


Abstract
Heme is a multifaceted molecule. While serving as a prosthetic group for many important proteins, elevated levels are toxic to cells. The complexity of this stimulus has shaped bacterial network evolution. However, only a small number of targets controlled by heme-responsive regulators have been described to date. Here, we performed chromatin affinity purification and sequencing to provide genome-wide insights into in vivo promoter occupancy of HrrA, the response regulator of the heme-regulated two-component system HrrSA of Corynebacterium glutamicum. Time-resolved profiling revealed dynamic binding of HrrA to more than 200 different genomic targets encoding proteins associated with heme biosynthesis, the respiratory chain, oxidative stress response and cell envelope remodeling. By repression of the extracytoplasmic function sigma factor sigC, which activates the cydABCD operon, HrrA prioritizes the expression of genes encoding the cytochrome bc1-aa3 supercomplex. This is also reflected by a significantly decreased activity of the cytochrome aa3 oxidase in the ΔhrrA mutant. Furthermore, our data reveal that HrrA also integrates the response to heme-induced oxidative stress by activating katA encoding the catalase. These data provide detailed insights in the systemic strategy that bacteria have evolved to respond to the versatile signaling molecule heme.
INTRODUCTION
Heme (iron bound protoporphyrin IX) is a versatile molecule that is synthesized and used by virtually all aerobic eukaryotic and prokaryotic cells (1). It serves as the prosthetic group of hemoglobins, hydroxylases, catalases, peroxidases and cytochromes (2) and is therefore essential for many cellular processes, such as electron transfer, respiration and oxygen metabolism (3). Furthermore, salvaged heme represents the most important iron source for a variety of pathogenic bacteria (4,5), and also non-pathogenic bacteria can meet their iron demand by degradation of environmental heme. This becomes evident from the diverse set of heme uptake systems and heme oxygenases that catalyze the degradation of the protoporphyrin ring to biliverdin and the concomitant release of carbon monoxide and iron (6).
While heme represents an essential cofactor for a variety of proteins, this molecule also exhibits severe toxicity at high concentrations. Therefore, organisms have evolved sophisticated regulatory networks to tightly control heme uptake, detoxification (including export), synthesis and degradation (4). Several heme-regulated transcription factors have been described, including the heme activator protein (Hap) 1, which is an activator of genes required for aerobic growth of the yeast Saccharomyces cerevisiae (7); the transcription factor BACH1 (BTB and CNC homology 1), conserved in mammalian cells (8,9); and the rhizobial Irr protein, which is a heme-regulated member of the Fur family of transcriptional regulators (10–12).
In Gram-positive bacteria, two-component systems (TCSs) appear to play a prevalent role in heme-responsive signaling (13,14), as exemplified by the heme sensor system HssRS of Staphylococcusaureus and Bacillusanthracis, which controls the expression of the hrtBA operon, encoding a heme efflux system in both species (15,16). Remarkably, several members of the Corynebacteriaceae family, including the human pathogen Corynebacterium diphtheriae and the biotechnological platform strain Corynebacterium glutamicum, have two paralogous TCSs, namely, HrrSA and ChrSA, dedicated to heme-responsive control of gene expression (17–20). The kinases HrrS and ChrS were recently shown to perceive transient changes in heme availability by direct intramembrane interactions with heme (21,22). Heme binding triggers autophosphorylation of the sensor kinase, followed by transfer of the phosphoryl group to the cognate response regulators HrrA and ChrA. In C. glutamicum, significant cross-phosphorylation was observed between the closely related systems; however, this crosstalk is proofread by a highly specific phosphatase activity of the kinases toward the cognate response regulators under non-inducing conditions (23). While the ChrSA system appears to be mainly involved in rapid activation of the HrtBA detoxification system (19), previous data suggest that HrrSA coordinates a homeostatic response to heme (18). In recent studies, six direct target operons have been described for HrrA, including genes encoding enzymes involved in heme synthesis (hemE, hemA and hemH), heme utilization (hmuO, encoding a heme oxygenase) and the ctaE-qcrCAB operon, encoding components of the heme-containing cytochrome bc1-aa3 supercomplex of the respiratory chain (18). Expression of hrrA as well as hmuO is, furthermore, repressed by the global iron-dependent regulator DtxR in C. glutamicum under conditions of sufficient iron supply (24,25) thereby linking iron and heme regulatory networks in this organism.
The branched electron transport chain of C. glutamicum consists of the cytochrome bc1-aa3 supercomplex (encoded by ctaD, the ctaCF operon, and the ctaE-qcrCAB operon) and the cytochrome bd oxidase, encoded by the first two genes of the cydABDC operon (26). Although both the cytochrome aa3 oxidase and the bd oxidase are involved in the establishment of a proton-motive force, the aa3 oxidase is an active proton pump that is responsible for the increased proton translocation number (6 H+/2 e−) of the cytochrome bc1-aa3 supercomplex compared to that of the bd oxidase (2 H+/2 e−) (26). The presence of the cytochrome bc1-aa3 supercomplex is a characteristic feature of almost all actinobacteria, because members of this phylum lack a soluble cytochrome c and instead harbor a diheme cytochrome c1 that directly shuttles electrons from the bc1 complex to the aa3 oxidase (27–32). Furthermore, both terminal oxidases differ in heme content, as the bc1-aa3 supercomplex harbors six heme molecules, while the bd oxidase harbors only three. Surprisingly, not much is known about the regulation of terminal oxidases in C. glutamicum. In addition to the described activation of the ctaE-qcr operon by HrrA, the hydrogen peroxide-sensitive regulator OxyR was described as a repressor of the cydABCD operon (33,34). Furthermore, the ECF sigma factor SigC (σC) activates expression of the cydABCD operon (33,35). For σC, a speculated stimulus is a defective electron transfer in the aa3 oxidase (35) and such a defect was observed under copper-deprivation or when heme a insertion was disturbed, which resulted in activation of the σC regulon (36,37).
Interestingly, the regulons of prokaryotic heme regulators described thus far comprise only a low number of direct target genes, which are mostly involved in heme export (e.g. hrtBA) or degradation (hmuO). This current picture of prokaryotic heme signaling, however, does not match the complexity of the cellular processes influenced by heme. In this study, we performed a time-resolved and genome-wide binding profiling of HrrA in C. glutamicum using chromatin affinity purification and sequencing (ChAP-Seq) of HrrA in C. glutamicum showing the transient HrrA promoter occupancy of more than 200 genomic targets in response to heme. The obtained results emphasize that HrrSA is a global regulator of heme homeostasis, which also integrates the response to oxidative stress and cell envelope remodeling. Transcriptome analysis (RNA-Seq) at different time points after heme induction revealed HrrA to be an important regulator of the respiratory chain by coordinating the expression of components of both quinol oxidation branches as well as menaquinol reduction. Remarkably, HrrA was found to prioritize the expression of operons encoding the cytochrome bc1-aa3 supercomplex by repressing sigC expression.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Supplementary Table S1. The C. glutamicum strain ATCC 13032 was used as wild-type (29) and cultivations were performed in liquid BHI (brain heart infusion, Difco BHI, BD, Heidelberg, Germany), as complex medium or CGXII (38) containing 2% (w/v) glucose as minimal medium. The cells were cultivated at 30°C; if appropriate, 25 μg/ml kanamycin was added. Escherichiacoli (DH5α and BL21 (DE3)) was cultivated in Lysogeny Broth (Difco LB, BD, Heidelberg, Germany) medium at 37°C in a rotary shaker and for selection, 50 μg/ml kanamycin was added to the medium.
Recombinant DNA work and cloning techniques
Cloning and other molecular methods were performed according to standard protocols (39). As template, chromosomal DNA of C. glutamicum ATCC 13032 was used for polymerase chain reaction (PCR) amplification of DNA fragments and was prepared as described previously (40). All sequencing and synthesis of oligonucleotides was performed by Eurofins Genomics (Ebersberg, Germany). For ChAP sequencing, the native hrrA was replaced with a twin-strep-tagged version of this gene using a two-step homologous recombination system. This system is based on the suicide vector pK19 mob-sacB (41,42), containing 500 bps flanking each site of the targeted sequence inside the C. glutamicum genome. The pK19mob-sacB hrrA-C-twinstrep plasmid was constructed using Gibson assembly of PCR products (primers indicated in Supplementary Table S2) and the cut pK19 vector (43).
ChAP-Seq — sample preparation
The preparation of DNA for ChAP sequencing was adapted from (44). The C. glutamicum strain ATCC 13032::hrrA-C-twinstrep was used for the time series experiment. A preculture was inoculated in liquid BHI medium from a fresh BHI agar plate and incubated for 8–10 h at 30°C in a rotary shaker. Subsequently, cells were transferred into a second preculture in CGXII medium containing 2 % (w/v) glucose and 0 μM FeSO4 to starve the cells from iron. Protocatechuic acid (PCA), which was added to the medium, allowed the uptake of trace amounts of iron. From an overnight culture, six main cultures were inoculated to an OD600 of 3.0 in 1 l CGXII medium containing 4 μM hemin as sole iron source. For the time point t = 0, the cells were added to 1 l fresh CGXII containing no additional iron source. After 0, 0.5, 2, 4, 9 and 24 h, cells corresponding to an OD600 of 3.0 in 1 l were harvested by centrifugation at 4°C, 5000 × g and washed once in 20 ml CGXII. Subsequently, the cell pellet was resuspended in 20 ml CGXII containing 1 % (v/v) formaldehyde to crosslink the regulator protein to the DNA. After incubation for 20 min at RT, the cross linking was stopped by addition of glycine (125 mM), followed by additional incubation of 5 min at RT. After that, the cells were washed three times in buffer A (100 mM Tris–HCl, 1 mM ethylenediaminetetraacetic acid (EDTA), pH = 8.0) and the pellets stored overnight at −80°C. For cell disruption, the pellet was resuspended in buffer A containing ‘cOmplete’ protease inhibitor cocktail (Roche, Germany) and disrupted using a French press cell (SLM Ainco, Spectronic Instruments, Rochester, NY, USA) five times at 207 MPa. The DNA was fragmented to ∼500 bp by sonication (Branson Sonifier 250, Branson Ultrasonics Corporation, CT, USA) and the supernatant was collected after ultra-centrifugation (150 000 × g, 4°C, 1 h). The DNA bound by the twin-Strep-tagged HrrA protein was purified using Strep-Tactin XT Superflow column material (IBA Lifesciences, Göttingen, Germany) according to the supplier's manual (applying the gravity flow protocol, 1.5 ml column volume). Washing of the column was performed with buffer W (100 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, pH 8,0) and the tagged protein was eluted with buffer E (100 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, pH 8.0, added 50 mM D-Biotin). After purification, 1 % (w/v) sodium dodecyl sulphate was added to the elution fractions and the samples were incubated overnight at 65°C. For the digestion of protein, 400 μg/ml Proteinase K (AppliChem GmbH, Darmstadt, Germany) was added and incubated for 3 h at 55°C. Subsequently, the DNA was purified as following: Roti-Phenol/Chloroform/Isoamyl alcohol (Carl Roth GmbH, Karlsruhe, Germany) was added to the samples in a 1:1 ratio and the organic phase was separated using Phase Lock Gel (PLG) tubes (VWR International GmbH, Darmstadt, Germany) according to the supplier's manual. Afterwards, the DNA was precipitated by adding ice-cold ethanol (to a conc. of 70 % (v/v) and centrifugation at 16 000 × g, 4°C for 10 min. The DNA was washed with ice-cold 70 % (v/v) ethanol, then dried for 3 h at 50°C and eluted in dH2O.
ChAP-Seq — sequencing and peak discovery
The obtained DNA fragments of each sample (up to 2 μg) were used for library preparation and indexing using the TruSeq DNA PCR-free sample preparation kit according to the manufacturer's instruction, yet skipping fragmentation of the DNA and omitting the DNA size selection steps (Illumina, Chesterford, UK). The resulting libraries were quantified using the KAPA library quant kit (Peqlab, Bonn, Germany) and normalized for pooling. Sequencing of pooled libraries was performed on a MiSeq (Illumina) using paired-end sequencing with a read-length of 2 × 150 bases. Data analysis and base calling were accomplished with the Illumina instrument software and stored as fastq output files. The sequencing data obtained for each sample were imported into CLC Genomics Workbench (Version 9, Qiagen Aarhus A/S, Aarhus, Denmark) for trimming and base quality filtering. The output was mapped to accession NC_003450.3 as C. glutamicum reference genome (29). Genomic coverage was convoluted with second order Gaussian kernel. The kernel was truncated at 4 sigmas (that is all kernel values positioned further then 4 sigmas from the center were set to zero) and expanded to the ‘expected peak width’. The expected peak width was estimated via the following procedure: (i) all the peaks higher than 3-fold mean coverage were detected. (ii) Points at which their coverage dropped below  of the maximal peak height were found and the distance between them was considered as a peak width. (iii) The ‘estimated peak width’ was set equal to the median peak width. The convolution profile was scanned in order to find points where first derivative changes its sign from positive to negative (Supplementary Figure S1). Each such point was considered as a potential peak and was assigned with a convolution score (that is convolution with second order Gaussian kernel centred at the peak position). Furthermore, we explored the distribution of the convolution scores. It appeared to resemble normal distribution, but with a heavy right tail. We assumed that this distribution is indeed bimodal of normal distribution (relatively low scores) representing ‘noise’ and a distribution of ‘signal’ (relatively high scores). We fit the Gaussian curve to the whole distribution (via optimize.fit function from SciPy package; http://www.scipy.org/) and set a score thresholds equal mean + 4 sigmas of the fitted distribution. Further filtering with this threshold provided estimated FDR (false discovery rate) of 0.004–0.013 depending on a sample. Filtered peaks were normalized to allow inter-sample comparisons. Sum of coverages of the detected peaks was negated from the total genomic coverage. The resulting difference was used as normalization coefficient; that is peak intensities were divided by this coefficient.
of the maximal peak height were found and the distance between them was considered as a peak width. (iii) The ‘estimated peak width’ was set equal to the median peak width. The convolution profile was scanned in order to find points where first derivative changes its sign from positive to negative (Supplementary Figure S1). Each such point was considered as a potential peak and was assigned with a convolution score (that is convolution with second order Gaussian kernel centred at the peak position). Furthermore, we explored the distribution of the convolution scores. It appeared to resemble normal distribution, but with a heavy right tail. We assumed that this distribution is indeed bimodal of normal distribution (relatively low scores) representing ‘noise’ and a distribution of ‘signal’ (relatively high scores). We fit the Gaussian curve to the whole distribution (via optimize.fit function from SciPy package; http://www.scipy.org/) and set a score thresholds equal mean + 4 sigmas of the fitted distribution. Further filtering with this threshold provided estimated FDR (false discovery rate) of 0.004–0.013 depending on a sample. Filtered peaks were normalized to allow inter-sample comparisons. Sum of coverages of the detected peaks was negated from the total genomic coverage. The resulting difference was used as normalization coefficient; that is peak intensities were divided by this coefficient.
ChAP-Seq — estimation of confidence intervals
To compare peak intensities between the samples, we assessed the significance levels of the detected intensity values by an extensive in silico simulation of ChAP-seq experiments along with further peak-detection analyses.
The simulation consisted of the following steps: The reads were artificially generated from C. glutamicum genome (NC_003450.3) with the error rate (number of nucleotide mismatches) equal to the average error rate of the real HrrA ChAP-seq reads (estimated from the mapping statistics). The reads were taken from randomly selected spots in the genome (simulation of the non-peak coverage) and from the regions of the detected HrrA binding peaks with the probabilities proportional to the original peak intensities. Thus, we tried to emulate the original binding architecture. We also added a small amount (10% of the total simulated reads) of the sequences heavily affected by mismatches (25% mismatches for the original C. glutamicum sequences), as we wanted to account for around 10% of the unmapped reads in the original HrrA ChAP-seq experiments. Finally, the simulated reads were subjected to the computational peak-detection pipeline with the same parameters as in the original analyses. As a result, we obtained the peak intensity values for the detected peaks.
In total, we simulated 200 ChAP-seq samples, each containing 1.14M reads (the average amount of reads in the original samples). For each of the detected peaks we estimated the variation of the reported peak intensity among all the simulations. That is, for each peak intensity we estimated 0.95 confidence interval, as a difference between 97.5 and 2.5 percentiles. We discovered a strong positive correlation (0.94 Pearson) between the width of the confidence intervals and mean intensity (Supplementary Figure S2A). Therefore, we then normalized the width of the confidence intervals to the mean intensity values. The normalized confidence interval width (NCIW) appears to be a convenient metric as it is similar for all peaks, weakly dependent on their intensity. However, for the strongest peaks (peak intensity > 10) the NCIW is limited by 0.2, while for the weaker ones by 0.28 (Supplementary Figure S2B). Then we convert NCIW upper limits to the minimum confident fold changes by the following rule: min_fold = (1+NCIW/2)/(1-NCIW/2). Thus, we conclude that for the stronger peaks minimum confident fold change (P-value < 0.05) is ∼1.23, while for the weaker peaks it is ∼1.33.
RNA-Seq — sample preparation
For RNA sequencing, C. glutamicum wild-type and the ΔhrrA mutant strain were cultivated under the same conditions as described for ChAP Sequencing. Both strains did not contain any plasmids and, hence, were cultivated without addition of antibiotics in biological duplicates. After 0 (no heme), 0.5 and 4 h, cells corresponding to an OD600 of 3 in 0.1 l were harvested in falcon tubes filled with ice by centrifugation at 4°C and 5000 × g for 10 min and the pellets were stored at −80°C. For the preparation of the RNA, the pellets were resuspended in 800 μl RTL buffer (QIAGEN GmbH, Hilden, Germany) and the cells disrupted by 3 × 30 s silica bead beating, 6000 rt/min (Precellys 24, VWR International GmbH, Darmstadt, Germany). After ultra-centrifugation (150 000 × g, 4°C, 1 h), the RNA was purified using the RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany) according to the supplier's manual. Subsequently, the ribosomal RNA was removed by running twice the workflow of the Ribo-Zero rRNA Removal Kit [Bacteria] (Illumina, CA, USA) in succession. Between steps, the depletion of rRNA as well as the mRNA quality was analysed using the TapeStation 4200 (Agilent Technologies Inc, Santa Clara, USA). After removal of rRNA, the fragmentation of RNA, cDNA strand synthesis and indexing was carried out using the TruSeq Stranded mRNA Library Prep Kit (Illumina, CA, USA) according to the supplier's manual. Afterward, the cDNA was purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, IN, USA). The resulting libraries were quantified using the KAPA library quant kit (Peqlab, Bonn, Germany) and normalized for pooling. Pooled libraries were sequenced on a MiSeq (Illumina, CA, USA) generating paired-end reads with a length of 2 × 75 bases. Data analysis and base calling were performed with the Illumina instrument software and stored as fastq output files.
RNA-Seq — analysis
Sequencing reads quality was explored with the FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) tool. Since reads appeared to be of a good quality and did not harbor significant fraction of adapters or over-represented sequences, no pre-processing was undertaken. Identical reads were collapsed with a custom script in order to prevent gene levels’ misquantification caused by PCR overamplification. Reads were mapped to the C. glutamicum genome (NC_003450.3) with Bowtie2 (45). Bowtie2 was run with the following parameters: bowtie2 -1 [path to the reads, 1st mate] -2 [path to the reads, 2nd mate] -S [path to the mappings] –phred33 –sensitive-local –local –score-min C,90 –rdg 9,5 –rfg 9,5 -a –no-unal -I 40 -X 400 –no-mixed –ignore-quals.
The reads mapped to multiple locations were split proportionally between parental genes. That is, if 3 reads are mapped to gene A and gene B, expression of gene A is 10 and expression of gene B is 5, then 2 reads will go to gene A and 1 read to gene B. For each C. glutamicum gene (46) we assigned an expression value equal to the average read coverage over the gene region. These expression values were then normalized to TPM (transcripts per million) values (47).
Furthermore, we analyzed which genes are significantly differentially expressed between conditions. We set combinatorial thresholds on normalized GEC (gene expression change) [|expr1-expr2|/(expr1+expr2)] and MGE (mean gene expression) [log2((expr1+expr2)/2)] where ‘expr1’ is gene expression for the first condition and ‘expr2’ for the second. Thresholds were set in a way to achieve maximal sensitivity while keeping FDR < 0.05. FDR was estimated as GECintra/(GECintra + GECinter); where GECintra is a number of genes passed the thresholds based on intrasample GEC (that is, gene expression change between the replicates for the same condition), GECinter is a number of genes passed the thresholds based on intersample GEC (that is, gene expression change between two different conditions). Threshold function for GEC was defined as: 1 | if MGE < C; 2**(-A*MGE) + B | if MGE ≥ C; where A, B, C are parameters to be adjusted. Parameters A, B, C were adjusted with genetic algorithm optimization approach to achieve maximal sensitivity in discovery of differentially expressed genes while keeping FDR below 0.05.
Measurement of cell-associated hemin
C. glutamicum was cultivated in 4 μM hemin as described above (see ChAP-Seq). To measure the cell-associated heme pool, CGXII minimal medium supplemented with 2% (w/v) glucose and 4 μM heme was inoculated to an OD600 of 3.5. Samples were taken 0.5, 2, 4, 9 and 24 h after addition of heme. Cells were harvested, resuspended in 100 mM Tris–HCl (pH 8) and adjusted to an OD600 of 100. Cells cultivated in 4 μM FeSO4 supplemented medium were taken as a control and harvested at the same time points. Absolute spectra of cells reduced with a spatula tip of sodium dithionite were measured at room temperature using the Jasco V560 with a silicon photodiode detector in combination with 5 mm light path cuvettes. Absorption values at 406 nm were normalized by subtracting the measured absorption values of Fe-cultivated cells.
Electrophoretic mobility shift assays (EMSA)
The promotor regions of HrrA target genes (100 bp) were chosen based on the ChAP-Seq analyses and covered the maximal HrrA peak area (for primers see Supplementary Table S2). For quantitative measurements, Cy3-labeled oligonucleotides were used for the generation of the DNA fragments. Before addition of the DNA, HrrA was phosphorylated by incubation for 60 min with MBP-HrrSΔ1-248 in a ratio of 2:1 and 5 mM adenosine triphosphate. Binding assays were performed in a total volume of 20 μl using 15 nM DNA and increasing HrrA concentrations (75 and 375 nM) for the qualitative analyses and 10 nM DNA with increasing HrrA concentrations from 5–1000 nM for quantitative analyses, respectively. The binding buffer contained 20 mM Tris–HCl (pH 7.5), 50 mM KCl, 10 mM MgCl2, 5% (v/v) glycerol, 0.5 mM EDTA and 0.005% (w/v) Triton X-100. After incubation for 20 min at room temperature, the reaction mixtures were loaded onto a 10 % native polyacrylamide gel and subsequently separated and documented using a Typhoon TrioTM scanner (GE healthcare). The band intensities of unbound DNA were quantified using Image Studio Lite (Licor, Bad Homburg, Germany). The band intensities were normalized to the lane containing no DNA and plotted against the HrrA concentration in log10 scale. Apparent Kd values were calculated based on at least three gels each using a sigmoidal fit and the software GraphPad Prism 8. For the sigmoidal fit, Y = 0 and Y = 1 were set as top and bottom constraints. The turning point of the curve was defined as the apparent Kd.
TMPD oxidase assay
C. glutamicum wild-type strain and the ΔhrrA mutant were cultivated to an OD600 of 4 in CGXII minimal with or without the addition of 4 μM hemin. Subsequently, cells were disrupted in a Precellys® homogenisator (VWR International GmbH, Darmstadt, Germany) using zirconia/silica-beads (Ø 0.1 mm, Roth, Karlsruhe) in 100 mM Tris–HCl (pH 7.5) buffer. Ultracentrifugation at 200 000 × g for 1 h was used for membrane isolation. The pellet was resuspended in 100 mM Tris×HCl buffer and the protein concentration was determined using a BCA assay. The N,N,N’,N’-Tetramethyl-p-phenylenediamine (TMPD) oxidase activity in the membrane faction was measured spectrophotometrically at 562 nm in a TECAN Reader (Thermo Fisher Scientific, Massachusetts, US) by injecting 200 μM TMPD (37). An extinction coefficient of 10.5 mM−1 cm−1 was used (48). One unit of activity was defined as 1 μmol of TMPD oxidized per minute. As a control for autooxidation, a sample containing only 100 mM Tris–HCl buffer was recorded after TMPD addition and substracted from the actual rates. Significance was evaluated by an unpaired t-test with a 95% confidence interval.
RESULTS
Genome-wide profiling of HrrA promoter occupancy
In previous studies, a number of direct HrrA target operons were described in C. glutamicum and C. diphtheriae, suggesting an important role of the HrrSA TCS in the control of heme homeostasis (17–20). It has to be noted, that the membrane embedded HrrS sensor kinase is also activated by endogenously synthesized heme (21) and that the addition of external heme leads to a boost of the HrrSA response. In this study, we investigated the genome-wide binding profile of HrrA using chromatin affinity purification of twin-Strep-tagged HrrA combined with DNA sequencing (ChAP-Seq). Importanty, qPCR experiments confirmed wild-type level expression of the twin-Strep-tagged version of HrrA.
To obtain insights into the stimulus-dependent DNA association and dissociation, C. glutamicum cells were grown in iron-depleted glucose minimal medium, and samples were obtained before (T0) and 0.5, 2, 4, 9 and 24 h after the addition of 4 μM hemin. HrrA was purified, and the bound DNA fragments were sequenced (Figure 1A). We obtained substantial enrichment of known HrrA targets in response to heme (e.g. after 0.5 h: 5-fold hmuO, 54-fold hemE, 105-fold ctaE; Figure 1B–D, respectively) and identified more than 200 previously unknown HrrA-bound regions in the C. glutamicum genome (Supplementary Table S3).
Figure 1.

Genome-wide profiling of HrrA binding in response to addition of external heme. (A) ChAP-Seq analysis of the Corynebacterium glutamicum strain ATCC 13032::hrrA-C-twinstrep grown in iron-depleted glucose minimal medium before and after addition of 4 μM hemin. The experimental approach is briefly depicted: cells were harvested at the indicated time points, twin-Strep tagged HrrA was purified and co-purified DNA was sequenced to identify HrrA genomic targets. This approach resulted in the identification of more than 200 genomic regions bound by HrrA upon addition of hemin after 30 min. Exemplarily shown is the HrrA binding to regions upstream of operons involved in (B) the respiratory chain (ctaE), (C) heme degradation (hmuO) and (D) heme biosynthesis (hemE).
As expected, the highest number of peaks was identified at the first time point after the heme pulse (0.5 h), with 199 peaks meeting our applied threshold (distance of <800 bp to the closest downstream or <200 bp to the closest upstream transcription start site (TSS)). In comparison, only 15 peaks showed a more than 2-fold enrichment before hemin addition (T0, Supplementary Table S3 and Figure S3). It has to be noted, that these 15 peaks detected at T0 appear to be specific HrrA targets, since none of them was detected in an input control sample. Overall, these data illustrate the fast and transient DNA binding by HrrA in response to heme. In general, the majority of the discovered HrrA binding sites were close to TSSs (Supplementary Figure S4). The binding of HrrA to 11 selected targets was confirmed by electrophoretic mobility shift assays (Supplementary Figure S5), and a palindromic binding motif was deduced (Figure 2B and Supplementary Figure S6).
Figure 2.

ChAP-Seq analysis revealed HrrA as a global regulator of heme homeostasis in Corynebacterium glutamicum. (A) HrrA binding in response to the addition of hemin. The bar plot reflects the average peak intensities among detected peaks in ChAP-Seq experiments (<800 bp to the next TSS). The binding was correlated with the amount of cell-associated hemin (dashed line), measured at corresponding time points by spectroscopy as described in ‘Materials and Methods’ section. (B) A binding motif was deduced from the sequences of the top 25 peaks (T0.5) using MEME v.5 analysis (http://meme-suite.org). (C) Pie chart presenting HrrA targets, which can be attributed to known functional categories (total of 272 genes, among which 128 encode proteins of unknown function, e.g. target genes within the CGP3 prophage region were excluded). For a complete overview of HrrA targets, see Supplementary Table S3. (D) Proportional behavior of the HrrA regulon. For each peak that passed the threshold (distance of <800 bp to the closest downstream or <200 to the closest upstream TSS) at time point A, the highest peak in the same region (±50 nt from the center of the peak) was selected for time point B and vice versa. Thus, ‘paired’ peaks for these two time points were obtained, and the Pearson correlation of the intensities of all paired peaks was calculated for all six time points. (E) Peak intensities of selected HrrA targets over time, as identified by ChAP-Seq.
The HrrA binding patterns depicted in Figure 1B–D are representative of many bound regions. Thirty minutes after the heme pulse, the average peak intensities increased ∼2.5-fold in comparison to those at T0 (Figure 2A). After 2 h of cultivation in hemin, the average peak intensity is declining and is, after 9 h, already below the starting level at T0 reaching a minimum in stationary phase (24 h). This is likely the result of the pre-cultivation and main cultivation under iron starvation conditions leading to a lowered intracellular heme pool. The dissociation of HrrA from its target promoters is, consequently, caused by rapid depletion of heme and a switch of HrrS from kinase to phosphatase state (23). Heme depletion was confirmed by spectroscopy of C. glutamicum cells (Figure 2A, dashed line) and was also obvious upon visual inspection (Supplementary Figure S7). Of all peaks, that passed our threshold, 128 were upstream of genes encoding hypothetical proteins, while 150 could be assigned to genes with known or predicted function (Figure 2C). Furthermore, we assessed the significance levels of HrrA binding changes between samples from different conditions or/and time-points. It turned out, that for the stronger peaks (peak intensity > 10) the minimum significant fold change (P-value < 0.05) is ∼1.23, while for the weaker peaks (peak intensity < 10) it is ∼1.33 (see ‘Materials and Methods’ section).
To analyze the synchronicity in the HrrA regulon, peak intensities were correlated over time. A relatively high correlation between peak intensities for the time points 0.5, 2 and 4 h (Figure 2D) showed that the system reacted proportionally for a majority of the binding sites and the strength of HrrA binding changed in response to heme availability. Relaxation of the system was observed after 9 h where peak intensities correlated well with T0.
The HrrSA TCS coordinates heme homeostasis by integrating the response to oxidative stress and cell envelope remodeling
Our dataset confirmed the binding of HrrA to all previously known targets, including genes encoding components of heme biosynthesis (hemE, hemH and hemA), degradation (hmuO) and export (hrtBA) pathways and heme-containing complexes of the respiratory chain (ctaE-qcrCAB operon and ctaD). A comprehensive overview of all identified HrrA targets is presented in Supplementary Table S3; selected target genes are listed in Table 1. Among the more than 180 novel targets identified in this study, we observed HrrA binding upstream of ctaB, which encodes a protoheme IX farnesyltransferase that catalyzes the conversion of heme b to heme o (26) and upstream of ctaC, which encodes subunit 2 of the cytochrome aa3 oxidase. Remarkably, HrrA binding was also observed upstream of the cydABDC operon, which encodes the cytochrome bd oxidase of the respiratory chain. Altogether, this set of target genes highlights the global role of the HrrSA system in heme-dependent coordination of both branches of the respiratory chain. The HrrA regulon appeared to cover also the aspect of cofactor supply for the respiratory chain, as several HrrA targets encode enzymes involved in menaquinone reduction (sdhCD, lldD and dld).
Table 1.
Selected target genes of HrrA
| Locus tag | Gene name | Annotation | Dist. TSSa | Peak intensityb | log2(ΔhrrA/wt)c T 0.5 h | log2(ΔhrrA/wt)c T 4 h |
|---|---|---|---|---|---|---|
| Heme homeostasis/metabolism | ||||||
| cg2445 | hmuO | Heme oxygenase | 43 | 5.4 | −3.1 | −3.8 |
| cg0516 | hemE | Uroporphyrinogen decarboxylase | 17 | 54 | 3.1 | 2.2 |
| cg0497 | hemA | Glutamyl-tRNA reductase | −162 | 13 | 0.7 | 1.0 |
| cg0517 | hemY | Protoporphyrinogen oxidase | 429 | 3.0 | 2.8 | 1.6 |
| cg2079 | hemQ | Putative chlorite dismutase-family protein, conserved | 19 | 2.8 | 1.8 | |
| cg3156 | htaD | Secreted heme transport-associated protein | −108 | 15 | −0.3 | −1.1 |
| cg1734 | hemH | Ferrochelatase | 21 | 41 | 4.0 | 2.2 |
| cg3247 | hrrA | Heme-dependent response regulator | 108 | 3.7 | n.d. | n.d. |
| cg2201 | chrS | Heme-dependent histidine kinase (chrSA operon) | 32 | 2.5 | −0.4 | 1.3 |
| cg2202 | hrtB | Heme exporter (hrtBA operon) | 78 | 2.5 | −1.0 | 4.3 |
| Respiratory chain | ||||||
| cg2406 | ctaE | Cytochrome aa3 oxidase, subunit 3 | 307 | 105 | −1.7 | −0.8 |
| cg2780 | ctaD | Cytochrome aa3 oxidase, subunit 1 | 197 | 36 | −1.1 | −0.9 |
| cg1301 | cydA | Cytochrome bd oxidase | 192 | 11 | −0.7 | −2.6 |
| cg2409 | ctaC | Cytochrome aa3 oxidase, subunit 2 | 47 | 22 | −1.4 | −1.0 |
| cg1773 | ctaB | Protoheme IX farnesyltransferase | 667 | 7.9 | 0.4 | −1.4 |
| cg0445 | sdhC | Succinate:menaquinone oxidoreductase, cytochrome b subunit | 83 | 38 | −1.7 | −1.6 |
| cg3226 | L-lactate permease, operon with lldD | 533 | 5.5 | −1.7 | 0.9 | |
| Glucose uptake | ||||||
| cg2121 | ptsH | Phosphocarrier protein HPr, general component of PTS | −70 | 2.1 | −1.2 | −0.3 |
| cg1537 | ptsG | Glucose-specific EIIABC component EIIGlc of PTS | 70 | 1.6 | −1.1 | −0.1 |
| cg2091 | ppgG | Polyphosphate glucokinase | 199 | 266 | 0.2 | −0.8 |
| cg0223 | iolT1 | Myo-Inositol transporter 1, alternative glucose uptake system | 73 | 2.0 | −1.0 | −0.7 |
| Signal transduction | ||||||
| cg0986 | amtR | Master regulator of nitrogen control, repressor, TetR-family | 366 | 1.8 | 0.3 | 0.1 |
| Cg2461 | benR | Transcriptional regulator, LuxR-family | 229 | 5.6 | −0.1 | −1.2 |
| cg2761 | cpdA | cAMP phosphodiesterase | 309 | 4.2 | 0.4 | −0.5 |
| cg0309 | sigC | Extracytoplasmid-function σ factor, control of branched quinol oxidation pathway | 29 | 19 | 2.1 | 0.6 |
| cg0444 | ramB | Transcriptional regulator, involved in acetate metabolism | 83 | 38 | −0.7 | −0.6 |
| cg2831 | ramA | Transcriptional regulator, acetate metabolism, LuxR-family | −10 | 2.1 | −0.5 | 0.6 |
| Oxidative stress | ||||||
| cg0310 | katA | Catalase | 132 | 19 | −0.7 | −1.2 |
| cg0831 | tusG | Trehalose uptake system, ABC-type, permease protein | −30 | 1.8 | 0.0 | −0.2 |
| cg1791 | gapA | Glyceraldehyde-3-phos. dehydrogenase, glycolysis | 86 | 3.9 | −0.3 | −0.4 |
| cg1069 | gapB | Glyceraldehyde-3-phos. dehydrogenase, gluconeogenesis | 175 | 2.4 | 1.6 | −0.1 |
| Cell envelope | ||||||
| cg2077 | aftC | arabinofuranosyltransferase | 271 | 3.0 | −0.3 | −0.2 |
| cg3323 | ino1 | D-myo-inositol-1-phosphate synthase | −46 | 4.2 | 1.7 | 0.6 |
| cg0337 | whcA | WhiB homolog, role in SigH-mediated oxidative stress response | −21 | 2.1 | −0.5 | −0.7 |
| cg0306 | lysC | Aspartate kinase | 32 | 13 | 0.7 | 0.1 |
| cg0422 | murA | UDP-N-acetylenolpyruvoylglucosamine reductase | 591 | 3.5 | −0.3 | −0.1 |
aDistance of the HrrA binding peak, identified via ChAP-Seq, to the start codon (TSS).
bThe corresponding peak intensity.
cRelative ratio of the transcript levels of the ΔhrrA deletion mutant compared to the wild-type (log2 fold change). The values are derived from a comparison between the two strains 0.5 and 4 h after hemin addition. The log2(ΔhrrA/wt) value for was not determined for the deleted hrrA gene (n.d.).
This table summarizes results from the HrrA ChAP-Seq analysis of the C. glutamicum strain ATCC 13032::hrrA-C-twinstrep and the transcriptome analysis of C. glutamicum wild-type and strain ΔhrrA (complete datasets are provided in Supplementary Tables S3 and 4, respectively). For both experiments, cells were grown on glucose minimal medium and 4 M heme (see ‘Materials and Methods’ section).
Besides the known heme biosynthesis targets (hemE, hemH and hemA), HrrA binding was also observed upstream of a gene (hemQ) encoding a putative dismutase-family protein. However, in actinobacteria, it was proposed that these proteins do not possess chlorite dismutase activity but instead are essential for heme synthesis (49). Furthermore, we observed binding of HrrA upstream of the chrSA operon encoding the second TCS involved in heme-dependent regulation in C. glutamicum. This finding therefore confirmed the previously postulated cross-regulation of these TCS at the level of transcription (24,50).
Furthermore, HrrA binding was also observed upstream of several genes involved in the oxidative stress response, including katA, encoding catalase, tusG, encoding a trehalose uptake system (51), and upstream of gapA and gapB (glyceraldehyde-3-phosphate dehydrogenase, glycolytic and gluconeogenetic, respectively) (52,53). In line with these findings, the phenotypic analysis of a C. glutamicum hrrA mutant revealed a significantly higher sensitivity to oxidative stress (treatment with H2O2) in comparison to the wild-type (Supplementary Figure S8). These findings suggest that the HrrSA system not only controls heme biosynthesis and degradation but also integrates the response to heme-induced oxidative stress.
A further important class of HrrA targets is represented by genes associated with the regulation or maintenance of the C. glutamicum cell envelope. The gene products of these previously unknown HrrA targets are, for instance, involved in the synthesis of peptidoglycan (murA), the peptidoglycan precursor meso-2,6-diaminopimelate (mDAP), inositol-derived lipids (ino1) and arabinogalactan (aftC). Furthermore, HrrA binding was revealed upstream of a number of genes encoding global transcriptional regulators (e.g. ramA, ramB and amtR), adding a further level of complexity to this systemic response to heme.
Temporal dynamics of promoter occupancy reveal hierarchy in the HrrA regulon
With the time-resolved and genome-wide analysis of HrrA binding, we were also able to visualize distinct binding patterns of HrrA in response to addition and depletion of heme. Consequently, we asked whether the binding patterns (ChAP-Seq coverage) could provide information regarding the apparent dissociation constant (Kd) of HrrA to specific genomic targets. We compared the in vivo binding patterns of HrrA to ctaE, hmuO and cydAB (Figure 1 ,2E and Supplementary Table S3). While a comparably high peak was observed upstream of the ctaE promoter—even before the addition of heme (T0)—the binding of HrrA to the promoter of hmuO occurred with apparently high stimulus dependency and appeared to be rather transient, as HrrA was fully dissociated from this promoter 9 h after the addition of hemin (Figure 1B and C).
Subsequently, we determined the in vitro affinity of phosphorylated HrrA to the promoter regions of ctaE, cydAB and hmuO (Table 2 and Supplementary Figure S9). Consistent with the ChAP-Seq data, we measured the highest affinity of HrrA to PctaEwith an apparent Kd of 125 nM. We therefore hypothesize that the ctaE promoter is a prime target that is constitutively activated by HrrA (Supplementary Table S3) to maintain high gene expression of the operon encoding the bc1–aa3 supercomplex. In line with this hypothesis, we also found a high HrrA binding peak upstream of the other operons encoding components of the bc1–aa3 supercomplex (ctaD and ctaCF, Table 1 and Supplementary Table S3).
Table 2.
Apparent Kdvalues of HrrA to the promoters of hmuO, ctaE, sigC and cydA
| Promoter | Function | Apparent Kd value (nM) | 95% confidence interval (nM) | R² | Peak intensity after hemin addition (ChAP-Seq) |
|---|---|---|---|---|---|
| PhmuO | Heme oxygenase | 196 | 182–212 | 0.95 | 10 |
| PctaE | Cytochrome aa3 oxidase | 125 | 117–132 | 0.97 | 53 |
| PsigC | ECF sigma factor σC | 271 | 247–299 | 0.96 | 25 |
| PcydA | Cytochrome bd oxidase | 350 | 318–386 | 0.96 | 18 |
The affinity of phosphorylated HrrA to the indicated regions was measured using purified protein in increasing concentrations and its ability to shift 10 nM DNA fragments of ∼100 bp size covering the maximal ChAP-Seq peak (for detailed information, see Supplementary Figure S9).
In contrast, we measured an almost 3-fold higher apparent Kd (350 nM) for PcydAB, which was consistent with the relatively transient binding pattern observed for this target. With an apparent Kd of 196 nM, the in vitro binding affinity of HrrA to the hmuO promoter was rather high considering the genomic coverage measured in the ChAP-Seq analysis. However, in vitro analysis does not account for the widespread interference among regulatory networks in vivo. In the particular example of hmuO, the pattern of HrrA binding was likely the result of interference with the global regulator of iron homeostasis, DtxR, which has previously been described to repress hmuO expression by binding to adjacent sites (54). Taken together, these results suggest that in vivo promoter occupancy is not only influenced by the binding affinity of the regulator to the particular target, but also significantly shaped by network interference. Consequently, high in vivo promoter occupancy indicates high binding affinity, but conclusions based on weakly bound regions may be confounded by competition with other binding factors.
HrrA activates the expression of genes encoding components of both branches of the quinol oxidation pathway
To evaluate how HrrA binding affects the expression of individual target genes, we analyzed the transcriptome (RNA-Seq) of the C. glutamicum wild-type strain (ATCC 13032) as well as a ΔhrrA mutant (Supplementary Table S4). Analogous to the ChAP-Seq experiments, RNA-Seq analysis was performed prior to the addition of heme (T0) as well as 0.5 and 4 h after the heme pulse (in medium containing no other iron source). The RNA-Seq analysis was performed in two independent biological replicates (for inter-replicate variation, see Supplementary Table S5).
At T0, before the addition of heme, already 212 genes showed a more that 2-fold altered expression level in ΔhrrA cells compared to wild-type cells (ΔhrrA/wt). Directly after the addition of heme (0.5 h), the expression of 309 genes changed more than 2-fold. (Supplementary Table S4 and Figure 3A, orange dots). Of these genes, 174 were upregulated and 135 were downregulated in the hrrA deletion strain. Four hours after addition of heme, only 167 genes exhibited a >2-fold increase or decrease (scatter plots for additional time points are presented in Supplementary Figure S10).
Figure 3.
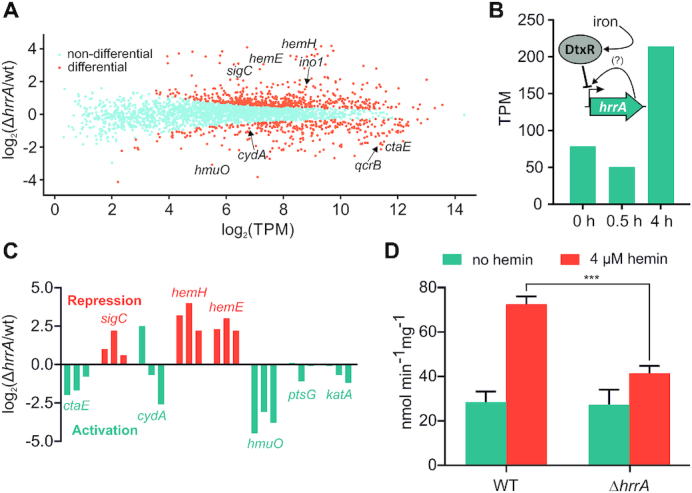
Differential gene expression analysis of wild-type Corynebacterium glutamicum and a ΔhrrA mutant. (A) Differential gene expression analysis (RNA-Seq) revealed 120 upregulated and 154 downregulated genes in the hrrA deletion strain compared to the wild-type (in transcripts per million, TPM) after 30 min of cultivation in iron-depleted glucose minimal medium containing 4 μM heme. (B) Expression levels of hrrA (TPM) 0, 0.5 and 4 h after the addition of hemin. A scheme depicts HrrA autoregulation and iron-dependent DtxR repression (24). (C) Impact of hrrA deletion on the transcript levels of six selected target genes at three different time points (0, 0.5, 4 h; orange: HrrA acts as a repressor, turquoise: HrrA acts as an activator). (D) Measurement of cytochrome aa3 oxidase activity using the TMPD oxidase assay in C. glutamicum wild-type and ΔhrrA grown with or without 4 μM heme.
The hrrA expression decreased after 0.5 h upon the addition of heme, which was likely caused by DtxR repression in response to increased intracellular iron levels (Figure 3B) (24). In contrast, after 4 h of cultivation, hrrA levels significantly increased, reflecting the depletion of heme as an alternative iron source and dissociation of DtxR. Furthermore, differential gene expression analysis revealed HrrA to be an activator of all genes encoding components of the respiratory chain (ctaE, ctaD, ctaF and cydAB) and as a repressor of heme biosynthesis (hemA, hemE and hemH) (Figure 3C). The impact on the cytochrome bc1–aa3 supercomplex was also confirmed by measuring the activity of the aa3 oxidase, which was about 2-fold reduced in a hrrA mutant in comparison to the wild-type when grown on heme (Figure 3D). Additionally, expression of lldD (L-lactate dehydrogenase) as well as sdhCD (succinate dehydrogenase) contributing to the reduced menaquinone pool was downregulated more than 3-fold upon deletion of hrrA. In addition to these considerable differences between the wild-type and the ΔhrrA mutant, we also observed decreased mRNA levels of genes involved in the oxidative stress response (e.g. katA) or cell envelope remodeling (e.g. murA) in the ΔhrrA mutant, suggesting HrrA to be an activator of these targets.
In some cases, promoter occupancy by HrrA did not result in altered expression levels of the particular target gene in a ΔhrrA mutant under the tested conditions (Table 1 and Supplementary Table S3). This finding is, however, not surprising considering the multiplicity of signals and regulators affecting gene expression. Under changing environmental conditions, transcription factor binding will not necessarily always be translated in an altered gene expression of the respective target. When we compare the results obtained from RNA-Seq and ChAP-Seq analysis, 269 genes out of 309 genes featuring an >2-fold change in gene expression did not show HrrA binding in their upstream promoter region. Looking at all HrrA targets (ChAP-Seq analysis) on a global scale, there is, nevertheless, a significantly higher impact on gene expression in a strain lacking hrrA for all targets bound by HrrA in comparison to non-targets (unbound, Supplementary Figure S11). Overall, 109 out of 228 HrrA targets featured a significantly altered gene expression in the hrrA mutant (64 increased and 55 decreased).
HrrA determines the prioritization of terminal cytochrome oxidases by repression of sigC
The results from ChAP-Seq and RNA-Seq experiments highlight the important role of HrrA in the control of the respiratory chain, including cofactor supply. Our data revealed that HrrA activates the expression of genes encoding the cytochrome bc1-aa3 supercomplex (ctaE-qcrCAB, ctaD, ctaCF) and of cydAB, encoding the cytochrome bd branch of the respiratory chain (Figure 4 and Supplementary Table S3). Remarkably, the mRNA profiles of the corresponding operons exhibited significantly delayed activation of cydAB in response to heme, which was abolished in the ΔhrrA mutant (Figure 4). In contrast, ctaE expression was significantly higher in wild-type cells, even before hemin addition (T0), but showed a further induction after stimulus addition (T 0.5 h, Supplementary Table S4). Notably, we also observed binding of HrrA upstream of sigC, encoding an ECF sigma factor that was shown to be involved in the activation of the cydABDC operon (35). The mRNA level of sigC increased more than 2-fold in the ΔhrrA mutant, indicating HrrA to be a repressor of this gene (Figure 4). Consistent with this hypothesis, sigC expression was slightly decreased in response to the addition of heme, which correlated with increased HrrA peak intensity (Figure 4F). Additionally, the higher cydAB expression, observed in the ΔhrrA strain before addition of stimulus (Figure 4B) is likely the effect of increased sigC expression (Figure 4C). Dissociation of HrrA from PsigC at a later time point (4 h after heme pulse) led to derepression of sigC coinciding with an increased expression of cydAB in the wild-type. Because cydAB levels were constitutively low in the ΔhrrA mutant in response to heme, we hypothesized that activation by HrrA together with an additional boost by SigC (Figure 5) leads to delayed activation of cydAB after the heme pulse. This regulation enables cells to channel most of the available heme pool into the more efficient cytochrome bc1–aa3 supercomplex. The lower apparent Kd of HrrA for the ctaE promoter (125 nM) compared to PcydAB (350 nM) or PsigC (270 μM) also reflects this prioritization of HrrA targets (Table 2). Consequently, this almost 3-fold decrease in affinity (apparent Kd) increases the threshold for HrrSA activity to control these targets.
Figure 4.

HrrA prioritizes the expression of genes encoding components of the bc1–aa3 supercomplex. Depicted are HrrA binding peaks as identified by ChAP-Seq analysis (Figures 1 and 2) in comparison to the normalized coverage of RNA-Seq results (wild-type and the ΔhrrA mutant) for the genomic loci of ctaE (A andD), sigC (B andE) and cydA (C andF). D–F: HrrA binding (max. peak intensities measured by ChAP-Seq experiments) and the mRNA levels (in transcripts per million, TPM) of the respective genes in the ΔhrrA strain as well as in wild-type Corynebacterium glutamicum cells 0, 0.5 and 4 h after the addition of hemin.
Figure 5.

Model of heme-responsive control of components of the respiratory chain by HrrSA. The results of this study reveal HrrSA as a global regulator of heme homeostasis coordinating the expression of genes involved in heme biosynthesis, oxidative stress responses, glucose uptake and cell envelope remodeling. Genes encoding the components of the branched respiratory chain of Corynebacterium glutamicum comprise an important part of the HrrA regulon. While HrrA acts as an activator of almost all components (ctaE-qcrCAB, ctaB, cydAB), it represses transcription of the sigC gene encoding an important sigma factor required for cydAB expression. This regulatory network architecture consequently confers prioritization to the synthesis of the more efficient proton pump, the cytochrome bc1–aa3 supercomplex. Bordered boxes, b, c, a, d: heme b, heme c, heme a, heme d.
HrrA activates PTS-dependent and -independent glucose uptake
Besides the activation of all components constituting the respiratory chain, ChAP-Seq experiments and transcriptome analysis revealed HrrA as a direct activator of genes encoding components of the phosphotransferase (PTS) system (ptsH and ptsG) and of iolT1 encoding inositol permease with a reported function as a PTS-independent glucose uptake system (55). Remarkably, the gene ppgK, encoding the polyphosphate glucokinase was among the targets with the highest HrrA peak and showed reduced expression in the 4 h sample (Table 1 and Supplementary Table S4). These results emphasize that cellular respiration and glucose uptake is coordinated via the HrrSA system in response to cellular heme levels.
DISCUSSION
In this work, we applied a genome-wide approach to study the ‘heme-responsive regulator’ HrrA in C. glutamicum and identified more than 200 genomic target regions of this response regulator. This intriguingly diverse set of target genes, encoding enzymes involved in heme biosynthesis, heme-containing proteins and components of the respiratory chain as well as proteins involved in oxidative stress response, glucose uptake and cell envelope remodeling, provided unprecedented insight into the systemic response to heme coordinated by the TCS HrrSA.
In Gram-positive bacteria, TCSs appear to play a central role in transient heme sensing, and heme-responsive systems have been described in several prominent pathogens, including C. diphtheriae, S. aureus and B. anthracis (15–18). However, for all prokaryotic heme regulatory systems, only a small number of target genes have been described to date, focusing on targets involved in degradation (hmuO (18,56)), heme export (hrtBA (19,57)) or heme biosynthesis (hemA (18,20)). Systems orthologous to HrrSA are present in almost all corynebacterial species and the high amino acid sequence identity shared by the response regulators (87%, between C. glutamicum and C. diphtheriae HrrA) suggests that the important role of HrrSA in the control of heme homeostasis is conserved. In many corynebacteria, including C. diphtheriae, control of heme homeostasis is shaped by the tight interplay of HrrSA with a second heme-dependent system, ChrSA. While the present study emphasized that HrrSA governs a large and complex homeostatic response, the only known target of the response regulator ChrA in C. glutamicum is the divergently located operon hrtBA encoding a heme export system. There is, however, also evidence for a cross-regulation between the TCSs, not only by cross-phosphorylation but also on the transcriptional level (23,24). In C. diphtheria, evidence for more overlap between the regulons of the TCSs has been provided, since both response regulators were shown to control a common set of target genes including hrtBA, hemA and hmuO (20,58). Genome-wide analysis of these systems have, however, not been performed so far and invitro protein–DNA interaction studies may not necessarily reflect the invivo promoter preferences of these highly similar systems.
Coping with heme stress
While being an essential cofactor for many proteins, heme causes severe toxicity to cells at high levels (4). In mammalian cells, the BACH1 regulator is inactivated by heme binding and plays a key role in maintaining the balance of the cellular heme pool (8,59). Heme oxygenases are targets of various heme-dependent regulators (18,60–61), and consistent with this principle, the mammalian HMOX1 gene, encoding an NADPH-dependent oxygenase, is regulated by BACH1 (59). Other identified BACH1 targets are involved in redox regulation, the cell cycle, and apoptosis as well as subcellular transport processes (9,62–63).
Although neither the regulator nor the constitution of the regulon is conserved, the responses of BACH1 and HrrSA share a similar logic. Analogous to eukaryotic BACH1, we observed HrrA-mediated activation of genes involved in the oxidative stress response, including katA, which appears to be required to counteract oxidative stress caused by elevated heme levels (Supplementary Figure S8).
Remarkably, HrrA binding was also observed upstream of both gapA and gapB, which encode glyceraldehyde-3-phosphate dehydrogenases (GapDHs) involved in glycolysis and gluconeogenesis, respectively. Previous studies in baker's yeast and mammalian cells have revealed that oxidative stress may block glycolysis by inhibiting GapDH (53,64). Furthermore, GapDH of C. diphtheriae was recently shown to be redox-controlled by S-mycothiolation (65). Slight activation of gapA by HrrA may thus counteract an impaired glycolytic flux under conditions of heme stress.
Furthermore, several HrrA targets play a role in the biosynthesis and remodeling of the corynebacterial cell envelope, including ino1, which is required for the synthesis of inositol-derived lipids (66), lysC, providing the peptidoglycan precursor meso-2,6-diaminopimelate (mDAP) and murA (Table 1). Taken together, these insights emphasize the important role of the HrrSA system in the control of heme stress responses.
From networks to function
Genome-wide analysis of regulatory networks may provide important hints toward the physiological function of genes. An example is provided by the HrrA-dependent regulation of cg2079 (hemQ), described in this study (Table 1). In actinobacteria, it was recently proposed that these proteins inherit an essential role in heme biosynthesis (49,67). The finding that HrrA binds to the promoter of this gene and represses its expression supports a role of HemQ in heme biosynthesis in C. glutamicum. Among the direct targets of HrrA are many further targets encoding proteins of unknown function, including several ABC transport systems with a potential role in heme uptake or export. Therefore, this dataset provides guidance for further functional analysis of these HrrA targets to decipher their role in heme homeostasis.
Coordinated control of the respiratory chain
Among the most significantly affected targets in the ΔhrrA mutant were many genes encoding components of the respiratory chain (26). These genes comprise all the genes constituting the cytochrome bc1-aa3 branch of the respiratory chain (ctaE-qcrCAB, ctaCF and ctaD) (68); genes encoding the cytochrome bd branch (cydAB (26)); ctaA (69) and ctaB (70), encoding enzymes responsible for heme a synthesis; and lldD and dld, encoding lactate dehydrogenases that contribute to the reduced menaquinone pool (26) (Figure 4; Supplementary Figure S12 and Table S3).
In a recent study, Toyoda and Inui described the ECF sigma factor σC to be an important regulator of both branches of the C. glutamicum respiratory chain. The ctaE-qcrCAB operon was shown to be significantly downregulated after σC overexpression due to binding of the sigma factor to the antisense strand of the promoter (35). Here, we demonstrated that this repression is counteracted by HrrA, which not only represses sigC but also activates ctaE-qcrCAB expression. While the two proteins have antagonistic effects on the expression of the supercomplex, both σC and HrrA positively regulate the cyd operon, encoding the cytochrome bd branch of the respiratory chain (Figure 5).
Interestingly, a hierarchy in the regulon was reflected by the differences in the apparent Kd values of HrrA with PcydA and PsigC, which were 2-fold lower than those with the promoter of ctaE. These findings were also consistent with the ChAP-Seq experiments, where the peaks upstream of ctaE and ctaD were among the highest peaks at T0 and after 0.5 h (Figure 4A). These data suggest that under conditions of sufficient heme supply, production of the cytochrome bc1–aa3 supercomplex is preferred, which is highly effective but requires the incorporation of six heme molecules (in contrast to only three molecules for the synthesis of the bd oxidase). Repression of sigC by HrrA and the relatively low affinity to the cydAB promoter results in delayed production of the bd branch. Under the applied aerobic conditions, available heme is thus first channeled to the cytochrome bc1–aa3 supercomplex before the cytochrome bd oxidase is used, which is less efficient but has a higher oxygen affinity. Remarkably, HrrA was also found to activate expression of genes involved in PTS-dependent (ptsH and ptsG) and -independent (iolT1) glucose uptake thereby ensuring a high glucose uptake rate under conditions of active cellular respiration.
Interference with other regulatory networks
Deletion of the hrrA gene led to more than 2-fold upregulation of 174 genes, while 135 genes were downregulated after the addition of heme. Several other genes were significantly affected but to a lesser extent. Remarkably, among the direct target genes controlled by HrrA, we identified several prominent global regulators, including the regulators of acetate metabolism ramA and ramB (71,72), and amtR encoding the master regulator of nitrogen control (73). Furthermore, cpdA encoding a cAMP phosphodiesterase playing a key role in the control of cellular cAMP levels in C. glutamicum (74) was found to be under direct control of HrrA. These examples illustrate the profound influence of HrrA on cellular networks and the systemic response cells have programmed to respond to heme availability.
CONCLUSION
Genome-wide analyses of targets controlled by prokaryotic transcription factors will change our view on many systems we believe to know. In this study, we provide an unprecedented insight into the systemic response to heme coordinated by the TCS HrrSA. Given the many properties of this molecule, the complexity of this response is actually not surprising but paves the way for further functional analysis of HrrA targets with so far unknown functions in heme homeostasis.
DATA AVAILABILITY
The custom-developed software used in this study is publicly available at GitHub repository under the link https://github.com/afilipch/afp.
All sequencing data were deposited in the GEO database under the accession numbers GSE121962 (ChAP-Seq) and GSE120924 (RNA-Seq).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Eva Davoudi for fruitful discussions and critical reading of the manuscript. We thank Helga Etterich for her practical help with the sequencing experiments.
Contributor Information
Marc Keppel, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Max Hünnefeld, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Andrei Filipchyk, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Ulrike Viets, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Cedric-Farhad Davoudi, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Aileen Krüger, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Christina Mack, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Eugen Pfeifer, Microbial Evolutionary Genomics, Institute Pasteur, 75015 Paris, France.
Tino Polen, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Meike Baumgart, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Michael Bott, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
Julia Frunzke, Institute of Bio- und Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation [FR 2759/4-1]; European Research Council [ERC-StG-2017, 757563]; Helmholtz Association [W2/W3-096]. Funding for open access charge: Forschungszentrum Jülich, European Research Council [757563].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ponka P. Cell biology of heme. Am. J. Med. Sci. 1999; 318:241–256. [DOI] [PubMed] [Google Scholar]
- 2. Layer G., Reichelt J., Jahn D., Heinz D.W.. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010; 19:1137–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ajioka R.S., Phillips J.D., Kushner J.P.. Biosynthesis of heme in mammals. Biochim. Biophys. Acta. 2006; 1763:723–736. [DOI] [PubMed] [Google Scholar]
- 4. Anzaldi L.L., Skaar E.P.. Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect. Immun. 2010; 78:4977–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang W., Wilks A.. Extracellular heme uptake and the challenge of bacterial cell membranes. Annu. Rev. Biochem. 2017; 86:799–823. [DOI] [PubMed] [Google Scholar]
- 6. Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid. Redox. Signal. 2002; 4:603–614. [DOI] [PubMed] [Google Scholar]
- 7. Hickman M.J., Winston F.. Heme levels switch the function of Hap1 of Saccharomyces cerevisiae between transcriptional activator and transcriptional repressor. Mol. Cell. Biol. 2007; 27:7414–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ogawa K., Sun J., Taketani S., Nakajima O., Nishitani C., Sassa S., Hayashi N., Yamamoto M., Shibahara S., Fujita H. et al.. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001; 20:2835–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warnatz H.J., Schmidt D., Manke T., Piccini I., Sultan M., Borodina T., Balzereit D., Wruck W., Soldatov A., Vingron M. et al.. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011; 286:23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qi Z., Hamza I., O’Brian M.R.. Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:13056–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qi Z., O’Brian M.R.. Interaction between the bacterial iron response regulator and ferrochelatase mediates genetic control of heme biosynthesis. Mol. Cell. 2002; 9:155–162. [DOI] [PubMed] [Google Scholar]
- 12. O’Brian M.R. Perception and homeostatic control of iron in the rhizobia and related bacteria. Annu. Rev. Microbiol. 2015; 69:229–245. [DOI] [PubMed] [Google Scholar]
- 13. Torres V.J., Stauff D.L., Pishchany G., Bezbradica J.S., Gordy L.E., Iturregui J., Anderson K.L., Dunman P.M., Joyce S., Skaar E.P.. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe. 2007; 1:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mike L.A., Choby J.E., Brinkman P.R., Olive L.Q., Dutter B.F., Ivan S.J., Gibbs C.M., Sulikowski G.A., Stauff D.L., Skaar E.P.. Two-component system cross-regulation integrates Bacillus anthracis response to heme and cell envelope stress. PLoS Pathog. 2014; 10:e1004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stauff D.L., Skaar E.P.. The heme sensor system of Staphylococcus aureus. Contrib. Microbiol. 2009; 16:120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stauff D.L., Skaar E.P.. Bacillus anthracis HssRS signalling to HrtAB regulates haem resistance during infection. Mol. Microbiol. 2009; 72:763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bibb L.A., Kunkle C.A., Schmitt M.P.. The ChrA-ChrS and HrrA-HrrS signal transduction systems are required for activation of the hmuO promoter and repression of the hemA promoter in Corynebacterium diphtheriae. Infect. Immun. 2007; 75:2421–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frunzke J., Gatgens C., Brocker M., Bott M.. Control of heme homeostasis in Corynebacterium glutamicum by the two-component system HrrSA. J. Bacteriol. 2011; 193:1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heyer A., Gatgens C., Hentschel E., Kalinowski J., Bott M., Frunzke J.. The two-component system ChrSA is crucial for haem tolerance and interferes with HrrSA in haem-dependent gene regulation in Corynebacterium glutamicum. Microbiology. 2012; 158:3020–3031. [DOI] [PubMed] [Google Scholar]
- 20. Burgos J.M., Schmitt M.P.. The ChrSA and HrrSA two-component systems are required for transcriptional regulation of the hemA promoter in Corynebacterium diphtheriae. J. Bacteriol. 2016; 198:2419–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keppel M., Davoudi E., Gätgens C., Frunzke J.. Membrane topology and heme binding of the histidine kinases HrrS and ChrS in Corynebacterium glutamicum. Front. Microbiol. 2018; 9:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ito Y., Nakagawa S., Komagata A., Ikeda-Saito M., Shiro Y., Nakamura H.. Heme-dependent autophosphorylation of a heme sensor kinase, ChrS, from Corynebacterium diphtheriae reconstituted in proteoliposomes. FEBS Lett. 2009; 583:2244–2248. [DOI] [PubMed] [Google Scholar]
- 23. Hentschel E., Mack C., Gätgens C., Bott M., Brocker M., Frunzke J.. Phosphatase activity of the histidine kinases ensures pathway specificity of the ChrSA and HrrSA two-component systems in Corynebacterium glutamicum. Mol. Microbiol. 2014; 92:1326–1342. [DOI] [PubMed] [Google Scholar]
- 24. Keppel M., Piepenbreier H., Gätgens C., Fritz G., Frunzke J.. Toxic but tasty—temporal dynamics and network architecture of heme-responsive two-component signaling in Corynebacterium glutamicum. Mol. Microbiol. 2019; 111:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wennerhold J., Bott M.. The DtxR regulon of Corynebacterium glutamicum. J. Bacteriol. 2006; 188:2907–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bott M., Niebisch A.. The respiratory chain of Corynebacterium glutamicum. J. Biotechnol. 2003; 104:129–153. [DOI] [PubMed] [Google Scholar]
- 27. Kao W.C., Kleinschroth T., Nitschke W., Baymann F., Neehaul Y., Hellwig P., Richers S., Vonck J., Bott M., Hunte C.. The obligate respiratory supercomplex from Actinobacteria. Biochim. Biophys. Acta. 2016; 1857:1705–1714. [DOI] [PubMed] [Google Scholar]
- 28. Niebisch A., Bott M.. Molecular analysis of the cytochrome bc1-aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch. Microbiol. 2001; 175:282–294. [DOI] [PubMed] [Google Scholar]
- 29. Kalinowski J., Bathe B., Bartels D., Bischoff N., Bott M., Burkovski A., Dusch N., Eggeling L., Eikmanns B.J., Gaigalat L. et al.. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 2003; 104:5–25. [DOI] [PubMed] [Google Scholar]
- 30. Ikeda M., Nakagawa S.. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 2003; 62:99–109. [DOI] [PubMed] [Google Scholar]
- 31. Sone N., Nagata K., Kojima H., Tajima J., Kodera Y., Kanamaru T., Noguchi S., Sakamoto J.. A novel hydrophobic diheme c-type cytochrome. Purification from Corynebacterium glutamicum and analysis of the QcrCBA operon encoding three subunit proteins of a putative cytochrome reductase complex. Biochim. Biophys. Acta. 2001; 1503:279–290. [DOI] [PubMed] [Google Scholar]
- 32. Niebisch A., Bott M.. Purification of a cytochrome bc-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. Identification of a fourth subunity of cytochrome aa3 oxidase and mutational analysis of diheme cytochrome c1. J. Biol. Chem. 2003; 278:4339–4346. [DOI] [PubMed] [Google Scholar]
- 33. Teramoto H., Inui M., Yukawa H.. OxyR acts as a transcriptional repressor of hydrogen peroxide-inducible antioxidant genes in Corynebacterium glutamicum R. FEBS J. 2013; 280:3298–3312. [DOI] [PubMed] [Google Scholar]
- 34. Milse J., Petri K., Ruckert C., Kalinowski J.. Transcriptional response of Corynebacterium glutamicum ATCC 13032 to hydrogen peroxide stress and characterization of the OxyR regulon. J. Biotechnol. 2014; 190:40–54. [DOI] [PubMed] [Google Scholar]
- 35. Toyoda K., Inui M.. The extracytoplasmic function sigma factor sigma(C) regulates expression of a branched quinol oxidation pathway in Corynebacterium glutamicum. Mol. Microbiol. 2016; 100:486–509. [DOI] [PubMed] [Google Scholar]
- 36. Morosov X., Davoudi C.F., Baumgart M., Brocker M., Bott M.. The copper-deprivation stimulon of Corynebacterium glutamicum comprises proteins for biogenesis of the actinobacterial cytochrome bc1-aa3 supercomplex. J. Biol. Chem. 2018; 293:15628–15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davoudi C.F., Ramp P., Baumgart M., Bott M.. Identification of Surf1 as an assembly factor of the cytochrome bc1-aa3 supercomplex of Actinobacteria. Biochim. Biophys. Acta Bioenerget. 2019; 1860:148033. [DOI] [PubMed] [Google Scholar]
- 38. Keilhauer C., Eggeling L., Sahm H.. Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J. Bacteriol. 1993; 175:5595–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J.F., Russell D.W.. Molecular Cloning: A Laboratory Manual. 2001; 3rd ednNY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 40. Eikmanns B.J., Thum-Schmitz N., Eggeling L., Ludtke K.U., Sahm H.. Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology. 1994; 140:1817–1828. [DOI] [PubMed] [Google Scholar]
- 41. Niebisch A., Bott M.. Molecular analysis of the cytochrome bc1-aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch. Microbiol. 2001; 175:282–294. [DOI] [PubMed] [Google Scholar]
- 42. Schäfer A., Tauch A., Jager W., Kalinowski J., Thierbach G., Pühler A.. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994; 145:69–73. [DOI] [PubMed] [Google Scholar]
- 43. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 44. Pfeifer E., Hünnefeld M., Popa O., Polen T., Kohlheyer D., Baumgart M., Frunzke J.. Silencing of cryptic prophages in Corynebacterium glutamicum. Nucleic Acids Res. 2016; 44:10117–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baumgart M., Unthan S., Kloss R., Radek A., Polen T., Tenhaef N., Muller M.F., Kuberl A., Siebert D., Bruhl N. et al.. Corynebacterium glutamicum Chassis C1*: Building and testing a novel platform host for synthetic biology and industrial biotechnology. ACS Synth. Biol. 2018; 7:132–144. [DOI] [PubMed] [Google Scholar]
- 47. Pachter L. Models for transcript quantification from RNA-Seq. Conference Proceedings. 2011; arXiv:13 May 2011,preprint: not peer reviewedhttps://arxiv.org/abs/1104.3889. [Google Scholar]
- 48. Sakamoto J., Shibata T., Mine T., Miyahara R., Torigoe T., Noguchi S., Matsushita K., Sone N.. Cytochrome c oxidase contains an extra charged amino acid cluster in a new type of respiratory chain in the amino-acid-producing Gram-positive bacterium Corynebacterium glutamicum. Microbiology. 2001; 147:2865–2871. [DOI] [PubMed] [Google Scholar]
- 49. Dailey H.A., Gerdes S.. HemQ: an iron-coproporphyrin oxidative decarboxylase for protoheme synthesis in Firmicutes and Actinobacteria. Arch. Biochem. Biophys. 2015; 574:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frunzke J., Gätgens C., Brocker M., Bott M.. Control of heme homeostasis in Corynebacterium glutamicum by the two-component system HrrSA. J. Bacteriol. 2011; 193:1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alvarez-Peral F.J., Zaragoza O., Pedreno Y., Arguelles J.C.. Protective role of trehalose during severe oxidative stress caused by hydrogen peroxide and the adaptive oxidative stress response in Candida albicans. Microbiology. 2002; 148:2599–2606. [DOI] [PubMed] [Google Scholar]
- 52. Kuehne A., Emmert H., Soehle J., Winnefeld M., Fischer F., Wenck H., Gallinat S., Terstegen L., Lucius R., Hildebrand J. et al.. Acute activation of oxidative pentose phosphate pathway as First-Line response to oxidative stress in human skin cells. Mol. Cell. 2015; 59:359–371. [DOI] [PubMed] [Google Scholar]
- 53. Ralser M., Wamelink M.M., Latkolik S., Jansen E.E., Lehrach H., Jakobs C.. Metabolic reconfiguration precedes transcriptional regulation in the antioxidant response. Nat. Biotechnol. 2009; 27:604–605. [DOI] [PubMed] [Google Scholar]
- 54. Wennerhold J., Bott M.. The DtxR regulon of Corynebacterium glutamicum. J. Bacteriol. 2006; 188:2907–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lindner S.N., Seibold G.M., Henrich A., Kramer R., Wendisch V.F.. Phosphotransferase system-independent glucose utilization in Corynebacterium glutamicum by inositol permeases and glucokinases. Appl. Environ. Microbiol. 2011; 77:3571–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bibb L.A., King N.D., Kunkle C.A., Schmitt M.P.. Analysis of a heme-dependent signal transduction system in Corynebacterium diphtheriae: deletion of the chrAS genes results in heme sensitivity and diminished heme-dependent activation of the hmuO promoter. Infect. Immun. 2005; 73:7406–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bibb L.A., Schmitt M.P.. The ABC transporter HrtAB confers resistance to hemin toxicity and is regulated in a hemin-dependent manner by the ChrAS two-component system in Corynebacterium diphtheriae. J. Bacteriol. 2010; 192:4606–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bibb L.A., Schmitt M.P.. The ABC transporter, HrtAB, confers resistance to hemin toxicity and is regulated in a hemin-dependent manner by the ChrAS two-component system in Corynebacterium diphtheriae. J. Bacteriol. 2010; 192:4606–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun J., Hoshino H., Takaku K., Nakajima O., Muto A., Suzuki H., Tashiro S., Takahashi S., Shibahara S., Alam J. et al.. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002; 21:5216–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ratliff M., Zhu W., Deshmukh R., Wilks A., Stojiljkovic I.. Homologues of neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J. Bacteriol. 2001; 183:6394–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schmitt M.P. Transcription of the Corynebacterium diphtheriae hmuO gene is regulated by iron and heme. Infect. Immun. 1997; 65:4634–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hintze K.J., Katoh Y., Igarashi K., Theil E.C.. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J. Biol. Chem. 2007; 282:34365–34371. [DOI] [PubMed] [Google Scholar]
- 63. Dhakshinamoorthy S., Jain A.K., Bloom D.A., Jaiswal A.K.. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005; 280:16891–16900. [DOI] [PubMed] [Google Scholar]
- 64. Kuehne A., Emmert H., Soehle J., Winnefeld M., Fischer F., Wenck H., Gallinat S., Terstegen L., Lucius R., Hildebrand J. et al.. Acute activation of oxidative pentose phosphate pathway as First-Line response to oxidative stress in human skin cells. Mol. Cell. 2015; 59:359–371. [DOI] [PubMed] [Google Scholar]
- 65. Hillion M., Imber M., Pedre B., Bernhardt J., Saleh M., Loi V.V., Maaß S., Becher D., Astolfi Rosado L., Adrian L. et al.. The glyceraldehyde-3-phosphate dehydrogenase GapDH of Corynebacterium diphtheriae is redox-controlled by protein S-mycothiolation under oxidative stress. Sci. Rep. 2017; 7:5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Baumgart M., Luder K., Grover S., Gatgens C., Besra G.S., Frunzke J.. IpsA, a novel LacI-type regulator, is required for inositol-derived lipid formation in Corynebacteria and Mycobacteria. BMC Biol. 2013; 11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dailey H.A., Dailey T.A., Gerdes S., Jahn D., Jahn M., O’Brian M.R., Warren M.J.. Prokaryotic heme Biosynthesis: Multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 2017; 81:e00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Niebisch A., Bott M.. Purification of a cytochrome bc-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. Identification of a fourth subunity of cytochrome aa3 oxidase and mutational analysis of diheme cytochrome c1. J. Biol. Chem. 2003; 278:4339–4346. [DOI] [PubMed] [Google Scholar]
- 69. Mueller J.P., Taber H.W.. Isolation and sequence of ctaA, a gene required for cytochrome aa3 biosynthesis and sporulation in Bacillus subtilis. J. Bacteriol. 1989; 171:4967–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Svensson B., Lubben M., Hederstedt L.. Bacillus subtilis CtaA and CtaB function in haem A biosynthesis. Mol. Microbiol. 1993; 10:193–201. [DOI] [PubMed] [Google Scholar]
- 71. Auchter M., Cramer A., Huser A., Ruckert C., Emer D., Schwarz P., Arndt A., Lange C., Kalinowski J., Wendisch V.F. et al.. RamA and RamB are global transcriptional regulators in Corynebacterium glutamicum and control genes for enzymes of the central metabolism. J. Biotechnol. 2011; 154:126–139. [DOI] [PubMed] [Google Scholar]
- 72. Shah A., Blombach B., Gauttam R., Eikmanns B.J.. The RamA regulon: complex regulatory interactions in relation to central metabolism in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2018; 102:5901–5910. [DOI] [PubMed] [Google Scholar]
- 73. Burkovski A. Nitrogen control in Corynebacterium glutamicum: proteins, mechanisms, signals. J. Microbiol. Biotechnol. 2007; 17:187–194. [PubMed] [Google Scholar]
- 74. Schulte J., Baumgart M., Bott M.. Identification of the cAMP phosphodiesterase CpdA as novel key player in cAMP-dependent regulation in Corynebacterium glutamicum. Mol. Microbiol. 2017; 103:534–552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom-developed software used in this study is publicly available at GitHub repository under the link https://github.com/afilipch/afp.
All sequencing data were deposited in the GEO database under the accession numbers GSE121962 (ChAP-Seq) and GSE120924 (RNA-Seq).