Oryza sativa.IRGSP-1.0.42 for rice and FB2014_03, R5.57 for fly). All coordinates for all insertions in each species are provided in Supplemental Tables S1–S5. Coordinates for insertions near tRNA and rRNA genes are provided in Supplemental Table S6.
Raw FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values of publicly available RNA-sequencing experiments for AGPv4 genes from maize were retrieved from the Maize Genetic Resource database (http://maize.plantbiology.msu.edu) (29). PCA analysis was performed based on the average FPKM values generated from RNAseq datasets of different maize tissues using the R package FactoMineR (41). The rice and Drosophila RNAseq data were retrieved from the Rice Expression Database and FlyAtlas 2, respectively (30,31). Expression levels for all genes used this analysis are available in Supplemental Table S7.
Meta-analysis of transposon distributions near transcription start sites (TSSs) and transcription termination sites (TTSs) of genes at various transcriptional landscapes
For a given gene set (all genes in a genome or a gene subset), each de novo and ancient TE insertion was classified as being either genic or intergenic and distances from transcriptional start sites (TSSs) and transcriptional termination sites (TTSs) were calculated for each insertion event. Genic insertions were plotted along the positive X-axis and intergenic insertions were plotted along the negative X-axis relative to the TSS and TTS in comparison to randomly selected genomic loci. A total 421 280 random insertions were in-silico generated on the maize chromosomes at a density of one insertion per 5 kb, which is comparable to the largest insertion dataset (SomaticMu). The densities of random insertions in both rice and Drosophila were set to be one insertion per 400 bp on average given that these genomes are relatively small and gene-rich. For each TE insertion or randomly selected locus within intergenic regions, its distance to the TSS and the TTS of both the nearest upstream gene and the nearest downstream gene were counted in the meta-profiling plots. In a number of gene rich regions, a small proportion of TE insertions or randomly distributed loci are <4 kb from both upstream and downstream genes, so they were counted twice. This would be expected to cause a mildly uneven distribution of a subset of the random selected loci.
The metaprofiles for intergenic and genic transposon insertions were plotted separately, using normalized insertion numbers in sliding 30-basepair (bp) windows centered on each position. In order to compare the enrichment of TEs near all annotated genes (or subsets of genes), normalization of the insertion numbers was performed by calculating the number of insertions per 30 bp window per 100 000 insertions per 10 000 genes at each position surrounding TSSs or TTSs. For both the TSSs and TTSs, genic transposon insertions were plotted along the positive X-axis while the intergenic insertions were plotted along negative X-axis.
For CHH islands near the 5′ or 3′ ends of genes, the relative position of these islands (which are 100 bp in length) and gene TSSs or TTSs was unified in such a way that the CHH islands were located on an interval [−50, 49] on X-axis of each plot, and adjacent genes were placed downstream of both 5′-end CHH and 3′-end CHH on the positive X-axis. Insertion numbers at each position are normalized to 100 000 insertions and 10 000 CHH islands. Given the repetitive nature of tRNA, and particularly rRNA genes, special care was taken to ensure that only independent insertions were counted by using polymorphisms between sequences flanking the insertions in these genes. The ∼30% of insertions into tRNA and rRNA genes that lacked sufficient polymorphism were not included in our analysis.
Independent RNAseq experiments were treated as replicates in each organism. For each experiment, genes were placed into 20 bins based on their relative level of expression, with bin 1 representing the lowest level of expression and bin 20, the highest. The percentage of TSS-associated insertions (<2 kb upstream of TSSs) in each bin were calculated for each RNAseq dataset, and the averaged percentages in all datasets were plotted along the X-axis. For each experiment, a bin represents a categorical level of gene expression. That is to say, a bin is not always the collection of identical genes in independent RNAseq experiments, but rather it contains a set of genes whose ranks based on their expression level fall in the same category in each particular experiment.
Sequencing-based transposon profiling and sequencing-based allele frequency analysis
Miseq-based Mu element profiling was performed as described previously using F1 hybrid progeny seedlings (42). The B73 parent was carried Mutator activity that had been introgressed into the B73 genetic background. The Mo17 parent lacked active Mu elements. Thus, all new insertions were into the B73 genome. Genomic DNA was extracted from 6-day-old seedlings of B73/Mo17 hybrid plants. Amplicon-based enrichment of Mu flanking DNA was then performed. The purified PCR products were subject to Miseq-based Wideseq pipeline at Purdue Genomics Core Facility (https://www.purdue.edu/hla/sites/genomics/wideseq-2/). Wideseq reads were mapped to the B73 reference genome as described previously (42). By identifying the Mu target site duplications (TSDs), a set of genes targeted by Mu insertions that segregated in hybrid progeny was obtained and those containing B73/Mo17 SNPs in their mRNA sequences were used for allele-specific expression analysis. Because new insertions were into the B73 genome, the effect of these insertions would be expected to be specific in all cases to the B73 allele. To quantify the allele frequency, we performed RT-PCR followed by Wideseq from the identical shoot tissues of the hybrid seedlings mentioned above. Total RNA was extracted using the RNA Extraction Kit (Zymo) and cDNAs were synthesized using Promega M-MLV Reverse Transcriptase. For a subset of 16 genes that carried SNPs, RNA fragments containing B73/Mo17 SNPs were amplified by RT-PCR. Primers used for this analysis are provided in Supplemental Table S8. The RT-PCR products were then sequenced by the ‘WideSeq’ pipeline. We also performed RNAseq on endosperms of four individual hybrid seeds. Preliminary processing of RNAseq reads and transcriptome mapping were carried out as described previously (43). As above, segregating Mu insertions were identified using Wideseq in these four hybrid individuals. For 16 genes with segregating Mu insertions, SNPs are available so that the allele frequency in the B73/Mo17 hybrid transcripts could be called. Fold changes of gene expression caused by Mu insertions for each gene were calculated by comparing the B73 allele frequency in individuals containing Mu insertions with those without Mu insertions which were further normalized using the Mo17 allele transcript frequency in plants that lacked an insertion in either B73 or Mo17.
RESULTS
Distribution of de novo transposons near TSSs and TTSs of host genes
To understand bona fide target preferences of transposons, we examined the target site distribution of nine de novo insertion datasets. These included the UniformMu (44), SomaticMu (this report) and Ds-GFP collections in maize (32,33), the Tos17, Ds and Spm collections in rice (34–36), as well as the P-element, Pb and Mb insertion collections in Drosophila (37,38). All TEs examined here are DNA transposons with the exception of Tos17, which is a low copy number LTR retrotransposon. UniformMu is primarily composed of germinally transmitted Mu insertions and SomaticMu elements are inferred to be derived primarily from somatic insertions due to the relative low number of reads obtained relative to the insertions that segregated in the families examined.
A comparative analysis of insertion profiles relative to randomly selected loci revealed dramatic similarities and differences between different elements in different species with respect to their association with TSSs or TTSs. The difference between the distribution pattern of TE insertions and that of randomly selected loci reveals a dramatic enrichment of TE insertions near TSSs or TTSs for some elements. Both the Mu element (UniformMu and SomaticMu) and P element insertions were vastly enriched near TSSs (peak shift < 50 bp), but were largely missing near TTSs, indicating a tight TSS-specific association with both transposases (Figure 1A, B, D, E, Supplementary Figure S1A, B, D, E). In addition, the distribution curves of Mu element insertions decrease rapidly upstream of the TSSs but are reduced more gradually downstream of the TSSs, particularly within 1 kb (Supplementary Figure S1A). Given the extreme bias in integration of Mu and P elements, we refer to these elements as precision-targeting elements. In contrast, enrichment of Ds and Pb insertions was observed near both the TSSs and TTSs, with much wider and lower peaks, suggesting less specificity than Mu and P elements (Figure 1A, B, D, E, Supplementary Figure S1A, B, D, E). There is no enrichment of Mb, Spm and Tos17 insertions near either TSSs or TTSs. Indeed, Tos17 insertions are actually somewhat enriched in the gene body relative to these sites (Figure 1C, F, Supplementary Figure S1C, F).
Figure 1.

Characterization of distribution of de novo transposon insertions that are near genes. The figure shows normalized numbers of insertions around TSSs and TTSs of annotated genes. The insertion numbers at each distance was smoothed by computing the mean in a 30-bp rolling window. For both TSS and TTS plots, normalized numbers of genic insertions were plotted on the positive X-axis coordinates and normalized numbers of intergenic insertions were plotted on the negative coordinates. Thus, positive values for both plots represent insertions into the gene body. Randomly selected loci were used as a background control to take into account the fact that although insertions were plotted up to 4 kilobase pairs (kb) away from the TSSs and TTSs, many gene bodies are shorter than 4 kb, and many genes are more or less than 4 kb downstream or downstream from a neighboring gene. Panels in the upper and bottom rows show metaprofiles of transposon insertions in maize (A, D), Drosophila (B, E) and rice (C, F) for regions surrounding the gene TSS and TTS, respectively.
To determine whether Mu and P element targeting is specific to Pol II-dependent transcription or is actually associated with any RNA polymerase, we examined the distribution of de novo Mu element and P element insertions in maize and Drosophila, respectively, near rRNA and tRNA genes, which are transcribed by RNA Pol I or III, respectively. To minimize Pol II TSS-associated TE enrichment, we filtered the rRNA and tRNA gene set in maize based on their distance to Pol II TSSs and obtained a list of 1610 genes over 5 kb away from the TSSs of any annotated genes transcribed by Pol II. Few transposon insertions are into the gene body of the 1610 rRNA and tRNA genes, as can be observed along the positive X-axis, likely due to the small size of these genes. In regions both downstream of TTSs and upstream of TSSs of these genes, UniformMu and SomaticMu resemble random selected genomic loci in their distribution except that the UniformMu shows a less smooth distribution curve than the SomaticMu (Figure 2A, B), which can be attributed to the fact that there are ∼3.5 times as many SomaticMu insertions as there are UniformMu insertions. Similar to Mu element insertions, P elements insert into or near the TSSs and TTSs of rRNA and tRNA genes at a frequency comparable to randomly selected loci (Supplementary Figure S2).
Figure 2.

Distribution of de novo Mu insertions near target sites of RNA Polymerases I, III, IV and V. (A and B) Metaprofiles of UniformMu and SomaticMu insertions surrounding TSS (A) and TTS (B) of tRNA and rRNA genes. The figure shows normalized numbers of insertions around TSSs and TTSs of annotated tRNA and rRNA genes. The insertion numbers at each distance were smoothed by computing the means in 30-bp rolling windows. Normalized numbers of genic insertions were plotted on the positive X-axis coordinates and those of intergenic insertions were plotted on the negative coordinates. (C and D) Distribution of UniformMu and SomaticMu insertions near CHH islands in maize located at 5′ (C) and 3′ ends (D) of maize genes. The 100-bp CHH islands are centered on position zero and range from −50 to 49 bp on the X-axis. Directions of CHH islands were unified so that nearby genes are located on the right of both 5′ and 3′ CHH islands.
Plants have two plant-specific RNA polymerases, Pol IV and Pol V, that are required for cytosine methylation in asymmetrical (CHH, where H is A, T or C) sequence contexts (45). A large number of CHH islands are located immediately upstream of the 5′ ends or downstream of the 3′ ends of genes in maize (46). We observed no enrichment of Mu element insertions in CHH islands; the vast majority of insertions are adjacent to the 5′ CHH islands, where Pol II transcripts are initiated at TSSs. These results indicate that Pol IV and Pol V transcription start or stop sites are not notable targets for Mu element insertions (Figure 2C, D).
Distribution of TE target sites near host genes with different expression levels
Given that the target sites of several TE families examined are TSS- or TTS-associated, we hypothesized that the transposases of some families are recruited to TSSs or TTSs in a manner that is dependent on the level of transcription. To test this hypothesis, we examined the correlation between transposon targeting frequency and relative expression levels of host genes. We extracted a subset of TSS-associated TE insertions that are located near (<2 kb) the TSSs and retrieved publicly available RNAseq datasets from 79 tissues in maize, 38 tissues in Drosophila and 59 tissues in rice (29–31). For each dataset, genes were binned into 20 equal sized groups based on ranked FPKM values in each RNAseq experiment, where bin 1 contains the lowest expressed 5% genes and bin 20 contains the highest expressed 5% genes. While randomly selected genomic loci (the control datasets) were evenly distributed near genes expressed at various levels, Mu and P elements preferentially target highly expressed genes, as indicated by the upward sloping curves (Figure 3A, B). Targeting frequency of Pb also positively correlates with gene expression, but to a lesser extent than that of P and Mu elements (Figure 3B). Interestingly, the distribution of Ds (in both maize and rice), Spm and Tos17 transposon insertion hotspots are all overrepresented in the medium expression bins (Figure 3A, C), suggesting that genes expressed at these levels are preferred targets for these elements. In contrast, the Mb elements, which show a mild enrichment at TTSs but not TSSs, actually target the lowest expression bins (Figure 3B).
Figure 3.

De novo transposon insertions near genes expressed at different levels. Percentage of transposon insertions distributed in each of the 20 expression bins were shown for maize (A), Drosophila (B) and rice (C) transposons. The figures show the correlation between TE targeting frequency with categorized gene expression levels. For each RNA-seq dataset, the whole annotated gene set was grouped to 20 expression bins from low to high independently, each containing an equal number of genes. Allocation of genes in each bin was independently performed for each RNA-seq experiment. Each RNA-seq dataset was regarded as one replicate and the averaged percentages in each categorized expression levels were calculated. Error bars represent standard deviations of the percentages in each bin based on independent RNAseq datasets.
Using the two available Mu insertion datasets, we tested the hypothesis that genes frequently targeted by precision-targeting TEs are associated with specialized biological functions or processes by performing gene ontology (GO) analysis of 4225 Mu element hotspot genes (>3 UniformMu and >10 SomaticMu insertions <2 kb from TSSs, Supplementary Table S1) using the online webserver agriGO (47). The seven GO terms enriched for Mu hotspot genes are related to a wide range of general, rather than specialized, biological processes and molecular functions (Supplementary Figure S3A). Genes categorized by these seven GO terms, on average, were expressed at markedly higher levels than the total gene set (Kolmogorov-Smirnov test, P values as indicated next to the box plots) (Supplementary Figure S3B), consistent with our observation that Mu elements preferentially target highly expressed genes. Moreover, in each of these seven gene sets, Mu hotspot genes are expressed at a significantly higher level than the non-hotspot genes in each set (Kolmogorov–Smirnov test) (Supplementary Figure S3C). These observations suggest that these GO terms are enriched not because they are associated with particular processes, but because they tend to express at higher levels than the average gene.
Distribution of TE insertions in meristematic and differentiated tissues
Given that the targeting frequency of several TE families associates with host gene expression levels in a collection of tissues, we hypothesized that tissues showing the strongest correlation between the two would be the tissues in which transposition occurs most frequently, assuming transposition has tissue-specificity. We explored the tissue specificity of TE transposition by using Mu and Ds elements in maize as examples.
To explore the major factors that contribute to transcriptome variation in different maize tissues, we first performed principle component analysis (PCA) on the above-mentioned 79 RNAseq datasets and found that the first principle component (17.8% of the variance) separated tissues of meristematic and differentiated identities well (Supplementary Figure S4A). The distribution curves of Mu and Ds element insertions in low-to-high expression bins showed distinct patterns in the six meristematic and six differentiated tissues (Figure 4A, Supplementary Figure S4B). The two curves differed most in the medium (bins 8–12, ranked between 35% and 60%) and highly (bins 16–20, ranked between 75% and 100%) expressed genes (Figure 4A). To address whether tissue-specific gene expression in meristematic and differentiated tissues associated with this shift of the distribution curve, we identified genes that were expressed at high levels in the meristem-enriched tissues and at medium levels in the differentiated tissues (meristematic-dominant genes), and those that were expressed at high levels in differentiated tissues and medium levels in meristem enriched tissues (differentiated-dominant genes). We obtained a set of 746 meristematic-dominant genes that were present in bins 16–20 in the majority (no less than five) of six meristematic tissues and in bins 8–12 in the majority (no less than five) of six differentiated tissues. We also obtained a set of 723 differentiated-dominant genes that were present in bins 16–20 in the majority (no less than five) of six differentiated tissues and in bins 8–12 in the majority (no less than five) of six meristematic tissues (Supplementary Figure S5). We found a much higher enrichment of Mu element insertions (both UniformMu and SomaticMu) that were near TSSs of the meristematic-dominant genes than were near TSSs of differentiated-dominant genes (Figure 4B). Such enrichment is specific to TSSs, but not TTSs, consistent with our previous observations (Figure 4C). We also found that Ds enrichment in the medium expression bins 8–12 is higher in meristematic tissues (Figure 4A), likely due to a preference for medium expressed genes by Ds elements. In line with this observation, we observed a lower level of Ds enrichment near TSSs, and to a lesser extent, TTSs in the meristematic-dominant gene set (Figure 4B, C), suggesting that Ds elements insert at a higher frequency in genes that express at a medium level in meristematic tissues (the differented-dominant gene set). These results suggest that both Mu and Ds elements insert most frequently in genes that express at targeted levels in meristematic or rapidly dividing cells. In the case of Mu, these are genes that express at a high level in those cells. In the case of Ds, it is genes that express at a medium level in those cells.
Figure 4.

Comparative analysis of the distribution of Mu and Ds elements in meristematic or differentiated tissues. (A) Percentage of random genomic loci, UniformMu, SomaticMu and Ds element insertions distributed in each of 20 expression bins in meristematic and differentiated tissues. An identical set of randomly selected loci that was used in Figure 1A (and 1D) was used as a background control. Error bars represent standard deviations of the percentages in each bin based on independent RNAseq datasets. (B and C) Metaprofiles of random genomic loci, UniformMu, SomaticMu and Ds elements in maize surrounding TSSs (B) and TTSs (C) of meristematic-dominant and differentiated-dominant gene sets, respectively. For both TSS and TTS plots, normalized numbers of genic insertions were plotted on the positive X-axis coordinates and normalized numbers of intergenic insertions were plotted on the negative coordinates. The insertion numbers at each distance were smoothed by computing the means in 30-bp rolling windows. An identical set of randomly selected loci used in Figure 1A (and 1D) was used as a background control.
Re-evaluation of the mutagenic capability of precision-targeting transposons
TE insertions in regions in or near genes may or may not disrupt host genes depending on where the TEs integrate. To evaluate the mutagenic capability of the precision-targeting transposons, we examined the enrichment of Mu and P elements in 5′ and 3′ proximal regions as well as sub-genic features of annotated maize and fly genes, respectively. Consistent with the meta-analysis above (Figure 1), insertions of both Mu and P element show strong enrichment in the 5′ ends of genes (particularly 200 bp upstream of the TSSs and 5′ UTRs) (Table 1). Mu element insertions are enriched in 5′ UTRs (64.4-fold for SomaticMu; 90.1-fold for UniformMu) relative to insertions into coding sequences (8.8-fold for SomaticMu; 7.0-fold for UniformMu) (all P values less than 1E−5, χ2 test). Enrichment of P elements at the 5′ ends of genes is coupled with a 4-fold lower frequency of P elements insertions into coding sequences relative to random chance. Indeed, out of a total of P element 18 213 insertions, only 729, or 4%, are inserted into coding sequences (CDSs).
Table 1.
A summary of the numbers and percentages of Mu and P elements insertions in or near genes. For random insertions, enrichment fold is defined as 1; values above 1 mean an enrichment of TE insertions, values below 1 mean a depletion of TE insertions
| Total (100%) | Genic | 500 bp upstream of TSS | 200 bp upstream of TSS | 5′ UTR | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Count | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | enrichment fold | P-value (χ² test) | |
| Random_Zm | 421280 | 32246 | 7.65% | 1 | NA | 3985 | 0.95% | 1 | NA | 1544 | 0.37% | 1 | NA | 1268 | 0.30% | 1 | NA |
| UniformMu_Zm | 92702 | 54040 | 58.29% | 7.62 | <1E−5 | 15976 | 17.23% | 18.22 | <1E−5 | 12103 | 13.06% | 35.62 | <1E−5 | 25142 | 27.12% | 90.11 | <1E−5 |
| SomaticMu_Zm | 320044 | 181776 | 56.80% | 7.42 | <1E−5 | 45582 | 14.24% | 15.06 | <1E−5 | 32739 | 10.23% | 27.91 | <1E−5 | 62028 | 19.38% | 64.39 | <1E−5 |
| Pack_MULE_Zm | 1369 | 183 | 13.37% | 1.75 | <1E−5 | 167 | 12.20% | 12.9 | <1E−5 | 94 | 6.87% | 18.73 | <1E−5 | 20 | 1.46% | 4.85 | <1E−5 |
| Random_Dm | 349280 | 222916 | 63.82% | 1 | NA | 13746 | 3.94% | 1 | NA | 6511 | 1.86% | 1 | NA | 11315 | 3.24% | 1 | NA |
| P_Dm | 18213 | 12586 | 69.10% | 1.08 | <1E−5 | 5485 | 30.12% | 7.65 | <1E−5 | 4409 | 24.21% | 12.99 | <1E−5 | 4578 | 25.14% | 7.76 | <1E−5 |
| Total (100%) | CDS | 3'UTR | 200 bp downstream of TTS | 500 bp downstream of TTS | |||||||||||||
| Count | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | Enrichment fold | P-value (χ² test) | Count | % of total | Enrichment fold | P-value (χ² test) | |
| Random_Zm | 421280 | 9188 | 2.18% | 1 | NA | 1889 | 0.45% | 1 | NA | 1508 | 0.36% | 1 | NA | 3781 | 0.90% | 1 | NA |
| UniformMu_Zm | 92702 | 14220 | 15.34% | 7.03 | < 1E-5 | 1471 | 1.59% | 3.54 | < 1E-5 | 1220 | 1.32% | 3.68 | <1E−5 | 2703 | 2.92% | 3.25 | <1E−5 |
| SomaticMu_Zm | 320044 | 61393 | 19.18% | 8.8 | < 1E-5 | 7046 | 2.20% | 4.91 | < 1E-5 | 5760 | 1.80% | 5.03 | <1E−5 | 12279 | 3.84% | 4.27 | <1E−5 |
| Pack_MULE_Zm | 1369 | 17 | 1.24% | 0.57 | 0.01947 | 25 | 1.83% | 4.07 | < 1E-5 | 16 | 1.17% | 3.27 | <1E−5 | 52 | 3.80% | 4.23 | <1E−5 |
| Random_Dm | 349280 | 56979 | 16.31% | 1 | NA | 18468 | 5.29% | 1 | NA | 5498 | 1.57% | 1 | NA | 10968 | 3.14% | 1 | NA |
| P_Dm | 18213 | 729 | 4.00% | 0.25 | < 1E-5 | 264 | 1.45% | 0.27 | < 1E-5 | 333 | 1.83% | 1.16 | 0.00849 | 967 | 5.31% | 1.69 | <1E−5 |
Assuming that TE insertion into 5′ end of genes are less deleterious than CDS insertions, we further examined the degree to which insertions of precision targeting TEs affect the expression of nearby genes. To do this, we evaluated the consequences of a list of de novo Mu element insertions, most of which are into promoter or 5′ UTRs, by experimentally testing the fold change of gene expression levels caused by Mu element insertions. A Mu-active maize line in the B73 background was crossed with a Mu-inactive line in the Mo17 background. The segregating Mu element insertions in the progeny were profiled using a Miseq-based amplicon-sequencing pipeline (42) and the B73-Mo17 SNPs were called for quantifying relative transcript levels of both parental alleles with and without Mu element insertions using deep sequencing (Supplementary Figure S6). A knockdown index was deduced by normalizing the observed ratio to that observed in genes that lacked Mu insertions in both genetic backgrounds for each insertion. We found that none of the four promoter insertions changed the expression of nearby genes. A quarter of 5′ UTR insertions (5 out of 20) caused knockout or strong knockdown effects and one of seven intronic insertions resulted in a knockout effect (Table 2). Collectively, of a total of 33 Mu insertions, all of which were within 200 bp of genes, only 11 significantly reduced gene expression, and only two eliminated completely expression. These results indicate that Mu element insertions near TSSs of host genes are often associated with quantitative and in many cases neglectable functional consequences on nearby gene expression.
Table 2.
Re-evaluation of mutagenic capability of Mu insertions by RNAseq and sequencing RT-PCR products via Miseq
| Affected_gene | Insertion_site | B73_insertion | Mo17_no_insertion | B73_no_insertion | Mo17_no_insertion | Knockdown_index | Consequence |
|---|---|---|---|---|---|---|---|
| Zm00001d020901 | five_prime_UTR | 0 | 247 | 461 | 519 | ∞ | knockout |
| Zm00001d027950 | intron | 2 | 517 | 221 | 285 | 200.5 | knockout |
| Zm00001d039733 | five_prime_UTR | 39 | 960 | 553 | 480 | 28.4 | knockdown |
| Zm00001d044446 | five_prime_UTR | 31 | 703 | 148 | 144 | 23.3 | knockdown |
| Zm00001d028712 | five_prime_UTR | 270 | 510 | 192 | 30 | 12.1 | knockdown |
| Zm00001d006460 | five_prime_UTR | 86 | 102 | 50 | 5 | 11.9 | knockdown |
| Zm00001d017424 | five_prime_UTR | 6 | 34 | 819 | 1065 | 4.4 | weak_knockdown |
| Zm00001d005587 | five_prime_UTR | 10 | 60 | 81 | 122 | 4 | weak_knockdown |
| Zm00001d023962 | five_prime_UTR | 78 | 488 | 181 | 463 | 2.4 | weak_knockdown |
| Zm00001d008642 | five_prime_UTR | 119 | 1235 | 361 | 1782 | 2.1 | weak_knockdown |
| Zm00001d006126 | intron | 83 | 68 | 144 | 50 | 2.4 | weak_knockdown |
| Zm00001d006768 | promoter -65 | 63 | 93 | 79 | 62 | 1.9 | unchanged |
| Zm00001d033167 | promoter-117 | 154 | 69 | 140 | 112 | 0.6 | unchanged |
| Zm00001d039683 | promoter-32 | 206 | 95 | 166 | 86 | 0.9 | unchanged |
| Zm00001d012812 | promoter-78 | 752 | 405 | 239 | 138 | 0.9 | unchanged |
| Zm00001d039156 | five_prime_UTR | 455 | 838 | 542 | 676 | 1.5 | unchanged |
| Zm00001d047761 | five_prime_UTR | 161 | 393 | 50 | 83 | 1.5 | unchanged |
| Zm00001d018461 | five_prime_UTR | 402 | 818 | 223 | 383 | 1.2 | unchanged |
| Zm00001d022153 | five_prime_UTR | 320 | 208 | 122 | 79 | 1 | unchanged |
| Zm00001d034667 | five_prime_UTR | 924 | 854 | 288 | 302 | 0.9 | unchanged |
| Zm00001d034191 | five_prime_UTR | 138 | 96 | 36 | 30 | 0.8 | unchanged |
| Zm00001d028784 | five_prime_UTR | 27 | 20 | 355 | 349 | 0.8 | unchanged |
| Zm00001d050081 | five_prime_UTR | 53 | 53 | 31 | 42 | 0.7 | unchanged |
| Zm00001d039253 | five_prime_UTR | 30 | 184 | 16 | 133 | 0.7 | unchanged |
| Zm00001d001788 | five_prime_UTR | 233 | 178 | 497 | 563 | 0.7 | unchanged |
| Zm00001d029856 | intron | 189 | 314 | 104 | 96 | 1.8 | unchanged |
| Zm00001d050163 | intron | 8 | 11 | 127 | 110 | 1.6 | unchanged |
| Zm00001d053452 | intron | 963 | 1324 | 236 | 310 | 1 | unchanged |
| Zm00001d049619 | intron | 70 | 260 | 251 | 1275 | 0.7 | unchanged |
| Zm00001d006610 | three_prime_UTR | 336 | 292 | 288 | 232 | 1.1 | unchanged |
| Zm00001d029059 | five_prime_UTR | 244 | 131 | 46 | 59 | 0.4 | unchanged/weak_activation |
| Zm00001d022122 | intron | 43 | 29 | 2 | 9 | 0.1 | unchanged/weak_activation |
Table 3.
Classification of integration strategies used by TE families analyzed in this study based on their distribution near genes and the association of TE targeting frequency with gene expression levels
| Strategy | Typical TE family | Host | Associated region | Expression levels of target genes |
|---|---|---|---|---|
| A | Mu | Maize | TSS | High |
| A | P | Drosophila | TSS | High |
| B | Ac/Ds | Maize, rice | TSS and TTS | Medium-to-high |
| B | Piggybac | Drosophila | TSS and TTS | Higher than Ac/Ds, lower than Mu and P |
| C | Minos | Drosophila | Not observed | Low-to-medium |
| C | Tnt1 | Medicago | Gene body | Low-to-medium |
| C | Tos17 | Rice | Gene body | Medium-to-high |
| C | Spm | Rice | Not observed | Medium-to-high |
Over a longer time-scale, purifying selection would be expected to purge insertion mutations that have only weak deleterious effects. To evaluate the selection pressures on older Mu element insertions, we examined a class of Mutator-like elements called Pack-MULEs, many of which are ancient insertions in the genomes that have diverged terminal inverted repeats (TIRs) (48). Profiling the distribution of 1358 Pack-MULEs in maize and 2959 Pack-MULEs in rice surrounding TSSs show that in both species, Pack-MULE occupancy peaks just upstream of TSSs and is reduced to nearly background levels >1 kb upstream of TSSs (Figure 5A, B). This is quite similar to our observation of de novo insertions in maize. However, there is a sharp decline of Pack-MULE insertions into gene bodies (Figure 5A, B), indicating selection pressure against older genic insertions into this region. Consistent with this observation, and consistent for selection against insertion into genes, underrepresentation of genic transposon insertions downstream of TSSs were found in other DNA transposon families in the maize genome, including hAT, Mariner, CACTA, Harbinger and Helitrons and LTR retrotransposons (Figure 5C-H). In contrast, we found that P element annotated in the genomes of wild D. melanogaster accessions exhibit an identical distribution of the de novo P element insertions (Figures 1B and 5I). Presumably, this is due to the fact that P elements have only been in the D. melanogaster genome for a relatively short period of time and are unlikely to be fixed or homozygous in wild populations (49).
Figure 5.

Metaprofiles of ancient transposons near the TSS of host genes. Normalized numbers of insertions around TSSs and TTSs of annotated genes. Genic insertions were plotted on the positive X-axis coordinates and intergenic insertions were plotted on the negative coordinates. The insertion numbers at each distance were smoothed by computing the means in 30-bp rolling windows. Identical sets of randomly selected loci used in Figure 1 were used as a background control.
DISCUSSION
It has been suggested that genomes resemble ecological systems, and that different TE families occupy distinct niches, presumably because there are multiple ways to be a successful genomic parasite (50). In each case, TE targeting represents a balance between successful amplification of the TE and minimization of the negative consequences of that amplification. In some cases, this results in TE insertions that rarely result in deleterious mutations. In others, it is likely that the costs of those mutations are outweighed by the benefits with respect to successful amplification.
Our comparative genomic analysis of multiple de novo transposon collections has revealed two types (A and B) of transcription-associated TE integration strategies (Figure 6A). The type-A precision-targeting strategy, employed by two of the most active plant and animal transposons, Mu and P elements (9), is characterized by a very tight association between TE integration and Pol II-dependent transcription initiation. These TEs are strongly and specifically enriched near annotated TSSs, particularly in genes that express at a high level. Further, in maize, insertions of Mu elements insert preferentially into genes expressing at a high level in the actively dividing cells that are most likely to give rise to germinal lineages.
Figure 6.
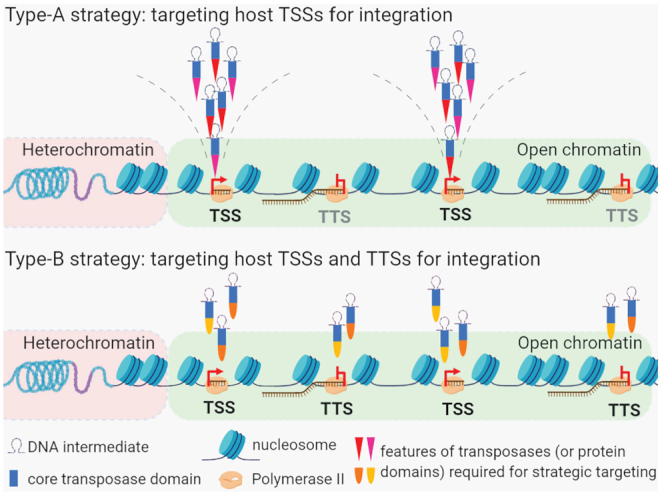
Classification of TE targeting strategies in context of host transcription. A proposed model depicting occupation of TEs at genomic locations with different expression characteristics. Type-A and type B transposons primarily target open chromatin regions where active transcription occurs. Integration of the type-A transposons associates with RNA Pol II transcription initiation. Factors that directly mediate transposase-recruitment to TSSs are yet to be identified. Targeting of the type-B transposons associates with both RNA Pol II transcription initiation and termination.
In contrast to type-A strategy, sites targeted by TE families employing the type-B strategy (Ds and Pb) are enriched at both TSSs and TTSs of medium expressed genes, and the enrichment levels of TE insertions near TSSs are lower for type-B than for type-A elements (Figure 6A, B). The type-A and type-B strategies have been named according to the single-peak and double-peak shapes of representative TE distribution curves near annotated genes reminiscent of the single- and double-hump nature of Arabian camels and Bactrian camels, respectively. Given that transposases encoded by the different TE families we have analyzed are distantly related phylogenetically (51), the transcription-associated type-A and type-B strategies suggest a process of convergent evolution among different TE families in plants and animals. Although it is formally possible that targeting of these different classes of TEs predated their divergence, we suggest that it is more likely that selection independently favored similar targeting strategies. To our knowledge, this is the first comparative study showing such relationships between TEs in both plants and animals.
Those TEs lacking an association with either Pol II transcription initiation or termination fall in the type-C group. Pol II-independent integration can be transcription-independent or transcription-associated. Reminiscent of the Sleeping Beauty transposons reported previously (52), Tos17 insertions do not show an association with either TSSs or TTSs, but are enriched in gene bodies of genes that express at a moderate level (Figure 3C, Supplementary Figure S1C and S1F). In contrast, Mb insertions show a mild association with TTSs, but not TSSs (Figure 1B and E) and actually exhibit a negative correlation between targeting frequencies and gene expression levels (Figure 3B). Spm also show targeting enrichment near genes that are expressed at medium-to-high levels, but this TE preferentially targets intergenic regions in host genomes. Overall, the type-C strategy may involve genome targeting mechanisms that are only indirectly related to host transcriptional activities.
Previous reports provided evidence for an ‘open-chromatin’ targeting model for some DNA transposons (12,53). Our results challenge the universality of this model because it does not fully account for the type-A integration strategy, although the insertion sites of precision-targeting type-A TEs certainly do co-localize to some extent with a set of chromatic modifications associated with open chromatin (12). The majority of open chromatin regions lie just upstream of TSSs and downstream of TTSs in both plants and animals (54,55). Occupancy of Pol II at or near both TSSs and TTSs has also been reported in both maize and Drosophila (56,57). The distribution of Mu and P element insertions is reminiscent of Pol II occupancy near TSSs, but not near TTSs, indicating that integration of the typical type-A TEs is strongly associated with transcription initiation, and not simply with the occupancy of Pol II. Further, we demonstrated that the type-A transposition strategy is specific to Pol II, but not other RNA polymerases like Pol I, Pol III or plant-specific Pol IV and Pol V (Figure 2). P integration near TSSs has been attributed to enrichment of replication origins in those regions, suggesting that an association with Pol II transcription initiation is indirect (13). The proposed model involves targeting of unfired replication origins by transposons in combination with homologous repair of excision sites following replication. While this model provides an attractive mechanism for increasing element copy number, an association with replication origins does not immediately account for the observed correlations of both P and Mu targeting with levels of gene expression (Figure 3A, B). Instead, the extreme specificity of P and Mu elements suggests that they are targeted via some form of tethering. This is consistent with the observation that type-A Tf1 retrotransposons in fission yeast are known to target the 5′ end of Pol II-transcribed genes via interaction between the integrase and the DNA binding protein Sap1, which causes replication fork arrest (26,27). Similarly, a comparative analysis of insertion site profiles has revealed that the Mouse Leukemia Virus (MLV) and the piggyBac transposon in human cell lines are targeted specifically to acetylated histones near TSSs via a tethering mechanism dictated by chromatin-bound bromodomain and extraterminal (BET) domain proteins that bind to acetylated H3 and H4 near TSSs (24). Future characterization of proteins or chromatin features associated with Mu and P element transposases will provide mechanistic insights into precision targeting of these elements as well.
Our analysis highlights two features that can facilitate TE survival and rapid proliferation: tissue-specific transposition and minimization of negative impacts on nearby gene function due to precision targeting. Certainly, tissue-specific transposition is true for P elements, which only express functional transposase in the germline (58). In plants, rapid and heritable amplification of TEs would also be facilitated by meristematic-tissue-specific transposition because actively dividing plant cells (particularly in floral tissues and meristems) are more likely to be transmitted to the next generation than those that are not. In this regard, we have observed clear targeting preferences of Mu and Ds elements for genes that express at targeted levels (highly expressed for Mu and expressed at a medium level for Ds) in meristem-enriched tissues (Figure 4B). Given that the very large number of somatic Mu insertions we have identified showed insertion preferences similar to germinally transmitted Mu insertions, Mu elements may be primarily avoiding insertions into genes that express at high levels primarily in terminally differentiated cells rather than targeting ‘germinal’ lineages.
The type-A (TSS-targeting) strategy employed by Mu and P elements endows these TEs with the capacity to exploit a permissive environment with respect to transcription of autonomous elements. This is particularly important for the survival of TEs in heterochromatin-rich genomes such as the maize genome. This strategy has the potential to cause deleterious effects on host gene expression and function. Indeed, we found that Mu insertions into CDS regions, which are most likely to be disruptive, were more frequent by 7–9-fold than random insertions. Despite of this, there was a much higher enrichment (64–90-fold) for Mu insertions near TSSs, and the majority of Mu insertions in promoters and 5′ UTRs have minimal to no effect on gene expression. This suggests that Mu elements are actually much less mutagenic than one might expect given their propensity to target genic regions because the reduction of host fitness is minimized due to a tight association between Mu element insertions and TSSs. Given this, and given that the vast majority of genic Mu element insertions are into the 5′UTR, we suggests that researchers who use Mu as a genetic resource treat these insertions with some caution, as they are unlikely to be knockouts. P elements also rarely insert into CDSs, likely because they are also precisely targeted to TSSs, although the effects caused by promoter and 5′ UTR targeting by P elements require future evaluation. Similarly, MITE TE insertions, although they tend to be into or near genes, also have a minimal effect on gene expression, although this may be in part due to their small size (59). Collectively, these data suggest that for some TEs selection has favored insertions that are into genes but that are minimally disruptive. This historical view of Mu and P elements as highly effective mutagens has likely been shaped by the fact that many of the insertion mutations caused by these elements were identified in screens for mutant phenotypes (60,61). More broadly, nearly all of the known active TEs in higher eukaryotes were first identified due to their mutagenic effects. It may well be that there are many additional active TEs in natural populations that have yet to be identified because they only rarely cause visible mutations.
Our analysis of older MULE insertions in both maize and rice suggests that 5′ UTR insertions of these elements are eventually purged from the genome, indicating that TEs that target 5′ UTRs are subject to purifying section in the long run. Interestingly, the purging appears to be much less efficient for proximal promoter MULE insertions, which are still present in high numbers in both species.
TE families occupy distinct genomic niches by employing distinct strategies for integration. This, in turn has influenced the degree to which TEs have affected host gene function and, ultimately, host genome evolution. TEs have also proved to be invaluable tools, both as mutagens and as transformation vectors (62). A deeper understanding of the ways in which TEs target particular regions of the genome for integration promise to make those tools both more effective and more precise.
DATA AVAILABILITY
The RNA-seq data of B73/Mo17 hybrids and Museq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) data bank, accession codes PRJNA556108 and GSE146647.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Phillip SanMiguel for helpful discussions on high-throughput-sequencing-based insertion profiling, Nathan Springer for sharing the CHH island data and Gurmukh Johal for providing the Mu-active line in the B73 background.
Authors contributions: D.L. and X.Z. conceived the study and designed the experiments. X.Z. performed most of the experiments. D.L. and X.Z. wrote the manuscript. X.Z. and M.Z. performed insertion profiling and analyzed the sequencing data. D.M. and M.Z. created the Mu insertion datasets and contributed to the Mu distribution analysis. The majority of the work was done when X.Z. was working at Purdue University. Data organization, manuscript preparation and submission were partly done after X.Z. moved to the AGIS. All authors read and approved the final manuscript.
Contributor Information
Xinyan Zhang, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China; Department of Botany and Plant Pathology, Purdue University, West Lafayette, IN 47907, USA.
Meixia Zhao, Department of Biology, Miami University, Oxford, OH 45056, USA.
Donald R McCarty, Horticultural Sciences Department, University of Florida, Gainesville, FL 32611, USA.
Damon Lisch, Department of Botany and Plant Pathology, Purdue University, West Lafayette, IN 47907, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [IOS-1237931]; Purdue Center for Plant Science SEED grant (to D.L.). Funding for open access charge: Lisch Startup Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wendel J.F., Jackson S.A., Meyers B.C., Wing R.A.. Evolution of plant genome architecture. Genome Biol. 2016; 17:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Naville M., Henriet S., Warren I., Sumic S., Reeve M., Volff J.N., Chourrout D.. Massive changes of genome size driven by expansions of non-autonomous transposable elements. Curr. Biol. 2019; 29:1161–1168. [DOI] [PubMed] [Google Scholar]
- 3. Zhang J., Yu C., Krishnaswamy L., Peterson T.. Transposable elements as catalysts for chromosome rearrangements. Methods Mol. Biol. 2011; 701:315–326. [DOI] [PubMed] [Google Scholar]
- 4. Lisch D. How important are transposons for plant evolution. Nat. Rev. Genet. 2013; 14:49–61. [DOI] [PubMed] [Google Scholar]
- 5. Chuong E.B., Elde N.C., Feschotte C.. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 2017; 18:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blumenstiel J.P., Chen X., He M., Bergman C.M.. An age-of-allele test of neutrality for transposable element insertions. Genetics. 2014; 196:523–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klattenhoff C., Xi H., Li C., Lee S., Xu J., Khurana J.S., Zhang F., Schultz N., Koppetsch B.S., Nowosielska A. et al.. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell. 2009; 138:1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Slotkin R.K., Martienssen R.. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007; 8:272–285. [DOI] [PubMed] [Google Scholar]
- 9. Huang C.R., Burns K.H., Boeke J.D.. Active transposition in genomes. Annu. Rev. Genet. 2012; 46:651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sultana T., Zamborlini A., Cristofari G., Lesage P.. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017; 18:292–308. [DOI] [PubMed] [Google Scholar]
- 11. Cowperthwaite M., Park W., Xu Z., Yan X., Maurais S.C., Dooner H.K.. Use of the transposon Ac as a gene-searching engine in the maize genome. Plant Cell. 2002; 14:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu S., Yeh C.T., Ji T., Ying K., Wu H., Tang H.M., Fu Y., Nettleton D., Schnable P.S.. Mu transposon insertion sites and meiotic recombination events co-localize with epigenetic marks for open chromatin across the maize genome. PLos Genet. 2009; 5:e1000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spradling A.C., Bellen H.J., Hoskins R.A.. Drosophila P elements preferentially transpose to replication origins. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:15948–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sultana T., van Essen D., Siol O., Bailly-Bechet M., Philippe C., Zine El Aabidine A., Pioger L., Nigumann P., Saccani S., Andrau J.C. et al.. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell. 2019; 74:555–570. [DOI] [PubMed] [Google Scholar]
- 15. Flasch D.A., Macia A., Sanchez L., Ljungman M., Heras S.R., Garcia-Perez J.L., Wilson T.E., Moran J.V.. Genome-wide de novo L1 retrotransposition connects endonuclease activity with replication. Cell. 2019; 177:837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheung S., Ma L., Chan P.H., Hu H.L., Mayor T., Chen H.T., Measday V.. Ty1 integrase interacts with RNA polymerase III-specific subcomplexes to promote insertion of Ty1 elements upstream of polymerase (Pol) III-transcribed genes. J. Biol. Chem. 2016; 291:6396–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bridier-Nahmias A., Tchalikian-Cosson A., Baller J.A., Menouni R., Fayol H., Flores A., Saib A., Werner M., Voytas D.F., Lesage P.. Retrotransposons. An RNA polymerase III subunit determines sites of retrotransposon integration. Science. 2015; 348:585–588. [DOI] [PubMed] [Google Scholar]
- 18. Baller J.A., Gao J., Stamenova R., Curcio M.J., Voytas D.F.. A nucleosomal surface defines an integration hotspot for the Saccharomyces cerevisiae Ty1 retrotransposon. Genome Res. 2012; 22:704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mularoni L., Zhou Y., Bowen T., Gangadharan S., Wheelan S.J., Boeke J.D.. Retrotransposon Ty1 integration targets specifically positioned asymmetric nucleosomal DNA segments in tRNA hotspots. Genome Res. 2012; 22:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sigman M.J., Slotkin R.K.. The first rule of plant transposable element silencing: location, location, location. Plant Cell. 2016; 28:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zamudio N., Bourc’his D.. Transposable elements in the mammalian germline: a comfortable niche or a deadly trap. Heredity (Edinb). 2010; 105:92–104. [DOI] [PubMed] [Google Scholar]
- 22. Tsukahara S., Kawabe A., Kobayashi A., Ito T., Aizu T., Shin-i T., Toyoda A., Fujiyama A., Tarutani Y., Kakutani T.. Centromere-targeted de novo integrations of an LTR retrotransposon of Arabidopsis lyrata. Genes Dev. 2012; 26:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie W., Gai X., Zhu Y., Zappulla D.C., Sternglanz R., Voytas D.F.. Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions between integrase and Sir4p. Mol. Cell. Biol. 2001; 21:6606–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gogol-Doring A., Ammar I., Gupta S., Bunse M., Miskey C., Chen W., Uckert W., Schulz T.F., Izsvak Z., Ivics Z.. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4(+) T cells. Mol. Ther. 2016; 24:592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yieh L., Kassavetis G., Geiduschek E.P., Sandmeyer S.B.. The Brf and TATA-binding protein subunits of the RNA polymerase III transcription factor IIIB mediate position-specific integration of the gypsy-like element, Ty3. J. Biol. Chem. 2000; 275:29800–29807. [DOI] [PubMed] [Google Scholar]
- 26. Jacobs J.Z., Rosado-Lugo J.D., Cranz-Mileva S., Ciccaglione K.M., Tournier V., Zaratiegui M.. Arrested replication forks guide retrotransposon integration. Science. 2015; 349:1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hickey A., Esnault C., Majumdar A., Chatterjee A.G., Iben J.R., McQueen P.G., Yang A.X., Mizuguchi T., Grewal S.I., Levin H.L.. Single-nucleotide-specific targeting of the Tf1 retrotransposon promoted by the DNA-binding protein Sap1 of Schizosaccharomyces pombe. Genetics. 2015; 201:905–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J., Greenblatt I.M., Dellaporta S.L.. Transposition of Ac from the P locus of maize into unreplicated chromosomal sites. Genetics. 1987; 117:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoopes G.M., Hamilton J.P., Wood J.C., Esteban E., Pasha A., Vaillancourt B., Provart N.J., Buell C.R.. An updated gene atlas for maize reveals organ-specific and stress-induced genes. Plant J. 2019; 97:1154–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leader D.P., Krause S.A., Pandit A., Davies S.A., Dow J.A.T.. FlyAtlas 2: a new version of the Drosophila melanogaster expression atlas with RNA-Seq, miRNA-Seq and sex-specific data. Nucleic Acids Res. 2018; 46:D809–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia L., Zou D., Sang J., Xu X., Yin H., Li M., Wu S., Hu S., Hao L., Zhang Z.. Rice Expression Database (RED): an integrated RNA-Seq-derived gene expression database for rice. J. Genet. Genomics. 2017; 44:235–241. [DOI] [PubMed] [Google Scholar]
- 32. McCarty D.R., Settles A.M., Suzuki M., Tan B.C., Latshaw S., Porch T., Robin K., Baier J., Avigne W., Lai J. et al.. Steady-state transposon mutagenesis in inbred maize. Plant J. 2005; 44:52–61. [DOI] [PubMed] [Google Scholar]
- 33. Li Y., Segal G., Wang Q., Dooner H.K.. Gene tagging with engineered Ds elements in maize. Methods Mol. Biol. 2013; 1057:83–99. [DOI] [PubMed] [Google Scholar]
- 34. Miyao A., Tanaka K., Murata K., Sawaki H., Takeda S., Abe K., Shinozuka Y., Onosato K., Hirochika H.. Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon-rich regions of the genome. Plant Cell. 2003; 15:1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Upadhyaya N.M., Zhu Q.H., Zhou X.R., Eamens A.L., Hoque M.S., Ramm K., Shivakkumar R., Smith K.F., Pan S.T., Li S. et al.. Dissociation (Ds) constructs, mapped Ds launch pads and a transiently-expressed transposase system suitable for localized insertional mutagenesis in rice. Theor. Appl. Genet. 2006; 112:1326–1341. [DOI] [PubMed] [Google Scholar]
- 36. Kumar C.S., Wing R.A., Sundaresan V.. Efficient insertional mutagenesis in rice using the maize En/Spm elements. Plant J. 2005; 44:879–892. [DOI] [PubMed] [Google Scholar]
- 37. Bellen H.J., Levis R.W., He Y.C., Carlson J.W., Evans-Holm M., Bae E., Kim J., Metaxakis A., Savakis C., Schulze K.L. et al.. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011; 188:731–U341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thibault S.T., Singer M.A., Miyazaki W.Y., Milash B., Dompe N.A., Singh C.M., Buchholz R., Demsky M., Fawcett R., Francis-Lang H.L. et al.. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat. Genet. 2004; 36:283–287. [DOI] [PubMed] [Google Scholar]
- 39. McCarty D.R., Latshaw S., Wu S., Suzuki M., Hunter C.T., Avigne W.T., Koch K.E.. Mu-seq: sequence-based mapping and identification of transposon induced mutations. PLoS One. 2013; 8:e77172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao D., Hamilton J.P., Vaillancourt B., Zhang W., Eizenga G.C., Cui Y., Jiang J., Buell C.R., Jiang N.. The unique epigenetic features of Pack-MULEs and their impact on chromosomal base composition and expression spectrum. Nucleic Acids Res. 2018; 46:2380–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lê S., Josse J., Husson F.. FactoMineR: an R package for multivariate analysis. 2008. 2008; 25:18. [Google Scholar]
- 42. Zhang X., Zhao M., Lisch D.. Cost-effective profiling of Mutator transposon insertions in maize by next-generation sequencing. Methods Mol. Biol. 2020; 2072:39–50. [DOI] [PubMed] [Google Scholar]
- 43. Zhang X., Zhu Y., Liu X., Hong X., Xu Y., Zhu P., Shen Y., Wu H., Ji Y., Wen X. et al.. Suppression of endogenous gene silencing by bidirectional cytoplasmic RNA decay in Arabidopsis. Science. 2015; 348:120–123. [DOI] [PubMed] [Google Scholar]
- 44. McCarty D.R., Suzuki M., Hunter C., Collins J., Avigne W.T., Koch K.E.. Genetic and molecular analyses of UniformMu transposon insertion lines. Methods Mol. Biol. 2013; 1057:157–166. [DOI] [PubMed] [Google Scholar]
- 45. Tucker S.L., Reece J., Ream T.S., Pikaard C.S.. Evolutionary history of plant multisubunit RNA polymerases IV and V: subunit origins via genome-wide and segmental gene duplications, retrotransposition, and lineage-specific subfunctionalization. Cold Spring Harb. Symp. Quant. Biol. 2010; 75:285–297. [DOI] [PubMed] [Google Scholar]
- 46. Li Q., Gent J.I., Zynda G., Song J., Makarevitch I., Hirsch C.D., Hirsch C.N., Dawe R.K., Madzima T.F., McGinnis K.M. et al.. RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z.. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017; 45:W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang N., Ferguson A.A., Slotkin R.K., Lisch D.. Pack-Mutator-like transposable elements (Pack-MULEs) induce directional modification of genes through biased insertion and DNA acquisition. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anxolabehere D., Kidwell M.G., Periquet G.. Molecular characteristics of diverse populations are consistent with the hypothesis of a recent invasion of Drosophila melanogaster by mobile P elements. Mol. Biol. Evol. 1988; 5:252–269. [DOI] [PubMed] [Google Scholar]
- 50. Brookfield J.F.Y. The ecology of the genome - Mobile DNA elements and their hosts. Nat. Rev. Genet. 2005; 6:128–136. [DOI] [PubMed] [Google Scholar]
- 51. Arkhipova I.R. Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mobile DNA-UK. 2017; 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Jong J., Akhtar W., Badhai J., Rust A.G., Rad R., Hilkens J., Berns A., van Lohuizen M., Wessels L.F., de Ridder J.. Chromatin landscapes of retroviral and transposon integration profiles. PLos Genet. 2014; 10:e1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gangadharan S., Mularoni L., Fain-Thornton J., Wheelan S.J., Craig N.L.. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21966–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maher K.A., Bajic M., Kajala K., Reynoso M., Pauluzzi G., West D.A., Zumstein K., Woodhouse M., Bubb K., Dorrity M.W. et al.. Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. Plant Cell. 2018; 30:15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodgers-Melnick E., Vera D.L., Bass H.W., Buckler E.S.. Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E3177–E3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lozano R., Booth G.T., Omar B.Y., Li B., Buckler E.S., Lis J.T., Jannink J.-L., del Carpio D.P.. RNA polymerase mapping in plants identifies enhancers enriched in causal variants. 2018; bioRxiv doi:24 July 2018, preprint: not peer reviewed 10.1101/376640. [DOI] [PMC free article] [PubMed]
- 57. Core L.J., Waterfall J.J., Gilchrist D.A., Fargo D.C., Kwak H., Adelman K., Lis J.T.. Defining the status of RNA polymerase at promoters. Cell Rep. 2012; 2:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laski F.A., Rio D.C., Rubin G.M.. Tissue-specificity of Drosophila P-element transposition is regulated at the level of messenger-RNA splicing. Cell. 1986; 44:7–19. [DOI] [PubMed] [Google Scholar]
- 59. Naito K., Zhang F., Tsukiyama T., Saito H., Hancock C.N., Richardson A.O., Okumoto Y., Tanisaka T., Wessler S.R.. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature. 2009; 461:1130–1134. [DOI] [PubMed] [Google Scholar]
- 60. Spradling A.C., Stern D., Beaton A., Rhem E.J., Laverty T., Mozden N., Misra S., Rubin G.M.. The berkeley drosophila genome project gene disruption project: single p-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999; 153:135–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Candela H., Hake S.. The art and design of genetic screens: maize. Nat. Rev. Genet. 2008; 9:192–203. [DOI] [PubMed] [Google Scholar]
- 62. Munoz-Lopez M., Garcia-Perez J.L.. DNA transposons: nature and applications in genomics. Curr. Genomics. 2010; 11:115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data of B73/Mo17 hybrids and Museq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) data bank, accession codes PRJNA556108 and GSE146647.