


Abstract
DNA damage and epigenetic marks are well established to have profound influences on genome stability and cell phenotype, yet there are few technologies to obtain high-resolution genomic maps of the many types of chemical modifications of DNA. Here we present Nick-seq for quantitative, sensitive, and accurate mapping of DNA modifications at single-nucleotide resolution across genomes. Pre-existing breaks are first blocked and DNA modifications are then converted enzymatically or chemically to strand-breaks for both 3′-extension by nick-translation to produce nuclease-resistant oligonucleotides and 3′-terminal transferase tailing. Following library preparation and next generation sequencing, the complementary datasets are mined with a custom workflow to increase sensitivity, specificity and accuracy of the map. The utility of Nick-seq is demonstrated with genomic maps of site-specific endonuclease strand-breaks in purified DNA from Eschericia coli, phosphorothioate epigenetics in Salmonella enterica Cerro 87, and oxidation-induced abasic sites in DNA from E. coli treated with a sublethal dose of hydrogen peroxide. Nick-seq applicability is demonstrated with strategies for >25 types of DNA modification and damage.
INTRODUCTION
Genomic DNA in all cells is continuously subjected to extensive chemical modification including damage caused by endogenous and exogenous stresses (1), intermediates in the repair of this damage (1), and enzymatically-mediated epigenetic modifications (2). These mechanisms and processes are well established to have profound influences on genome stability and cell phenotype, with dysregulation causing many human diseases (1,2). While whole-genome sequencing is now commonplace in the post-genomic era, with the mutational consequences of DNA damage readily mapped across genomes, technologies to accurately and quantitatively localize DNA modifications in genomes are limited and highly specialized. A summary of existing methods for DNA modification mapping is presented in Table 1. For example, single-nucleotide-resolution genomic maps can be obtained for 5-methylcytidine (5mC) and 5-hydromethylcytidine (5hmC) epigenetic marks by a combination of bisulfite derivatization and next generation sequencing (NGS) (3), while nucleobase methylation and phosphorothioate modifications can be mapped using single-molecule real-time (SMRT) (4) and nanopore (5) sequencing technologies. Lower-resolution maps of specific types of DNA damage and modifications can be achieved by enrichment-based sequencing methods such as chromatin immunoprecipitation (ChIP) or chemical labelling coupled with NGS (6,7), for example. However, all of these techniques are limited to specific modifications, are poorly quantitative, or suffer from low resolution, low sensitivity, and lack of specificity. Here, we describe Nick-seq, a general method for highly sensitive and quantitative genomic mapping at single-nucleotide resolution for any type of DNA modification or damage that can be converted to a strand-break.
Table 1.
Summary of existing methods for genomic mapping of DNA modifications
| Modifications | Method | Resol’n | Method | Basis | Limitation | Ref |
|---|---|---|---|---|---|---|
| 5mC | Bisulfite reaction based | 1 nt | BS-seq | Differential deamination of C and 5mC to U | Specific to 5mC | (22) |
| 5mC, 5hmC | oxBS-Seq | 5hmC converted to uracil | Specific to 5hmC | (23) | ||
| 6mA, 4mC, 5mC et al. | Third generation sequencing | 1 nt | SMRT sequencing | Detection during sequencing | Sensitivity, specificity | (4,27) |
| 5mC; 6mA | Nanopore sequencing | Detection during sequencing | Noise, sensitivity, specificity | (42) | ||
| Methylation status of CpG islands | DNA microarray based | Low | CpG island microarray | Restriction enzyme digestion; microarray | Methylation-sensitive restriction enzymes | (20) |
| Methylation | MeDIP-on-Chip | IP, hybridize of DNA on microarray | Modification-specific antibody | (21) | ||
| 5mC and 5hmC | Chemical-labeling for NGS sequencing | 1 nt | TAPS | Oxidation of 5mC/5hmC to 5caC, reduce to dihydrouracil | Specific to 5mC and 5hmC | (24) |
| 8-oxoG | ∼150 nt | OG-seq | Derivatize 8-oxoG with biotin | Specific to 8-oxoG | (6) | |
| AP sites | 1 nt | snAP-seq | Chemical labelling of AP site with biotin | Specific to AP sites | (25) | |
| 8-oxoG | 1 nt | Click-code-Seq | BER excision; click chemistry | DNA 3′-OH end required | (31) | |
| UV-induced DNA lesions | Antibody-enrichment for NGS sequencing | 1 nt | XR-seq | Damaged DNA IP, NGS | Lesion-specific antibody | (26) |
| 8-oxoG | ∼150 nt | OxiDIP-Seq | 8-oxoG IP for NGS sequencing | 8-oxoG -specific antibody | (12) | |
| 5ghmC, 5hmC | Modif’n-dependent restriction enzyme | 3∼5 nt | Aba-seq | AbaSI recognizes 5ghmC and 5hmC | Limited to a narrow spectrum of modifications | (29) |
| Uracil, pyrimidine dimer | BER enzyme excision | <1 nt | Excision-seq | BER enzyme excision, NGS | Very low sensitivty; low-resolution | (11) |
| Single-strand breaks | TdT-labeling | 1 nt | SSiNGLe | TdT-labeling of strand break, NGS | T modific’n not detected; limited to SSB only | (36) |
| Single-strand breaks | Nick translation | ?>1 nt | SSB-seq | Nick translation with biotin dNTP, IP, NGS | Uncontrolled nuclease S1 trimming; SSB only | (35) |
| Double-strand breaks | TdT-labeling | ?>10 nt | DSB-seq | TdT-tailing with labeled dNTP, IP, NGS | T modific’n not detected; nuclease S1 trimming; SSB only | (35) |
MATERIALS AND METHODS
Materials
Nicking endonucleases Nb.BsmI, Nb.BspQI, Nb.BsrDI, endonuclease IV, DNA polymerase I, OneTaq DNA polymerase, dNTPs, Nci I, exonuclease III and RecJf were purchased from New England Biolabs. All DNA oligos were synthesized by Integrated Device Technology, Inc. ddNTPs and α-thio-dNTPs were purchased from TriLink BioTech. Agilent Bioanalyzer 2100 was used for size analysis of DNA fragments. Other chemicals were of molecular biology grade. All cell lines used in this work are readily available from the authors.
Cell growth and preparation of DNA
The PT-containing strain Salmonella enterica serovar Cerro 87 and its genomic DNA were prepared as described previously (8). Escherichia coli DH10B was used for nicking enzyme and H2O2-induced DNA damage mapping studies. A single colony of E. coli DH10B was grown in 5 ml LB medium overnight at 37°C. Cells (1 ml) were harvested by centrifuge at ambient temperature (unless indicated otherwise) and resuspended and diluted with fresh LB medium to a starting optical density at 600 nm (OD600) of 0.1, followed by growth at 37°C, 230 rpm until OD600 = 0.8 for DNA extraction or H2O2 treatment. Diluted H2O2 solution (10 μl) was added to the culture with a final concentration 0.1, 0.5, 1 and 2 mM. As un-exposed control, 10 μl sterile water was used instead of H2O2. After sitting at ambient temperature for 30 min, 10 μl of the cells were used for lethal dose (LD) analysis by counting the colony formation unit on LB agar plate. The remaining cells were harvested for DNA extraction with an OMEGA bacterial genomic DNA or plasmid isolation kit following the manufacture's protocol.
Mapping of modification/damage sites on DNA by NT-dependent method
These studies were initiated by random fragmentation of purified genomic DNA (1 μg) in each of three separate digestions with NciI, or HindIII and XhoI, or SalI, XbaI and NdeI. RNase A was also added to each reaction to remove contaminating RNA. After digestion, the DNA was purified using a Qiagen PCR Purification Kit. The three purified DNA samples were mixed for the blocking step. Blocking of pre-existing strand-break sites was achieved in a reaction mixture (40 μl) containing 4 μl of reaction buffer (NEBcutsmart buffer), 1 μl of shrimp alkaline phosphatase (NEB), and 1 μg of template genomic DNA, with incubation at 37°C for 30 min to remove phosphate at 3′ end of the strand-breaks. The phosphatase was then inactivated by heating at 70°C for 10 min. After cooling, 2 μl of ddNTPs (2.5 mM each, TriLink) and 1 μl of DNA polymerase I (10 U, NEB) was added to the reaction with incubation at 37°C for 40 min to block any pre-existing strand-break sites. Shrimp alkaline phosphatase (1 μl) was then added at 37°C for 30 min to degraded excess ddNTPs and the reaction was terminated by heating at 75°C for 10 min. Following de-salting using a DyeEx column (QIAGEN), the DNA was ready for one of the following nick creation or conversion procedures.
Nicking E. coli DH10B genomic DNA with Nb. BsmI and Nb. BsrDI was accomplished in a 50 μl reaction mixture containing 1 μl Nb. BsmI (10 U, NEB), 1 μl Nb. BsrDI (10 U, NEB), 1 μg genomic DNA, and 1× NEBcutsmart buffer incubated at 65°C for 1 h. The reaction was terminated by heating at 80°C for 20 min and cooled down to 4°C at the rate of 0.1°C/s. The reaction product was used for NT or TdT reactions as described below with no further purification.
For mapping PT modifications, 40 μl of blocked DNA from S. enterica was mixed with 5 μl of dibasic sodium phosphate buffer (500 mM, pH 9.0) and 2 μl of iodine solution (0.1 N, FLUKA). After incubation at 65°C for 5 min and cooling to 4°C, the reaction product was purified using a DyeEX column (QIAGEN) to remove salts and iodine. The purified product was treated with shrimp alkaline phosphatase by adding 5 μl of NEBcutsmart buffer and 1 μl of phosphatase to remove 3′-phosphates arising from iodine cleavage. After incubation at 37°C for 20 min and 75°C for another 10 min, the product was kept on ice for the following NT or TdT reactions with no additional purification.
For mapping H2O2-induced AP sites, genomic DNA was extracted from H2O2-treated E. coli and the AP sites converted to strand-breaks in a 50 μl reaction mixture containing 1 μg genomic DNA, endonuclease IV (20 U), and 1X NEBcutsmart buffer, with incubation at 37°C for 60 min. The reaction mixture was then kept on ice and used for the following NT- or TdT- reactions with no further inactivation or purification.
Following splitting of the nicked sample into two portions for NT- and TdT- reactions, the NT- reaction was achieved by further splitting the DNA sample into two parts: one for NT-reaction and the other as a negative control. NT-reaction was performed in a 50 μl reaction system containing 2 μl of α-thio-dNTPs (2.5 mM each, TRILINK), 1× of NEBcutsmart buffer, 2 μl of DNA polymerase I, and the DNA template. The negative control consisted of H2O instead of DNA polymerase I. The reaction mixture was incubated at 15°C for 90 min and then terminated by heating at 75°C for 20 min. The product was ready of template DNA digestion after the purification by DyeEx. The template DNA digestion reaction was performed in a 50 μl reaction system containing 200 U of exonuclease III, 5 μl NEBcutsmart buffer and DNA sample by incubating at 37°C for 60 min. The DNA was then denatured by heating at 95°C for 3 min and crashing on ice. RecJf (60 U) was then added to the reaction mixture with incubation at 37°C for 60 min. For some DNA samples with high complexity of structure and/or modifications, digestion with an additional 60 units of RecJf might be necessary. After digestion, the enzymes were inactivated by incubation at 80°C for 10 min. The DNA product was then purified using a Zymo Oligo Clean & Concentrator kit (Zymo) following the manufacturer's protocol. The purified product was ready for Illumina library preparation.
Illumina library preparation was performed by the Clontech SMART ChIp-seq kit (Clontech) by following the manufacturer's protocol. Twelve cycles were used in the final step of PCR amplification. The PCR product of each sample was combined with its corresponding negative control and then size selected using AMPure XP beads (NEB). The purified library was submitted to Illumina NextSeq 500 instrument for 75 bp paired-end sequencing.
Mapping of modification and damage sites by TdT-dependent method
Using the other half of the DNA sample, the steps of DNA fragmentation, blocking and nick conversion are the same as described above in NT-method. Nick-converted DNA (100 ng) was denatured by heating at 95°C for 3 min in 20 μl of H2O, followed by adding A poly(T) tail to the ssDNA in a 30 μl reaction system containing 3 μl DNA SMART buffer (Clontech), 1 μl terminal deoxynucleotidyl transferase (TdT, Clontech) and 1 μl DNA SMART T-Tailing Mix (Clontech) by incubating at 37°C for 20 min and terminating the reaction at 70°C for 10 min. The primer annealing and template switching reaction was then performed with the Clontech SMART ChIp-seq kit (Clontech) by following the manufacturer's protocol. The final step of PCR was perfomed using the Illumina primers provided in ChIp-seq kit and 12 cycles were used for amplification. The PCR product of each sample, with unique sequencing barcode, was combined with its corresponding negative control and then size selected using AMPure XP beads (NEB). The purified library was submitted to Illumina NextSeq 500 instrument for 75 bp paired-end sequencing.
Data analysis
Sequencing results were processed on the Galaxy web platform (https://usegalaxy.org/). Initially, the paired-end reads were pre-processed by Trim Galore! to remove adapters, as well as trimming the first 3 bp on the 5′ end of read 1. All the reads were aligned to the corresponding genome using Bowtie 2. A custom method for peak calling of sequencing data was developed with BamTools, BEDTools and Rstudio. Briefly, the BamTools results were filtered based on R1 (selected for NT data) or R2 (selected for TdT data). The 5′ coverage (experiment sample and controls) or full coverage (controls) on each position were calculated based on the filtered BamTools results by BEDTools (positive and negative strand separately). For both NT or TdT data on each strand, three ‘.tabular’ files containing the genome position and their corresponding read coverage (sample_coverage_5.tabular, control_coverage_5.tabular, control_coverage_full.tabular) were prepared using R. These data were used to normalize the read coverage by the sequencing depth and then calculate the read coverage ratio of specific position compared to its up- and downstream position in the same sample and the same sample in negative controls. Three ratios were calculated at each position by RStudio for modification site calling: coverage of position N (sample)/coverage of position N – 1(sample), coverage of position N(sample)/coverage of position N + 1(sample), and coverage of position N(sample)/coverage of position N(control). Positions with a ratio >1 were retained using the following R scripts: TdT_positive_strand.R TdT_negative_strand.R NT_positive_strand.R NT_negative_strand.R From these datasets, the intersection of the datasets from the NT and TdT methods were calculated using the following R scripts: TdT_positive+NT_negative.R TdT_negative+NT_positive.R The output files (CSV files; Excel format) contain the read coverage ratio information for the putative nick sites. The ratio cutoffs can be varied in the Excel spreadsheet as needed. For example, for site-specific nicking by Nb. BsmI and Nb. BsrDI, we determined that a ratio >2 was adequate to capture nearly all sites, while for variable sites (PT) or unknown samples (H2O2), the ratio was increased to 5–10.
RESULTS
Nick-seq design and data processing workflow
As shown in the Nick-seq method concept and workflow in Figure 1A, purified genomic DNA is first subjected to sequencing-compatible fragmentation and the resulting 3′-OH ends are blocked with dideoxyNTPs. The DNA modification is then converted to a strand-break by enzymatic or chemical treatment, followed by capture of the 3′- and 5′-ends of resulting strand-breaks using two complementary strategies. One portion of DNA is subjected to nick translation (NT) with α-thio-dNTPs to generate 100–200 nt phosphorothioate (PT)-containing oligonucleotides that are resistant to subsequent hydrolysis of the bulk of the genomic DNA by exonuclease III and RecJf. The purified PT-protected fragments are used to generate an NGS library with the modification of interest positioned at the 5′-end of the PT-labeled fragment. A second portion of the same DNA sample is used for terminal transferase (TdT)-dependent poly(dT) tailing of the 3′-end of the strand-break, with the tail used to create a sequencing library by reverse transcriptase template switching (9). Subsequent NGS positions the modification of interest 5′-end of the poly(dT) tail.
Figure 1.
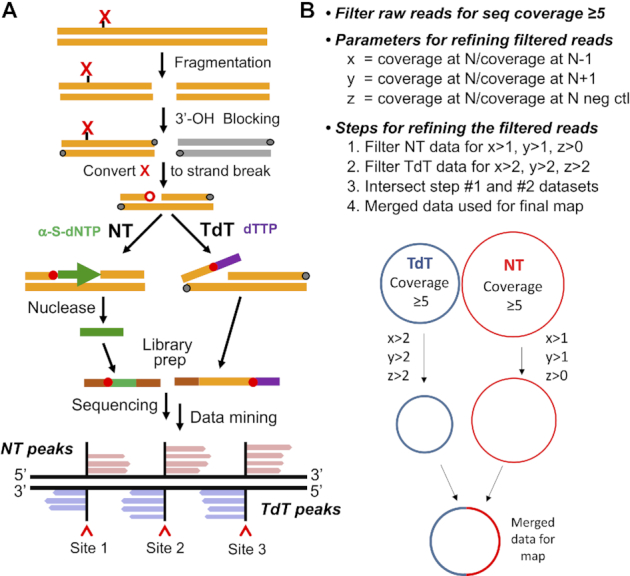
Overview of Nick-seq and data analysis workflow. (A) Nick-seq library preparation. Briefly, genomic DNA is first subjected to sequencing-compatible fragmentation; the resulting 3′-OH ends are blocked with dideoxyNTPs; the DNA modification is converted to a strand-break by enzymatic or chemical treatment; capture of the 3′- and 5′-ends of resulting strand-breaks using two complementary strategies: one portion of DNA is subjected to nick translation (NT) with α-thio-dNTPs to generate phosphorothioate (PT)-containing oligonucleotides that are resistant to subsequent hydrolysis of the bulk of the genomic DNA by exonuclease III and RecJf. The purified PT-protected fragments are used to generate an NGS library with the modification of interest positioned at the 5′-end of the PT-labeled fragment. A second portion of the same DNA sample is used for terminal transferase (TdT)-dependent poly(dT) tailing of the 3′-end of the strand-break, with the tail used to create a sequencing library by reverse transcriptase template switching (9). Subsequent NGS positions the modification of interest 5′-end of the poly(dT) tail. (B) Processing of the Nick-seq data includes: raw NGS reads are aligned to the reference genome for read coverage calculation; the genome sites with reads coverage ≥5 are then filtered for nick site calling with three parameters: x = the read coverage at position N/coverage at N – 1; y = coverage at position N/coverage at N + 1; z = coverage at position N/coverage at N of negative control sample. The site N is defined as a nick site if its x > 1, y > 1, z > 1 for NT reads and x > 2, y > 2, z > 2 for TdT reads.
The workflow for sequencing data processing (Figure 1B) uses the NT-derived reads as the primary dataset for developing a preliminary modification map, with TdT-derived reads as complementary corrective data. This hybrid approach exploits the fact that NT is agnostic to the base identify at the damage site but generates a high background of false positive sites, while TdT cannot be used with modifications occurring at dT due to loss of the poly(dT) tail during data analysis. The TdT reads are used to correct NT false-positive reads. For example, if NT maps a strand-break at T 1000 in the genome, then TdT reads are examined for a strand-break at position 999 and 1001. This one-nucleotide shift accommodates poly(dT) tail removal during data processing and validates the NT map. If TdT does not call a strand-break at 999 or 1001, then the NT result is considered a false positive. In other cases, if the NT-detected site occurs at a G, C or A, then TdT validates the same site. The use of both methods increases the sensitivity and specificity of the resulting genomic map.
Method validation by quantitative mapping of genomic DNA strand breaks
We validated Nick-seq by mapping DNA single-strand-breaks caused by the site-specific endonuclease Nb.BsmI, which cuts at G/CATTC motifs of which there are 2681 in the E. coli genome. Purified E. coli DNA was treated with Nb.BsmI and the Nick-seq-processed library sequenced using the Illumina NextSeq platform with an average of 107 raw sequencing reads for each sample (Figure 2A). Paired-end sequencing confirmed that >80% of reads uniquely aligned to the E. coli genome (Supplementary Table S1). For subsequent reads enrichment (Figure 1B), we calculated position-wise coverage values using the 5′-end of sequencing reads (NT read 1, TdT read 2) and used these values to define Nick-seq peaks as having >5 reads and 2-times more reads than sites located one-nucleotide up- and downstream. We then calculated the coverage ratio of the peaks relative to corresponding sites in an untreated DNA control. To identify the optimal minimal coverage ratio, we varied the ratio and calculated the number of identified sites at each ratio value (Figure 2B). As the coverage ratio increased from 2 to 7 for the combined TdT and NT data, the number of identified sites decreased from 92% to 59% of the 2681 expected sites (‘sensitivity’), while the accuracy (the number of identified sites divided by the number of expected sites; ‘specificity’) only increased from 98% to 99.5%. To maximize sensitivity, we chose a coverage ratio of 2, which allowed identification of 2462 (97.5%) of the predicted Nb.BsmI sites (Figure 2C, Supplementary Table S2). Another 1% of called sites (10) occurred in sequences differing from the consensus by one nucleotide. These sites showed lower average sequencing coverage (75 versus 1318) and likely represent Nb.BsmI ‘star’ activity. A second validation experiment with Nb.BsrDI, which cuts at N/CATTGC, showed no evident 3′-end sequence bias for DNA break site detection (Supplementary Table S3, Figure S1). These control studies showed that Nick-seq has high accuracy and sensitivity for single-nucleotide genomic mapping of DNA strand-breaks.
Figure 2.

Nick-seq validation. (A) Mapping single-strand breaks produced by Nb.BsmI in E. coli genomic DNA. Middle panel: Representative view of sequencing reads distributed in one genomic region. Red and green peaks mark reads mapped to forward and reverse strands of the genome, respectively. Lower panel: Amplification of the genomic region surrounding one peak, with read pile ups for TdT and NT sequencing converging on the site of the strand-break. (B) Nb.BsmI mapping data were used to define data processing parameters for accuracy and sensitivity of Nick-seq. Coverage ratios (the ratio of the peaks relative to corresponding sites in an untreated DNA control) were calculated for sequencing data performed with TdT alone (blue line) or the combination of TdT and NT (orange line). The sensitivity and specificity for detection of site-specific strand-breaks was then plotted for ratios ranging from 2 to 7. In general, higher coverage ratios yield greater accuracy but lower sensitivity, and the combination of TdT and NT provided significantly greater specificity. (C) With a coverage ratio of 2, Nick-seq identified 2462 (97.5%) of the 2681 predicted Nb.BsmI sites. Among the 62 (2.5%) ‘false-positive’ sites, 27 (1%) of them occurred in sequences differing from the consensus by one nucleotide. These sites showed lower average sequencing coverage (75 versus 1318) and likely represent Nb.BsmI ‘star’ activity.
Genomic mapping of DNA phosphorothioate modification by Nick-seq
The validated Nick-seq was applied to map the naturally-occurring phosphorothioate (PT) DNA modifications in Salmonella enterica serovar Cerro 87, using iodine to oxidize PTs to produce DNA strand-breaks (8) (Figure 3A). Using SMRT sequencing, we previously established that PTs occur as bistranded modifications at 10–15% of the 40,701 GAAC/GTTC motifs in the genome of E. coli B7A, which shares a nearly identical Dnd modification system as S. enterica Cerro 87 (8). Nick-seq recognized 12 239 PT sites in S. enterica, of which 11 684 (96%) occurred among the 32 795 possible GPSAAC/GPSTTC sites (37%), with 8568 (73%) modified on both strands and 27% modified on one strand (Figure 3B; data for PT sites on the S. enterica genome are detailed in Supplementary Table S4). This agrees with our previous observations using the orthogonal SMRT sequencing method (8). In addition to GAAC/GTTC motifs, Nick-seq also revealed less abundant PTs at GPSTAC (168), GPSATC (11), and GPSAAAC or GPSAAAAC sites (12), with half of GPSTAC and GPSATC sites modified on only one strand. These results indicate that Nick-seq has a higher sensitivity to detect PT modifications than SMRT sequencing (8).
Figure 3.
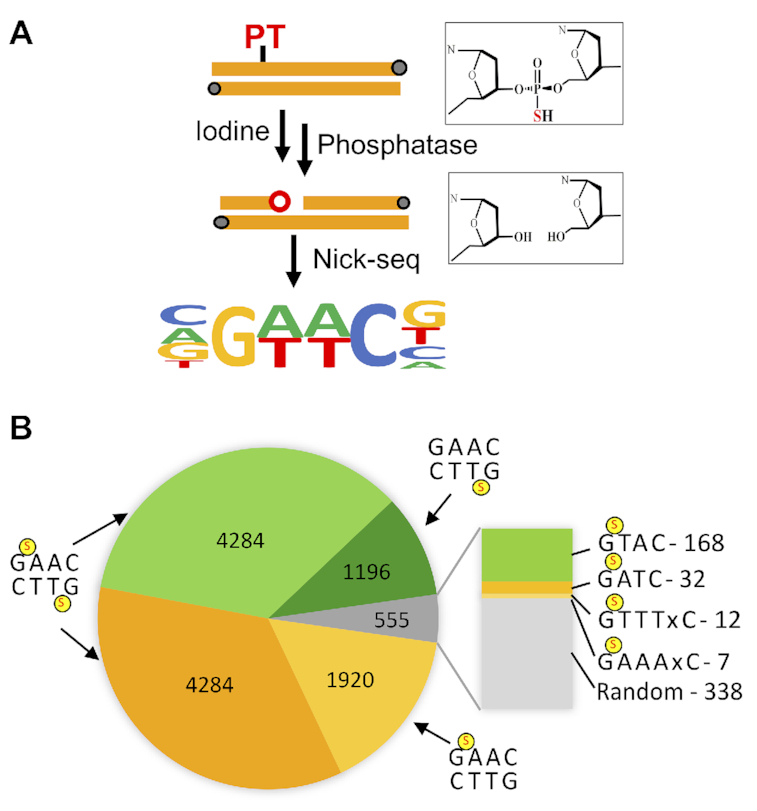
Mapping PTs across the S. enterica genome by Nick-seq. (A) Schematic showing the iodine cleavage method for converting PTs to strand-breaks. (B) the number of PT modification sites identified by Nick-seq in different sequence motifs. A total of 11 684 breaks were detected at the expected GAAC/GTTC motif, with single- and double-strand PTs denoted by a red ‘S’. The blow up shows minor modification motifs.
Nick-seq reveals H2O2-induced oxidative DNA damage sites on genome
Finally, we applied Nick-seq to DNA modifications not previously subjected to genomic mapping: oxidatively-induced abasic sites. Apurinic and apyrimidinic (AP) sites represent a prevalent and toxic form of DNA damage that blocks DNA replication and transcription (13). AP sites arise as intermediates in base excision DNA repair, in which damaged bases are excised by DNA glycosylases and the resulting AP sites are cleaved by AP endonucleases (14). AP sites can also arise by oxidation of DNA on both the nucleobase and 2′-deoxyribose moieties (15), as well as by demethylation of m5C epigenetic marks (16). In spite of the importance of AP sites, little is known about their formation, persistence, and distribution in genomic DNA. Here, we used Nick-seq to profile AP sites in E. coli exposed to hydrogen peroxide (H2O2) at a non-lethal dose of 0.2 mM (the LD50 for E. coli is ∼5 mM) (Figure 4A). Following DNA purification, AP sites were expressed as strand-breaks using endonuclease IV (EndoIV), which cleaves both native and oxidized AP sites (15,17). Nick-seq identified 1,519 EndoIV-sensitive sites in the genome (Figure 4B), as well as 82 sites in an endogenous plasmid (Figure 4C), with an unexposed sample of cells showing 11 and 8 sites, respectively. Data for AP sites in the E. coli genome and plasmid are detailed in Supplementary Tables S5 and S6. Considering the nucleobase precursor of the AP site, there was a weak preference for thymine (33%) followed by adenine (25%), cytosine (24%), and guanine (18%), with a similar distribution in the plasmid. This suggests that H2O2-derived DNA oxidizing agents either do not selectively oxidize guanine as predicted (18) or that the predominant form of damage is DNA sugar oxidation. However, there was a more pronounced sequence context effect. Analysis of the sequences 15 bp up- and down-stream of the AP sites revealed a strong preference for cytosine (47%) at –1 relative to the AP sites (Supplementary Figure S2). The distribution of AP sites was also non-random (Figure 4D and E). We observed the AP sites on the plasmid clustering in three regions related to DNA replication and transcription: the F1 origin, pUC origin, and AmpR gene (Figure 4E, Table 2). AP site clustering near DNA replication sites was observed previously by immunostaining (19), suggesting that the transcriptionally active and single-strand DNA are vulnerable to oxidatively-induced AP sites. We tested this by analyzing the distribution of AP sites in the E. coli genome relative to origins of replication (OriC), coding sequences, and non-coding sequences (Supplementary Table S5). While there was an average of 0.32 AP sites/kbp (1519 AP per 4 686 000 bp), the 20 kbp region around OriC showed 0.70 AP sites/kbp. Nick-seq also revealed 1401 AP sites in the 4.1 × 106 bp coding sequence region (0.34 AP/kb), and 118 AP sites (0.20 AP/kb) in the 0.58 × 106 bp non-coding region. These results suggest a preference for AP sites in DNA undergoing replication or transcription during H2O2 stress.
Figure 4.

Application of Nick-seq to quantify abasic sites. (A) generated by H2O2 exposure in E. coli. Cells were treated with a non-lethal dose of H2O2 (0.2 mM) and AP sites in isolated DNA were converted to strand-breaks with EndoIV, followed by Nick-seq mapping. (B) Detection of H2O2-induced EndoIV- sensitive DNA damage sites in E. coli. genomic DNA by Nick-seq. Data for AP sites in the E. coli genome are detailed in Supplementary Table S5. (C) Detection of H2O2-induced EndoIV- sensitive DNA damage sites in a plasmid maintained in this strain of E. coli. Data for AP sites in the E. coli genome are detailed in Supplementary Table S6. (D) The circos plot shows the locations of EndoIV-sensitive sites in E.coli genomic DNA. Outward from the center, circles represent: 0 and 0.2 mM H2O2 induced EndoIV-specific DNA damage sites. (E) The distribution of EndoIV-sensitive sites in the plasmid. Outward from the center, circles represent: 0 and 0.2 mM H2O2 induced EndoIV-specific DNA damage sites.
Table 2.
Frequency of AP sites distributed on an endogenous plasmid in H2O2-treated E. coli
| H2O2 | Functional region | Start position | End position | Size (bp) | # AP sites | Frequency | Sum of frequency |
|---|---|---|---|---|---|---|---|
| 0 mM | F1(+) Origin | 135 | 441 | 306 | 1 | 0.33% | 0.35% |
| LacZ(α) | 460 | 816 | 356 | 1 | 0.28% | ||
| LacZ Promoter | 817 | 938 | 121 | 0 | 0% | ||
| pUC Origin | 1158 | 1825 | 667 | 3 | 0.45% | ||
| AmpR Gene | 1976 | 2833 | 857 | 3 | 0.35% | ||
| Others | 654 | 0 | 0% | 0% | |||
| Average | 2961 | 8 | 0.27% | 0.27% | |||
| 0.2 mM | F1(+) Origin | 135 | 441 | 306 | 22 | 7.2% | 3.2% |
| LacZ(α) | 460 | 816 | 356 | 11 | 3.1% | ||
| LacZ Promoter | 817 | 938 | 121 | 2 | 1.6% | ||
| pUC Origin | 1158 | 1825 | 667 | 14 | 2.1% | ||
| AmpR Gene | 1976 | 2833 | 857 | 25 | 2.9% | ||
| Others | 654 | 8 | 1.2% | 1.2% | |||
| Average | 2961 | 82 | 2.8% | 2.8% |
DISCUSSION
The ability to map DNA damage and physiological modifications at single nucleotide-resolution across genomes is critical to understanding the biological function of epigenetic marks and the mutational and genomic instability consequences of DNA lesions caused by both endogenous and exogenous genotoxic agents. As summarized in Table 1, the wide recognition of the impact of genome mapping is evident in the growing number of methods that couple DNA sequencing technology with chemical derivatisation of the DNA modifications, enrichment of modified DNA fragments, or deconvolution sequencing signals to identify the modifications. Resolution at the level of hundreds of nucleotides can be achieved by fragmenting the DNA followed by affinity purification of modification-containing fragments using antibodies or other affinity reagents (6,12,20,21). To locate a modified base at single-nucleotide resolution, a selective chemical transformation of the modified nucleotides must be applied, with subsequent DNA processing that preserves the location of the modification at the exact 5′- or 3′-end of the final DNA fragment in the sequencing library (22–26). For example, bisulfite selectively deaminates C, but not 5mC, to T, with routine DNA sequencing revealing the C as a T mutation and 5mC retained as C, which can be confirmed by parallel sequencing of untreated DNA (22). Similarly, 5hmC can be oxidized to 5-formylcytidine (5fC) that is then converted to U by bisulfite, with deconvolution by comparison of the C-to-U transition in the genomic maps (23). Single-nucleotide resolution can also be achieved with third-generation sequencing technologies such as SMRT and nanopore sequencing, although both approaches suffer from low sensitivity due to the need to distinguish modification-specific signals from noise, with many modifications not producing a detectable signal (4,27).
Considering the fact that many types of DNA modifications can be converted to strand-breaks with modification-dependent restriction endonucleases (MRE) (28), DNA base excision repair (BER) enzymes (10) and their associated AP endonucleases (14), and nucleotide excision repair (10) enzymes, several groups have attempted to detect DNA lesion-derived strand breaks instead of the lesion itself. For example, the MRE AbaSI cleaves DNA on either side of 5-glucosylhydroxymethylcytosine (5ghmC) and 5hmC to facilitate mapping (29). However, the limited availability and specificity of MREs makes them highly specialized tools for modification mapping.
Both BER and NER enzymes excise DNA damage products with predictable precision necessary for single-nucleotide resolution mapping. For example, the Burrows group succeeded in detecting DNA lesions on synthesized oligonucleotides by installation of either the dNaM or d5SICS (marker nucleotide) at the lesion site after processing with a BER enzyme followed by Nanopore sequencing to detect the dNaM or d5SICS (30). Another method developed by Hesselberth and coworkers, Excision-seq, applies BER enzymes coupled with NGS to map several types of DNA damage on a genomic scale (11). Excision-seq comprises two approaches: a ‘pre-digestion’ method relying on closely-spaced BER enzyme-sensitive damage to create 5′ and 3′ ends for double-strand ligation. This requires very high, biologically-irrelevant levels of the damage product. The ‘post-digestion’ method in Excision-seq uses pre-sheared, BER-treated genomic DNA to destroy the damage-containing DNA fragments, which allows identification of undamaged regions of the genome. Thus, this method suffers from low resolution and low sensitivity, with a PCR step that could confound the sequencing if PCR-blocking modifications are present. The Sturla group mapped 8-oxoG in the yeast genomic DNA using a method they termed Click-code-Seq. Here, the free 3′-OH generated by BER enzyme excision was labelled with a synthetic O-3′-propargyl-modified 2′-deoxyribonucleotide (prop-dGTP) to generate a 3′-alkynyl-modified DNA that can be ligated and enriched by a 5′-azido- and biotin-modified code sequence using a copper(I)-catalyzed click reaction and biotin-avidin affinity purification (31). Both Excision-seq and Click-code-seq methods could be adapted to analyze other types of DNA damage with appropriate BER and NER enzymes. The main limitation of using DNA repair enzymes to express damage as strand-breaks, including Nick-seq, lies in the relatively broad specificity of the enzymes for different chemical classes of damage (Table 3) (32), as well as dual enzymatic functions. For example, E. coli FAPY DNA glycosylase (Fpg) recognizes a variety of oxidized purine base products, and contains an AP endonuclease activity (33,34). Similarly, E. coli NTH recognizes oxidized pyrimidine nucleobase lesions (34). However, the catalytic efficiency and specificity of DNA repair enzymes is high enough to confidently map relatively narrow classes of damage products using different sets of enzymes.
Table 3.
DNA modifications and damage amenable to Nick-seq mapping
| DNA modification | Base excision enzymea | Additional processingb | Reference |
|---|---|---|---|
| DNA repair intermediates (abasic sites, strand-breaks) | None | Endo IV (NEB) | (15) |
| Bulky adducts formed by benzo[a]pyrene, acrolein, aflatoxin, et al. | UvrABC | None | (43) |
| 8-Oxoguanine, formamidopyrimidine | FPG, hOGG1 (NEB) | None | (44) |
| UV photodimers (TT, TC, CC) | T4 Endo V (NEB) | Endo IV (NEB) for 3′-UA | (45) |
| 1,N6-Ethenoadenine, hypoxanthine | hAAG (NEB) | Endo IV (NEB) for AP | (45) |
| Uracil | UDG (NEB) | Endo IV (NEB) for AP | (45) |
| A:8-oxoG mispair | MutY (RD) | None | (45) |
| Thymine glycol, 5-hydroxyuracil, 6-dihydroxythymine, 5-hydroxycytosine, urea | Endo III (NEB) | Endo IV (NEB) for AP | (46) |
| T:G mispair | TDG (RD) | Endo IV (NEB) for AP | (45) |
| N3-Methylcytosine, N1-methyladenine, N3-methyladenine, N7‐methylguanine | AlkC, AlkD | Endo IV (NEB) for AP | (47) |
| Hypoxanthine, xanthine | E. coli EndoV (NEB) | Endo IV (NEB) for AP | (48) |
| Single-strand nicking endonucleases | >20 types (NEB) | None | (49) |
| 5-Methylcytosine | TET, TDG | Endo IV (NEB) for AP | (50) |
| 5-Methylcytosine | DEMETER, ROS1, DML2 and DML3 | Endo IV (NEB) for AP | (51) |
| 5-Methylcytosine | Bisulfite seq + TDG (RD) | Endo IV (NEB) for AP | (52) |
| 5-Hydroxymethyl-cytosine | TET/bisulfite seq. + TDG (RD) | Endo IV (NEB) for AP | (52) |
| DNase I footprinting | DNase I (NEB) | None | (53) |
aEnzyme suppliers in parentheses: NEB, New England Biolabs; RD, R&D Systems; other abbreviations: TDG, thymine DNA glycosylase; Endo, endonuclease; UDG, uracil DNA glycosylase.
bProcessing to remove 3′-α, β-unsaturated aldehydes (3′-UA) or abasic (AP) sites for Nick-seq library preparation (46).
Two groups have reported methods similar to Nick-seq for genome-wide mapping of single-strand breaks (Table 1). Baranello et al. (35) used both TdT-tailing and NT to label 3′-hydroxyl-ended double- (DSB-seq) and single-strand breaks (SSB-seq), respectively, with biotinylated and digoxigenin-labeled nucleotides, respectively, for affinity enrichment of the labelled DNA fragments. In addition to a lack of quantitative validation, the DSB-seq method cannot be used to map single-strand breaks occurring at Ts due to the polyA-tailing problem noted earlier. Furthermore, for both DSB-seq and SSB-seq, an imprecise exonuclease trimming step to remove the biotin and digoxigenin tags confounds subsequent localization of the break site (35). These methods are thus potentially useful for genomically ballparking strand-breaks but not for quantitative single-nucleotide resolution.
The other method, SSiNGLe, is quite different from Nick-seq and is limited solely to mapping single-strand breaks with 3′-hydroxyl groups (36). Unfortunately, the method cannot be applied to any other form of DNA modification or damage and cannot be used for single-strand breaks with ‘dirty 3′-ends’ as occur in DNA repair intermediates (e.g. 3′-[4-hydroxy-5-phospho-2-pentenal] or 3′-phosphate) and strand-breaks caused by DNA oxidation (e.g. 3′-formylphosphate and others) (37). The requirement for micrococcal nuclease fragmentation of the DNA in formaldehyde-fixed nuclei prior to DNA purification further restricts the method since chemical or enzymatic conversion of DNA modifications to single-strand breaks will be biased by the presence of chromatin proteins, as occurs in chromatin footprinting methods that use chemicals or ionizing radiation (e.g. see (38–40). In fact, the DNA fragmentation with micrococcal nuclease relies on the presence of nucleosomes to limit the fragmentation to the ∼150 nt periodicity of nucleosome-bound DNA (40). Furthermore, the accuracy of SSiNGLe for single-nucleotide resolution is relatively low, with only 87–95% of the breaks mapping to ± 1 nt of the true cutting site of the single-strand endonuclease Nt.BbvCI used to validate the method (36), with no mention of the number of missed consensus sites. The combination of NT and TdT labelling in Nick-seq significantly increases the accuracy of the method, with 98% of the exact cutting sites accurately called. Finally, SSiNGLe cannot be used to map single-strand breaks occurring at Ts due to the polyA-tailing problem noted earlier.
Compared to existing methods (Table 1), Nick-seq provides a highly sensitive and quantitative general approach to mapping a variety of epigenetic marks and DNA damage products (Table 3) at single-nucleotide resolution in any genome. While the two complementary sequencing strategies—NT and TdT tailing—can be used individually for genomic mapping of DNA strand breaks, the pairing of the two approaches increases the sensitivity and accuracy of the mapping. Each library preparation approach complements the deficiencies of the other: NT generates a nuclease-resistant PT-labeled oligonucleotide that obviates the TdT problem of assigning break sites at thymines, while the TdT library preparation produces lower background sequencing signals than NT. We believe that the relatively high background associated with NT mapping resulted from the use of exonuclease III (3′→5′ direction) combined with RecJf (5′→3′) for hydrolysis of genomic DNA and release of the phosphorothioate-containing oligonucleotides generated by NT with α-thio-dNTPs. Nuclease P1 is another nuclease that is inhibited by naturally-occurring Rp configuration PT linkages and we have successfully used it for DNA hydrolysis to release PT-containing dinucleotides in our chromatography-coupled mass spectrometry method for quantifying PTs (8). Indeed, we initially used nuclease P1 in the development of the NT method and it showed higher efficiency for removal of genomic DNA and lower background sequencing noise than the ExoIII/RecJf combination. However, batch to batch contamination of commercial preparations of nuclease P1 with PT-cleaving nucleases obviated the use of this nuclease in Nick-seq.
Nick-seq was shown to be highly sensitive and accurate for mapping DNA breaks and modifications at single-nucleotide resolution. The method detected 2462 of the 2681 Nb.BsmI sites in the E. coli genome, which amounts to >97% specificity and <1% false positives. In the mapping of PT modifications in the S. enterica genome, Nick-seq revealed 2- to 3-times the density of modification sites, with ∼30% of GAAC/GTTC motifs modified, compared to our previous studies of SMRT PT mapping in E. coli B7A (10–15% modified), which possesses an identical Dnd protein system that targets GAAC/GTTC (8). This is not surprising since SMRT sequencing technology is relatively insensitive to PT modifications compared to DNA methylation and relies on sampling statistics to call a particular modification site (8). Interestingly, among the extra detected sites in S. enterica, PTs were found in GATC sites well-known for Dam-mediated methylation (G6mATC). This agrees with our previous observation that d(GPS6mA) dinucleotides was detected at low levels in mass spectrometric analyses of this E.coli strain. Finally, while our observation of AP sites enriched in sites of replication and transcription is consistent with other published studies (19), the use of Nick-seq to map H2O2-induced AP sites revealed an unexpected formation of AP sites at roughly equal proportions at T, C, A and G, which is not consistent with the idea – based on studies in purified DNA – that G should be the most reactive site for oxidation (18). This is not altogether surprising since AP sites can arise by both 2′-deoxyribose and nucleobase oxidation in DNA, with our results revealing new features AP site formation in vivo. It will be especially informative to compare the AP site maps to maps of DNA glycosylase-sensitive lesions at both purines and pyrimidines (Table 3).
As detailed in Table 3, there are numerous potential applications of Nick-seq for genomic mapping of DNA modifications, including a variety of DNA damage products, DNA repair intermediates (abasic sites, strand-breaks), and epigenetic marks (e.g. 5-methylcytosine, 5-hydroxymethylcytosine). The method can also be used to characterize restriction break sites in many restriction-modification systems (41) and mapping genomic landmarks by DNase I footprinting (40), for example. Nick-seq thus provides a widely-applicable, efficient, label-free approach to quantitative mapping of DNA damage and modifications in genomes.
DATA AVAILABILITY
Custom scripts for processing the sequencing data are described in Methods and are available at https://github.com/BoCao2019/Nick-seq: .gitignore, NT_negative_strand.R, NT_positive_strand.R, TdT_negative+NT_positive.R, TdT_negative_strand.R, TdT_positive+NT_negative.R, and TdT_positive_strand.R.
Sequencing data has been deposited in NCBI GEO database under accession numbers GSE138070, GSE138173 and GSE138476.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the MIT BioMicro Center, MIT Center for Environmental Health Science, Singapore-MIT Alliance for Research and Technology (SMART) for use of facilities.
Notes
Present address: Chen Gu, Merck Research Laboratories, Merck & Co., Inc., Boston, MA 02115, USA.
Contributor Information
Bo Cao, College of Life Sciences, Qufu Normal University, Qufu, Shandong 273165, China; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Singapore-MIT Alliance for Research and Technology, Antimicrobial Drug Resistance Interdisciplinary Research Group, Singapore 138602, Singapore; State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic and Developmental Sciences, and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Xiaolin Wu, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Singapore-MIT Alliance for Research and Technology, Antimicrobial Drug Resistance Interdisciplinary Research Group, Singapore 138602, Singapore; Key Laboratory of Combinatorial Biosynthesis and Drug Discovery, Ministry of Education and School of Pharmaceutical Sciences, Wuhan University, Wuhan, Hubei 430071, China.
Jieliang Zhou, KK Research Center, KK Women's and Children's Hospital, 229899, Singapore.
Hang Wu, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; School of Life Sciences, Anhui University, Hefei, Anhui 230601, China.
Lili Liu, College of Life Sciences, Qufu Normal University, Qufu, Shandong 273165, China.
Qinghua Zhang, College of Life Sciences, Qufu Normal University, Qufu, Shandong 273165, China.
Michael S DeMott, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Chen Gu, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Lianrong Wang, Key Laboratory of Combinatorial Biosynthesis and Drug Discovery, Ministry of Education and School of Pharmaceutical Sciences, Wuhan University, Wuhan, Hubei 430071, China.
Delin You, State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic and Developmental Sciences, and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China.
Peter C Dedon, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Singapore-MIT Alliance for Research and Technology, Antimicrobial Drug Resistance Interdisciplinary Research Group, Singapore 138602, Singapore; Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31630002]; National Science Foundation of the USA [CHE-1709364], National Research Foundation of Singapore through the SMART Infectious Disease and Antimicrobial IRGs, National Institute of Environmental Health Sciences [R01-ES031576, P30-ES002109]; Fundamental Research Funds for the Central Universities of China [2015306020202]; X.W. was supported by China Scholarship Council fellowship [201606270163]. Funding for open access charge: NIH and NSF.
Conflict of interest statement. B.C., M.S.D. and P.C.D. are co-inventors on a PCT patent (PCT/US2019/013714) and US Patent (US 2019/0284624 A1) relating to the published work.
REFERENCES
- 1. Roos W.P., Thomas A.D., Kaina B.. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 2016; 16:20–33. [DOI] [PubMed] [Google Scholar]
- 2. Chen Y., Hong T., Wang S., Mo J., Tian T., Zhou X.. Epigenetic modification of nucleic acids: from basic studies to medical applications. Chem. Soc. Rev. 2017; 46:2844–2872. [DOI] [PubMed] [Google Scholar]
- 3. Li Q., Hermanson P.J., Springer N.M.. Detection of DNA methylation by Whole-Genome bisulfite sequencing. Methods Mol. Biol. 2018; 1676:185–196. [DOI] [PubMed] [Google Scholar]
- 4. Clark T.A., Spittle K.E., Turner S.W., Korlach J.. Direct detection and sequencing of damaged DNA bases. Genome Integr. 2011; 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schibel A.E., An N., Jin Q., Fleming A.M., Burrows C.J., White H.S.. Nanopore detection of 8-oxo-7,8-dihydro-2′-deoxyguanosine in immobilized single-stranded DNA via adduct formation to the DNA damage site. J. Am. Chem. Soc. 2010; 132:17992–17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ding Y., Fleming A.M., Burrows C.J.. Sequencing the mouse genome for the oxidatively modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017; 139:2569–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grimm C., Adjaye J.. Analysis of the methylome of human embryonic stem cells employing methylated DNA immunoprecipitation coupled to next-generation sequencing. Methods Mol. Biol. 2012; 873:281–295. [DOI] [PubMed] [Google Scholar]
- 8. Cao B., Chen C., DeMott M.S., Cheng Q.X., Clark T.A., Xiong X.L., Zheng X.Q., Butty V., Levine S.S., Yuan G. et al.. Genomic mapping of phosphorothioates reveals partial modification of short consensus sequences. Nat. Commun. 2014; 5:3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu Y.Y., Machleder E.M., Chenchik A., Li R., Siebert P.D.. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. BioTechniques. 2001; 30:892–897. [DOI] [PubMed] [Google Scholar]
- 10. Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S.. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004; 73:39–85. [DOI] [PubMed] [Google Scholar]
- 11. Bryan D.S., Ransom M., Adane B., York K., Hesselberth J.R.. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014; 24:1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amente S., Di Palo G., Scala G., Castrignano T., Gorini F., Cocozza S., Moresano A., Pucci P., Ma B., Stepanov I. et al.. Genome-wide mapping of 8-oxo-7,8-dihydro-2-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019; 47:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clauson C.L., Oestreich K.J., Austin J.W., Doetsch P.W.. Abasic sites and strand breaks in DNA cause transcriptional mutagenesis in Escherichia coli. Proc. Natl Acad. Sci. U.S.A. 2010; 107:3657–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dianov G.L., Sleeth K.M., Dianova I.I., Allinson S.L.. Repair of abasic sites in DNA. Mutat. Res. 2003; 531:157–163. [DOI] [PubMed] [Google Scholar]
- 15. Greenberg M.M., Weledji Y.N., Kim J., Bales B.C.. Repair of oxidized abasic sites by exonuclease III, endonuclease IV, and endonuclease III. Biochemistry. 2004; 43:8178–8183. [DOI] [PubMed] [Google Scholar]
- 16. Wu X., Zhang Y.. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017; 18:517–534. [DOI] [PubMed] [Google Scholar]
- 17. Xu Y.J., Kim E.Y., Demple B.. Excision of C-4′-oxidized deoxyribose lesions from double-stranded DNA by human apurinic/apyrimidinic endonuclease (Ape1 protein) and DNA polymerase beta. J. Biol. Chem. 1998; 273:28837–28844. [DOI] [PubMed] [Google Scholar]
- 18. Dedon P.C., Tannenbaum S.R.. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004; 423:12–22. [DOI] [PubMed] [Google Scholar]
- 19. Chastain P.D. II, Nakamura J., Rao S., Chu H., Ibrahim J.G., Swenberg J.A., Kaufman D.G.. Abasic sites preferentially form at regions undergoing DNA replication. FASEB J. 2010; 24:3674–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schumacher A., Kapranov P., Kaminsky Z., Flanagan J., Assadzadeh A., Yau P., Virtanen C., Winegarden N., Cheng J., Gingeras T. et al.. Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res. 2006; 34:528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu Y.W., Huang R.L., Lai H.C.. MeDIP-on-Chip for methylation profiling. Methods Mol. Biol. 2015; 1249:281–290. [DOI] [PubMed] [Google Scholar]
- 22. Yong W.S., Hsu F.M., Chen P.Y.. Profiling genome-wide DNA methylation. Epigen Chrom. 2016; 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Booth M.J., Branco M.R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S.. Quantitative sequencing of 5-Methylcytosine and 5-Hydroxymethylcytosine at Single-Base resolution. Science. 2012; 336:934–937. [DOI] [PubMed] [Google Scholar]
- 24. Liu Y., Siejka-Zielinska P., Velikova G., Bi Y., Yuan F., Tomkova M., Bai C., Chen L., Schuster-Bockler B., Song C.X.. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019; 37:424–429. [DOI] [PubMed] [Google Scholar]
- 25. Liu Z.J., Cuesta S.M., van Delft P., Balasubramanian S.. Sequencing abasic sites in DNA at single-nucleotide resolution. Nat. Chem. 2019; 11:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li W.T., Adebali O., Yang Y.Y., Selby C.P., Sancar A.. Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc. Natl Acad. Sci. U.S.A. 2018; 115:E3408–E3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beaulaurier J., Schadt E.E., Fang G.. Deciphering bacterial epigenomes using modern sequencing technologies. Nat Rev Genet. 2019; 20:157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loenen W.A., Raleigh E.A.. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 2014; 42:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sun Z.Y., Terragni J., Borgaro J.G., Liu Y.W., Yu L., Guan S.X., Wang H., Sun D.P., Cheng X.D., Zhu Z.Y. et al.. High-Resolution enzymatic mapping of genomic 5-Hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013; 3:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riedl J., Ding Y., Fleming A.M., Burrows C.J.. Identification of DNA lesions using a third base pair for amplification and nanopore sequencing. Nat. Commun. 2015; 6:8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu J.Z., McKeague M., Sturla S.J.. Nucleotide-resolution genome-wide mapping of oxidative DNA damage by click-code-Seq. J. Am. Chem. Soc. 2018; 140:9783–9787. [DOI] [PubMed] [Google Scholar]
- 32. O’Brien P.J. Catalytic promiscuity and the divergent evolution of DNA repair enzymes. Chem. Rev. 2006; 106:720–752. [DOI] [PubMed] [Google Scholar]
- 33. Tchou J., Kasai H., Shibutani S., Chung M.H., Laval J., Grollman A.P., Nishimura S.. 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boiteux S. Properties and biological functions of the NTH and FPG proteins of Escherichia coli: two DNA glycosylases that repair oxidative damage in DNA. J. Photochem. Photobiol. B. 1993; 19:87–96. [DOI] [PubMed] [Google Scholar]
- 35. Baranello L., Kouzine F., Wojtowicz D., Cui K., Przytycka T.M., Zhao K., Levens D.. DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci. 2014; 15:13111–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cao H., Salazar-Garcia L., Gao F., Wahlestedt T., Wu C.L., Han X., Cai Y., Xu D., Wang F., Tang L. et al.. Novel approach reveals genomic landscapes of single-strand DNA breaks with nucleotide resolution in human cells. Nat. Commun. 2019; 10:5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dedon P.C. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem. Res. Toxicol. 2008; 21:206–219. [DOI] [PubMed] [Google Scholar]
- 38. Hayes J.J., Tullius T.D., Wolffe A.P.. The structure of DNA in a nucleosome. Proc. Natl Acad. Sci. U.S.A. 1990; 87:7405–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Staynov D.Z., Crane-Robinson C.. Footprinting of linker histones H5 and H1 on the nucleosome. EMBO J. 1988; 7:3685–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sollner-Webb B., Melchior W. Jr, Felsenfeld G.. DNase I, Dnase II and staphylococcal nuclease cut at different, yet symmetrically located, sites in the nucleosome core. Cell. 1978; 14:611–627. [DOI] [PubMed] [Google Scholar]
- 41. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q., Fang L., Yu G.L., Wang D.P., Xiao C.L., Wang K.. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019; 10:2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feng Z., Hu W., Hu Y., Tang M.S.. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:15404–15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sidorenko V.S., Grollman A.P., Jaruga P., Dizdaroglu M., Zharkov D.O.. Substrate specificity and excision kinetics of natural polymorphic variants and phosphomimetic mutants of human 8-oxoguanine-DNA glycosylase. FEBS J. 2009; 276:5149–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wallace S.S. Base excision repair: a critical player in many games. DNA Repair. 2014; 19:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ide H. DNA substrates containing defined oxidative base lesions and their application to study substrate specificities of base excision repair enzymes. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:207–221. [DOI] [PubMed] [Google Scholar]
- 47. Alseth I., Rognes T., Lindback T., Solberg I., Robertsen K., Kristiansen K.I., Mainieri D., Lillehagen L., Kolsto A.B., Bjoras M.. A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol. Microbiol. 2006; 59:1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vongchampa V., Dong M., Gingipalli L., Dedon P.. Stability of 2′-deoxyxanthosine in DNA. Nucleic Acids Res. 2003; 31:1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chan S.H., Stoddard B.L., Xu S.Y.. Natural and engineered nicking endonucleases–from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res. 2011; 39:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weber A.R., Krawczyk C., Robertson A.B., Kusnierczyk A., Vagbo C.B., Schuermann D., Klungland A., Schar P.. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 2016; 7:10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee J., Jang H., Shin H., Choi W.L., Mok Y.G., Huh J.H.. AP endonucleases process 5-methylcytosine excision intermediates during active DNA demethylation in Arabidopsis. Nucleic Acids Res. 2014; 42:11408–11418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu M., Hon G.C., Szulwach K.E., Song C.X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B. et al.. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012; 149:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sung M.H., Baek S., Hager G.L.. Genome-wide footprinting: ready for prime time. Nat. Methods. 2016; 13:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Custom scripts for processing the sequencing data are described in Methods and are available at https://github.com/BoCao2019/Nick-seq: .gitignore, NT_negative_strand.R, NT_positive_strand.R, TdT_negative+NT_positive.R, TdT_negative_strand.R, TdT_positive+NT_negative.R, and TdT_positive_strand.R.
Sequencing data has been deposited in NCBI GEO database under accession numbers GSE138070, GSE138173 and GSE138476.