

Abstract
A radical clock system was developed to investigate single-electron transfer (SET) in the reactions of organomagnesium reagents with carbonyl compounds. The fluorenylcyclopropyl radical clock was selected because it is the fastest known radical clock. Additions of Grignard reagents to aldehydes or methyl ketones provided no evidence for ring-opened products that would indicate reaction through SET. Additions of some Grignard reagents to aromatic ketones, however, resulted in the formation of ring-opened products, suggesting SET.
Graphical Abstract

Introduction
Considering the important role played by the addition of Grignard reagents to carbonyl compounds in synthetic organic chemistry, substantial attention has been directed to understand the mechanisms of these reactions.1–12 In general, two mechanistic pathways are considered for these reactions. One pathway involves concerted formation of the new carbon–carbon bond through two-electron transition states, likely involving a dimeric reagent.2–4,12–14 Alternatively, single-electron transfer (SET) pathways have been invoked for some additions, such as the additions to benzophenone and fluorenone.6,13–17 Recent ab initio molecular dynamics simulations (AIMD) and quantum calculations addressed the relative importance of these two pathways for additions of methylmagnesium reagents to acetaldehyde.1 These computations confirmed experimental studies that indicate that additions of Grignard reagents to alkyl aldehydes do not proceed via radical intermediates.4 They also suggest that, with fluorenone, the SET pathway is accelerated, and the concerted pathway is decelerated, likely due to steric destabilization in the transition state.
In this Article, we provide evidence that, for most additions of Grignard reagents and related organometallic nucleophiles to a wide range of carbonyl compounds, the reactions proceed via mechanisms involving concerted, two-electron pathways. We used the fastest-known radical clock to evaluate these mechanisms. Furthermore, the highly sterically hindered substrate should disfavor the concerted addition and thus be particularly sensitive to possible SET pathways.1 In all cases, additions of Grignard reagents to aldehydes and alkyl ketones proceeded by concerted, two-electron pathways. SET was only observed in the case of aryl ketones, and then only predictably with a pentafluorophenyl ketone, which likely has a lower-lying π*CO orbital than the other substrates, and is thus more prone to SET.1,18
The fluorenylcyclopropyl radical clock was chosen because it opens an order of magnitude faster than the more commonly used 2,2-diphenylcyclopropylcarbinyl radical clock.19 The presence of any radical intermediates in the course of a reaction is likely to be revealed by the presence of radical coupling products, although addition to the fluorenyl radical intermediate could lead to the formation of multiple products.20 Probes bearing the fluorenylcyclopropyl radical clock have not been used extensively due to synthetic challenges posed by the high reactivity of synthetic intermediates.21,22
Results and Discussions
Four radical clocks bearing carbonyl compounds were synthesized to explore the mechanism of the addition of organometallic reagents to electronically differentiated carbonyl groups. Three radical clocks were synthesized in three steps from 9-fluorenone hydrazone (1, Scheme 1).23,24 Additionally, a perfluorophenyl ketone (4) was synthesized from aldehyde 5 in two additional steps. These synthetic steps provide some mechanistic information: no ring-opened products formed from single-electron transfer processes were observed in the addition of a magnesium amide to ester 2, in the reduction of ester 2 with LiAlH4, in the oxidation of cyclopropylmethyl alcohols by the Dess–Martin periodinane (DMP),25 or in the additions of Grignard reagents to Weinreb amide 4.26
Scheme 1.

Synthesis of radical clock probes
Additions of Grignard reagents to the radical clock bearing a formyl group (3) provided no evidence for SET processes in the carbon–carbon bond forming step of the mechanism (Table 1). The crude reaction mixtures were carefully analyzed by HPLC and NMR spectroscopy for evidence of minor ring-opened products that would indicate that SET processes occurred. In most cases, only the 1,2-addition product was formed, and the reaction mixtures were clean. The relative stereochemical configuration of alcohol 8d was established by X-ray crystallographic analysis of the minor diastereomer formed in this addition. The stereochemistry for the other alcohols formed in additions to aldehyde 3 were tentatively assigned by analogy to 8d. In the addition of tertbutylmagnesium chloride to aldehyde 3, some 1,2-reduction product was observed. This product was also not formed by an SET process because it was formed without ring opening.27,28
Table 1.
Additions of Grignard reagents to an aldehyde bearing radical clock
 | ||||
|---|---|---|---|---|
| R–MgX | temp (°C) | Yield (%) | d.r.a | Product |
| Me–MgCl | 20 | 73 | 58:42 | 8a |
| n-Pr–MgCl | 20 | 81 | 53:47 | 8b |
| H2C=CH–MgBr | 20 | 68 | 57:43 | 8c |
| Ph–MgCl | 20 | 81 | 64:36 | 8d |
| Bn–MgCl | 20 | 78 | 75:25 | 8e |
| t-Bu–MgCl | 20 | 33b | >99:1 | 8f |
| H2C=CHCH2–MgCl | −78 | 79 | 66:34 | 8g |
| Me2C=CHCH2–MgCl | −78 | 48c | 65:35d | 8h |
Ratios determined by 1H NMR or 13C NMR spectroscopy.
1,2-Reduction product (10%) was also isolated.
The minor diastereomer formed in this addition was not isolated by column chromatography.
Diastereomeric ratio for the major regioisomer of addition (γ-addition); the regioisomeric ratio of γ-addition:α-addition was 90:10; the diastereomeric ratio of the α-addition product was >99:1.
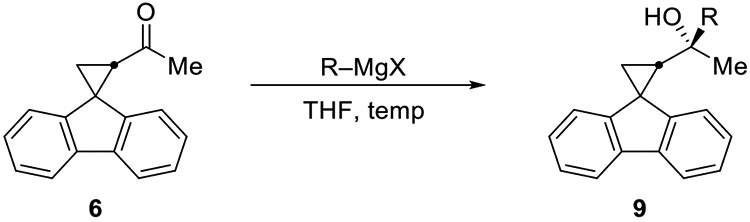
No evidence for radical mechanisms was observed in additions to the methyl ketone 6, either. Most Grignard reagents added in high yield with no evidence of either enolization or ring-opened products (Table 2). Additions to ketone 6 were highly diastereoselective, favoring the product predicted by the Felkin–Anh stereochemical model.29–32 The relative stereochemical configuration of alcohol 9d was determined by X-ray crystallographic analysis. For the addition of tert-butylmagnesium chloride to sterically hindered ketone 6, however, no product was formed, potentially because the ketone had enolized. No product was formed, however, in the presence of CeCl3, which was added to minimize enolization.33
Table 2.
Additions of Grignard reagents to a methyl ketone bearing radical clock (6)
 | ||||
|---|---|---|---|---|
| R–MgX | temp (°C) | Yield (%) | d.r.a | Product |
| Me–MgCl | 20 | 87 | – | 9a |
| n-Pr–MgCl | 20 | 70 | >99:1 | 9b |
| H2C=CH–MgBr | 20 | 70 | >99:1 | 9c |
| Ph–MgCl | 20 | 62 | >99:1 | 9d |
| Bn–MgCl | 20 | 88 | >99:1 | 9e |
| H2C=CHCH2–MgCl | −78 | 94 | >99:1 | 9f |
| Me2C=CHCH2–MgCl | −78 | 84 | >99:1b | 9g |
Ratios determined by 1H NMR spectroscopy.
Diastereomeric ratio for the major regioisomer of addition (γ-addition); the regioisomeric ratio of γ-addition:α-addition was 79:21; the diastereomeric ratio of the α-addition product was >99:1.
Even additions of many Grignard reagents to the phenyl ketone 7 showed no evidence of ring-opening (Table 3). These products were formed in moderate yields; conversion was much lower than with the previous radical clock substrates, likely due to the considerable steric congestion at the carbonyl group. Additions to this substrate would only occur at elevated temperatures, except for the additions of the more reactive allylic magnesium reagents.3 Similarly, the Barbier addition of n-propyl bromide to ketone 7 resulted in only addition product 10b (eq 1).
 |
(1) |
Table 3.
Addition of Grignard reagents to a phenyl ketone bearing radical clock
 | ||||
|---|---|---|---|---|
| R–MgX | temp (°C) | Yield (%) | d.r.a | Product |
| Me–MgCl | 20 | 72 | >99:1 | 10a |
| n-Pr–MgCl | 20 | 53c | >99:1 | 10b |
| H2C=CH–MgBr | 20 | 67c | >99:1 | 10c |
| Ph–MgCl | 66 | 74 | – | 10d |
| Bn–MgCl | 20 | 68 | >99:1 | 10e |
| H2C=CHCH2–MgCl | −78 | 82 | >99:1 | 10f |
| Me2C=CHCH2–MgCl | −78 | 84 | >99:1 | 10g |
Ratios determined by 1H NMR spectroscopy.
Diastereomeric ratio for the major regioisomer of addition (γ-addition); the regioisomeric ratio of γ-addition:α-addition was 89:11; the diastereomeric ratio of the α-addition product was >99:1.
Some (<20%) 1,2-reduction product (8d) was detected in the unpurified 1H NMR spectrum, but this product was not isolated by column chromatography.
Addition of tert-butylmagnesium chloride to radical clock 7, in contrast, resulted in a complex mixture of products (eq 2). Analysis of the unpurified reaction mixture by NMR spectroscopy and HPLC indicated that at least 20 distinct compounds were formed. One ring-opened product in the resulting mixture was identified as ring-opened ketone 11; an authentic sample of this ketone was synthesized by hydrogenation of radical clock 7 (eq 3). LC-MS analysis (ESI) of the reaction mixture from eq 2 provided evidence for multiple ring-opened products as well as ring-closed addition products (Scheme 2). These compounds, however, could not be isolated from the reaction mixture due to the small amount of each that was formed, so the identity of these products cannot be firmly established. The formation of multiple products, and the formation of products such as 15 and 17, are consistent with outcomes of reactions of fluorenone with Grignard reagents, which are believed to involve SET reactions.20 We also observed that, in contrast to the reactions illustrated in Table 3, addition of tert-butylmagnesium chloride to aryl ketone 7 (eq 2) was accompanied by a rapid color change, leading to a bright orange reaction mixture. This color corresponds to the color expected had fluorenyl radical or fluorenyl anion intermediates been formed,34,35 which would only be expected had the three-membered ring opened. Taken together, these observations suggest that the formation of complex mixtures of products corresponds to ring opening that would accompany SET pathways.
 |
(2) |
 |
(3) |
Scheme 2.

Possible ring-opened and ring-closed products that correspond to ions detected by LC-MS (ESI) in the unpurified reaction mixture of the addition of t-Bu–MgCl to ketone 7
Experiments with benzylmagnesium chloride provide additional evidence that the formation of ring-opened products are associated with SET pathways. Just as with the addition of tert-butylmagnesium chloride to phenyl ketone 7, addition of benzylmagnesium chloride to a radical clock bearing a perfluorophenyl group (5, eq 4) gave a complex mixture of products, as evidenced by NMR spectroscopy and HPLC. SET processes have been proposed for the additions of benzylic Grignard reagents to aromatic ketones,17 and perfluorophenyl-substituted carbonyl compounds are particularly prone to reactions by SET.6,18 Therefore, in a combination of reagents that are prone to react by SET, complex mixtures of products were formed, indicating that the presence of multiple products is evidence for a pathway involving radicals. By contrast, the reaction of benzylmagnesium chloride with phenyl ketone 7 provided only the addition product without the formation of ring-opened products (Table 3), suggesting that radicals were not involved in that reaction. The difference in reactivity between the two ketones 5 and 7 with the same Grignard reagent suggests that the one-electron and two-electron pathways may have similar activation energies. The electron-withdrawing nature of the perfluorophenyl group results in further stabilization of ketyl intermediates formed in the course of a reaction,6,18 thus favoring ring opening.
 |
(4) |
Addition of n-propylmagnesium chloride to ketone 5 resulted in the formation of 1,2-reduction product 20 and a complex mixture of products, indicating ring opening had occurred (eq 5). The formation of reduction product in the course of this reaction is likely due to the increased steric hindrance on the carbonyl group.16 In this case, two reaction pathways, a two-electron reduction and SET, are competitive, but, the two-electron addition pathway does not compete. Conversely, addition of allylmagnesium chloride to ketone 5 occurred without ring-opening to provide alcohol 21 in high yield (eq 6). No reduction product was detected in this reaction mixture, likely due to the high reactivity of allylmagnesium reagents with carbonyl compounds.2,3,36,37
 |
(5) |
 |
(6) |
Addition of isopropylmagnesium chloride to some radical clocks resulted in ring-opening. Addition of isopropylmagnesium chloride to fluorinated ketone 5 resulted in 1,2-reduction product 20 and a complex mixture of products, indicating that ring-opening occurred (eq 7). This result is consistent with the observation that secondary Grignard reagents can add to some carbonyl compounds through SET.6,17,38 In contrast, addition of isopropylmagnesium chloride to the phenyl ketone 7 led to the formation of a small amount (6%) of 1,2-reduction product 8d at 20 °C (eq 8). Addition of isopropylmagnesium chloride-lithium chloride complex (turbo Grignard)39 to ketone 7, however, resulted in 1,2-reduction product 8d and a complex mixture that indicated the formation of radical intermediates (eq 9). There is some evidence that suggests i-Pr–MgCl•LiCl reacts through a radical mechanism in magnesium–halogen exchange reactions,40 but previous studies have not explored the possibility of an SET mechanism for i-Pr–MgCl•LiCl in carbon–carbon bond forming reactions. The fact that i-Pr–MgCl•LiCl is more prone to react by SET than isopropylmagnesium chloride provides evidence that the magnesium–halogen exchange reaction may involve an SET pathway.
 |
(7) |
 |
(8) |
 |
(9) |
Radical clocks were also used to investigate the mechanism of the reactions of other organometallic reagents with carbonyl compounds. For these reactions, radical clocks 3 and 7 were used because they most resemble substrates regularly used by synthetic chemists. Additions of organolithium reagents to phenyl ketone 7 provided no evidence of ring-opened products (Table 4). Both methyllithium and phenyllithium added to ketone 7 in high yield. Addition of lithium dimethylcuprate, a reagent that is generally useful for chelation-controlled additions,41 to aldehyde 3 provided alcohol 8a as a single diastereomer in moderate yield with no evidence of ring-opened products (eq 10). Similarly, addition of lithium diphenylcuprate to aldehyde 3 led to formation of only alcohol 8d as a single diastereomer (eq 11). Reductions of radical clock 7 with diisobutylaluminum hydride (DIBAL-H) and LiAlH4 also provided no evidence of ring-opened products (Table 5).42
 |
(10) |
 |
(11) |
Table 4.
Additions of alkyllithium reagents to phenyl ketone 7
 | |||
|---|---|---|---|
| R | yield (%) | d.r.a | Product |
| Me | 88 | >99:1 | 10a |
| Ph | 95 | – | 10d |
Ratios determined by 1H NMR spectroscopy.
Table 5.
Reductions of phenyl ketone 7
 | ||
|---|---|---|
| reducing agent | yield (%) | dra |
| i-BU2AlH | 90 | >99:1 |
| LiAlH4 | 81 | >99:1 |
Ratios determined by 1H NMR spectroscopy.
Additions of allyl chromium reagents under catalytic Nozaki–Hiyama–Kishi conditions43 to radical clocks 3 and 7 provided evidence that the carbon–carbon bond-forming step of these reactions involves SET.44 In the addition of allyl bromide to aldehyde 3, a complex mixture of products was formed, indicating SET had occurred (eq 12). Reaction under the same conditions with ketone 7 led to the formation of ketone 11 (eq 13). No other products were detected by NMR spectroscopy, HPLC, or LC-MS. When ketone 7 was mixed with the metal reagents without allyl bromide, a small amount of ring-opened ketone 11 was formed, suggesting that this product could be a byproduct of the reduction of CrCl3 with manganese (eq 14).
 |
(12) |
 |
(13) |
 |
(14) |
Conclusions
In summary, additions of Grignard reagents to aldehydes and alkyl ketones appear to proceed by concerted, two-electron pathways (Table 6). Even in these hindered substrates that should favor single-electron transfer pathways, evidence of SET was not observed in the reactions of organomagnesium or organolithium reagents. Only the reactions of organochromium reagents with an alkyl aldehyde proceeded through SET pathways. In the case of phenyl ketones, only with tert-butylmagnesium chloride was single-electron transfer the dominant pathway. With the most electron-deficient pentafluorophenyl ketone, SET could be observed even with primary alkylmagnesium reagents but not with allylmagnesium chloride. As a result, single-electron transfer pathways are unlikely to contribute to product formation in the reactions of most Grignard reagents with most carbonyl compounds.
Table 6.
Summary of reaction mechanisms observed in reactions with radical clock probes
 |
 |
 |
 |
|
|---|---|---|---|---|
| n-Pr–MgCl | 2 e− | 2 e− | 2 e− | 2 e− and SET |
| i-Pr–MgCl | – | – | 2 e− | 2 e− and SET |
| t-Bu–MgCl | 2 e− | NR | SET | – |
| Bn–MgCl | 2 e− | 2 e− | 2 e− | SET |
| Allyl–MgCl | 2 e− | 2 e− | 2 e− | 2 e− |
| Turbo Grignard | – | – | 2 e− and SET | – |
| Me-Li | – | – | 2 e− | – |
| Ph-Li | – | – | 2 e− | – |
| Me2CuLi | 2 e− | – | – | – |
| Ph2CuLi | 2 e− | – | – | – |
| i-Bu2AlH | – | – | 2 e− | – |
| LiAlH4 | – | – | 2 e− | – |
| Allyl–CrCl2 | SET | – | NR | – |
NR = no reaction
Experimental Section
General Procedures.
1H and 13C NMR spectra were obtained at room temperature using Bruker AVIII-400 (400 MHz and 100 MHz, respectively), AVIIIHD-400 (400 MHz and 100 MHz, respectively), and AV-600 (600 MHz and 150 MHz, respectively) spectrometers. 19F NMR spectra were obtained at room temperature using a Bruker AVIII-400 (377 MHz) spectrometer. Spectroscopic data are reported as follows: chemical shifts are reported in ppm on the δ scale, 1H and 13C NMR spectra are internally referenced to tetramethylsilane (1H NMR: CDCl3 δ 0.00; 13C NMR: CDCl3 δ 0.00; 1H NMR: (CD3)2SO δ 0.00; 13C NMR: (CD3)2SO δ 0.00), 19F NMR spectra are externally referenced to trifluorotoluene (19F NMR: CDCl3 δ −63.72), multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. Ratios of products were obtained from 1H NMR or 13C NMR integrations45 using diagnostic peaks in the unpurified reaction mixture. One-pulse 1H spectra were taken when determining product ratios. Multiplicities of carbon peaks were determined using HSQC experiments. Unpurified reaction mixtures were analyzed by HPLC using a Shimadzu LC-2010A equipped with an ES Industries Chromegasphere SI60 silica adsorption normal phase column (4.6 × 250 mm, 60 Å pore size, 5 μm particle size). LC-MS analyses were performed using an Agilent 1260 Infinity HPLC System, MS-Agilent 1100 Series equipped with a Phenomenex Kinetex C18 reverse phase column (3 × 50 mm, 100 Å pore size, 2.6 μm particle size). Infrared (IR) spectra were recorded using a Thermo Nicolet AVATAR Fourier Transform IR spectrometer using attenuated total reflectance (ATR). High-resolution mass spectra were acquired on an Agilent 6224 Accurate-Mass time-of-flight spectrometer and were obtained using peak matching. The ionization sources used were either atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI), as indicated. Liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on silica gel (SiO2) 60 (230–400 mesh). Tetrahydrofuran, diethyl ether, dichloromethane, and methanol were dried and degassed using a solvent purification system before use. All dry reactions were run under a nitrogen atmosphere in glassware that had been flame-dried under reduced pressure. Unless otherwise noted, all reagents and substrates were commercially available. Hydrazone 1,46 iodosobenzene,47 and prenylmagnesium chloride2 were prepared using known procedures. In most cases, the concentrations of commercially available reagents were assumed to be near the concentrations reported by the suppliers because an excess of the reagent was used.
Spiro[cyclopropane-1,9’-fluorene]-2-carboxylic acid methyl ester (2).
A reported procedure23 was adapted to prepare ester 2. To hydrazone 1 (10.05 g, 51.7 mmol) was added methyl acrylate (30.0 mL, 331 mmol) and nickel(II) hydroxide (1.51 g, 16.3 mmol). The mixture was heated to 80 °C in a silicone oil bath then iodosobenzene (13.43 g, 61.0 mmol) was added portionwise over 20 min. After 2 h, the reaction mixture was cooled to room temperature and filtered through Celite. The Celite was rinsed with CH2Cl2 (100 mL) and the combined filtrate was concentrated in vacuo to provide ester 2. Ester 2 was used without purification (12.31 g, 95% unpurified yield). When performed on a smaller scale (6.1 mmol hydrazone 1), the product was purified by column chromatography (5:95 EtOAc:hexanes) to afford ester 2 as an orange solid (1.254 g, 82%): mp = 97–99 °C; 1H NMR (400 MHz, CDCl3) δ 7.82–7.79 (m, 2H), 7.58 (d, J = 7.7, 1H), 7.40–7.36 (m, 2H), 7.32–7.26 (m, 2H), 7.04 (d, J = 7.5, 1H), 3.63 (s, 3H), 2.77–2.73 (m, 1H), 2.47–2.44 (dd, J = 7.5, 5.2, 1H), 2.16–2.12 (dd, J = 8.4, 5.2, 1H); 1H NMR (400 MHz, (CD3)2SO) δ 7.96–7.92 (m, 2H), 7.43–7.39 (m, 3H), 7.36–7.27 (m, 3H), 3.57 (s, 3H), 2.91–2.87 (m, 1H), 2.33–2.25 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 170.0 (C), 146.4 (C), 142.5 (C), 141.0 (C), 139.8 (C), 127.2 (CH), 127.0 (CH), 126.9 (CH), 122.9 (CH), 119.90 (CH), 119.87 (CH), 118.7 (CH), 52.0 (CH3), 37.1 (C), 32.7 (CH), 20.9 (CH2); 13C{1H} NMR (100 MHz, (CD3)2SO) δ 169.1 (C), 145.8 (C), 142.1 (C), 140.3 (C), 138.8 (C), 127.3 (CH), 127.02 (CH), 126.95 (CH), 126.8 (CH), 122.0 (CH), 120.1 (CH), 119.8 (CH), 119.6 (CH), 51.8 (CH3), 36.3 (C), 32.2 (CH), 20.2 (CH2); IR (ATR) 3037, 2955, 1728, 1478, 1201, 937 cm−1; HRMS (APCI) m / z calcd for C17H15O2 (M + H)+ 251.1067, found 251.1066.
Spiro[cyclopropane-1,9’-fluorene]-2-carbaldehyde (3).
To a cooled suspension (0 °C) of LiAlH4 (1.59 g, 41.9 mmol) in Et2O (50 mL) was added a solution of ester 2 (6.302 g, unpurified) in Et2O (150 mL) over 30 min. The reaction mixture was warmed to 20 °C over 1 h. After 16 h, H2O (10 mL) and NaOH (30 mL, 1.0 M in H2O) were slowly added over 1 h and the resulting mixture was stirred for an additional hour. MgSO4 (about 1 g) was added and the mixture was stirred for 1 h, filtered through Celite, and concentrated in vacuo. The resulting alcohol was used without purification (5.155 g, 92% unpurified yield). When performed on a smaller scale (2.8 mmol ester 2), the product was purified by column chromatography (5:95 EtOAc:hexanes) to afford alcohol S1 as a white solid (0.377 g, 61%): mp = 94–96 °C; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 7.6, 1H), 7.82 (d, J = 7.4, 1H), 7.42–7.28 (m, 5H), 7.05 (d, J = 7.3, 1H), 4.19–4.15 (dd, J = 12.1, 6.2, 1H), 4.00–3.95 (dd, J = 12.1, 8.7, 1H), 2.36–2.28 (m, 1H), 2.01–1.98 (dd, J = 9.0, 5.1, 1H), 1.80–1.77 (dd, J = 7.4, 5.2, 1H), 1.14 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.1 (C), 141.1 (C), 139.1 (C), 127.1 (CH), 126.7 (CH), 126.5 (CH), 126.2 (CH), 121.1 (CH), 120.4 (CH), 119.8 (CH), 118.5 (CH), 62.0 (CH2), 33.5 (C), 33.5 (CH), 21.8 (CH2); IR (ATR) 3303, 3065, 3008, 2928, 1443, 1049 cm−1; HRMS (ESI) m / z calcd for C16H13 ((M + H) – H2O)+ 205.1012, found 205.1018. Anal. Calcd for C16H14O: C, 86.45; H, 6.35. Found: C, 86.33; H, 6.32. To a solution of spiro[cyclopropane-1,9’-fluoren]-2-ylmethanol (5.155 g, 23.2 mmol) in CH2Cl2 (130 mL) was added Dess–Martin periodinane (12.31 g, 29.0 mmol). After 12 h, 1:1 saturated aqueous NaHCO3:saturated aqueous Na2S2O3 (200 mL) was added and the resulting mixture was stirred for an additional 24 h. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The resulting solid was recrystallized from 10:90 EtOAc:hexanes and the filtrate was concentrated in vacuo to afford aldehyde 3 as a brown solid (4.654 g, 81% over two steps): mp = 48–50 °C; 1H NMR (400 MHz, CDCl3) δ 9.69 (d, J = 4.1, 1H), 7.83–7.79 (m, 2H), 7.42–7.29 (m, 5H), 7.08 (d, J = 7.5, 1H), 2.92–2.87 (m, 1H), 2.66–2.63 (dd, J = 7.3, 5.6, 1H), 2.30–2.27 (dd, J = 8.3, 5.5, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 197.2 (CH), 145.8 (C), 142.0 (C), 141.2 (C), 140.0 (C), 127.54 (CH), 127.51 (CH), 127.1 (CH), 122.9 (CH), 120.4 (CH), 120.2 (CH), 118.9 (CH), 40.6 (CH), 39.9 (C), 20.8 (CH2); 13C{1H} NMR (100 MHz, (CD3)2SO) δ 198.5 (C), 144.7 (C), 141.3 (C), 139.7 (C), 138.3 (C), 126.8 (CH), 126.6 (CH), 126.5 (CH), 126.3 (CH), 122.3 (CH), 119.8 (CH), 119.4 (CH), 119.2 (CH), 19.9 (CH2); IR (ATR) 3064, 2848, 2748, 1694, 1448, 1090 cm−1; HRMS (ESI) m / z calcd for C16H13O (M + H)+ 221.0961, found 221.0971.
(R*)-(Perfluorophenyl)((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (20) and (R*)-(perfluorophenyl)((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (20’).
A reported procedure48 was adapted to prepare alcohols 20 and 20’. To a cooled (−78 °C) solution of bromopentafluorobenzene (490 μL, 3.9 mmol) in THF (20 mL) was added isopropylmagnesium chloride (2.0 mL, 2.0 M in THF, 3.9 mmol). After 30 min, a solution of aldehyde 3 (0.668 g, 3.0 mmol) in THF (10 mL) was added by cannula to the reaction mixture. The mixture was stirred for an additional hour at −78 °C before warming to 20 °C. After 16 h, H2O (5 mL) and HCl (20 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed alcohol 20 was formed as a 78:22 mixture of diastereomers (20:20’). Purification by flash chromatography (10:90 to 25:75 EtOAc:hexanes) afforded alcohol 20 as a colorless oil (0.476 g, 40%) and alcohol 20’ as an orange oil (0.102 g, 9%).
Major diastereomer 20:
1H NMR (600 MHz, CDCl3) δ 7.92–7.91 (m, 1H), 7.85–7.84 (d, J = 7.5, 1H), 7.47–7.45 (m, 1H), 7.41–7.32 (m, 4H), 7.05–7.04 (m, 1H), 5.42–5.39 (dd, J = 10.2, 4.4, 1H), 2.64–2.59 (m, 1H), 1.95–1.93 (dd, J = 9.2, 5.6, 1H), 1.83 (d, J = 4.8, 1H), 1.78–1.76 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.1 (C), 146.2–143.7 (br d, JC–F = 251.5, CF), 142.9 (C), 142.1–139.6 (br d, JC–F = 254.4, CF), 141.4 (C), 139.2 (C), 139.0–136.5 (br d, JC–F = 253.3, CF), 127.3 (CH), 127.08 (CH), 127.06 (CH), 126.7 (CH), 121.0 (CH), 120.7 (CH), 120.0 (CH), 118.6 (CH), 115.9 (m, C), 65.5 (CH), 36.4 (br s, CH), 34.9 (C), 20.4 (CH2); 19F{1H} NMR (377 MHz, CDCl3) δ–143.95 (dd, J = 22.3, 7.3, 2F), −155.58 (t, J = 20.5, 1F), −162.4 (m, 2F); IR (ATR) 3614, 3072, 2974, 1522, 1447, 988 cm−1; HRMS (APCI) m / z calcd for C22H12F5 ((M + H) − H2O)+ 371.0854, found 371.0855.
Minor diastereomer 20’:
1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 7.4, 1H), 7.75 (d, J = 7.4, 1H), 7.38–7.27 (m, 5H), 7.02 (d, J = 7.4, 1H), 5.32–5.29 (m, 1H), 2.65–2.59 (m, 1H), 2.46 (d, J = 6.9, 1H), 2.13–2.10 (dd, J = 8.9, 5.4, 1H), 2.05–2.02 (dd, J = 6.9, 5.9, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.1 (C), 145.8–143.3 (br d, JC–F = 250.0, CF), 142.8 (C), 142.0–139.4 (br d, JC–F = 253.8, CF), 141.0 (C), 139.4 (C), 138.6–136.1(br d, JC–F = 252.9, CF), 127.1 (CH), 126.8 (CH), 126.55 (CH), 126.49 (CH), 121.6 (CH), 120.3 (CH), 119.8 (CH), 118.5 (CH), 116.1 (m, C), 65.0 (CH), 35.2 (m, CH), 34.5 (C), 21.6 (CH2); 19F{1H} NMR (377 MHz, CDCl3) δ −143.57 (dd, J = 22.3, 6.9, 2F), −155.53 (t, J = 21.1, 1F), −162.52 (m, 2F).
(Perfluorophenyl)(spiro[cyclopropane-1,9’-fluoren]-2-yl)methanone (5).
To a solution of alcohol 20 (0.476 g, 1.22 mmol) in CH2Cl2 (12 mL) was added Dess–Martin periodinane (0.643 g, 1.52 mmol). After 16 h, 1:1 saturated aqueous NaHCO3:saturated aqueous Na2S2O3 (20 mL) was added and the resulting mixture was stirred for an additional 4 h. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded ketone 5 as a yellow solid (0.266 g, 56%): mp = 100–103 °C; 1H NMR (600 MHz, CDCl3) δ 7.81–7.79 (m, 2H), 7.43–7.37 (m, 3H), 7.33–7.32 (m, 1H), 7.28–7.25 (m, 1H), 7.14–7.12 (m, 1H), 3.37–3.34 (m, 1H), 2.87–2.85 (dd, J = 7.5, 5.4, 1H), 2.39–2.37 (dd, J = 7.7, 5.4, 1H); 13C{1H} NMR (150 MHz, CDCl3) δ 187.4 (C), 145.7 (C), 145.5–143.8 (br d, JC–F = 267.5, CF), 143.8–142.0 (br d, JC–F = 258.2, CF), 141.0 (C), 140.9 (C), 140.1 (C), 138.3–136.6 (br d, JC–F = 255.7, CF), 127.6 (CH), 127.3 (CH), 127.1 (CH), 123.0 (CH), 120.1 (CH), 120.0 (CH), 118.6 (CH), 115.2 (m, C), 42.8 (CH), 42.4 (C), 21.2 (CH2); 19F{1H} NMR (377 MHz, CDCl3) δ − 141.76 (m, 2F), −149.91 (tt, J = 20.6, 4.1, 1F), −161.08 (m, 2F); IR (ATR) 2971, 1690, 1487, 1156, 993 cm−1; HRMS (APCI) m / z calcd for C22H12F5O (M + H)+ 387.0803, found 387.0793.
N-Methoxy-N-methylspiro[cyclopropane-1,9’-fluorene]-2-carboxamide (4).
A reported procedure24 was adapted to prepare Weinreb amide 4. To a cooled (−20 °C) solution of N,O-dimethylhydroxylamine hydrochloride (9.21 g, 94.4 mmol) and ester 2 (12.32 g, 49.3 mmol) in THF (76 mmol) was added isopropylmagnesium chloride (55.0 mL, 2.0 M in Et2O, 110 mmol) dropwise over 1 h. After the full volume of isopropylmagnesium chloride was added, saturated aqueous NH4Cl (50 mL) was added and the mixture was warmed to 20 °C. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Weinreb amide 4 was used without purification (12.53 g, 92% unpurified yield). When performed on a smaller scale (4.0 mmol ester 2), the reaction was purified by column chromatography (25:75 EtOAc:hexanes) to afford Weinreb amide 4 as a colorless oil (0.734 g, 66%): 1H NMR (400 MHz, CDCl3) δ 7.81–7.79 (m, 2H), 7.46–7.44 (m, 1H), 7.40–7.25 (m, 4H), 7.12 (d, J = 7.3, 1H), 3.24–3.19 (m, 1H), 3.12 (br s, 3H), 2.91 (br s, 3H), 2.56–2.53 (dd, J = 7.7, 5.2, 1H), 2.10–2.07 (dd, J = 8.4, 5.2, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.8 (C), 146.7 (C), 143.2 (C), 140.5 (C), 139.6 (C), 127.2 (CH), 127.1 (CH), 126.8 (CH), 122.3 (CH), 119.9 (CH), 119.8 (CH), 118.7 (CH), 61.1 (CH3), 36.2 (C), 32.6 (CH3), 31.6 (CH), 19.4 (CH2); IR (ATR) 2934, 1652, 1449, 1079, 909 cm−1; HRMS (APCI) m / z calcd for C18H18NO2 (M + H)+ 280.1332, found 280.1341.
1-(Spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-one (6).
To a cooled (0 °C) solution of Weinreb amide 4 (12.32 g, 44.1 mmol) in Et2O (200 mL) was added methylmagnesium bromide (21.0 mL, 3.0 M in Et2O, 63 mmol) dropwise over 40 min. After an additional 20 min, H2O (50 mL) and HCl (150 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The resulting solid was recrystallized from 5:95 EtOAc:hexanes to provide ketone 6 as a yellow solid (6.25 g, 51% over three steps from hydrazone 1): mp = 75–78 °C; 1H NMR (400 MHz, CDCl3) δ 7.82–7.79 (m, 2H), 7.42–7.32 (m, 4H), 7.28–7.24 (m, 1H), 7.11 (d, J = 7.5, 1H), 3.05–3.01 (m, 1H), 2.58–2.55 (dd, J = 7.6, 5.1, 1H), 2.11–2.07 (dd, J = 7.9, 5.1, 1H), 2.05 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 202.3 (C), 146.6 (C), 142.0 (C), 140.6 (C), 139.9 (C), 127.12 (CH), 127.06 (CH), 127.04 (CH), 127.01 (CH), 122.7 (CH), 119.9 (CH), 119.8 (CH), 118.6 (CH), 40.7 (CH), 38.8 (C), 31.7 (CH3), 20.5 (CH2); IR (ATR) 2992, 1698, 1438, 1171, 955, 789 cm−1; HRMS (APCI) m / z calcd for C17H15O (M + H)+ 235.1117, found 235.1112. Anal. Calcd for C17H14O: C, 87.15; H, 6.02. Found: C, 87.14; H, 6.17.
Phenyl(spiro[cyclopropane-1,9’-fluoren]-2-yl)methanone (7).
To a cooled (0 °C) solution of Weinreb amide 4 (12.53 g, 44.9 mmol) in Et2O (200 mL) was added phenylmagnesium bromide (21.0 mL, 3.0 M in Et2O, 63 mmol) dropwise over 40 min. After an additional 20 min, H2O (50 mL) and HCl (150 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The resulting solid was filtered through a 5 cm plug of silica with 5:95 EtOAc:hexanes (100 mL). The filtrate was concentrated in vacuo then recrystallized from EtOH to provide ketone 7 as a yellow solid (9.50 g, 62% over three steps from hydrazone 1): mp = 104–105 °C; 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.5, 1H), 7.76 (d, J = 7.2, 1H), 7.62–7.60 (m, 2H), 7.47–7.37 (m, 3H), 7.30–7.23 (m, 5H), 7.18–7.14 (m, 1H), 3.61–3.57 (m, 1H), 2.82–2.79 (dd, J = 7.6, 5.1, 1H), 2.26–2.23 (dd, J = 8.0, 5.1, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 194.5 (C), 146.2 (C), 142.2 (C), 140.5 (C), 140.2 (C), 137.7 (C), 132.9 (CH), 128.4 (CH), 128.1 (CH), 127.3 (CH), 127.1 (CH), 127.0 (CH), 126.9 (CH), 122.2 (CH), 120.0 (CH), 119.8 (CH), 118.7 (CH), 39.1 (C), 37.8 (CH), 20.0 (CH2); IR (ATR) 2998, 1662, 1448, 1223, 1008, 728 cm−1; HRMS (APCI) m / z calcd for C22H17O (M + H)+ 297.1274, found 297.1274.
Representative procedure for additions of Grignard reagents to carbonyl compounds ((R*)-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (8a) and (R*)-1-((S*)-spiro-[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (8a’)).
To a solution of aldehyde 3 (0.112 g, 0.51 mmol) in THF (5 mL) was added methylmagnesium chloride (250 μL, 3.0 M in THF, 0.75 mmol) dropwise over 5 min. After 16 h, H2O (5 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 13C{1H} NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8a was formed as a 58:42 mixture of diastereomers (8a:8a’). Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded the major diastereomer alcohol 8a as a yellow solid (0.058 g, 38%) and the minor diastereomer 8a’ as a yellow oil (0.053 g, 35%).
Major Diastereomer 8a:
mp = 142–145 °C; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 7.5, 1H), 7.82 (d, J = 7.5, 1H), 7.42–7.38 (m, 1H), 7.37–7.29 (m, 4H), 7.03 (d, J = 7.1, 1H), 4.17–4.13 (m, 1H), 2.14–2.07 (m, 1H), 1.99–1.95 (dd, J = 9.1, 5.1, 1H), 1.75–1.72 (dd, J = 7.3, 5.1, 1H), 1.45 (d, J = 6.2, 3H), 1.23 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.0 (C), 141.3 (C), 138.9 (C), 127.1 (CH), 126.8 (CH), 126.5 (CH), 126.1 (CH), 120.9 (CH), 120.5 (CH), 119.8 (CH), 118.5 (CH), 67.9 (CH), 40.1 (CH), 33.9 (C), 22.3 (CH3), 22.1 (CH2); IR (ATR) 3397, 3063, 2969, 1447, 1008, 782 cm−1; HRMS (APCI) m / z calcd for C17H15 ((M + H) − H2O)+ 219.1168, found 219.1172.
Minor Diastereomer 8a’:
1H NMR (400 MHz, CDCl3) δ 7.86–7.80 (m, 2H), 7.39–7.27 (m, 4H), 7.23–7.21 (m, 1H), 7.04 (d, J = 7.3, 1H), 4.14–4.07 (m, 1H), 2.03–1.96 (m, 2H), 1.88–1.84 (m, 1H), 1.69 (br s, 1H), 0.99 (d, J = 6.2, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.2 (C), 144.2 (C), 141.1 (C), 139.2 (C), 127.0 (CH), 126.4 (CH), 126.2 (CH), 126.1 (CH), 121.6 (CH), 120.2 (CH), 119.7 (CH), 118.6 (CH), 68.2 (CH), 38.2 (CH), 34.1 (C), 23.3 (CH3), 22.3 (CH2).
(R*)-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)butan-1-ol (8b) and (R*)-1-((S*)-spiro-[cyclopropane-1,9’-fluoren]-2-yl)butan-1-ol (8b’).
Alcohols 8b and 8b’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.110 g, 0.50 mmol) and n-propylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 13C{1H} NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8b was formed as a 53:47 mixture of diastereomers (8b:8b’). Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded the major diastereomer alcohol 8b as a colorless oil (0.053 g, 40%) and the minor diastereomer 8b’ as a yellow oil (0.054 g, 41%).
Major diastereomer 8b:
1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 7.5, 1H), 7.81 (d, J = 7.6, 1H), 7.41–7.36 (m, 1H), 7.34–7.24 (m, 4H), 7.03 (d, J = 7.2, 1H), 3.99–3.93 (m, 1H), 2.12–2.05 (m, 1H), 2.00–1.96 (dd, J = 9.0, 5.0, 1H), 1.78–1.72 (m, 3H), 1.64–1.50 (m, 2H), 1.18 (br s, 1H), 1.02 (t, J = 7.3, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.2 (C), 144.1 (C), 141.3 (C), 139.0 (C), 127.1 (CH), 126.8 (CH), 126.5 (CH), 126.1 (CH), 120.9 (CH), 120.4 (CH), 119.8 (CH), 118.5 (CH), 71.4 (CH), 39.1 (CH2), 39.0 (CH), 33.2 (C), 22.5 (CH2), 19.0 (CH2), 14.3 (CH3); IR (ATR) 3433, 2956, 2869, 1447, 1094, 729 cm−1; HRMS (APCI) m / z calcd for C19H19 ((M + H) − H2O)+ 247.1481, found 247.1484. Anal. Calcd for C19H20O: C, 86.32; H, 7.63. Found: C, 85.11; H, 7.51.
Minor diastereomer 8b’:
1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.6, 1H), 7.81 (d, J = 7.4, 1H), 7.39–7.27 (m, 4H), 7.22–7.21 (m, 1H), 7.03 (d, J = 7.4, 1H), 3.96–3.91 (m, 1H), 2.02–1.94 (m, 2H), 1.87–1.83 (m, 1H), 1.67 (br s, 1H), 1.43–1.33 (m, 1H), 1.18–0.99 (m, 3H), 0.66–0.62 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.2 (C), 144.5 (C), 141.0 (C), 139.2 (C), 127.0 (CH), 126.3 (CH), 126.2 (CH), 126.1 (CH), 121.7 (CH), 120.2 (CH), 119.7 (CH), 118.5 (CH), 71.0 (CH), 39.7 (CH2), 37.3 (CH), 34.3 (C), 21.6 (CH2), 18.1 (CH2), 13.8 (CH3).
(R*)-1-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)prop-2-en-1-ol (8c) and (R*)-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)prop-2-en-1-ol (8c’).
Alcohols 8c and 8c’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.110 g, 0.50 mmol) and vinylmagnesium bromide (750 μL, 1.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8c was formed as a 57:43 mixture of diastereomers (8c:8c’). Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded the major diastereomer alcohol 8c as a yellow solid (0.048 g, 39%) and the minor diastereomer 8c’ as a yellow oil (0.036 g, 29%).
Major Diastereomer 8c:
mp = 99–101 °C; 1H NMR (400 MHz, CDCl3) δ 7.88–7.86 (m, 1H), 7.82–7.80 (m, 1H), 7.43–7.27 (m, 5H), 7.03 (d, J = 7.3, 1H), 6.15–6.07 (ddd, J = 17.2, 10.5, 5.5, 1H), 5.45–5.41 (m, 1H), 5.26–5.23 (m, 1H), 4.46–4.43 (dd, J = 9.6, 5.5, 1H), 2.17–2.11 (m, 1H), 2.02–1.98 (dd, J = 9.1, 5.2, 1H), 1.80–1.77 (dd, J = 7.4, 5.3, 1H), 1.35 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.9 (C), 143.9 (C), 141.3 (C), 139.0 (C), 138.8 (CH), 127.1 (CH), 126.8 (CH), 126.6 (CH), 126.2 (CH), 121.0 (CH), 120.5 (CH), 119.8 (CH), 118.6 (CH), 115.2 (CH2), 72.4 (CH), 38.1 (CH), 33.9 (C), 21.7 (CH2); IR (ATR) 3422, 3040, 2991, 1447, 1044, 758 cm−1; HRMS (APCI) m / z calcd for C18H15 ((M + H) − H2O)+ 231.1168, found 231.1171. Anal. Calcd for C18H16O: C, 87.06; H, 6.49. Found: C, 86.90; H, 6.63.
Minor Diastereomer 8c’:
1H NMR (400 MHz, CDCl3) δ 7.86–7.80 (m, 2H), 7.39–7.26 (m, 5H), 7.04–7.03 (m, 1H), 5.64–5.56 (ddd, J = 17.3, 10.6, 4.9, 1H), 4.93–4.88 (m, 1H), 4.83–4.79 (m, 1H), 4.47–4.44 (m, 1H), 2.07–2.00 (m, 2H), 1.96–1.92 (m, 1H), 1.83 (br s, 1H); 1H NMR (400 MHz, (CD3)2SO) δ 7.94–7.89 (m, 2H), 7.39–7.28 (m, 5H), 7.17 (d, J = 7.2, 1H), 5.45–5.37 (m, 1H), 5.26 (d, J= 5.2, 1H), 4.73–4.63 (m, 2H), 4.30–4.25 (m, 1H), 2.02–1.88 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.0 (C), 144.2 (C), 141.1 (C), 139.3 (CH), 139.2 (C), 127.0 (CH), 126.3 (CH), 126.2 (CH), 121.6 (CH), 120.2 (CH), 119.8 (CH), 118.6 (CH), 114.4 (CH2), 71.8 (CH), 36.2 (CH), 34.0 (C), 21.6 (CH2); 13C{1H} NMR (100 MHz, (CD3)2SO) δ 148.6 (C), 144.9 (C), 141.4 (CH), 140.7 (C), 139.0 (C), 127.5 (CH), 126.8 (CH), 126.5 (CH), 126.4 (CH), 122.5 (CH), 120.6 (CH), 120.2 (CH), 119.5 (CH), 113.4 (CH2), 70.2 (CH), 37.3 (CH), 34.3 (C), 22.4 (CH2).
(R*)-Phenyl((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (8d) and (R*)-phenyl-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (8d’).
Alcohols 8d and 8d’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.101 g, 0.49 mmol) and phenylmagnesium chloride (375 μL, 2.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8d was formed as a 64:36 mixture of diastereomers (8d:8d’). Purification by flash chromatography (10:90 to 25:75 EtOAc:hexanes) afforded the major diastereomer alcohol 8d as a yellow oil (0.082 g, 55%) and the minor diastereomer 8d’ as a white solid (0.038 g, 26%). The relative stereochemistry of the minor diastereomer 8d’ was assigned by X-ray crystallography.
Major diastereomer 8d:
1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 7.5, 1H), 7.85 (d, J = 7.5, 1H), 7.56 (d, J = 7.4, 2H), 7.49–7.41 (m, 4H), 7.39–7.35 (m, 3H), 7.32–7.28 (m, 1H), 7.01 (d, J = 7.5, 1H), 5.03 (d, J = 9.9, 1H), 2.39–2.32 (m, 1H), 2.05–2.02 (m, 1H), 1.97 (dd, J = 7.4, 5.4, 1H), 1.57 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.8 (C), 143.8 (C), 142.9 (C), 141.3 (C), 139.1 (C), 128.6 (CH), 127.8 (CH), 127.2 (CH), 126.9 (CH), 126.8 (CH), 126.3 (CH), 125.8 (CH), 121.0 (CH), 120.6 (CH), 119.9 (CH), 118.6 (CH), 73.8 (CH), 40.4 (CH), 34.5 (C), 22.5 (CH2); IR (ATR) 3559, 3061, 1446, 1022, 763 cm−1; HRMS (APCI) m / z calcd for C22H17 ((M + H) − H2O)+ 281.1325, found 281.1325. Anal. Calcd for C22H18O: C, 88.56; H, 6.08. Found: C, 88.49; H, 5.88.
Minor diastereomer 8d’:
mp = 125–128 °C; 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.5, 1H), 7.78 (d, J = 7.5, 1H), 7.41–7.24 (m, 5H), 7.11–7.08 (m, 3H), 7.02–6.98 (m, 3H), 5.06–5.03 (dd, J = 8.8, 3.4, 1H), 2.30 (q, J = 8.4, 1H), 2.10–2.08 (m, 2H), 2.07 (s, 1H); 1H NMR (400 MHz, (CD3)2SO) δ 7.94 (d, J = 7.3, 1H), 7.88 (d, J = 7.4, 1H), 7.52 (d, J = 7.3, 1H), 7.39–7.24 (m, 4H), 7.16 (d, J = 7.4, 1H), 7.04–6.98 (m, 3H), 6.87–6.85 (m, 2H), 5.65 (d, J = 5.0, 1H), 4.88–4.84 (dd, J = 9.1, 5.0, 1H), 2.22–2.15 (m, 1H), 2.10–2.04 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.8 (C), 144.4 (C), 143.5 (C), 141.1 (C), 139.3 (C), 128.6 (CH), 127.4 (CH), 127.0 (CH), 126.4 (CH), 126.2 (CH), 125.3 (CH), 121.9 (CH), 120.3 (CH), 119.8 (CH), 118.6 (CH), 72.2 (CH), 38.1 (CH), 34.8 (C), 21.4 (CH2); 13C{1H} NMR (100 MHz, (CD3)2SO) δ 147.8 (C), 145.0 (C), 144.6 (C), 140.1 (C), 138.4 (C), 127.7 (CH), 126.9 (CH), 126.4 (CH), 126.3 (CH), 126.0 (CH), 125.9 (CH), 125.1 (CH), 122.2 (CH), 120.2 (CH), 119.6 (CH), 119.0 (CH), 70.0 (CH), 38.8 (CH), 34.5 (C), 21.8 (CH2).
(R*)-2-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (8e) and (R*)-2-phenyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (8e’).
Alcohols 8e and 8e’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.110 g, 0.50 mmol) and benzylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 13C{1H} NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8e was formed as a 75:25 mixture of diastereomers (8e:8e’). Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded the major diastereomer alcohol 8e as a yellow solid (0.098 g, 63%) and the minor diastereomer 8e’ as a yellow oil (0.024 g, 15%).
Major diastereomer 8e:
mp = 72–75 °C; 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.6, 1H), 7.80 (d, J = 7.3, 1H), 7.39–7.27 (m, 10H), 7.02 (d, J = 7.3, 1H), 4.21–4.14 (m, 1H), 3.07 (d, J = 6.3, 2H), 2.17–2.10 (m, 1H), 1.91–1.88 (dd, J = 9.1, 5.3, 1H), 1.60–1.57 (dd, J = 7.4, 5.3, 1H), 1.28 (d, J = 2.4, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.0 (C), 141.2 (C), 139.0 (C), 138.2 (C), 129.6 (CH), 128.5 (CH), 127.1 (CH), 126.8 (CH), 126.5 (CH), 126.1 (CH), 121.0 (CH), 120.4 (CH), 119.8 (CH), 118.5 (CH), 72.7 (CH), 43.4 (CH2), 38.3 (CH), 33.4 (C), 22.3 (CH2); IR (ATR) 2988, 2901, 1446, 1077, 728 cm−1; HRMS (APCI) m / z calcd for C23H19 ((M + H) − H2O)+ 295.1481, found 295.1485.
Minor diastereomer 8e’:
1H NMR (400 MHz, CDCl3) δ 7.85–7.80 (m, 2H), 7.41–7.27 (m, 5H), 7.11–7.09 (m, 3H), 7.02 (d, J = 7.4, 1H), 6.86–6.84 (m, 2H), 4.15–4.10 (m, 1H), 2.47 (d, J = 6.1, 2H), 2.04–1.98 (m, 2H), 1.93–1.87 (m, 1H), 1.79 (d, J = 3.4, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.1 (C), 141.2 (C), 139.2 (C), 137.3 (C), 129.3 (CH), 128.3 (CH), 127.0 (CH), 126.4 (CH), 126.35 (CH), 126.31 (CH), 126.2 (CH), 121.6 (CH), 120.4 (CH), 119.7 (CH), 118.6 (CH), 72.5 (CH), 43.6 (CH2), 36.2 (CH), 34.0 (C), 21.9 (CH2).
(R*)-2,2-Dimethyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)propan-1-ol (8f).
Alcohol 8f was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.110 g, 0.50 mmol) and tert-butylmagnesium chloride (450 μL, 1.7 M solution in THF, 0.77 mmol) in THF (5 mL) at 20 °C for 16 h. 13C{1H} NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8f was formed as a single diastereomer. 13C{1H} NMR spectroscopic analysis of the unpurified reaction mixture also showed formation of 1,2-reduction product. Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded alcohol 8f as an orange solid (0.046 g, 33%) and the 1,2-reduction product as a white solid (0.011 g, 10%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) for the 1,2-reduction product match the data reported for the product obtained through the reduction of ester 2 by LiAlH4.
Alcohol 8f:
mp = 88–90 °C; 1H NMR (400 MHz, CDCl3) δ 7.83–7.78 (m, 2H), 7.38–7.27 (m, 4H), 7.24–7.22 (m, 1H), 3.59 (d, J = 9.2, 1H), 2.08–2.02 (m, 1H), 1.96–1.93 (dd, J = 9.1, 4.8, 1H), 1.83–1.80 (dd, J = 7.7, 4.8, 1H), 1.65 (br s, 1H), 0.60 (s, 9H); 1H NMR (400 MHz, (CD3)2SO) δ 7.93–7.87 (m, 2H), 7.37–7.28 (m, 5H), 7.22–7.20 (m, 1H), 4.85 (br s, 1H), 3.41 (d, J = 8.6, 1H), 2.02–1.92 (m, 2H), 1.88–1.85 (dd, J = 7.5, 4.1, 1H), 0.47 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.8 (C), 145.1 (C), 140.8 (C), 139.3 (C), 127.0 (CH), 126.17 (CH), 126.15 (CH), 126.13 (CH), 122.1 (CH), 120.2 (CH), 119.7 (CH), 118.5 (CH), 35.6 (C), 35.3 (C), 33.5 (CH), 25.2 (CH3), 20.1 (CH2); 13C{1H} NMR (100 MHz, (CD3)2SO) δ 147.4 (C), 144.9 (C), 139.5 (C), 137.9 (C), 126.4 (CH), 125.4 (CH), 125.22 (CH), 125.21 (CH), 121.7 (CH), 119.6 (CH), 119.1 (CH), 118.3 (CH), 73.9 (CH), 35.0 (C), 34.3 (C), 33.6 (CH), 24.7 (CH3), 19.9 (CH2); IR (ATR) 3327, 2957, 2867, 1447, 1050, 728 cm−1; HRMS (APCI) m / z calcd for C20H21 ((M + H) − H2O)+ 261.1638, found 261.1638.
(R*)-1-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (8g) and (R*)-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (8g’).
Alcohols 8g and 8g’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.111 g, 0.50 mmol) and allylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8g was formed as a 66:34 mixture of diastereomers (8g:8g’). Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded alcohol 8g and 8g’ as a white solid with a diastereomeric ratio of 84:16 (8g:8g’) (0.068 g, 52%) and alcohol 8g’ as an yellow oil (0.035 g, 27%).
Major Diastereomer 8g:
mp = 72–74 °C; 1H NMR (400 MHz, CDCl3) δ 7.89–7.87 (m, 1H), 7.83–7.81 (m, 1H), 7.42–7.29 (m, 5H), 7.05–7.03 (m, 1H), 6.05–5.95 (m, 1H), 5.25–5.21 (m, 1H), 5.19–5.16 (m, 1H), 4.06–4.00 (m, 1H), 2.62–2.49 (m, 2H), 2.15–2.09 (m, 1H), 2.01–1.97 (dd, J = 9.1, 5.1, 1H), 1.78–1.75 (dd, J = 7.4, 5.1, 1H), 1.30 (d, J = 2.2, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.1 (C), 141.3 (C), 139.0 (C), 134.4 (CH), 127.1 (CH), 126.8 (CH), 126.6 (CH), 126.2 (CH), 121.0 (CH), 120.5 (CH), 119.8 (CH), 118.5 (CH), 117.8 (CH2), 71.2 (CH), 41.4 (CH2), 38.2 (CH), 33.4 (C), 22.3 (CH2); IR (ATR) 3580, 2970, 1447, 1098, 908, 743 cm−1; HRMS (APCI) m / z calcd for C19H17 ((M + H) − H2O)+ 245.1325, found 245.1327.
Minor Diastereomer 8g’:
1H NMR (400 MHz, CDCl3) δ 7.86–7.84 (m, 1H), 7.82–7.80 (m, 1H), 7.39–7.28 (m, 4H), 7.24–7.22 (m, 1H), 7.04–7.02 (m, 1H), 5.60–5.49 (m, 1H), 4.94–4.86 (m, 2H), 4.00–3.95 (m, 1H), 2.05–1.98 (m, 4H), 1.91–1.88 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.1 (C), 144.3 (C), 141.1 (C), 139.2 (C), 133.7 (CH), 127.0 (CH), 126.4 (CH), 126.3 (CH), 126.1 (CH), 121.6 (CH), 120.2 (CH), 119.7 (CH), 118.5 (CH), 118.4 (CH2), 70.4 (CH), 41.8 (CH2), 36.3 (CH), 34.0 (C), 21.9 (CH2).
(R*)-2,2-Dimethyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (8h), (R*)-2,2-dimethyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (8h’) and (R*)-4-methyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)pent-3-en-1-ol (8h’).
Alcohols 8h, 8h’, and 8h” were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using aldehyde 3 (0.111 g, 0.50 mmol) and prenylmagnesium chloride (2.2 mL, 0.34 M solution in THF, 0.75 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that the products were formed as a 90:10 mixture of regioisomers. The minor regioisomer 8h” was identified by a multiplet at δ 5.42−5.37 ppm corresponding to the vinyl proton. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8h was formed as a 65:35 mixture of diastereomers (8h:8h’) and that alcohol 8h” was formed as one diastereomer. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 8h as a yellow solid (0.070 g, 48%).
Major Diastereomer 8h:
mp = 49–52 °C; 1H NMR (400 MHz, CDCl3) δ 7.87–7.86 (m, 1H), 7.82–7.80 (m, 1H), 7.41–7.28 (m, 5H), 7.05–7.04 (m, 1H), 6.10–6.03 (dd, J = 17.6, 10.8, 1H), 5.16–5.13 (dd, J= 11.7, 1.4, 1H), 5.11–5.10 (dd, J = 4.9, 1.3, 1H), 3.66 (d, J = 9.9, 1H), 2.15–2.08 (m, 1H), 2.05–2.02 (dd, J = 8.9, 5.0, 1H), 1.83–1.80 (dd, J = 7.3, 5.0, 1H), 1.21 (s, 3H), 1.20 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.3 (C), 144.9 (CH), 144.1 (C), 141.2 (C), 139.1 (C), 127.0 (CH), 126.9 (CH), 126.5 (CH), 126.1 (CH), 121.0 (CH), 120.4 (CH), 119.8 (CH), 118.5 (CH), 112.8 (CH2), 78.3 (CH), 41.9 (C), 35.2 (CH), 32.5 (C), 24.5 (CH2), 24.2 (CH3), 22.8 (CH3); IR (ATR) 3576, 3063, 2962, 1447, 1053, 728 cm−1; HRMS (APCI) m / z calcd for C21H21 ((M + H) − H 2O)+ 273.1638, found 273.1641.
Minor Diastereomer 8h’:
1H NMR (400 MHz, CDCl3, diagnostic peaks) δ 5.71–5.64 (dd, J = 17.5, 10.8, 1H), 4.95–4.92 (dd, J = 10.8, 1.1, 1H), 4.89–4.84 (dd, J = 17.5, 1.1, 1H), 3.62 (d, J = 8.9, 1H), 0.68 (s, 3H), 0.59 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, diagnostic peaks) δ 147.8, 75.7, 41.9, 35.4.
2-(Spiro[cyclopropane-1,9’-fluoren]-2-yl)propan-2-ol (9a).
Alcohol 9a was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.118 g, 0.50 mmol) and methylmagnesium chloride (250 μL, 3.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16h. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9a as a white solid (0.102 g, 87%): mp = 88–89 °C; 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 7.7, 1H), 7.81–7.77 (dd, J = 10.4, 7.6, 2H), 7.32–7.22 (m, 4H), 6.97 (d, J = 7.5, 1H), 2.16–2.13 (dd, J = 8.3, 4.3, 1H), 2.04–2.00 (m, 1H), 1.79–1.76 (dd, J = 9.5, 4.3, 1H), 1.47 (s, 3H), 1.03 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.5 (C), 145.1 (C), 141.0 (C), 139.0 (C), 126.6 (CH), 126.2 (CH), 125.8 (CH), 125.6 (CH), 124.7 (CH), 119.7 (CH), 119.5 (CH), 118.3 (CH), 70.0 (C), 43.4 (CH), 34.6 (C), 33.1 (CH3), 29.0 (CH3), 19.8 (CH2); IR (ATR) 3482, 2996, 2974, 1447, 733 cm−1; HRMS (APCI) m / z calcd for C18H17 ((M + H) − H2O)+ 233.1325, found 233.1326. Anal. Calcd for C18H18O: C, 86.36; H, 7.25. Found: C, 86.09; H, 7.19.
(R*)-2-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)pentan-2-ol (9b).
Alcohol 9b was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.118 g, 0.50 mmol) and n-propylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 9b was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9b as a white solid (0.099 g, 70%): mp = 66–68 °C; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.7, 1H), 7.83–7.78 (m, 2H), 7.34–7.22 (m, 4H), 7.00 (d, J = 7.4, 1H), 2.18–2.14 (dd, J = 8.3, 4.3, 1H), 2.02–1.97 (m, 1H), 1.82–1.78 (dd, J = 9.5, 4.3, 1H), 1.73–1.49 (m, 4H), 1.39 (s, 1H), 1.02–1.00 (m, 3H), 0.98 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.6 (C), 145.3 (C), 141.0 (C), 139.0 (C), 126.6 (CH), 126.2 (CH), 125.7 (CH), 125.6 (CH), 124.8 (CH), 119.7 (CH), 119.5 (CH), 118.3 (CH), 71.8 (C), 48.3 (CH2), 42.5 (CH), 33.9 (C), 27.4 (CH3), 20.3 (CH2), 17.0 (CH2), 14.6 (CH3); IR (ATR) 3558, 2964, 2927, 1449, 1050, 730 cm−1; HRMS (APCI) m / z calcd for C20H21 ((M + H) − H2O)+ 261.1638, found 261.1640. Anal. Calcd for C20H22O: C, 86.29; H, 7.97. Found: C, 86.59; H, 8.15.
(R*)-2-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-2-ol (9c).
Alcohol 9c was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.118 g, 0.50 mmol) and vinylmagnesium chloride (750 μL, 1.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 9c was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9c as a yellow solid (0.093 g, 70%): mp = 48–50 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 7.6, 1H), 7.83–7.78 (m, 2H), 7.34–7.21 (m, 4H), 6.99 (d, J = 7.4, 1H), 6.18–6.11 (dd, J = 17.3, 10.6, 1H), 5.36–5.32 (d, J = 17.3, 1H), 5.15–5.12 (d, J = 10.6, 1H), 2.19–2.16 (dd, J = 8.2, 4.1, 1H), 2.13–2.09 (m, 1H), 1.82–1.79 (dd, J = 9.1, 4.0, 1H), 1.58 (br s, 1H), 1.12 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.3 (C), 146.2 (CH), 144.8 (C), 141.1 (C), 139.0 (C), 126.7 (CH), 126.2 (CH), 125.9 (CH), 125.8 (CH), 124.9 (CH), 119.7 (CH), 119.5 (CH), 118.4 (CH), 111.6 (CH2), 71.9 (C), 42.1 (CH), 34.6 (C), 27.7 (CH3), 19.9 (CH2); IR (ATR) 3370, 2981, 2865, 1446, 1055, 732 cm−1; HRMS (APCI) m / z calcd for C19H17 ((M + H) − H2O)+ 245.1325, found 245.1329.
(R*)-1-Phenyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (9d).
Alcohol 9d was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.115 g, 0.49 mmol) and phenylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 9d was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9d as a white solid (0.097 g, 62%). The relative stereochemistry was assigned by X-ray crystallography. mp = 83–85 °C; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 7.7, 1H), 7.86–7.81 (dd, J = 14.7, 7.5, 2H), 7.64–7.61 (m, 2H), 7.44–7.40 (m, 2H), 7.37–7.24 (m, 5H), 6.96 (d, J = 7.6, 1H), 2.45–2.42 (dd, J = 8.4, 4.5, 1H), 2.29–2.25 (m, 1H), 1.87–1.84 (dd, J = 9.5, 4.5, 1H), 1.80 (s, 1H), 1.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.4 (C), 149.2 (C), 144.8 (C), 141.2 (C), 139.0 (C), 128.5 (CH), 127.3 (CH), 126.7 (CH), 126.3 (CH), 125.91 (CH), 125.86 (CH), 125.0 (CH), 124.8 (CH), 119.8 (CH), 119.5 (CH), 118.5 (CH), 73.1 (C), 44.8 (CH), 35.5 (C), 28.8 (CH3), 20.3 (CH2); IR (ATR) 3696, 2981, 2844, 1449, 1178, 1033 cm−1; HRMS (APCI) m / z calcd for C23H19 ((M + H) − H2O)+ 295.1481, found 295.1478.
(R*)-1-Phenyl-2-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)propan-2-ol (9e).
Alcohol 9e was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.119 g, 0.51 mmol) and benzylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 9e was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9e as a yellow solid (0.145 g, 88%): mp = 96–99 °C; 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.8, 1H), 7.79–7.75 (m, 2H), 7.34–7.24 (m, 8H), 7.20–7.16 (m, 1H), 6.98 (d, J = 7.2, 1H), 3.04 (d, J = 13.1, 1H), 2.85 (d, J = 13.1, 1H), 2.13–2.08 (m, 1H), 2.00–1.97 (dd, J = 8.4, 4.6, 1H), 1.70–1.66 (dd, J = 9.5, 4.5, 1H), 1.56 (s, 1H), 0.97 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.5 (C), 145.0 (C), 141.0 (C), 139.0 (C), 136.7 (C), 130.6 (CH), 128.3 (CH), 126.8 (CH), 126.6 (CH), 126.1 (CH), 125.8 (CH), 125.6 (CH), 125.0 (CH), 119.6 (CH), 119.5 (CH), 118.3 (CH), 71.4 (C), 51.2 (CH2), 42.6 (CH), 34.0 (C), 26.9 (CH3), 20.2 (CH2); IR (ATR) 3563, 2981, 1448, 1100, 774 cm−1; HRMS (APCI) m / z calcd for C24H21 ((M + H) − H2O)+ 309.1638, found 309.1634.
(R*)-2-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)pent-4-en-2-ol (9f).
Alcohol 9f was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.119 g, 0.51 mmol) and allylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 9f was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9f as a yellow solid (0.131 g, 94%): mp = 92–94 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 7.6, 1H), 7.78 (t, J = 8.1, 2H), 7.32–7.23 (m, 4H), 6.97 (d, J = 7.4, 1H), 6.06–5.96 (m, 1H), 5.25–5.19 (m, 2H), 2.52–2.47 (dd, J = 13.4, 6.6, 1H), 2.36–2.31 (dd, J = 13.4, 8.5, 1H), 2.10–2.07 (dd, J = 8.2, 4.0, 1H), 2.05–2.00 (m, 1H), 1.80 (s, 1H), 1.78–1.75 (dd, J = 9.1, 4.0, 1H), 0.98 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.4 (C), 145.1 (C), 140.9 (C), 139.1 (C), 133.4 (CH), 126.6 (CH), 126.2 (CH), 125.8 (CH), 125.6 (CH), 125.0 (CH), 119.8 (CH2), 119.6 (CH), 119.5 (CH), 118.3 (CH), 70.7 (C), 50.1 (CH2), 42.2 (CH), 33.8 (C), 27.0 (CH3), 20.4 (CH2); IR (ATR) 3564, 3022, 2989, 1445, 781 cm−1; HRMS (APCI) m / z calcd for C20H19 ((M + H) − H2O)+ 259.1481, found 259.1483. Anal. Calcd for C20H20O: C, 86.92; H, 7.29. Found: C, 87.19; H, 7.39.
(R*)-3,3-Dimethyl-2-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)pent-4-en-2-ol (9g) and (R*)-5-methyl-2-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)hex-4-en-2-ol (9g’).
Alcohols 9g and 9g’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 6 (0.117 g, 0.50 mmol) and prenylmagnesium chloride (2.2 mL, 0.34 M solution in THF, 0.75 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed an 82:18 mixture of regioisomers (9g:9g’). The minor regioisomer 9g’ was identified by a multiplet at δ 5.41−5.38 ppm corresponding to the vinyl proton. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that both alcohol 9g and alcohol 9g’ were formed as single diastereomers (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 9g as a white solid (0.122 g, 84%): mp = 77–80 °C; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 7.7, 1H), 7.80 (t, J = 7.4, 2H), 7.35–7.21 (m, 4H), 7.01 (d, J = 7.2, 1H), 6.15–6.08 (m, 1H), 5.20 (s, 1H), 5.18–5.15 (dd, J = 7.6, 1.2, 1H), 2.27–2.22 (m, 1H), 2.19–2.16 (dd, J = 8.6, 4.2, 1H), 1.82–1.78 (dd, J = 9.3, 4.2, 1H), 1.68 (s, 1H), 1.19 (s, 3H), 1.18 (s, 3H), 0.92 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.8 (C), 145.3 (CH), 144.9 (C), 141.0 (C), 139.1 (C), 126.6 (CH), 126.2 (CH), 125.8 (CH), 125.6 (CH), 125.2 (CH), 119.6 (CH), 119.5 (CH), 118.3 (CH), 114.5 (CH2), 74.7 (C), 46.2 (C), 39.5 (CH2), 33.6 (C), 22.71 (CH3), 22.67 (CH3), 21.9 (CH), 21.8 (CH3); IR (ATR) 3581, 3060, 2970, 1447, 1098, 908, 743 cm−1; HRMS (APCI) m / z calcd for C22H23 ((M + H) − H2O)+ 287.1794, found 287.1797. Anal. Calcd for C22H24O: C, 86.80; H, 7.95. Found: C, 86.52; H, 7.90.
Reaction of ketone 6 with tert-butylmagnesium chloride.
The reaction of ketone 6 with tertbutylmagnesium chloride was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 6 (0.046 g, 0.20 mmol) and tert-butylmagnesium chloride (150 μL, 0.30 mmol) in THF (2 mL) at 20 °C for 4 h. Analysis of the 1H NMR and 13C{1H} NMR spectra of the unpurified reaction mixture revealed only starting material was present (see Analytical Data).
(R*)-1-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (10a).
Alcohol 10a was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.149 g, 0.50 mmol) and methylmagnesium chloride (250 μL, 3.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10a was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10a as a yellow solid (0.112 g, 72%): mp = 104–106 °C; 1H NMR (400 MHz, CDCl3) δ 7.75–7.73 (m, 1H), 7.67–7.61 (m, 2H), 7.34–7.27 (m, 2H), 7.17–7.13 (m, 1H), 7.08–7.02 (m, 4H), 6.98–6.94 (m, 3H), 2.51–2.47 (m, 1H), 2.32–2.29 (dd, J = 8.3, 4.6, 1H), 1.97–1.94 (dd, J = 9.6, 4.6, 1H), 1.90 (s, 1H), 1.78 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.0 (C), 146.9 (C), 144.4 (C), 140.9 (C), 139.3 (C), 127.7 (CH), 126.7 (CH), 126.6 (CH), 126.0 (CH), 125.9 (CH), 125.5 (CH), 124.7 (CH), 124.2 (CH), 119.53 (CH), 119.50 (CH), 118.3 (CH), 73.9 (C), 42.7 (CH), 35.5 (C), 34.5 (CH3), 19.5 (CH2); IR (ATR) 3539, 3036, 2996, 1449, 1111, 736 cm−1; HRMS (APCI) m / z calcd for C23H19 ((M + H) − H2O)+ 295.1481, found 295.1485. Anal. Calcd for C23H20O: C, 88.43; H, 6.45. Found: C, 88.45; H, 6.28.
(R*)-1-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)butan-1-ol (10b).
Alcohol 10b was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.149 g, 0.50 mmol) and n-propylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10b was formed as a single diastereomer (dr > 99:1). Analysis of the 1H NMR spectrum of the unpurified reaction mixture also revealed the formation of 1,2-reduction product 8d (20% conversion). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10b as a yellow solid (0.091 g, 53%): mp = 78–80 °C; 1H NMR (400 MHz, CDCl3) δ 7.73–7.71 (m, 1H), 7.62–7.60 (dd, J = 7.8, 2.6, 2H), 7.35–7.29 (m, 2H), 7.12–7.08 (m, 2H), 7.01–6.99 (m, 3H), 6.93–6.89 (m, 3H), 2.57–2.52 (m, 1H), 2.32–2.29 (dd, J = 8.2, 4.6, 1H), 1.98–1.94 (m, 4H), 1.50–1.33 (m, 2H), 0.93 (t, J = 7.3, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.1 (C), 146.0 (C), 144.5 (C), 140.6 (C), 139.4 (C), 127.4 (CH), 126.6 (CH), 126.2 (CH), 125.9 (CH), 125.8 (CH), 125.3 (CH), 124.9 (CH), 124.2 (CH), 119.5 (CH), 119.3 (CH), 118.2 (CH), 75.9 (C), 50.1 (CH2), 41.6 (CH), 34.7 (C), 20.1 (CH2), 17.3 (CH2), 14.4 (CH3); IR (ATR) 3568, 2958, 2870, 1448, 1033, 759 cm−1; HRMS (APCI) m / z calcd for C25H23 ((M + H) − H2O)+ 323.1794, found 323.1796. Anal. Calcd for C25H24O: C, 88.20; H, 7.11. Found: C, 87.93; H, 7.12.
(R*)-1-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)prop-2-en-1-ol (10c).
Alcohol 10c was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.141 g, 0.48 mmol) and vinylmagnesium chloride (750 μL, 1.0 M solution in THF, 0.75 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10c was formed as a single diastereomer (dr > 99:1). Analysis of the 1H NMR spectrum of the unpurified reaction mixture also revealed the formation of 1,2-reduction product 8d (10% conversion). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10c as a white solid (0.103 g, 67%): mp = 118–120 °C; 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 7.4, 1H), 7.72–7.68 (m, 2H), 7.36–7.28 (m, 2H), 7.20–7.17 (m, 1H), 7.09–7.05 (m, 4H), 7.01–6.98 (m, 3H), 6.31–6.24 (dd, J = 17.1, 10.5, 1H), 5.40 (d, J = 17.1, 1H), 5.23 (d, J = 10.5, 1H), 2.56–2.52 (m, 1H), 2.32–2.29 (dd, J = 8.3, 4.7, 1H), 2.03 (s, 1H), 2.00–1.96 (dd, J = 9.6, 4.7, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.8 (C), 145.3 (CH), 144.6 (C), 144.3 (C), 141.0 (C), 139.3 (C), 127.9 (CH), 127.0 (CH), 126.8 (CH), 126.1 (CH), 126.0 (CH), 125.73 (CH), 125.65 (CH), 124.6 (CH), 119.60 (CH), 119.59 (CH), 118.4 (CH), 112.2 (CH2), 76.4 (C), 41.3 (CH), 35.7 (C), 19.5 (CH2); IR (ATR) 3560, 3062, 1480, 1167, 992 cm−1; HRMS (APCI) m / z calcd for C24H19 ((M + H) − H2O)+ 307.1481, found 307.1483.
Diphenyl(spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (10d).
Alcohol 10d was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.150 g, 0.51 mmol) and phenylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 66 °C in a silicone oil bath for 16 h. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10d as a white solid (0.141 g, 74%): mp = 144–147 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 7.5, 1H), 7.71 (d, J = 7.8, 2H), 7.41–7.39 (m, 2H), 7.36–7.31 (m, 3H), 7.29–7.25 (m, 2H), 7.19–7.15 (m, 1H), 7.08–7.03 (m, 2H), 6.97–6.94 (m, 5H), 2.68–2.64 (m, 1H), 2.53–2.50 (dd, J = 8.4, 4.6, 1H), 2.29 (s, 1H), 1.96–1.93 (dd, J = 9.5, 4.6, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.1 (C), 148.9 (C), 145.9 (C), 144.3 (C), 141.1 (C), 139.2 (C), 128.2 (CH), 127.8 (CH), 127.10 (CH), 127.08 (CH), 126.8 (CH), 126.6 (CH), 126.10 (CH), 126.08 (CH), 126.01 (CH), 125.7 (CH), 124.4 (CH), 119.7 (CH), 119.6 (CH), 118.5 (CH), 78.4 (C), 43.7 (CH), 36.6 (C), 19.0 (CH2); IR (ATR) 3565, 2989, 1445, 1068, 724 cm−1; HRMS (APCI) m / z calcd for C28H21 ((M + H) − H2O)+ 257.1638, found 257.1632.
(R*)-1,2-Diphenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (10e).
Alcohol 10e was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.151 g, 0.50 mmol) and methylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10e was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10e as a yellow solid (0.134 g, 68%): mp = 106–108 °C; 1H NMR (400 MHz, CDCl3) δ 7.70–7.68 (m, 1H), 7.61–7.55 (dd, J = 15.1, 7.6, 2H), 7.33–7.26 (m, 5H), 7.09–7.03 (m, 4H), 6.98–6.92 (m, 3H), 6.89–6.87 (m, 3H), 3.28 (d, J = 13.2, 1H), 3.20 (d, J = 13.2, 1H), 2.70–2.66 (m, 1H), 2.21 (s, 1H), 2.09–2.06 (dd, J = 8.1, 4.8, 1H), 1.82–1.78 (dd, J = 9.6, 4.7, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.0 (C), 145.6 (C), 144.3 (C), 140.6 (C), 139.5 (C), 136.0 (C), 130.7 (CH), 128.4 (CH), 127.4 (CH), 127.2 (CH), 126.6 (CH), 126.4 (CH), 126.0 (CH), 125.8 (CH), 125.4 (CH), 125.1 (CH), 124.5 (CH), 119.6 (CH), 119.3 (CH), 118.2 (CH), 75.4 (C), 53.8 (CH2), 41.5 (CH), 34.7 (C), 20.3 (CH2); IR (ATR) 3569, 3031, 1495, 1447, 1027, 735 cm−1; HRMS (APCI) m / z calcd for C29H23 ((M + H) − H2O)+ 371.1794, found 371.1800. Anal. Calcd for: C29H24O: C, 89.66; H, 6.23. Found: C, 89.52; H, 6.19.
(R*)-1-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (10f).
Alcohol 10f was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.149 g, 0.50 mmol) and allylmagnesium chloride (380 μL, 2.0 M solution in THF, 0.76 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10f was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10f as a yellow oil (0.139 g, 82%): 1H NMR (400 MHz, CDCl3) δ 7.72–7.70 (m, 1H), 7.67 (d, J = 7.8, 1H), 7.59 (d, J = 7.5, 1H), 7.35–7.28 (m, 2H), 7.13–7.07 (m, 2H), 7.05–6.97 (m, 3H), 6.93–6.86 (m, 3H), 5.93–5.82 (m, 1H), 5.31 (m, 1H), 5.28 (m, 1H), 2.81–2.75 (dd, J = 13.5, 6.9, 1H), 2.70–2.65 (dd, J = 13.5, 8.1, 1H), 2.61–2.57 (dd, J = 9.4, 8.2, 1H), 2.44 (s, 1H), 2.29–2.25 (dd, J = 8.0, 4.6, 1H), 1.96–1.92 (dd, J = 9.5, 4.6, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.0 (C), 145.8 (C), 144.3 (C), 140.5 (C), 139.5 (C), 133.2 (CH), 127.4 (CH), 126.6 (CH), 126.3 (CH), 125.9 (CH), 125.8 (CH), 125.3 (CH), 124.9 (CH), 124.5 (CH), 120.7 (CH2), 119.5 (CH), 119.2 (CH), 118.2 (CH), 74.2 (C), 52.5 (CH2), 41.3 (CH), 34.6 (C), 20.5 (CH2); IR (ATR) 3549, 3061, 1447, 1070, 733 cm−1; HRMS (APCI) m / z calcd for C25H21 ((M + H) − H2O)+ 321.1638, found 321.1641. Anal. Calcd for C25H22O: C, 88.72; H, 6.55. Found: C, 88.82; H, 6.53.
(R*)-2,2-Dimethyl-1-phenyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (10g) and (R*)-4-methyl-1-phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)pent-3-en-1-ol (10g’).
Alcohols 10g and 10g’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 7 (0.148 g, 0.50 mmol) and prenylmagnesium chloride (2.2 mL, 0.34 M solution in THF, 0.75 mmol) in THF (5 mL) at −78 °C for 30 min. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed an 82:18 mixture of regioisomers (10g:10g’). The minor regioisomer 10g’ was identified by a multiplet at δ 5.42−5.38 ppm corresponding to the vinyl proton. Alcohol 10g was formed as single diastereomer (>99:1) as determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10g as a yellow oil (0.153 g, 84%): 1H NMR (400 MHz, CDCl3) δ 7.72–7.69 (m, 1H), 7.51–7.47 (dd, J = 11.7, 7.7, 2H), 7.35–7.33 (m, 2H), 7.16–7.13 (m, 1H), 7.00–6.97 (m, 1H), 6.92–6.90 (m, 2H), 6.87–6.83 (m, 1H), 6.79–6.75 (m, 3H), 6.07–6.00 (dd, J = 17.3, 11.0, 1H), 5.30–5.29 (m, 1H), 5.27–5.24 (m, 1H), 2.90–2.86 (dd, J = 9.3, 8.3, 1H), 2.49 (s, 1H), 2.27–2.24 (dd, J = 8.1, 4.6, 1H), 1.94–1.90 (dd, J = 9.5, 4.6, 1H), 1.14 (s, 3H), 1.07 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 149.1 (C), 144.7 (CH), 144.2 (C), 142.5 (C), 140.2 (C), 139.6 (C), 127.2 (CH), 126.5 (CH), 126.0 (CH), 125.91 (CH), 125.85 (CH), 125.5 (CH), 125.0 (CH), 124.4 (CH), 119.5 (CH), 118.8 (CH), 118.0 (CH), 115.2 (CH2), 78.0 (C), 47.0 (C), 38.8 (CH), 34.5 (C), 23.2 (CH3), 21.8 (CH2), 21.5 (CH3); IR (ATR) 3541, 2975, 1447, 1071, 728 cm−1; HRMS (APCI) m / z calcd for C27H25 ((M + H) − H2O)+ 349.1951, found 349.1953. Anal. Calcd for C27H26O: C, 88.48; H, 7.15; Found: C, 88.21; H, 7.10.
Reaction of ketone 7 with tert-butylmagnesium chloride.
The reaction of ketone 7 with tertbutylmagnesium chloride was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 7 (0.150 g, 0.50 mmol) and tertbutylmagnesium chloride (380 μL, 2.0 M in THF, 0.76 mmol) in THF (5 mL) at −78 °C for 2 h. Analysis of the HPLC, LC-MS, 1H NMR, and 13C{1H} NMR spectra of the unpurified reaction mixture revealed the formation of a complex mixture of products (see Analytical Data). Analysis of the unpurified LC-MS suggested the formation of ketone 11, which was confirmed by hydrogenation of ketone 7 to create an authentic sample of ketone 11 (as discussed in Section V).
Reaction of ketone 5 with benzylmagnesium chloride.
The reaction of ketone 5 with benzylmagnesium chloride was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 5 (0.20 g, 0.05 mmol) and benzylmagnesium chloride (38 μL, 2.0 M in THF, 0.075 mmol) in THF (0.5 mL) at 20 °C for 16 h. Analysis of the HPLC, 1H NMR, 13C{1H} NMR, 19F{1H} NMR spectra of the unpurified reaction mixture revealed the formation of a complex mixture of products (see Analytical Data).
(R*)-(Perfluorophenyl)((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (20) and (R*)-(perfluorophenyl)((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (20’).
Alcohols 20 and 20’ were prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 5 (0.20 g, 0.05 mmol) and n-propylmagnesium chloride (40 μL, 2.0 M solution in THF, 0.08 mmol) in THF (0.5 mL) at 20 °C for 16 h. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed the formation of a complex mixture of products including 1,2-reduction products 20 and 20’. Alcohols 20 and 20’ were formed as a 79:21 mixture of diastereomers. Purification by flash chromatography (5:95 to 25:75 EtOAc:hexanes) afforded alcohol 20 as a yellow oil (0.007 g, 38%) and alcohol 20’ as a yellow oil (0.003 g, 16%). The spectroscopic data (1H NMR, 13C{1H} NMR, 19F{1H} NMR, HSQC, and HRMS) for alcohol 20 and 20’ match the data reported for the products obtained through the addition of C6F5MgBr to aldehyde 3 (Section II).
1-(Perfluorophenyl)-1-(spiro[cyclopropane-1,9’-fluoren]-2-yl)but-3-en-1-ol (21).
Alcohol 21 was prepared using the representative procedure for additions of Grignard reagents to carbonyl compounds using ketone 5 (0.020 g, 0.05 mmol) and allylmagnesium chloride (40 μL, 2.0 M solution in THF, 0.08 mmol) in THF (0.5 mL). 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 21 was formed as a single diastereomer (dr > 99:1). Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 21 as a white solid (0.020 g, 93%): mp = 69–72 °C; 1H NMR (400 MHz, CDCl3) δ 7.72–7.70 (m, 1H), 7.66 (d, J = 7.3, 1H), 7.62 (d, J = 7.5, 1H), 7.34–7.28 (m, 2H), 7.17–7.13 (m, 1H), 7.08–7.04 (m, 2H), 5.77–5.67 (m, 1H), 5.36–5.30 (m, 2H), 3.13–3.11 (m, 1H), 2.83 (d, J = 7.5, 2H), 2.69–2.63 (m, 1H), 2.52–2.48 (m, 1H), 1.98–1.94 (dd, J = 9.5, 4.9, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.3 (C), 145.3–142.9 (br d, JC–F = 250.9, CF), 143.4 (C), 140.5 (C), 140.4–138.0 (br d, JC–F = 247.0, CF), 139.0 (C), 138.3–135.9 (br d, JC–F = 239.0, CF), 131.1 (CH), 126.8 (CH), 126.1 (CH), 125.9 (CH), 125.6 (CH), 123.4 (CH), 122.4 (CH2), 119.6 (CH), 119.4 (CH), 118.9 (m, C), 118.1 (CH), 74.7 (C), 49.4 (CH2), 41.5–41.4 (t, J = 4.2, CH), 34.8 (C), 19.5 (CH2); 19F{1H} NMR (377 MHz, CDCl3) δ −141.7 (d, J = 19.6, 2F), −157.6 (m, 1F), −164.0 (m, 2F); IR (ATR) 3568, 3065, 1523, 1095, 986 cm−1; HRMS (APCI) m / z calcd for C25H16F5 ((M + H) − H2O)+ 411.1167, found 411.1164.
Reaction of ketone 5 with isopropylmagnesium chloride.
The reaction of ketone 5 with isopropylmagnesium chloride was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 5 (0.039 g, 0.10 mmol) and isopropylmagnesium chloride (75 μL, 2.0 M in THF, 0.15 mmol) in THF (1 mL) at 20 °C for 16 h. Analysis of the HPLC, LC-MS, 1H NMR, 13C{1H} NMR, and 19F{1H} NMR spectra of the unpurified reaction mixture revealed the formation of a complex mixture of products including 1,2-reduction products 20 and 20’ (see Analytical Data). Alcohols 20 and 20’ were formed as a 62:38 mixture of diastereomers.
Reaction of ketone 7 with isopropylmagnesium chloride.
The reaction of ketone 7 with isopropylmagnesium chloride was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 7 (0.030 g, 0.10 mmol) and isopropylmagnesium chloride (75 μL, 2.0 M in THF, 0.15 mmol) in THF (1 mL) at 20 °C for 16 h. Analysis of the HPLC, 1H NMR, and 13C{1H} NMR spectra of the unpurified reaction mixture revealed the presence of starting material and 1,2-reduction product 8d (about 6% conversion, see Analytical Data).
Reaction of ketone 5 with isopropylmagnesium chloride-lithium chloride complex.
The reaction of ketone 5 with isopropylmagnesium chloride-lithium chloride complex was performed following the general procedure for the additions of Grignard reagents to carbonyl compounds using ketone 5 (0.059 g, 0.20 mmol) and isopropylmagnesium chloride-lithium chloride complex (230 μL, 1.3 M in THF, 0.30 mmol) in THF (2 mL) at 20 °C for 16 h. Analysis of the HPLC, 1H NMR, and 13C{1H} NMR spectra of the unpurified reaction mixture revealed the formation of a complex mixture of products including some conversion to 1,2-reduction product 8d (see Analytical Data).
Reaction of ketone 6 with tert-butylmagnesium chloride in the presence of cerium chloride.
A reported procedure33 was adapted to perform the reaction of ketone 6 with tert-butylmagnesium chloride in the presence of cerium chloride. Cerium chloride heptahydrate (0.116 g, 0.31 mmol) was heated to 150 °C in a silicone oil bath for 1 h under reduced pressure (0.08 torr) with vigorous stirring. After an additional hour at 150 °C and reduced pressure, the flask was backfilled with nitrogen gas and slowly cooled to 0 °C over 1 h. THF (2 mL) was added and the resulting solution was vigorously stirred. After 30 min, the mixture was warmed to 20 °C and stirred for an additional 16 h. The solution was cooled to 0 °C and tert-butylmagnesium chloride was added dropwise. After an hour, ketone 6 (1 mL, 0.2 M in THF, 0.2 mmol) was added dropwise to the stirring solution of tert-butylmagnesium chloride and cerium chloride. After 6 h at 0 °C, H2O (1 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. LC-MS analysis of the unpurified reaction mixture revealed only starting material was present.
(R*)-1-Phenyl-1-((R*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)butan-1-ol (10b).
A reported procedure4 was adapted to prepare alcohol 10b. To a solution of ketone 7 (0.030 g, 0.10 mmol) and 1-bromopropane (45 μL, 0.50 mmol) in THF (1 mL) was added magnesium turnings (0.247 g, 1.0 mmol) in one portion. After 16h, H2O (2 mL) was added followed by the dropwise addition of HCl (20 mL, 1.0 M in H2O) over 2 h. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (4 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10b was formed as one diastereomer. Analysis of the 1H NMR spectrum of the unpurified reaction mixture also revealed the formation of 1,2-reduction product 8d (14% conversion). Purification by flash chromatography (5:95 EtOAc:hexanes) afforded alcohol 10b as a yellow oil (0.028 g, 81%). The spectroscopic data (1H NMR, 13C{1H} NMR, HSQC, and HRMS) match the data reported for the product obtained through the addition of n-propylmagnesium chloride to ketone 7 (Section III).
(R*)-1-Phenyl-1-((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (10a).
To a cooled (−78 °C) solution of ketone 7 (0.148 g, 0.50 mmol) in Et2O (5 mL) was added methyllithium (500 μL, 1.5 M in Et2O, 0.75 mmol) dropwise over 5 min. After 1 h, the solution was warmed to 20 °C. After 16 h, H2O (5 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 10a was formed as one diastereomer. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10a as a yellow solid (0.137 g, 88%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the product obtained through the addition of methylmagnesium chloride to ketone 7 (Section III).
Diphenyl(spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (10d).
To a cooled (−78 °C) solution of ketone 7 (0.059 g, 0.20 mmol) in Et2O (2 mL) was added phenyllithium (160 μL, 1.9 M in n-Bu2O, 0.30 mmol) dropwise over 5 min. After 1 h, the solution was warmed to 20 °C. After an additional 16 h, H2O (5 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 10d as a white solid (0.071 g, 95%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the product obtained through the addition of phenylmagnesium chloride to ketone 7.
(R*)-1-((R*)-Spiro[cyclopropane-1,9’-fluoren]-2-yl)ethan-1-ol (8a).
A reported procedure49 was adapted to prepare alcohol 8a. To a cooled (0 °C) suspension of copper iodide (0.077 g, 0.41 mmol) in Et2O (2 mL) in a Schlenk flask was added methyllithium (500 μL, 1.6 M in Et2O, 0.80 mmol). After 15 min, aldehyde 3 (0.045 g, 0.20 mmol) was added in one portion. The resulting mixture was stirred for 1 h at 0 °C before warming to 20 °C over 1 h. After an additional 16 h, H2O (1 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8a was formed as one diastereomer. Purification by flash chromatography (5:95 EtOAc:hexanes) afforded alcohol 8a as a yellow solid (0.032 g, 68%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the major product 8a obtained through the addition of methylmagnesium chloride to aldehyde 3.
(R*)-Phenyl((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (8d).
A reported procedure49 was adapted to prepare alcohol 8d. To a cooled (0 °C) suspension of copper iodide (0.077 g, 0.41 mmol) in Et2O (2 mL) in a Schlenk flask was added phenyllithium (500 μL, 1.6 M in Et2O, 0.80 mmol). After 15 min, aldehyde 3 (0.045 g, 0.20 mmol) was added in one portion. The resulting mixture was stirred for 1 h at 0 °C before warming to 20 °C over 1 h. After an additional 16 h, H2O (1 mL) and HCl (10 mL, 1.0 M in H2O) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8d was formed as one diastereomer. Purification by flash chromatography (5:95 EtOAc:hexanes) afforded alcohol 8d as a yellow oil (0.044 g, 74%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the major product 8d obtained through the addition of phenylmagnesium chloride to aldehyde 3.
(R*)-Phenyl((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (8d).
To a cooled (0 °C) solution of ketone 7 (0.030 g, 0.10 mmol) in Et2O (1 mL) was added i-Bu2AlH (125 μL, 1.2 M in PhMe, 0.15 mmol) dropwise over 5 min. After 4 h, H2O (1 mL) and HCl (5 mL, 1.0 M in H2O) were added and the mixture was warmed to 20 °C. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed that alcohol 8d was formed as one diastereomer. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 8d as a yellow oil (0.027 g, 90%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the major product 8d obtained through the addition of phenylmagnesium chloride to aldehyde 3.
(R*)-Phenyl((S*)-spiro[cyclopropane-1,9’-fluoren]-2-yl)methanol (8d).
To a cooled (0 °C) solution of ketone 7 (0.030 g, 0.10 mmol) in Et2O (1 mL) was added LiAlH4 (0.010 g, 0.25 mmol) in one portion. After 4 h, H2O (1 mL) and HCl (5 mL, 1.0 M in H2O) were added and the mixture was warmed to 20 °C. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR spectroscopic analysis of the unpurified reaction mixture revealed alcohol 8d as the only diastereomer formed. Purification by flash chromatography (10:90 EtOAc:hexanes) afforded alcohol 8d as a yellow oil (0.024 g, 81%). The spectroscopic data (1H NMR, 13C{1H} NMR, and HSQC) match the data reported for the major product 8d obtained through the addition of phenylmagnesium chloride to aldehyde 3.
Catalytic Nozaki–Hiyama–Kishi reaction with aldehyde 3 and allyl bromide.
A reported procedure43 was adapted for the reaction of aldehyde 3 with allyl bromide under catalytic Nozaki–Hiyama–Kishi conditions. To a suspension of CrCl3 (0.004 g, 0.02 mmol) and manganese powder (0.028 g, 0.51 mmol) in THF (1 mL) in a Schlenk flask was added aldehyde 3 (0.045 g, 0.20 mmol), allyl bromide (35 μL, 0.40 mmol), and trimethylsilyl chloride (60 μL, 0.47 mmol). After 16 h, H2O (10 mL) was added and the resulting mixture was stirred for an additional 4 h. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were filtered through Celite then concentrated in vacuo. Analysis of the HPLC, 1H NMR, and 13C{1H} NMR spectra of the unpurified reaction mixture revealed the formation of a complex mixture of products (see Analytical Data).
Catalytic Nozaki–Hiyama–Kishi reaction with ketone 7 and allyl bromide.
A reported procedure43 was adapted for the reaction of ketone 7 with allyl bromide under catalytic Nozaki–Hiyama–Kishi conditions. To a suspension of CrCl3 (0.003 g, 0.02 mmol) and manganese powder (0.028 g, 0.51 mmol) in THF (1 mL) in a Schlenk flask was added ketone 7 (0.060 g, 0.20 mmol), allyl bromide (35 μL, 0.40 mmol), and trimethylsilyl chloride (60 μL, 0.47 mmol). After 16 h, H2O (10 mL) was added and the resulting mixture was stirred for an additional 4 h. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were filtered through Celite then concentrated in vacuo. Analysis of the 13C{1H} NMR spectra of the unpurified reaction mixture revealed the formation of ketone 11 in 40% conversion.
Control experiment for the catalytic Nozaki–Hiyama–Kishi reaction with ketone 7.
A reported procedure43 was adapted for the reaction of ketone 7 under catalytic Nozaki–Hiyama–Kishi conditions. To a suspension of CrCl3 (0.002 g, 0.01 mmol) and manganese powder (0.014 g, 0.26 mmol) in THF (0.5 mL) in a Schlenk flask was added ketone 7 (0.029 g, 0.10 mmol) and trimethylsilyl chloride (30 μL, 0.24 mmol). After 16 h, H2O (10 mL) was added and the resulting mixture was stirred for an additional 4 h. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were filtered through Celite then concentrated in vacuo. Analysis of the 13C{1H} NMR spectra of the unpurified reaction mixture revealed the formation of ketone 11 in 7% conversion.
3-(9H-Fluoren-9-yl)-1-phenylpropan-1-one (11).
To a solution of ketone 7 (0.155 g, 0.52 mmol) in EtOH was added palladium (10%) on carbon (0.010 g, 0.10 mmol). Pressurized with H2 (4 bar), then stirred for 16 h. An aliquot (about 0.1 mL) of the reaction mixture was filtered through a syringe filter, diluted with MeOH, and analyzed by LC-MS to monitor conversion. The reaction mixture was then pressurized with H2 (4 bar) and stirred for an additional 24 h. After 24 h, the reaction mixture was filtered through Celite, rinsed with CH2Cl2 (20 mL), and concentrated in vacuo. Spectroscopic analysis (1H NMR, 13C{1H} NMR, and HPLC) of the unpurified reaction mixture revealed formation of ketone 11 in 94% conversion. 1H NMR (400 MHz, CDCl3, diagnostic peaks) δ 4.17–4.15 (m, 1H), 2.61–2.56 (m, 2H), 2.52–2.48 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3, diagnostic peaks) δ 200.2, 46.3, 33.2, 26.4; HRMS (APCI) m / z calcd for C22H19O (M + H)+ 299.1430, found 299.1431.
Supplementary Material
ACKNOWLEDGMENT
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research (57206-ND1). Additional support was provided by the National Institutes of Health, National Institute of General Medical Sciences (GM-129286). NDB thanks the NYU Department of Chemistry for support in the form of a Margaret Strauss Kramer Fellowship. We thank Dr. Chin Lin (NYU) for assistance with NMR spectroscopy and mass spectrometry. We also thank the Molecular Design Institute of NYU for purchasing a single-crystal diffractometer, and Dr. Chunhua Hu (NYU) for his assistance with data collection and structure determination. The Shared Instrumentation Facility in the Department of Chemistry was constructed through the support of the National Center for Research Resources, National Institutes of Health, under Research Facilities Improvement Award Number C06 RR-16572-01. The cryogenic probe for the 600 MHz NMR was acquired through the support of the National Institute of Health S10 grant under Award Number OD016343.
Footnotes
Supporting Information
Stereochemical correlations, X-ray structures, selected HPLC traces, selected LC-MS traces, 1H NMR, 13C{1H} NMR, and 19F{1H} NMR spectroscopic data (PDF) X-ray data for the minor diastereomer of alcohol 8d (CIF) X-ray data for the alcohol 9d (CIF)
The authors declare no competing financial interest.
References
- (1).Peltzer RM; Gauss J; Eisenstein O; Cascella M The Grignard Reaction – Unraveling a Chemical Puzzle. J. Am. Chem. Soc 2020, 142, 2984–2994. [DOI] [PubMed] [Google Scholar]
- (2).Bartolo ND; Woerpel KA Mechanistic Insight into Additions of Allylic Grignard Reagents to Carbonyl Compounds. J. Org. Chem 2018, 83, 10197–10206. [DOI] [PubMed] [Google Scholar]
- (3).Bartolo ND; Read JA; Valentín EM; Woerpel KA Reactions of Allylmagnesium Halides with Carbonyl Compounds: Reactivity, Structure, and Mechanism. Synthesis 2017, 49, 3237–3246. [Google Scholar]
- (4).Otte DAL; Woerpel KA Evidence that Additions of Grignard Reagents to Aliphatic Aldehydes Do Not Involve Single-Electron-Transfer Processes. Org. Lett 2015, 17, 3906–3909. [DOI] [PubMed] [Google Scholar]
- (5).Osztrovszky G; Holm T; Madsen R Ultrafast Grignard Addition Reactions in the Presence of Water. Org. Biomol. Chem 2010, 8, 3402–3404. [DOI] [PubMed] [Google Scholar]
- (6).Hoffmann RW; Holzer B Concerted and Stepwise Grignard Additions, Probed with a Chiral Grignard Reagent. Chem. Commun 2001, 491–492. [DOI] [PubMed] [Google Scholar]
- (7).Holm T Use of Competition Kinetics with Fast Reactions of Grignard Reagents. J. Org. Chem 2000, 65, 1188–1192. [DOI] [PubMed] [Google Scholar]
- (8).Holm T; Dahlbom R; Iversen T; Berg J-E; Pilotti A-M; Anthonsen T Mechanism of the Grignard Addition Reaction. XII. The Reversibility of the Addition of Allylic Grignard Reagents to Di-t-butyl Ketone. Acta Chem. Scand 1976, 30b, 985–990. [Google Scholar]
- (9).Holm T Mechanism of the Grignard Addition Reaction. XI. Electrode Kinetics and Chemical Reactivity of Grignard Reagents. Acta Chem. Scand 1974, 28b, 809–812. [Google Scholar]
- (10).Holm T; Crossland I Mechanism of the Grignard Addition Reaction. VIII. Reaction Rates and Product Distribution for the Reactions of t-Butylmagnesium Chloride and Methylmagnesium Bromide with Substituted Benzophenones. Acta Chem. Scand 1971, 25, 59–69. [Google Scholar]
- (11).Felkin H; Frajerman C Competitive Rate Studies for Some Reactions Involving the Allyl, Butenyl, αγ-Dimethylallyl, and Propyl Grignard Reagents. The Mechanism of the Reactions of Allylic Grignard Reagents with Electrophilic Substrates. Tetrahedron Lett. 1970, 11, 1045–1048. [Google Scholar]
- (12).Ashby EC; Duke RB; Neumann HM Concerning the Addition of Grignard Reagents to Ketones. Evidence for a Termolecular Mechanism. J. Am. Chem. Soc 1967, 89, 1964–1965. [Google Scholar]
- (13).Ashby EC; Laemmle J; Neumann HM Mechanisms of Grignard Reagent Addition to Ketones. Acc. Chem. Res 1974, 7, 272–280. [Google Scholar]
- (14).Ashby EC; Lopp IG; Buhler JD Mechanisms of Grignard Reactions with Ketones. Polar vs. Single Electron Transfer Pathways. J. Am. Chem. Soc 1975, 97, 1964–1966. [Google Scholar]
- (15).Ashby EC Single-Electron Transfer, a Major Reaction Pathway in Organic Chemistry. An Answer to Recent Criticisms. Acc. Chem. Res 1988, 21, 414–421. [Google Scholar]
- (16).Ashby EC; Bowers JR Organometallic Reaction Mechanisms. 17. Nature of Alkyl Transfer in Reactions of Grignard Reagents with Ketones. Evidence for Radical Intermediates in the Formation of 1,2-Addition Product Involving Tertiary and Primary Grignard Reagents. J. Am. Chem. Soc 1981, 103, 2242–2250. [Google Scholar]
- (17).Ashby EC; Goel AB Direct Evidence Supporting a Single Electron Transfer Pathway in the Reduction of Ketones by Primary, Secondary, and Tertiary Grignard Reagents. J. Am. Chem. Soc 1981, 103, 4983–4985. [Google Scholar]
- (18).Sheppard WA Pentafluorophenyl Group. Electronic Effect as a Substituent. J. Am. Chem. Soc 1970, 92, 5419–5422. [Google Scholar]
- (19).Choi SY; Toy PH; Newcomb M Picosecond Radical Kinetics. Fast Ring Openings of Secondary and Tertiary trans-2-Phenylcyclopropylcarbinyl Radicals. J. Org. Chem 1998, 63, 8609–8613. [Google Scholar]
- (20).Lund T; Pedersen ML; Frandsen LA Does the Reaction between Fluorenone and Grignard Reagents Involve Free Fluorenone Anion Radicals? Tetrahedron Lett. 1994, 35, 9225–9226. [Google Scholar]
- (21).Martin-Esker AA; Johnson CC; Horner JH; Newcomb M Picosecond Radical Kinetics. Fast Ring Openings of Constrained, Aryl-Substituted Cyclopropylcarbinyl Radicals. J. Am. Chem. Soc 1994, 116, 9174–9181. [Google Scholar]
- (22).Nevill SM; Pincock JA The Design of Radical Clocks to Probe the Reactivity of the Intermediates in Arylmethyl Ester Photochemistry. Can. J. Chem 1997, 75, 232–247. [Google Scholar]
- (23).Liu H; Wei Y; Cai C Hypervalent-Iodine(III) Oxidation of Hydrazones to Diazo Compounds and One-Pot Nickel(II)-Catalyzed Cyclopropanation. New J. Chem 2016, 40, 674–678. [Google Scholar]
- (24).Williams JM; Jobson RB; Yasuda N; Marchesini G; Dolling U-H; Grabowski EJJ A New General Method for Preparation of N-Methoxy-N-methylamides. Application in Direct Conversion of an Ester to a Ketone. Tetrahedron Lett. 1995, 36, 5461–5464. [Google Scholar]
- (25).Dess DB; Martin JC Readily Accessible 12-I-5 Oxidant for the Cconversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem 1983, 48, 4155–4156. [Google Scholar]
- (26).Nahm S; Weinreb SM N-Methoxy-N-methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- (27).Conant JB; Webb CN; Mendum WC Trimethylacetaldehyde and Dimethylethylacetaldehyde. J. Am. Chem. Soc 1929, 51, 1246–1255. [Google Scholar]
- (28).Conant JB; Blatt AH The Action of the Grignard Reagent on Highly Branched Carbonyl Compounds. J. Am. Chem. Soc 1929, 51, 1227–1236. [Google Scholar]
- (29).Chérest M; Felkin H; Prudent N Torsional Strain Involving Partial Bonds. The Stereochemistry of the Lithium Aluminium Hydride Reduction of Some Simple Open-Chain Ketones. Tetrahedron Lett. 1968, 9, 2199–2204. [Google Scholar]
- (30).Chérest M; Felkin H Torsional Strain Involving Partial Bonds. The Steric Course of the Reaction Between Allyl Magnesium Bromide and 4-t-Butyl-cyclohexanone. Tetrahedron Lett. 1968, 9, 2205–2208. [Google Scholar]
- (31).Anh NT; Eisenstein O; Lefour JM; Dâu M-ETH Orbital Factors and Asymmetric Induction. J. Am. Chem. Soc 1973, 95, 6146–6147. [Google Scholar]
- (32).Anh NT; Eisenstein O Theoretical Interpretation of 1–2 Asymmetric Induction. The Importance of Antiperiplanarity. Nouv. J. Chim 1977, 1, 61–70. [Google Scholar]
- (33).Imamoto T; Takiyama N; Nakamura K; Hatajima T; Kamiya Y Reactions of Carbonyl Compounds with Grignard Reagents in the Presence of Cerium Chloride. J. Am. Chem. Soc 1989, 111, 4392–4398. [Google Scholar]
- (34).Janssen MJ; de Jong J Anions of Cyclopentadithiophenes: New Heteroaromatic Systems. Recl. Trav. Chim. Pays-Bas 1967, 86, 1246–1248. [Google Scholar]
- (35).Jiao H; Schleyer P.v. R. ; Mo Y; McAllister MA; Tidwell TT Magnetic Evidence for the Aromaticity and Antiaromaticity of Charged Fluorenyl, Indenyl, and Cyclopentadienyl Systems. J. Am. Chem. Soc 1997, 119, 7075–7083. [Google Scholar]
- (36).Read JA; Woerpel KA Allylmagnesium Halides Do Not React Chemoselectively Because Reaction Rates Approach the Diffusion Limit. J. Org. Chem 2017, 82, 2300–2305. [DOI] [PubMed] [Google Scholar]
- (37).Bartolo ND; Read JA; Valentín EM; Woerpel KA Reactions of Allylmagnesium Reagents with Carbonyl Compounds and Compounds with C═N Double Bonds: Their Diastereoselectivities Generally Cannot Be Analyzed Using the Felkin–Anh and Chelation-Control Models. Chem. Rev 2020, 120, 1513–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hoffmann RW The Quest for Chiral Grignard Reagents. Chem. Soc. Rev 2003, 32, 225–230. [DOI] [PubMed] [Google Scholar]
- (39).Krasovskiy A; Knochel P A LiCl‐Mediated Br/Mg Exchange Reaction for the Preparation of Functionalized Aryl‐ and Heteroarylmagnesium Compounds from Organic Bromides. Angew. Chem. Int. Ed 2004, 43, 3333–3336. [DOI] [PubMed] [Google Scholar]
- (40).Blasberg F; Bolte M; Wagner M; Lerner H-W An Approach to Pin Down the Solid-State Structure of the “Turbo Grignard”. Organometallics 2012, 31, 1001–1005. [Google Scholar]
- (41).Still WC; Schneider JA Chelation-Controlled Nucleophilic Additions. 2. A Highly Effective System for Asymmetric Induction in the Reaction of Organometallics with β-Alkoxyaldehydes. Tetrahedron Lett. 1980, 21, 1035–1038. [Google Scholar]
- (42).Sodium borohydride was unable to reduce this ketone, likely due to the steric hindrance of the cyclopropyl fluorenyl group on the ketone.
- (43).Fürstner A; Shi N Nozaki–Hiyama–Kishi Reactions Catalytic in Chromium. J. Am. Chem. Soc 1996, 118, 12349–12357. [Google Scholar]
- (44).Mulzer J; Strecker AR; Kattner L A Mechanistic Study of the Hiyama–Nozaki Allylation: Evidence for Radical Intermediates. Tetrahedron Lett. 2004, 45, 8867–8870. [Google Scholar]
- (45).Otte DAL; Borchmann DE; Lin C; Weck M; Woerpel KA 13C NMR Spectroscopy for the Quantitative Determination of Compound Ratios and Polymer End Groups. Org. Lett 2014, 16, 1566–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang Z; Gui C; Zhao E; Wang J; Li X; Qin A; Zhao Z; Yu Z; Tang BZ Specific Fluorescence Probes for Lipid Droplets Based on Simple AIEgens. ACS Appl. Mater. Interfaces 2016, 8, 10193–10200. [DOI] [PubMed] [Google Scholar]
- (47).Saltzman H; Sharefkin JG Iodosobenzene. Org. Synth 1963, 43, 60. [Google Scholar]
- (48).Abarbri M; Dehmel F; Knochel P Bromine-Magnesium-Exchange as a General Tool for the Preparation of Polyfunctional Aryl and Heteroaryl Magnesium-Reagents. Tetrahedron Lett. 1999, 40, 7449–7453. [Google Scholar]
- (49).Djurdjevic S; Green JR Nicholas Reactions in the Synthesis of Dicobalt Dibenzocyclooctyne Complexes. Org. Lett 2013, 15, 5468–5471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.