


Abstract
A key aim in exploiting CRISPR–Cas is gRNA engineering to introduce additional functionalities, ranging from individual nucleotide changes that increase efficiency of on-target binding to the inclusion of larger functional RNA aptamers or ribonucleoproteins (RNPs). Cas9–gRNA interactions are crucial for complex assembly, but several distinct regions of the gRNA are amenable to modification. We used in vitro ensemble and single-molecule assays to assess the impact of gRNA structural alterations on RNP complex formation, R-loop dynamics, and endonuclease activity. Our results indicate that RNP formation was unaffected by any of our modifications. R-loop formation and DNA cleavage activity were also essentially unaffected by modification of the Upper Stem, first Hairpin and 3′ end. In contrast, we found that 5′ additions of only two or three nucleotides could reduce R-loop formation and cleavage activity of the RuvC domain relative to a single nucleotide addition. Such modifications are a common by-product of in vitro transcribed gRNA. We also observed that addition of a 20 nt RNA hairpin to the 5′ end of a gRNA still supported RNP formation but produced a stable ∼9 bp R-loop that could not activate DNA cleavage. Consideration of these observations will assist in successful gRNA design.
INTRODUCTION
The microbial CRISPR–Cas systems, and in particular type II-A Cas9, have found widespread utility as tools for site-specific DNA and RNA recognition (1,2). They can be readily programmed by changing the spacer sequence of a crRNA that recognises a DNA target protospacer by forming an R-loop, binding to the target strand (TS) through Watson-Crick base pairing (3). DNA recognition also requires binding of a protospacer adjacent motif (PAM), e.g. Streptococcus pyogenes (Sp) Cas9 binds 5′-NGG-3′ while Streptococcus thermophilus DGCC7710 CRISPR3 (St3) Cas9 binds 5′-NGGNG-3′ (4,5). Cas9 evolved to use a trans-activating RNA (tracrRNA) that base pairs via an anti-repeat sequence to the repeat sequence of individual crRNAs (4,6). The single gRNA more commonly used in tool applications is a fusion of the tracrRNA and crRNA through a short RNA loop between the repeat-anti-repeat sequences in the Upper Stem (Figure 1) (4). An important area of research has been the manipulation of the gRNA sequence and structure, through introduction of extra or modified nucleotides, and/or the addition of folded RNA aptamers (7,8). Using a combination of ensemble and single-molecule biochemical assays, we examined how gRNA modification affects ribonucleoprotein (RNP) assembly, R-loop stability and DNA cleavage. Our results indicate that SpCas9 R-loop formation and DNA cleavage activity is acutely sensitive to changes to the 5′ end of the RNA, even when the addition is only two nucleotides.
Figure 1.
Streptococcus pyogenes Cas9 gRNA structure. (A) The structure of SpCas9 (surface render in blue) in complex with gRNA (black) and target DNA (grey) from PDB 4OO8 (9). The gRNA and protospacer DNA structures are shown separately with structural features labelled: spacer (black); Lower Stem (green); Bulge (orange); Upper Stem (blue); Nexus (beige); Hairpins (purple). (B) (left) gRNA schematic as used in other figures with colouring consistent with panel B. (right) gRNA heatmap, based on Briner et al. (10), showing gRNA regions which tolerate (purple), partially tolerate (orange) and do not tolerate (grey) modification. (C) Schematic of the magnetic tweezers assay. See main text for details.
The highly-folded Lower and Upper Stems, Nexus and Hairpin regions of the gRNA make extensive contacts with positively-charged surfaces of Cas9 (Figure 1A): Cas9 folds around the RNA/DNA duplex and the Lower Stem-Bulge-Upper Stem region using the recognition (REC) and nuclease (NUC) lobes; much of the hairpin regions of the gRNA contact a positively-charged region on the Cas9 outer surface; the gRNA provides most of the interactions between the REC and NUC lobes which themselves do not form extensive protein-protein interactions. Removing structures with extensive Cas9-contacts, such as the Bulge and Nexus, abolished Cas9 function while mutations were tolerated in other regions of the gRNA, particularly those not directly contacted by Cas9 such as the Upper Stem (9). An extensive study by Briner et al. confirmed the importance of the Bulge and Nexus and also highlighted the possibility of modifying the 5′ and 3′ termini, Upper Stem and first Hairpin (10). These studies can be summarised as a theorised ‘heatmap’ of modification tolerance, Figure 1B.
Modification of the 5′ end of gRNAs is a familiar by-product of the everyday use of Cas9 in gene editing. T7 bacteriophage RNA polymerase is commonly used for in vitro transcription (IVT) of gRNAs. The T7 promoter has been minimised for in vitro use; whereas naturally occurring initiation sequences have up to 3 guanines in the +1, +2 and +3 positions (11), the minimal requirement for initiation is one guanine in the +1 position resulting in a single guanine at the 5′ end of the spacer sequence. Nonetheless, two or more guanines are often recommended to ensure higher yields of transcribed gRNA. Commonly used mammalian promoters for cellular gRNA transcription have similar initiation requirements; gRNAs expressed from the U6 promoter necessitates a G or A in the (+1) position for high expression. By careful choice of protospacer target sequence, these nucleotides can be incorporated into the spacer and thus R-loop DNA–RNA hybrid. However, in many cases this is not feasible, and the additional nucleotides become an unpaired overhang. There are additional problems with T7-based IVT due to non-templated effects, such as nucleotide additions to the 3′ of the transcript (12,13) or templated effects, where the transcript folds back on itself in cis to prime extension (14,15).
Unpaired nucleotides at the 5′ end of the spacer sequence can influence SpCas9 activity in a protein and target site-dependent manner (16,17). For WT Cas9, one or two additional 5′ guanines lead to increased specificity due to reduced unwinding promiscuity. However, two 5′ guanines caused a decrease in on-target activity. For the enhanced SniperCas9, one unpaired 5′ guanine had a positive effect in increasing the sensitivity to mismatches but two unpaired 5′ guanines lowered specificity. Other engineered Cas9s showed either reduced on-target activity (eCas9 and HypaCas9) or increased promiscuity (Cas9-HF1). These pleiotropic effects are most likely due to the observed interactions of the docked RuvC domain with the 5′ end of the RNA (9,17). Unpaired 5′ nucleotides could produce distortion of the DNA:RNA hybrid in this region that may influence R-loop formation, and possibly DNA cleavage.
There are also numerous examples of gRNA that have had additional functional RNA structures appended [(reviewed in (7)], including ribozymes at the 5′ end (16,18), modification of the upper stem with RNA aptamer binding effectors to colocalise fluorescent proteins (19), and 3′-fusion of viral RNA scaffolds to recruit a variety of transcriptional activators, suppressors, or protein modifiers for reprogramming gene expression (20). It is perhaps surprising given the sensitivity to even a single unpaired nucleotide that larger RNA structures can successfully accommodated at the 5′ end of Cas9 gRNAs [e.g. (21)]. Although using gRNAs as RNA-mediated scaffolds has been broadly shown to allow functionality in the downstream applications, generally in cells, little work has been done to understand the impacts of adding RNA scaffolds to gRNA on CRISPR RNP complex assembly, R-loop formation/dissociation, and downstream activation of nuclease function (where used). Knowledge of how gRNA-fusions affect all Cas9 activities will contribute to the rationale design of these structures.
We previously developed a single-molecule magnetic tweezers (MT) assay to monitor R-loop formation by St3Cas9 with its natural crRNA:tracrRNA (22). We used this assay here with both St3Cas9 and SpCas9 to explore the effects of gRNA modifications on R-loop formation (Figure 1C). We additionally used a FRET-based assay to measure formation of the RNP with each gRNA (23), and the effect on the rate of dsDNA cleavage using a supercoiled plasmid substrate where we can monitor formation of nicked intermediate and cleavage product without R-loop formation being rate-limiting (24). The modifications had no measurable differences in RNP formation as judged by the FRET assay. However, two or three unpaired 5′ guanine or uracil nucleotides resulted in inhibition of cleavage of the non-target strand by the RuvC nuclease and in some cases alterations in the R-loop formation dynamics. Our results are consistent with complex and variable effects that are influenced by RNA and/or local DNA sequence.
We additionally tested the effects of a 20 nt structured RNA hairpin [the putative mitochondrial targeting RNA aptamer, RP – (25)] when added to the 5′ end, Stem Loop, first Hairpin or 3′ end of the gRNA of SpCas9. For all RP modifications, there was no measurable differences in RNP formation as judged by the FRET assay. Except for the 5′ modification, the RP hairpin also had only moderate effects on R-loop formation dynamics and no measurable effects on the DNA cleavage kinetics. In contrast, the addition of the RP structure to the 5′ end resulted in formation of a stable half-sized R-loop that did not activate DNA cleavage. We propose that the structured RNA cannot be threaded into a full-length R-loop but can still produce a stable structure but where the RuvC/HNH domains are not docked in an active conformation. These observations highlight that modifications to other parts of the gRNA, most prudently the 3′ end, will be more likely to be successful if DNA cleavage is required but that 5′ modification could be used where just DNA binding is required.
MATERIALS and METHODS
gRNAs
IVT dsDNA templates were amplified by PCR from the relevant plasmid (see Supplementary Table S2 for sequences): pD1301i-SP1 was generated by cloning the 1025–1044 pSP1 spacer sequence into pD1301i (Atum Bio); the pEX-A2 family of plasmids were supplied by Eurofins; pCRISPR3 was from (26); pX330 was a gift from Feng Zhang (Addgene plasmid # 42230; http://n2t.net/addgene:42230; RRID:Addgene_42230) (27). Primers were designed for IVT that amplified the RNA spacer and structural component and introduced the T7 promoter upstream of the gRNA sequence. IVT was performed with HiScribe T7 High Yield RNA synthesis kit (New England BioLabs) as per the manufacturer's instructions. RNAs were purified by either phenol-chloroform extraction or with RNA Clean and Concentrator Columns (Zymo Research) and eluted in DEPC treated H2O. Synthetic crRNAs and gRNAs were supplied by Metabion and IDT, respectively (Supplementary Figure S1). gRNA concentrations were calculated by taking A260 with a DeNovix DS-11 spectrophotometer.
Cas9 protein
Wild type St3Cas9 was expressed and purified as published previously (6). St3Cas9(D31A), St3Cas9(N891A) and St3dCas9 variants were engineered from pBAD-Cas9 plasmid (6) using a Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific), and expressed and purified as for WT St3Cas9. SpCas9 was either supplied by New England Biolabs or the SpCas9 encoding gene was cloned into pBAD (6) from pMJ806 (4) and expressed/purified as for WT St3Cas9.
A plasmid to express SpCas9hinge (23) was produced by cloning five synthetic gene fragments in pE-SUMO (Life Sensors, PA) using NEBuilder HiFi DNA Assembly (New England Biolabs). For purification, Escherichia coli BL21 Rosetta 2 (DE3) cells were transformed with pSUMOCas9hinge and grown overnight on LB with 50 μg/ml ampicillin. 500 ml LB was inoculated with a single colony and cells were grown at 37°C in a 2.5 l flask at 250 rpm for 16 h. Once grown, bacteria were pelleted by centrifugation for 20 min at 4000 × g, 4°C, and supernatant discarded. Pellets could be frozen in liquid nitrogen and stored at –20°C at this point. The bacterial pellet was resuspended in 25 ml sonication buffer [50 mM Tris, pH 8.0, 500 mM NaCl, 1 mM β-mercaptoethanol, 5 mM MgCl2, 0.5 mM EDTA, Roche cOmplete ULTRA protease inhibitor] and lysed by sonication 2 × 2 min (10 s on/off) at 75% maximum power. Total lysate was cleared by ultracentrifugation for 40 min at 37 000 × g, 4°C and supernatant was dialysed at 4°C against 1L HisWash1 [50 mM Tris, pH 9.0, 500 mM NaCl, 1 mM β-mercaptoethanol, 30 mM imidazole] in Snakeskin 10 000 MWCO Dialysis tubing (Thermo Scientific). Sample was filtered through a 0.45 μm nitrocellulose filter and loaded onto a 5 ml HisWash1-equilibrated HisTrap HP column (GE Healthcare). 2 ml fractions were collected through a linear gradient over 100 ml up to 100% HisElution [50 mM Tris, pH 9.0, 500 mM NaCl, 1 mM β-mercaptoethanol, 500 mM imidazole]. Cas9hinge eluted in the range of 120–170 mM imidazole. Pooled fractions were dialysed at 4°C against HisWash2 [50 mM Tris, pH 9.0, 200 mM NaCl, 1 mM β-mercaptoethanol] and total protein concentration estimated using a DeNovix DS-11 spectrophotometer. The His tag-SUMO was cleaved overnight with SUMO protease (1 mg SUMO protease was added per mg Cas9 protein) in Snakeskin dialysis tubing, dialysed against 2 l HisWash2 at 4°C. After 15 h, an appropriate amount of NaCl and imidazole were added to give a final concentration of 30 mM imidazole, and the cleaved sample was loaded onto a HisWash2 equilibrated 5 ml HisTrap HP column (GE Healthcare). The flow-through was collected as 2 ml fractions, and bound tag and SUMO protease eluted with HisElution. Cas9-containing fractions were concentrated and exchanged into GF buffer [20 mM Tris, pH 7.5, 200 mM KCl, 1 mM TCEP, 10% (v/v) glycerol] with Amicon Ultra-15 50 kDa cut-off centrifugal filter units (Millipore), and frozen with liquid nitrogen and stored at -80°C.
SpCas9hinge was either single labelled with Cy3 only or double labelled with Cy3 and Cy5 using maleimide monoreactive dyes (Amersham, GE Healthcare) (23). 10 μM SpCas9hinge and 25 μl dye in GF buffer were incubated for 2 h at room temperature, and overnight at 4°C. The reaction was quenched with 10 mM DTT and free dye was separated by size exclusion chromatography on a HiLoad Superdex 200 16/60 column (GE Healthcare). Fractions contained labelled protein were pooled and concentrated with Amicon Ultra-15 50 kDa cut-off centrifugal filter units (Millipore), frozen with liquid nitrogen and stored at –80°C. To assess labelling efficiency, proteins were diluted and scanned in a quartz cuvette with a Cary 60 UV-Vis spectrophotometer (Agilent). Extinction coefficients for Cas9, Cy3 and Cy5 at 280 nm (protein), 552 nm (Cy3) and 650 nm (Cy5) were used to calculate the protein concentration and labelling efficiency.
Ribonucleoprotein assembly
Unless otherwise stated, for RNP complex assembly, equal concentrations of Cas9 and gRNA were mixed in SB buffer [10 mM Tris–Cl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.1 mM DTT, 5 μg/ml BSA] for binding assays, or RB buffer [10 mM Tris–Cl, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.1 mM DTT, 5 μg/ml BSA] for cleavage assays, and incubated at 37°C for 1 h, and then at 4°C.
FRET measurements of gRNA loading
100 nM gRNA was assembled with 50 nM SpCas9hinge labelled with Cy3 or Cy3/Cy5 on ice. Following a 10 min incubation at room temperature, measurements were collected using a FluoroLog Spectrophotometer (Horiba) in a 130 μl Quartz cuvette (Hellma); 5 mm slit widths, 1 s integration time. For each RNA, the sample was excited at 530 and 630 nm and spectra collected from 550 to 800 nm and 650 to 800 nm respectively. (Ratio)A, calculated as explained in Supplementary Figure S4 as a proxy measure for FRET (23).
Single molecule magnetic tweezers assay
The magnetic tweezers assays used a commercial PicoTwist microscope (Fleurieux sur L’Arbresle, France) equipped with a 60 Hz Jai CV-A10 GE camera (28). DNA molecules were tethered to 1 μm MyOne paramagnetic beads (Invitrogen) and the glass coverslip of the flow cell as previously described (22). Topologically-constrained DNA [pSP1, (26)] were identified from rotation curves at 0.3 pN and the rotational zero reference (Rot0) set. R-loop dynamics were measured in Buffer SB at 25°C using magnet rotations of 10 turns s−1. The R-loop size in turns was estimated by comparing the slope of the rotation curves at 1 turn s−1 when an R-loop was trapped in negative torque with the equivalent slopes in the absence of enzyme as described in van Aelst et al. (24). Each trace in a reference set of curves collected in the absence of enzyme (a total of 20) was compared to each rotation curve when an R-loop was trapped in the presence of enzyme (a total of 22). Torque values were calculated using software described in reference (22).
DNA cleavage assays
For each cleavage reaction, 3 nM plasmid substrate pSP1 in RB buffer was pre-heated at 20°C for 5 min. The reaction was started by addition of 50 nM assembled Cas9 RNP and incubated for the time period specified. The reaction was quenched by adding 0.5 volumes of STEB [0.1 M Tris (pH 7.5), 0.2 M EDTA, 40% (w/v) sucrose, 0.4 mg/ml bromophenol blue] and incubating at 80°C for 5 min. Samples were separated by agarose gel electrophoresis on a 1.5% (w/v) agarose gel in 1× TAE [40 mM Tris-acetate, 1 mM EDTA, 10 μg/ml ethidium bromide] at 2 V/cm overnight (16 h) and visualised by UV irradiation. DNA bands containing supercoiled, linear or open circle DNA were excised and placed into scintillation vials. 0.5 ml sodium perchlorate was added to each gel slice, and tubes were incubated at 67°C for 2 h to melt the agarose. The vials were cooled to room temperature and 10 ml Hionic-Fluor Scintillation Cocktail (Perkin Elmer) added to each vial and shaken thoroughly. Each vial was counted in a Tri-Carb Trio 3100TR Liquid Scintillation Counter for 10 min. Where indicated, the cleavage data was fitted to the models in Supplementary Figure S6 using numerical integration in Berkeley Madonna (https://berkeley-madonna.myshopify.com/).
RESULTS
Similar R-loop formation dynamics of wild type and nuclease mutants of Streptococcus thermophilus and Streptococcus pyogenes Cas9 using crRNA:tracrRNA
In our previous MT study of R-loop formation, we utilised the type II-A St3Cas9 and its natural crRNA:tracrRNA (22). Here we repeated the MT assay (Figure 1C), to compare R-loop formation by St3Cas9 with SpCas9 using their corresponding crRNA:tracrRNA (Supplementary Figures S1 and S2, Tables S1 and S2). These are highly related Type II-A Cas9s (60% identity, 73% similarity) (29), which recognise similar PAMs (Supplementary Figure S2A, Figure 2A) and can even exchange their crRNA:tracrRNA and retain activity (10), so we anticipated similar dynamics. crRNAs were synthesised using phosphoramadite chemistry and thus the 5′ end was paired in the R-loop (Supplementary Figure S2A). tracrRNAs were synthesised by in vitro transcription (IVT) using T7 RNA polymerase, resulting in 3 unpaired 5′ guanines (Supplementary Figures S1 and S2A). These are situated at the end of the Upper Stem where they are free of Cas9 interaction. We also measured the R-loop formation using the nuclease dead (D31A, N891A) and nicking variants (RuvC mutant D31A and HNH mutant N891A) of St3Cas9 (5). Because some restriction enzymes mutated within their nuclease active sites have tighter DNA binding activity than WT enzymes (e.g. 30), we wanted to test if there were similar effects on R-loop stability with the Cas9 nuclease mutants.
Figure 2.

The effects of unpaired guanines at the 5′ end of gRNA on Streptococcus pyogenes Cas9. (A) Schematic of R-loop formation. SpCas9 (grey) binds the PAM (grey) and the in vitro transcribed gRNA (black) forms a 20 bp R-loop at the protospacer sequence at 1025–1044 bp of pSP1 (blue). The sequences of the DNA and gRNA spacer sequence are shown. Unpaired 5′ guanines arising from IVT are shown in red. (B) FRET-based gRNA loading assay. (left) Assay schematic. SpCas9hinge was randomly labelled at E945C and D435C with Cy3 and Cy5. In the Apo state (top), there is high acceptor fluorescence which decreases upon gRNA binding (bottom) as fluorophores are moved apart. (right) Comparative (ratio)A (N = 3 ± SD) for loading gRNAs with 1, 2 or 3 unpaired 5′ guanines (light grey) relative to Apo Cas9 (dark grey). (C) Example MT traces of R-loop cycling (at 10 turns s−1) to measure R-loop formation (red arrows) and dissociation (blue arrows). Raw (grey) and 2 Hz smoothed data (black) are shown. Each trace represents measurements on the same single DNA. (D) Inverted cumulative probability distributions for the R-loop formation and dissociation times for each gRNA. Solid lines are single exponential fits. (E) Mean R-loop formation and dissociation times and standard error (N = 36–43, Supplementary Table S3) from the fits in panel D. (F) Supercoiled plasmid cleavage assay. Assay schematic (top). The first strand of supercoiled DNA (SC, dark blue) is cleaved to give an open circle (OC, light blue) intermediate, followed by second strand cleavage resulting in linear DNA (LIN, green). These three forms of DNA can be separated by agarose gel electrophoresis (middle). Tritiated-DNA bands are cut out from agarose gel and quantified by scintillation counting (bottom, N = 3 ± SD). The kinetic model (Materials and Methods, Supplementary Figure S6) was simultaneously fitted by numerical integration to each repeat separately, and the kinetic constants averaged (Supplementary Table S4). Solid lines are simulations using the average values. x-axes are split as follows: 0–100 s and 101–300 s (G); 0–350 s and 360–600 s (GG); and, 0–700 s and 710 - 1200 s (GGG). (G) Comparison of the appearance of LIN product and fitted profiles for each gRNA taken from panel F. (H) Average apparent rate constants for the first and second strand cleavage calculated from the fits in panel F (N = 3–4 ± SD) (Supplementary Table S4).
Linear DNA derived from pSP1 with a shared protospacer and overlapping St3 (5′-NGGNG-3′) and Spy (5′-NGG-3′) PAMs (Supplementary Figure S2A), was tethered between a glass coverslip and a paramagnetic bead (Figure 1C). The bead position above the glass surface was monitored at 60 Hz using video microscopy. Using a pair of permanent magnets, the DNA was first negatively supercoiled, favouring DNA unwinding and R-loop formation (Figure 1C). This caused the bead height to lower. The binding and unwinding of the DNA by Cas9 caused a reduction in DNA twist which was compensated by an increase in twist and writhe elsewhere in the DNA. This reduced the negative supercoils and the bead height increased. The magnets were then rotated rapidly to form positive supercoils, conditions favouring DNA rewinding and R-loop dissociation (Figure 1C). The increase in DNA twist that accompanied R-loop dissociation was compensated by a reduction in DNA twist/writhe. This reduced the positive supercoils and the bead height increased. The reaction buffer contained EDTA to prevent DNA cleavage so that this cycle could be repeated ad infinitum. The force was varied to explore the effect of torque, as required.
Each of the enzymes showed the characteristic changes in bead height corresponding to formation and dissociation of a 20 bp R-loop (Supplementary Figure S2B). R-loop formation was irreversible over several hours and could only be dissociated by positive torque. We observed previously that R-loop formation times were dependent on St3Cas9 concentration whereas dissociation times were not. The same pattern was observed here using the WT and mutant St3Cas9 and WT SpCas9 (Supplementary Figures S2C, S3A and S3B, and Supplementary Table S3). Elevated concentrations of SpCas9 were required compared to the St3 enzymes. This difference was variable between SpCas9 preparations from different sources, most likely due to specific activity variations; subsequent assays (below) used commercial SpCas9 preparations from New England Biolabs that gave more consistent results.
For all the Cas9s, R-loop formation times were independent of negative torque (Supplementary Figures S2D, S3C and S3D, and Supplementary Table S3); previously observed for WT St3Cas9, we suspect this is due to the second order dependence on protein concentration masking the expected torque dependence. The R-loop dissociation times showed a dependence on positive torque; dissociation times becomes shorter as positive torque increased (Supplementary Figure S2D). There was little difference between the times observed within experimental error, suggesting that WT SpCas9 and WT and mutant St3Cas9s have similar R-loop stabilities. The RuvC mutant St3Cas9(D31A) had a slightly shallower torque dependence, but this is not likely to be significant given experimental uncertainty and DNA-to-DNA variation in the assay.
5′ unpaired nucleotides in Streptococcus pyogenes Cas9 gRNA do not have any effect on ribonucleoprotein complex formation
To study the effects of RNA modifications, we first used IVT gRNA with SpCas9 (Figure 2A, Supplementary Figure S1, Tables S1 and S2). Since the number of 5′ guanines can affect transcription efficiency, we produced gRNAs with 1, 2 or 3 unpaired 5′ guanines (1025-G, 1025-GG and 1025-GGG, respectively). The gRNAs target the same protospacer on pSP1 as in Supplementary Figure S2. In general, sufficient gRNA could be generated by in vitro transcription even with a single guanine at the +1 position.
Cas9 loading of gRNAs was followed using a Förster resonance energy transfer (FRET)-based assay developed by Sternberg et al. [Materials and Methods, (23,31)]. In the SpCas9hinge mutant, the natural cysteines are mutated to serine, and positions E945 and D435 are mutated to cysteine for fluorophore labelling (Materials and Methods). In the Apo state, the labelled residues are close (21 Å), resulting in high Cy5 acceptor fluorescence (Figure 2B). As a gRNA is loaded, the labelled residues move ∼60 Å, resulting in a corresponding decrease in acceptor fluorescence relative to donor fluorescence. This change scales with increasing molar ratio of gRNA to Cas9 (Supplementary Figure S4A). The ratio between the Cy5 peak as excited by Cy3 fluorescence relative to direct excitation with 630 nm light, ‘(ratio)A’ (Supplementary Figures S4B and S4C), was used as a proxy readout for energy transfer (23). In the Apo state, where there is high donor-acceptor energy transfer, (ratio)A was 0.430. Conversely, (ratio)A was lower (0.145–0.173) for each of the three IVT gRNA (Figure 2B). The similar range suggests that there is little or no difference in RNP loading with 1, 2 or 3 additional guanines at the 5′ end of the gRNA.
5′ unpaired nucleotides in Streptococcus pyogenes Cas9 gRNA have only modest effects on R-loop dissociation kinetics
The three gRNA were then tested in the MT assay using the same DNA molecule. Each RNP tested showed DNA length changes consistent with R-loop formation and dissociation events as expected for a 20 bp R-loop (Figure 2C). The R-loop formation and dissociation times and calculated time constants are shown in Figure 2D and E, and in Supplementary Table S3. Within error the formation times were similar for the three gRNAs. We cannot directly compare the data to Supplementary Figure S2 as the Cas9 preparations and concentrations were different. The dissociation times for 1025-G and 1025-GG were similar to the value observed with SpCas9 and the crRNA:tracrRNA in Supplementary Figure S2C,D (the dissociation times being independent of concentration). For 1025-GGG, the dissociation time was ∼2-fold longer. Any rearrangement in RuvC and or in the DNA:RNA hybrid to accommodate three unpaired 5′ guanines does not measurably affect the R-loop formation kinetics but does stabilises the structure moderately once formed.
Unpaired 5′ nucleotides in Streptococcus pyogenes Cas9 gRNA have significant effects on the rates of DNA cleavage
Based on the modest effects on R-loop dynamics observed in Figure 2D and E, we expected that nuclease activity would be only moderately affected. DNA cleavage was measured using 3H-labelled plasmid DNA separated by agarose gel electrophoresis and the DNA bands quantified by scintillation counting (Materials and Methods) (Figure 2F). Negatively supercoiled DNA has the advantage in nuclease activity measurements that R-loop formation is supported by topology and is unlikely to be rate-limiting as it might be on unconstrained linear DNA (24). Additionally, the nicked intermediate (open circle, OC) is readily separated from supercoiled plasmid substrate (SC) and linear product (LIN). The data was fitted to an A-to-B-to-C reaction (Supplementary Figure S6A) to yield apparent rate constants for the first (ka) and second (kb) strand cleavages (Materials and Methods, Figure 2H, Supplementary Table S4). The cleavage assays shown in Figure 2F do not go to completion due to low specific activity of the SpCas9 preparation; this does not affect the observed rate constants. More complete DNA cleavage was observed in Figures 3 and 6 with other SpCas9 preparations. The appearance of the linear product with each of the gRNAs is compared in Figure 2G.
Figure 3.
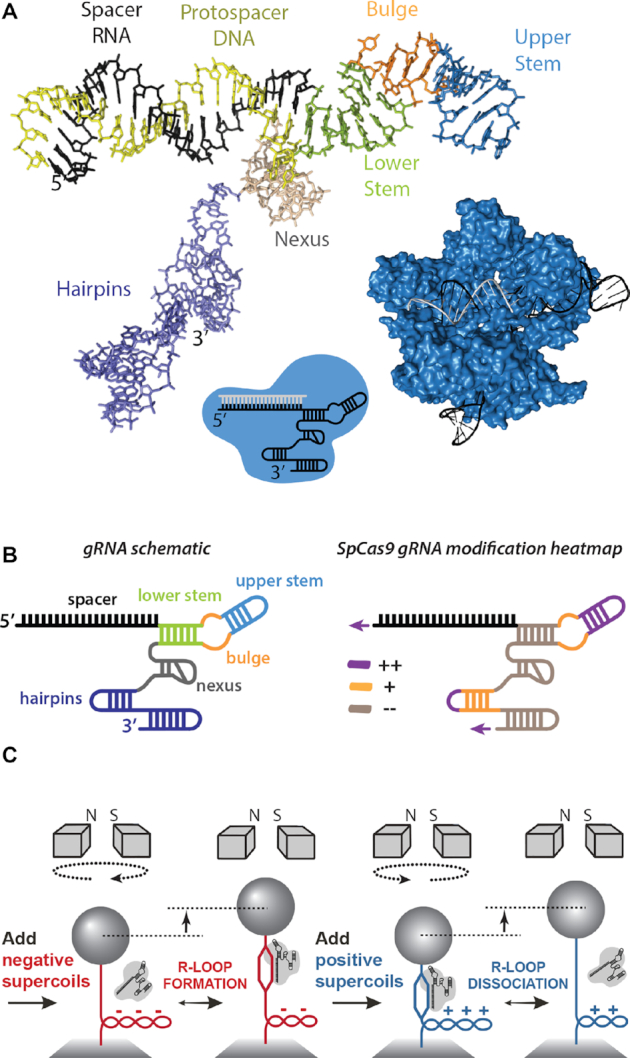
The effects of unpaired bases at the 5′ end of gRNA on Streptococcus pyogenes Cas9 at an alternative protospacer sequence. (A) Sequence of the 20 bp R-loop at the protospacer sequence at 1166–1147 bp of pSP1 (green). 5′ unpaired bases are shown in red for each of the gRNAs. (B) Quantified supercoiled plasmid cleavage assays from gels shown in Supplementary Figure S7A. Except for 1166-G for which a satisfactory fit was not possible, the kinetic model (Materials and Methods, Supplementary Figure S6) was simultaneously fitted by numerical integration to each repeat separately, and the kinetic constants averaged (Supplementary Table S4). Solid lines are simulations using the average values except for the 1166-G data where linear interpolations between the points (grey lines) are shown to guide the eye. x-axes are split as 0–65 s and 75–300 s. (C) Comparison of the appearance of LIN product and fitted profiles for each gRNA taken from panel B. x-axis split as 0–65 s and 75–300 s. (D) Average apparent rate constants for the first and second strand cleavage calculated from the fits in panel B (N = 3 ± SD) (Supplementary Table S4). (E) The alternative kinetic model that incorporates R-loop formation (Supplementary Figure S6) was simultaneously fitted by numerical integration to each repeat separately of the 1166-GGG data and the kinetic constants averaged (Supplementary Table S4). Solid lines are simulations using the average values. Ka was fixed at 0.14 s−1 based on the average value in panel D. x-axis split as 0–65 s and 75–300 s. (F) Example MT traces showing R-loop formation events (red arrows). Raw (grey) and 2 Hz smoothed data (black) are shown. Each trace represents measurements on the same single DNA.
Figure 6.
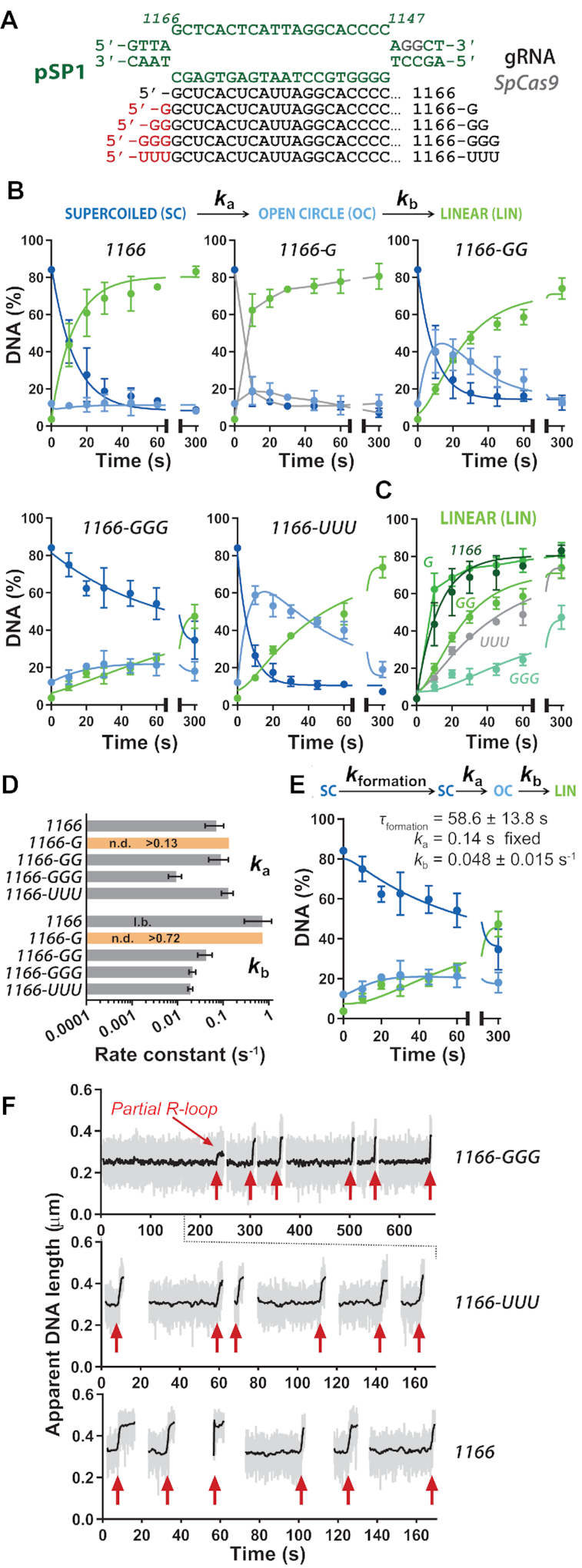
5′ RP modification of gRNA blocks DNA cleavage by Streptococcus pyogenes Cas9. (A) Quantified supercoiled plasmid cleavage assays from gels shown in Supplementary Figure S7C. The kinetic model (Materials and Methods, Supplementary Figure S6) was simultaneously fitted by numerical integration to each repeat separately, and the kinetic constants averaged (Supplementary Table S4). Solid lines are simulations using the average values. x-axes are split as 0–65 s and 75–300 s. (B) Cartoon of the 5′ RP gRNAs modified to include 1, 2, 4 or 8 uracils. Agarose gel of pSP1 cleavage reactions using SpCas9 and the gRNAs, as indicated. (C) Average apparent rate constants for the first and second strand cleavage calculated from the fits in panel A (N = 3 ± SD) (Supplementary Table S4) or estimated from panel B. (D) Model for partial R-loop formation by 5′ RP gRNA. See text for further details.
The rate constants for the first strand cleavage were comparable, with the 1025-GG and 1025-GGG gRNA being only ∼2-fold slower than the 1025-G gRNA (Figure 2H, Supplementary Table S4). The rates were similar to those observed for St3Cas9 with crRNA:tracrRNA on plasmid DNA (22). Surprisingly however, there was a marked difference in the rate of the second strand cleavage (Figure 2F, G), with the rate constant (kb) for 1025-GG and 1025-GGG being one or two orders of magnitude slower than 1025-G, respectively (Figure 2H, Supplementary Table S4). The moderate increase in R-loop stability observed in Figure 2E thus corresponds with a slower second strand cleavage. However, the events may not be directly correlated since 1025-GG has largely identical R-loop dynamics to 1025-G yet quite different second strand cleavage kinetics.
In their recent study, Okafor et al. suggested that effects of unpaired 5′ nucleotides can be sequence context specific (17). To explore this, we targeted an alternative protospacer sequence on pSP1, where the 20th nucleotide on the protospacer DNA target strand is a cytosine that can base pair with a terminal 5′ guanine on the gRNA spacer (Figure 3A). We used synthetic gRNAs rather than IVT gRNAs (Supplementary Table S1), so that we could explore nucleotides other than guanine at the 5′ end. We tested the effect of 0, 1, 2 or 3 unpaired 5′ guanines (1166, 1166-G, 1166-GG and 1166-GGG, respectively), and three unpaired 5′ uracils (1166-UUU), to explore whether the inhibitory effect seen in Figures 2F–H was specific to the bulkier purine.
Plasmid cleavage assays using the 1166-family of gRNAs (Supplementary Figure S7A), were quantified and fitted (Figure 3B), with the apparent cleavage rate constants presented in Figure 3D and Supplementary Table S4. The appearance of the linear product with each of the gRNAs is compared in Figure 3C. DNA cleavage with 1166-G was too fast to fit reliably with the model. 1166 (and 1166-G) did not produce any measurable increase in nicked intermediate, due to the second strand cleavage being faster than the first strand cleavage. This difference from the data in Figure 2F highlights that the relative rates of cleavage the first and second strands can vary with protospacer/gRNA sequence.
Apart from 1166-GGG, the apparent rate constants for first strand cleavage were similar (Figure 3D). However, as observed with the 1025 sequence, addition of 2 or more unpaired 5′ nucleotides resulted in >10-fold slower apparent rates constants for second strand cleavage relative to the 1166 gRNA. The addition of 3 unpaired 5′ uracils was more inhibitory than 2 unpaired 5′ guanines, reducing the second strand cleavage rate by >2-fold, suggesting that the inhibitory effect on cleavage is not specific to guanines and scales with length. However, the effect of 3 unpaired 5′ guanine residues had a markedly slower second strand cleavage (Figure 3C); the fitted kinetic values suggest this is because the first strand cleavage was also slow (Figure 3D, Supplementary Table S4).
An additional explanation for the cleavage profile with the 1166-GGG gRNA in Figure 3B is that the apparent cleavage rate constants are being influenced by slower R-loop formation kinetics using this gRNA compared to the others. A rate limiting R-loop formation step can mask subsequent DNA cleavage steps (24). To consider this, the 1166-GGG data was re-fitted with a model that incorporated R-loop formation as a one-step process (kformation) prior to first strand cleavage (Supplementary Figure S6B). Where kformation is rate-limiting, a fitted value for ka cannot be returned based on the cleavage data alone. To investigate whether the kinetic profiles we observe could be explained by a slow R-loop formation, we fitted the data using ka as a fixed values based on the average value for the other 1166-based gRNA in Figure 3D (0.14 s−1), and kformation and kb were allowed to float. This model could also describe the data equally well (Figure 3E), with an apparent kb similar to that for 1166-GG but with a slow apparent R-loop formation. Given the similar fits for both models, we note that we cannot exclude that both kformation and ka are slower with 1166-GGG.
To test whether the three unpaired 5′ guanines on 1166-GGG gRNA were changing the R-loop formation kinetics, we repeated the MT assay using 1166-GGG, 1166-UUU and 1166 gRNAs (Figure 3F, only R-loop formation events shown). R-loop formation events were markedly slower using 1166-GGG. Note that we cannot directly compare R-loop formation times in the tweezers to the apparent kformation value from the DNA cleavage assays as the SpCas9 RNP concentrations are different and R-loop formation has a second-order component (Supplementary Figure S2).
Slow second strand cleavage is due to the influence of the unpaired 5′ nucleotides on the activity of the RuvC nuclease domain
Okafor et al. (17) proposed that due to close interaction between the RuvC nuclease domain and the 5′ end of the spacer RNA, unpaired residues may alter Cas9 activity. The slower rate of second strand cleavage observed here with both the 1025 and 1166 gRNAs (Figures 2 and 3) could therefore be due to misalignment of the RuvC nuclease that slows cleavage of the non-target strand. To explore this, we compared the DNA cleavage profiles using st3Cas9, st3Cas9(D31A) and st3Cas9(N891A) with the St3-specific 1025-G and 1025-GGG gRNAs (Figure 4, Supplementary Figures S1 and S7B, Table S1).
Figure 4.
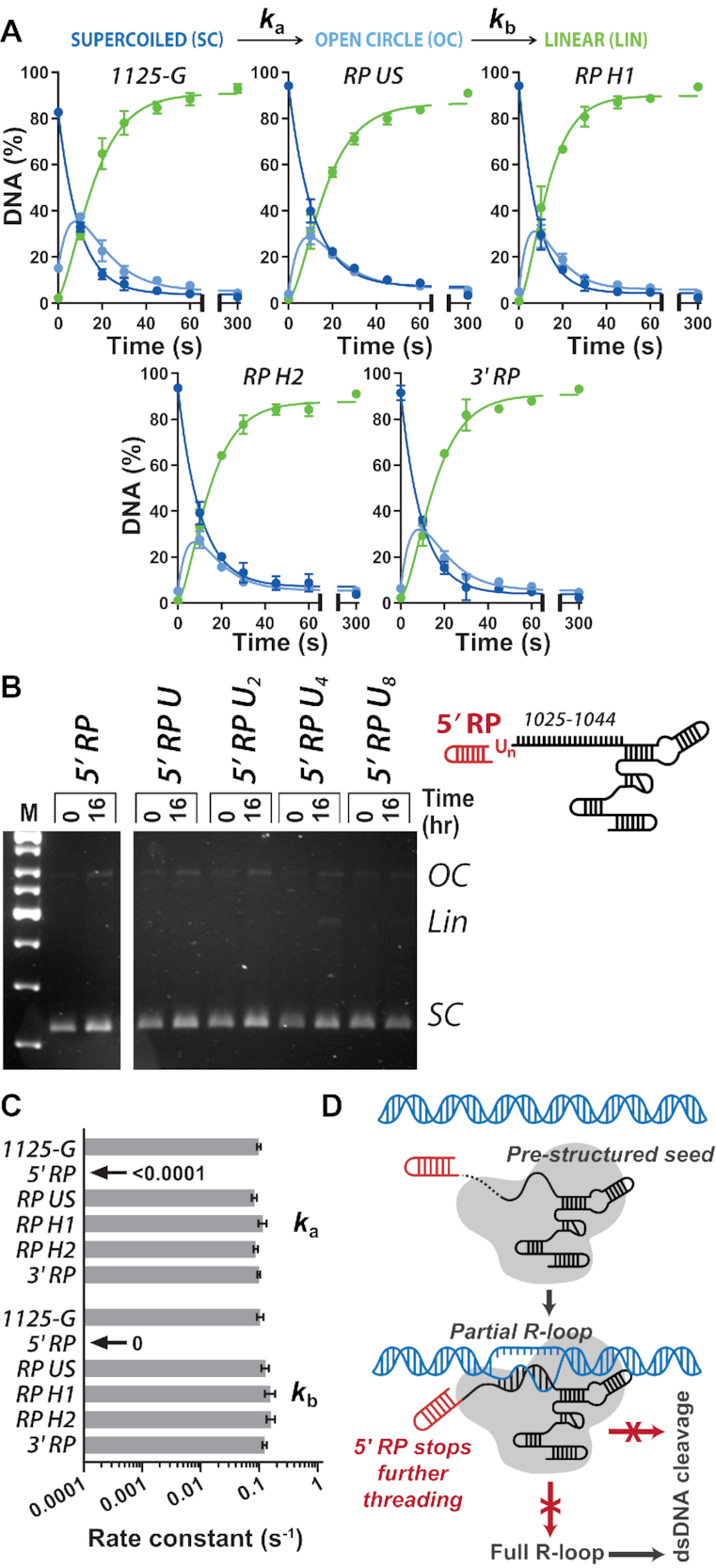
Unpaired bases at the 5′ end of gRNA slow the rate of DNA cleavage by the RuvC nuclease domain but not by the HNH nuclease domain. Quantified supercoiled plasmid cleavage assays from gels shown in Supplementary Figure S7B using Streptococcus thermophilus DGCC7710 CRISPR3 Cas9 WT (A), RuvC mutant D31A (B), or HNH mutant N891A (C). For clarity, only the disappearance of supercoiled (SC) DNA and appearance of linear (LIN) product for WT are compared to the appearance of nicked (OC) product for the nicking mutants. x-axes are split as 0–65 s and 75–300 s.
As observed with SpCas9, the first strand cleavage by StCas9 was unaffected by the number of unpaired 5′ guanines but there was an inhibition of second strand cleavage with 1025-GGG relative to 1025-G (Figure 4A). For St3Cas9(D31A), where only the HNH nuclease is active, the rate of appearance of nicked product was similar with either gRNA (Figure 4B). In contrast, for St3Cas9(N891A), where only the RuvC nuclease is active, there was a marked inhibition of the rate of appearance of nicked product using 1025-GGG. This result is consistent with the slower second strand cleavage being due to disruption of docking of the RuvC nuclease due to the three unpaired guanine residues. We note that we cannot distinguish based on our data between an ordered strand cleavage model (HNH cutting the target strand first, followed by RuvC cutting the non-target strand second) and a random cleavage model (24).
A structured RNA hairpin at the 5′ end of a gRNA can produce shorter hyperstable R-loops that do not support DNA cleavage
To explore the effect of adding a structured RNA aptamer to SpCas9 gRNA, we utilised a 20 bp RP hairpin (Figure 5A) derived from a stem loop structure in the nuclear RNaseP RNP, that can direct import of RNA into mitochondria (25,32). A library of gRNAs was designed in which the RP hairpin was added at one of four gRNA locations (Figure 5A, Supplementary Figure S1): the 5′ end (5′ RP); the Upper Stem (RP US); two alternative positions in the first Hairpin (RP H1 and RP H2); and the 3′ end (3′ RP). gRNAs were synthesised by IVT (Supplementary Table S2); this incorporated a single unpaired 5′ guanine in all gRNAs and a variable run of uracils at the 3′ end. We chose to target the 1025 protospacer sequence in pSP1 as in Figures 2 and 4.
Figure 5.

The effect of the RP RNA hairpin on R-loop dynamics. (A) Sequence of the RP hairpin and schematics of Streptococcus pyogenes Cas9 (grey) and the in vitro transcribed gRNAs (black) (sequences in Supplementary Figure S1). (B) FRET-based gRNA loading assay. Comparative (ratio)A for loading gRNAs as shown (light grey) relative to Apo Cas9 (dark grey) (N = 3 ± SD). (C) Example MT traces of R-loop cycling (at 10 turns s−1) to measure R-loop formation (red arrows) and dissociation (blue arrows). Raw (grey) and 2 Hz smoothed data (black) are shown. (D) Comparison of an R-loop formation event with 3′ RP (red) and 3′ RP (black) gRNA. (E) cumulative probability of R-loop size in turns estimated from R-loop cycling experiments in Supplementary Figure S8 (Materials and Methods). Grey bars are the standard error for each estimation. (F) Average R-loop size in turns (±SD) for each of the gRNAs. The average turns for 1025-G, RP US, RP H1, RP H2 and 3′ RP was assumed to estimate a full length R-loop and used to set R-loop size in bp. Significance test results in Supplementary Table S5. (G, H) Inverted cumulative probability distributions for the R-loop formation and dissociation times for each gRNA. Solid lines are single exponential fits or double exponential fits (5′ RP formation; 5′ RP and RP H2 dissociation). (I) Mean R-loop formation and dissociation times and standard error (N = 19 to 43, Supplementary Table S3) from the fits in panel D. For 5′ RP formation events, and 5′ RP and RP H2 dissociation events, the light and dark bars are the two constants from a double exponential fit with the percentage shown for the amplitude of the slower events. For the dissociation times, the x-axis is split as 0–1 s and 2–10 s.
The loading of the modified gRNA library and SpCas9 into RNPs was measured using the FRET-based SpCas9hinge assay described above (23), and compared to 1025-G (Figure 5B, Supplementary Figure S5). The gRNA RP library gave similar relative acceptor fluorescence changes in the (ratio)A range 0.104–0.154, comparable to the value seen with 1025-G (0.145). We conclude that RP modifications do not have a significant impact on the Cas9 structure with respect to REC lobe positioning during gRNA loading.
The MT assay was used to assess the effect of the RP hairpin on R-loop formation and dissociation (Figure 5C). In all cases, stable R-loops were observed that could be reversed by application of positive torque. It was noticeable that the change in bead height produced by the 5′ RP gRNA was smaller than the characteristic 20 bp R-loop change with the other gRNAs; R-loop events from 5′ RP and 3′ RP are compared in Figure 5D. Rotation curves in the absence and presence of SpCas9 were used to quantify the shift in the number of DNA turns that are captured by R-loop formation (Supplementary Figure S8, Figure 5E) (24). The 5′ RP gRNA produced an R-loop that captured only ∼0.78 DNA turns compared to the average values for the other gRNAs of 1.65–2.06 turns (Figure 5F); the results of a multiple comparison significance test are shown in Supplementary Table S5. The significantly smaller number of turns captured by the 5′ RP gRNA relative to all the other gRNAs suggest that it can only support formation of a stable ∼9 bp R-loop, a size which corresponds to that expected for the seed region.
The significance tests indicate a possible increased number of turns captured by RP Hairpin 2 (i.e. a slightly larger R-loop) compared to RP Hairpin 1 and, to a lesser extent, 1025-G, but not compared to RP Upper Stem or 3′ RP where the turns are similar within confidence limits (Supplementary Table S5, Figure 5E, F). Given that the differences are close to the error limits of our R-loop size estimation method, we suggest that RP Hairpin 2 forms a standard 20 bp R-loop.
The R-loop formation and dissociation times were calculated at single negative and positive torque values (Figure 5G–I, Supplementary Table S3). The formation times were all relatively slow; for 5′ RP the data had a double exponential distribution, with ∼61% of events being very slow (∼36 s) and 39% of events being markedly faster (∼3.5 s). The dissociation times for RP Upper Stem, RP Hairpin 1 and 3′ RP were all faster than 1025-G, indicating a moderate loss of R-loop stability. For both 5′ RP and RP Hairpin 2, the dissociation times were biphasic, with a proportion of events being slower to dissociate. These may indicate abnormal R-loop structures that are hyperstabilised. Alternatively, the R-loop state could be hyperstabilised by a transient RP hairpin-protein interaction.
Given the alterations to the R-loop dynamics and size, we expected that DNA cleavage kinetics would also be alerted for at least some of these modified gRNAs. However, for RP Upper Stem, RP Hairpin 1, RP Hairpin 2 and 3′ RP, the DNA cleavage profiles and the apparent rate constants for the first and second strand cleavages were very similar to 1025-G (Figure 6A and C, Supplementary Figure S7C, Table S4). In comparison, only a low percentage of DNA nicking was observed over 16 h using 5′ RP gRNA (Figure 6B and C). We reasoned that the inhibition by RP may be because the structured hairpin is too close to the DNA:RNA hybrid. However, adding uracil residues between the RP hairpin and spacer sequence to give more flexibility (Supplementary Table S2), did not activate DNA cleavage to measurable levels (Figure 6B), although the 5′ RP U4 gRNA did support formation of a small amount of linear DNA after 16 h incubation. Despite being relatively stable (Figure 5I), the shorter R-loop produced by the 5′ RP gRNA cannot activate DNA cleavage over a reasonable timescale.
DISCUSSION
gRNA engineering allows a wide range of enhancements to CRISPR–Cas activity (7). Chemical synthesis of gRNAs has become routine, and modification of nucleotides and addition of functional groups have increased nuclease-resistance and/or enhanced nuclear localization. Additionally, fusion of extra RNA sequences/structures to gRNA has been exploited to add new functionality to CRISPR–Cas. However, our single-molecule and ensemble measurements of gRNA loading, R-loop formation and DNA cleavage indicate that modified gRNAs can have significant inhibitory effects on DNA cleavage in a target sequence-dependent manner, and can influence the size and stability of the R-loop. These possible changes in CRISPR–Cas activity, which may be dependent on the spacer sequence and/or local sequence of the protospacer, need to be considered when testing any new designs.
The 5′ modification of a Cas9 gRNA with a 20 nt RP hairpin had the striking effect of preventing DNA cleavage due, most likely, to the formation of an incomplete R-loop corresponding to the seed sequence. Previous single molecule data suggested a critical cut off in R-loop length at ∼10–11 bp below which the R-loop could not form and above which R-loop stability was similar (22). Consistent with the prevailing model for activation of the Cas9 nuclease activity (23,33), this size of RNA:DNA hybrid will not engage the HNH domain. Very low levels of DNA cleavage were only observed here over very long timescales; this may reflect non-specific cleavage activity or a very slow formation of a full-length R-loop that was not observed in the MT experiments.
In contrast, fusion of the RP hairpin at the Upper Stem, Hairpin and 3′ end had much less drastic effects on R-loop formation and no measurable effect on DNA cleavage. These observations highlight that, for some sequences at least, modifications to other parts of the gRNA will be more likely to be successful. Consistent with this, these gRNA features have been shown to accommodate RNA aptamer scaffolds with a range of sequences and sizes, e.g. (19,34–37). Often gRNA fusions are used to provide a binding sequence to colocalise a protein or fluorescent label and are hence used with dCas9 rather than nuclease active WT Cas9. Thus, DNA cleavage activity is not always assessed, and one cannot rule out that atypical R-loop structures may be produced.
A possible explanation for the abridged R-loop zipping using 5′ RP is suggested in Figure 6D. Following the formation of the DNA:RNA hybrid in the seed region, the 5′ end of the gRNA may need to thread around the target DNA strand. This threading may be inhibited by the addition of a bulky structured RNA. Other studies have successfully modified the 5′ end of gRNA and retained cleavage activity (a proxy for complete R-loop formation), e.g. (20,21,38). However, in many cases the fusions were not highly structured. The type I Cascade complex can thread a 3′ crRNA hairpin when forming its 33 bp R-loop (22,39), but this is a quite different structural complex to Cas9. It may be that the effect seen here is unique to the RP hairpin and that empirical design can be used to select for RNA structures that have the desired effect and to avoid unwanted ones. There may also be protospacer specific effects, as observed for the short 5′ modifications (17), which means that empirical testing is important unless general rules can be established. The unusual activity of the 5′ RP-modified gRNA used here could be exploited to form stable R-loops without activating DNA cleavage. Multiplex expression of unmodified and modified gRNAs could allow for simultaneous site-specific editing and site-specific binding at different locations by WT Cas9.
The more surprising observation from our study was that two or more unpaired nucleotides at the 5′ end of two different Cas9 gRNAs can have significant effects on the activity of the RuvC nuclease domain while, in one case, having only relatively modest effects on R-loop stability. The reduced activity of second strand cleavage was observed using both in vitro transcribed and synthetic gRNAs (1025 versus 1166, Figures 2–4), suggesting that transcript variations due to IVT (12–15), are not the principal cause of the effect. As noted by Okafor et al. (17), because there are close interactions of the docked RuvC domain with the 5′ end of the RNA (9), 5′ modifications may result in rearrangements in the RuvC domain that slows, but does not completely abrogate, the cleavage of the second strand. Consistent with this, we observed that DNA nicking by the RuvC nuclease was affected by 3 unpaired 5′ guanines whereas DNA nicking by the HNH nuclease was not (Figure 4).
Our observations highlight a potential inefficiency of using gRNA with more than one unpaired nucleotide at the 5′ end if the DNA cleavage activity of Cas9 is critical to the application. However, the extent of the effect varied with DNA/RNA sequence, as was also noted by Okafor et al. (17). For example, the addition of 3 unpaired 5′ guanines to the 1025 gRNA slowed the second strand cleavage rate, while the same addition to the 1166 gRNA slowed both R-loop formation and second strand cleavage. Additionally, in the single molecule traces with 1166-GGG we also observed occasional events that were smaller than expected for a 20 bp R-loop (partial R-loop marked in Figure 3F). This suggest that even small 5′ modifications can, in some sequence contexts, inhibit the completion of R-loop zipping possibly for the same reason as shown in Figure 6D.
The possibility of undesirable effects of unpaired 5′ nucleotides with a given gRNA can be avoided. IVT is still relatively efficient with a single guanine which for the 1166-G sequence had minimal effect on cleavage efficiency compared to 1166 without any 5′ modification. Similarly, the eukaryotic U6 promoters typically exploited when expressing gRNAs from a gene in cell culture only add a single unpaired nucleotide. By careful selection of protospacer sequences and selection of sites using online tools (40), the base can be incorporated as part of the spacer RNA:DNA hybrid. Alternatively, a hammerhead ribozyme domain can be added to the 5′ end which can remove the mismatched guanine prior to RNP formation and CRISPR activity (16). For RNP transfection-based methods, the price of commercial RNA synthesis using phosphoramidites has decreased significantly in recent years and unmatched 5′ guanines can be excluded from the design.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Laura Lee, Ciaran Guy, Carlos Martinez Santiago and Jia Qi Cheng Zhang for help with experiments and data analysis.
Contributor Information
Grace Mullally, DNA-Protein Interactions Unit, School of Biochemistry, University of Bristol, Bristol BS8 1TD, UK; BrisSynBio, Life Sciences Building, Tyndall Avenue, University of Bristol, Bristol BS8 1TQ, UK.
Kara van Aelst, DNA-Protein Interactions Unit, School of Biochemistry, University of Bristol, Bristol BS8 1TD, UK.
Mohsin M Naqvi, DNA-Protein Interactions Unit, School of Biochemistry, University of Bristol, Bristol BS8 1TD, UK.
Fiona M Diffin, DNA-Protein Interactions Unit, School of Biochemistry, University of Bristol, Bristol BS8 1TD, UK.
Tautvydas Karvelis, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania.
Giedrius Gasiunas, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania; CasZyme, Vilnius, Lithuania.
Virginijus Siksnys, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania.
Mark D Szczelkun, DNA-Protein Interactions Unit, School of Biochemistry, University of Bristol, Bristol BS8 1TD, UK; BrisSynBio, Life Sciences Building, Tyndall Avenue, University of Bristol, Bristol BS8 1TQ, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
BBSRC/EPSRC through the BrisSynBio Synthetic Biology Research Centre [BB/L01386x1]; BBSRC [BB/L000873, BB/S001239/1]; South West Biosciences Doctoral Training Partnership; European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme [788405]. Funding for open access charge: Research Councils UK block grant to University of Bristol.
Conflict of interest statement. G.G. is an employee of CasZyme. V.S. is a chairman of and has financial interest in CasZyme.
REFERENCES
- 1. Wang H., La Russa M., Qi L.S.. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 2016; 85:227–264. [DOI] [PubMed] [Google Scholar]
- 2. Carroll D. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 2014; 83:409–439. [DOI] [PubMed] [Google Scholar]
- 3. Jiang F., Doudna J.A.. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017; 46:505–529. [DOI] [PubMed] [Google Scholar]
- 4. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gasiunas G., Barrangou R., Horvath P., Siksnys V.. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. U.S.A. 2012; 109:E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karvelis T., Gasiunas G., Miksys A., Barrangou R., Horvath P., Siksnys V.. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013; 10:841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moon S.B., Kim D.Y., Ko J.H., Kim J.S., Kim Y.S.. Improving CRISPR genome editing by engineering guide RNAs. Trends Biotechnol. 2019; 37:870–881. [DOI] [PubMed] [Google Scholar]
- 8. Filippova J., Matveeva A., Zhuravlev E., Stepanov G.. Guide RNA modification as a way to improve CRISPR/Cas9-based genome-editing systems. Biochimie. 2019; 167:49–60. [DOI] [PubMed] [Google Scholar]
- 9. Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O.. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014; 156:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Briner A.E., Donohoue P.D., Gomaa A.A., Selle K., Slorach E.M., Nye C.H., Haurwitz R.E., Beisel C.L., May A.P., Barrangou R.. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol. Cell. 2014; 56:333–339. [DOI] [PubMed] [Google Scholar]
- 11. Milligan J.F., Uhlenbeck O.C.. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989; 180:51–62. [DOI] [PubMed] [Google Scholar]
- 12. Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C.. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987; 15:8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krupp G. RNA synthesis: strategies for the use of bacteriophage RNA polymerases. Gene. 1988; 72:75–89. [DOI] [PubMed] [Google Scholar]
- 14. Triana-Alonso F.J., Dabrowski M., Wadzack J., Nierhaus K.H.. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J. Biol. Chem. 1995; 270:6298–6307. [DOI] [PubMed] [Google Scholar]
- 15. Gholamalipour Y., Karunanayake Mudiyanselage A., Martin C.T.. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-Seq analyses. Nucleic Acids Res. 2018; 46:9253–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim S., Bae T., Hwang J., Kim J.S.. Rescue of high-specificity Cas9 variants using sgRNAs with matched 5′ nucleotides. Genome Biol. 2017; 18:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okafor I.C., Singh D., Wang Y., Jung M., Wang H., Mallon J., Bailey S., Lee J.K., Ha T.. Single molecule analysis of effects of non-canonical guide RNAs and specificity-enhancing mutations on Cas9-induced DNA unwinding. Nucleic Acids Res. 2019; 47:11880–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang W., Hu J.H., Liu D.R.. Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat. Commun. 2017; 8:15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong Y., Lu G., Duan J., Liu W., Zhang Y.. Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging. Genome Biol. 2018; 19:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S. et al.. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015; 160:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kocak D.D., Josephs E.A., Bhandarkar V., Adkar S.S., Kwon J.B., Gersbach C.A.. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019; 37:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R.. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl Acad. Sci. U.S.A. 2014; 111:9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sternberg S.H., LaFrance B., Kaplan M., Doudna J.A.. Conformational control of DNA target cleavage by CRISPR–Cas9. Nature. 2015; 527:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Aelst K., Martinez-Santiago C.J., Cross S.J., Szczelkun M.D.. The effect of DNA topology on observed rates of r-Loop formation and DNA strand cleavage by CRISPR–Cas12a. Genes (Basel). 2019; 10:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang G., Chen H.W., Oktay Y., Zhang J., Allen E.L., Smith G.M., Fan K.C., Hong J.S., French S.W., McCaffery J.M. et al.. PNPASE regulates RNA import into mitochondria. Cell. 2010; 142:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sapranauskas R., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V.. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011; 39:9275–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lionnet T., Allemand J.F., Revyakin A., Strick T.R., Saleh O.A., Bensimon D., Croquette V.. Magnetic trap construction. Cold Spring Harb. Protoc. 2012; 2012:133–138. [DOI] [PubMed] [Google Scholar]
- 29. Hao M., Cui Y., Qu X.. Analysis of CRISPR–Cas System in Streptococcus thermophilus and Its Application. Front. Microbiol. 2018; 9:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wright D.J., King K., Modrich P.. The negative charge of Glu-111 is required to activate the cleavage center of EcoRI endonuclease. J. Biol. Chem. 1989; 264:11816–11821. [PubMed] [Google Scholar]
- 31. Dagdas Y.S., Chen J.S., Sternberg S.H., Doudna J.A., Yildiz A.. A conformational checkpoint between DNA binding and cleavage by CRISPR–Cas9. Sci Adv. 2017; 3:eaao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang G., Shimada E., Koehler C.M., Teitell M.A.. PNPASE and RNA trafficking into mitochondria. Biochim. Biophys. Acta. 2012; 1819:998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang F., Taylor D.W., Chen J.S., Kornfeld J.E., Zhou K., Thompson A.J., Nogales E., Doudna J.A.. Structures of a CRISPR–Cas9 R-loop complex primed for DNA cleavage. Science. 2016; 351:867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palumbo C.M., Gutierrez-Bujari J.M., O’Geen H., Segal D.J., Beal P.. Versatile 3′ functionalization of CRISPR single guide RNA. ChemBioChem. 2020; doi:10.1002/cbic.201900736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma H., Tu L.C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T.. CRISPR–Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016; 214:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shechner D.M., Hacisuleyman E., Younger S.T., Rinn J.L.. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods. 2015; 12:664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu Y., Rocha P.P., Luo V.M., Raviram R., Deng Y., Mazzoni E.O., Skok J.A.. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016; 7:11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E. et al.. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018; 36:536–539. [DOI] [PubMed] [Google Scholar]
- 39. Xiao Y., Luo M., Dolan A.E., Liao M., Ke A.. Structure basis for RNA-guided DNA degradation by cascade and Cas3. Science. 2018; 361:eaat0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dhanjal J.K., Radhakrishnan N., Sundar D.. CRISPcut: a novel tool for designing optimal sgRNAs for CRISPR/Cas9 based experiments in human cells. Genomics. 2019; 111:560–566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.