

Abstract
We report the development of palladium(0)-catalyzed syn-selective 1,2-carboboration and -silylation reactions of alkenes containing cleavable directing groups. With B2pin2 or PhMe2Si–Bpin as nucleophiles and aryl/alkenyl triflates as electrophiles, a broad range of mono-, di-, tri- and tetrasubstituted alkenes are compatible in these transformations. We further describe a directed dearomative 1,2-carboboration of electron-rich heteroarenes by employing this approach. Through use of a removable chiral directing group, we demonstrate the viability of achieving stereoinduction in Heck-type alkene 1,2-difunctionalization. This work introduces new avenues to access highly functionalized boronates and silanes with precise regio- and stereocontrol.
Keywords: carboboration, borylation, palladium, directing group, alkene
Graphical Abstract
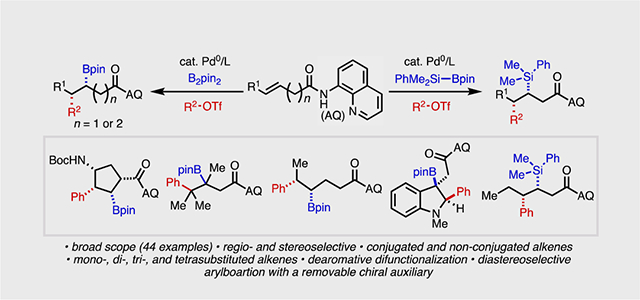
Alkene Functionalization. A method to affect syn-1,2-carboboration and -silylation of alkenes and heterocycles via Pd(0)/Pd(II) catalysis is described. Strategic use of a chiral directing groups allows for stereocontrol in the key 1,2-migratory insertion step.
Introduction
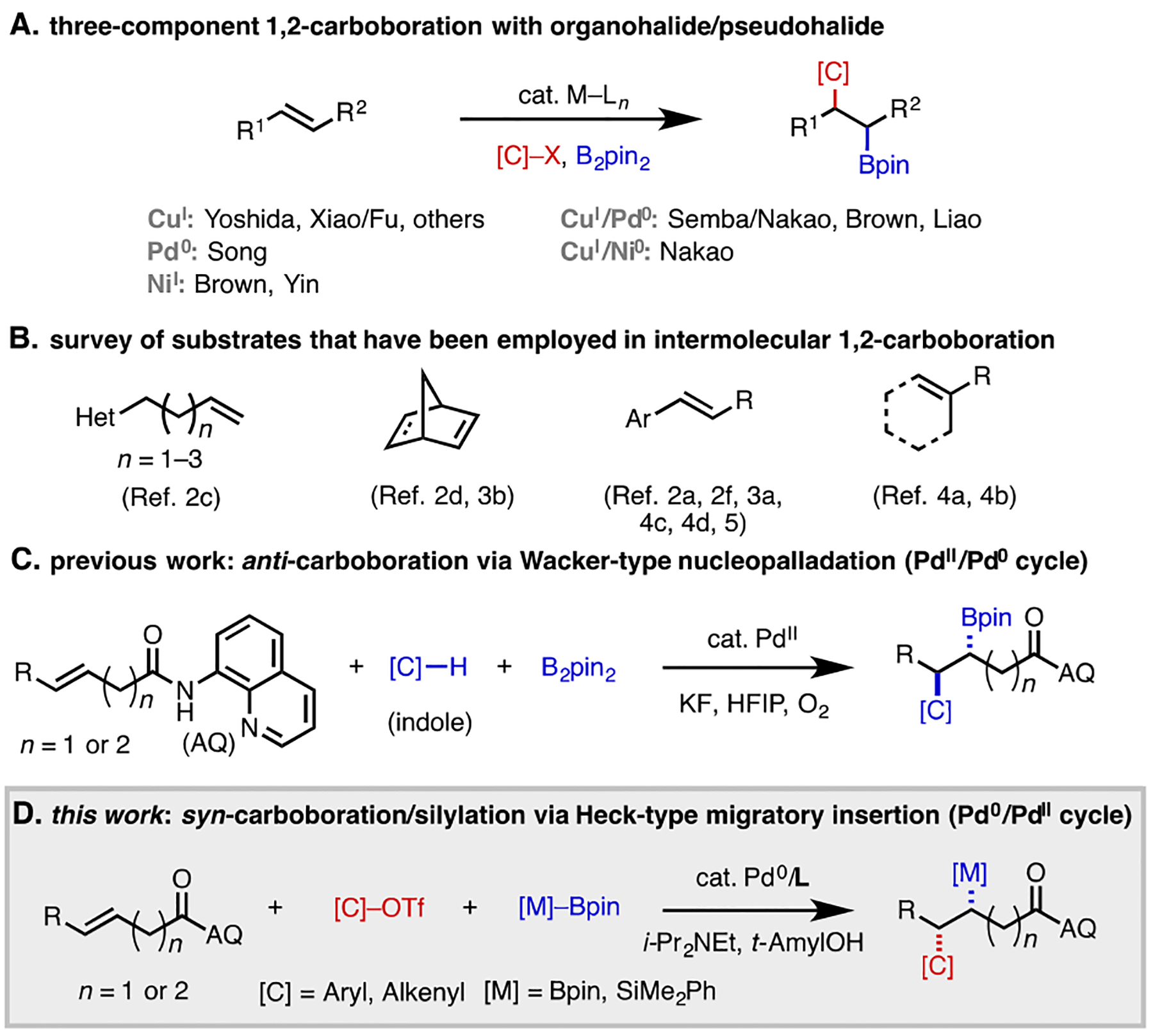
Metal-catalyzed 1,2-carboboration of alkenes is a powerful means of simultaneously forming a C(sp3)–C and a C(sp3)–B bond in a single step with multiple levels of selectivity control.[1] Indeed, successful examples of catalytic 1,2-carboboration have been developed with copper,[2] palladium,[3] nickel,[4] and dual catalyst systems[5]. Of the various coupling partners that can be engaged in 1,2-carboboration, organohalides and pseudohalides are particularly important, given the structural diversity and widespread availability of these electrophiles (Scheme 1A). In this area, classical limitations have included the scope of compatible alkenes and associated issues with regiocontrol; indeed, the vast majority of successful examples involve activated alkenes, such as styrenes[2a,f,3a,4c,d,5] or norbornenes (Scheme 1B).[2d,3b] Previously, Xiao and Fu developed a Cu-catalyzed regiodivergent 1,2-alkylboration of non-conjugated terminal alkenes containing a proximal heteroatom.[2c] Recently, Brown has developed an elegant series of nickel-catalyzed 1,2-arylboration methods capable of functionalizing mono-, di- and tri- substituted non-conjugated alkenes[4a,b] without formation of competitive chain-walking products.[6,7] These reactions are believed to proceed via a NiI–Bpin intermediate, which generally delivers the Bpin group to the less substituted carbon atom. With trisubstituted alkenes, the reactions are highly regioselective, whereas with non-symmetric 1,2-disubstituted alkenes and terminal alkenes, regiomeric ratios (r.r.) are variable and controlled by a combination of steric and electronic factors.
Scheme 1.
Background and Project Synopsis.
To complement these approaches, we reasoned that a substrate-directed strategy involving palladium catalysis could enhance reactivity with hindered substrates (e.g., tetrasubstituted alkenes[8]) and provide a means of controlling regioselectivity in a manner that is independent of the alkene substitution pattern. Previously, our lab[9] and others[10] have developed a toolkit of hydro- and difunctionalization reactions of alkenyl carbonyl compounds bearing the 8-aminoquinoline[11] (AQ) directing group. As part of this effort, we described an anti-selective 1,2-carboboration of alkenyl carbonyl compounds that initiates via Wacker-type carbopalladation (Scheme 1C).[3c–e] Though useful in its own right, this method is limited in terms of the carbogenic groups that can be introduced (indole-type nucleophiles) and the alkene scope (terminal and 1,2-disubstituted alkenes). We hypothesized that these issues could be overcome by developing a catalytic system that instead initiates via Heck-type 1,2-migratory insertion.[9d,f,10b,e] Based on this idea, herein we describe a Heck-type directed 1,2-carboboration that proceeds via a Pd(0)/Pd(II) redox manifold (Scheme 1D). In addition to tolerating essentially all possible alkene substitution patterns, the protocol can also be applied in directed dearomative 1,2-carboboration of electron-rich heterocycles. The generality of this directed 1,2-difunctionalization approach via Pd(0)/Pd(II) catalysis is demonstrated through the realization of a 1,2-carbosilylation method by using PhMe2Si–Bpin[12] in place of B2Pin2. Lastly, stereoinduction by use of a chiral directing group is shown.
Results and Discussion
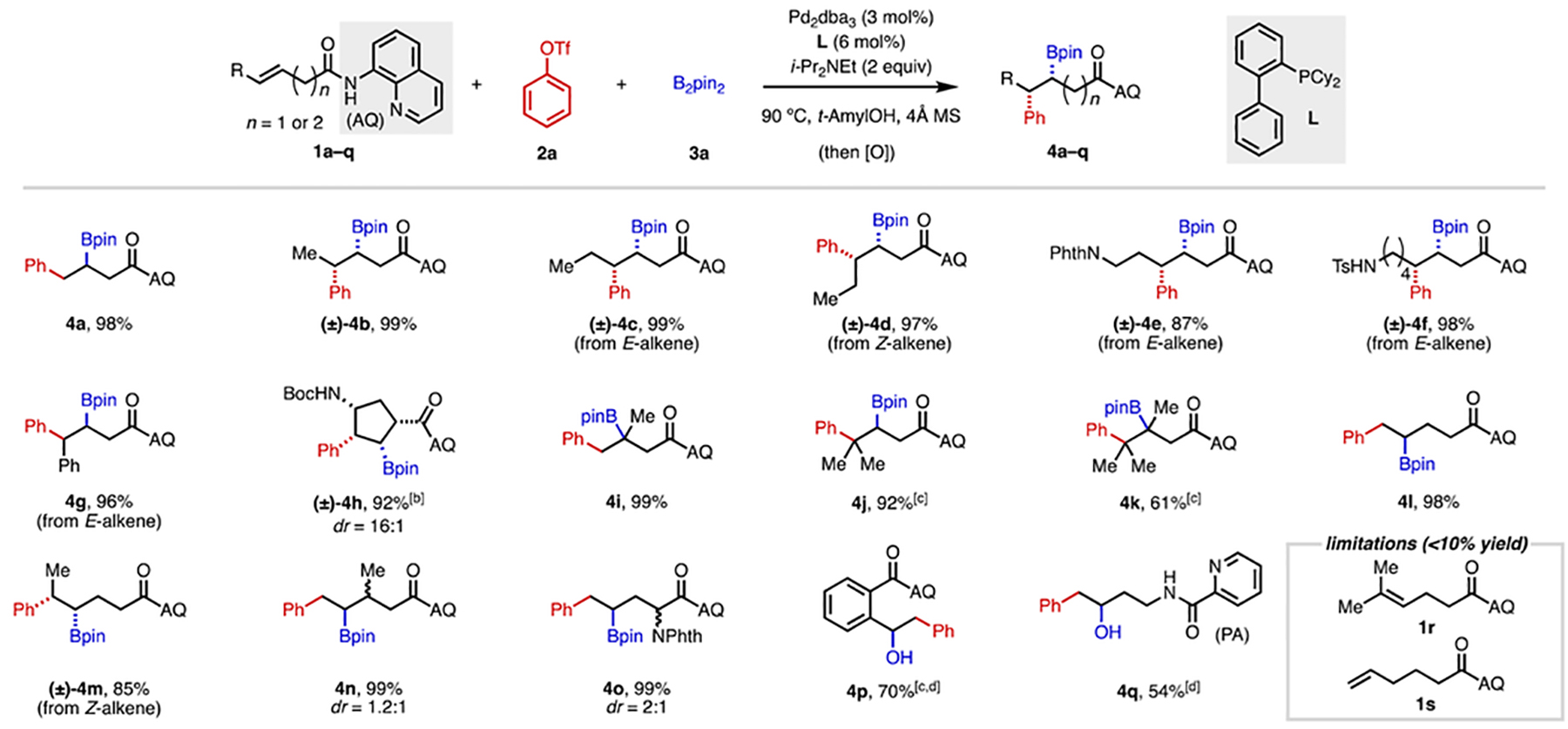
To initiate our study, we first selected 8-aminoquinoline (AQ)-masked 3-butenoic acid (1a) and (E)-3-hexenoic acid (1c) as our pilot alkene substrates, phenyl triflate (2a) as the electrophile, and B2pin2 (3a) as the boron-based nucleophile. We elected to use a catalyst system consisting of Pd2dba3 and a phosphine ligand.[10b] We hypothesized that the phosphine ligand would coordinate to Pd(0) and promote oxidative addition to the aryl triflate. After extensive screening of different ligands, bases, and reaction solvents (see SI for details), we identified an optimal combination: i-Pr2NEt as base and t-AmylOH as solvent with a Buchwald-type ligand,[13] such as RuPhos, XPhos, or Cy-JohnPhos (L). Under these conditions syn-1,2-arylborylated products 4a and 4c could be isolated in nearly quantitative yield without observable formation of regio- and stereoisomers based on 1H NMR.
We then tested the alkene scope of this Pd(0)-catalyzed 1,2-arylboration (Table 1). Pleasingly, we found that a remarkably broad collection of alkenes were reactive. For β,γ-unsaturated carbonyl compounds, which react via five-membered palladacycles, the reaction was largely insensitive to the alkene substitution pattern. Di-, tri-, and even tetrasubstituted alkenes were all competent, leading to a variety of secondary and tertiary alkyl boronates in excellent yields (4a–k). These results stand in contrast to our previously developed directed 1,2-difunctionalization reactions involving Wacker-type nucleopalladation,[3c,9b,c,e] which are highly sensitive to steric hindrance and are incompatible with tri- and tetrasubstituted alkenes. Substrate 1h, which is derived from Vince lactam,[14] underwent arylboration smoothly to give product 4h, which bears four substituents on the same face of the cyclopentane ring. Furthermore, from tetrasubstituted alkene 1k, the resulting product contains two contiguous quaternary centers, a motif that is inaccessible using existing arylboration methods.
Table 1.
Alkene Scope of 1,2-Arylboration[a]
|
Reaction conditions: 1a–q (0.05 mmol or 0.1 mmol), 2a (1.5 equiv), 3a (2 equiv), Pd2dba3 (3 mol%), L (6 mol%), i-Pr2NEt (2 equiv), 4Å MS (15–30 mg), 90 °C, N2, 38–44 h. Percentages refer to isolated yields. Unless otherwise noted, diastereomeric ratio (dr) was found to be >30:1 in all cases. [b] 2a (3 equiv), 3a (3 equiv), Pd2dba3 (5 mol%), L (10 mol%), i-Pr2NEt (3 equiv), 100 °C. [c] 2a (2 equiv), 3a (3 equiv), Pd2dba3 (4.5 mol%), L (9 mol%), i-Pr2NEt (3 equiv). [d] The final product was oxidized to the corresponding alcohol with NaBO3•4H2O (5 equiv) for ease of isolation.
The reaction also works reasonably well for substrates containing one additional methylene spacer between the C=C bond and the carbonyl groups (4l–p), which react via six-membered palladacycles. However, with these substrates, the reaction is more sensitive to steric hindrance. With 1,2-disubstituted alkene 1m, the product (4m) was obtained in 85% yield, which is slightly diminished compared to analogous earlier examples (4b–4d). With trisubstituted alkene 1r, no reaction was observed. We attribute this pattern of attenuated reactivity to less favorable kinetics and thermodynamics for 1,2-migratory insertion to form a six-membered palladacycle versus a five-membered palladacycle. Substrates with even more distal alkenes, including terminal alkene 1s, were unreactive under the standard conditions. Interestingly, an alkenyl amine substrate containing a picolinamide (PA) directing group also underwent 1,2-arylboration in moderate yield (4q). Importantly, across all of these examples, the phenyl group was reliably delivered to position distal to the AQ group, and the Bpin group moiety was installed proximal to the AQ group, irrespective of alkene substitution pattern, illustrating a unique aspect of this directed 1,2-carboboration compared to other approaches.[1–5]
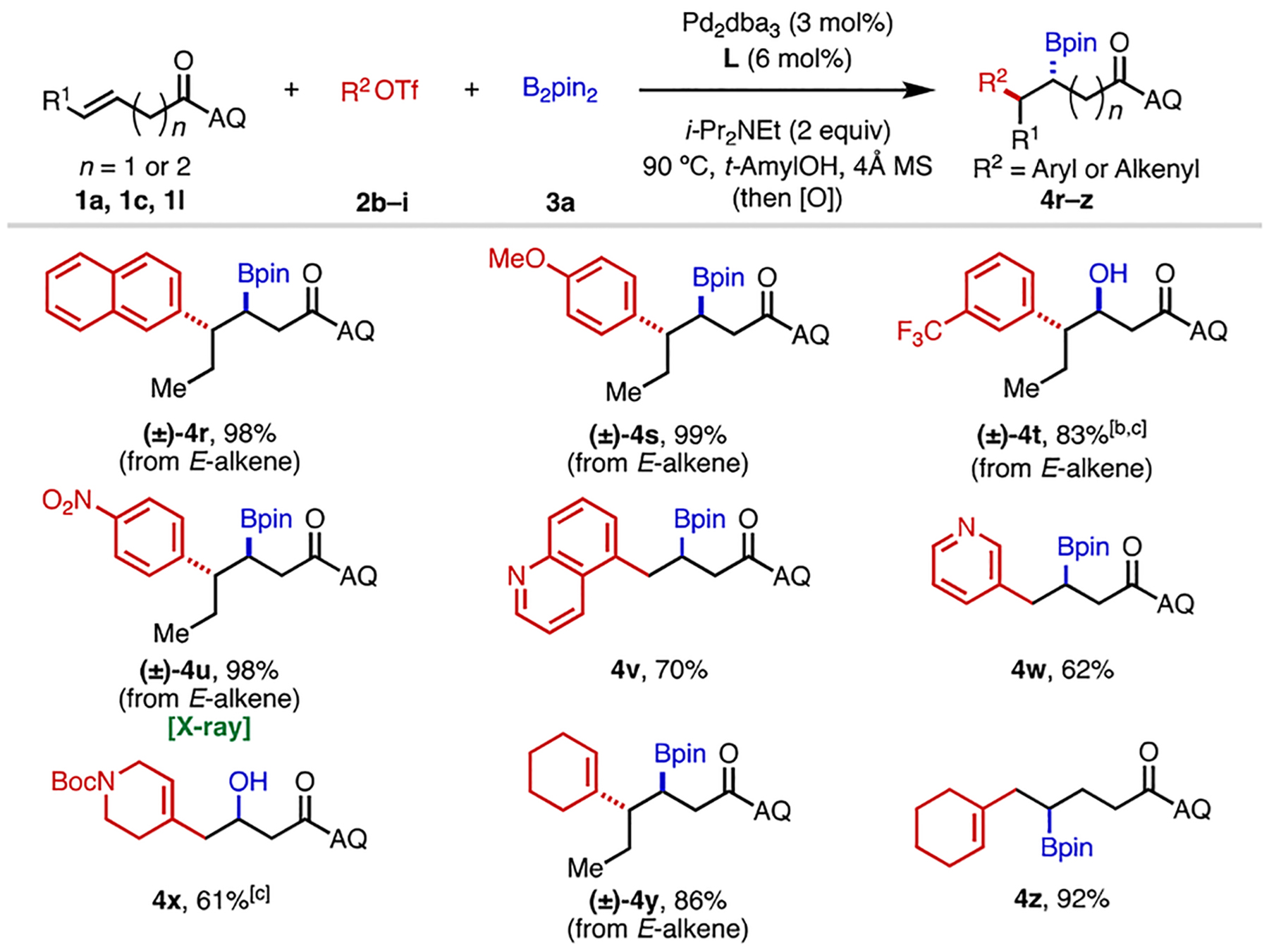
Next, we investigated the compatibility of carbon electrophiles in this transformation (Table 2). An array of aryl triflates containing substituents with different electronic properties on the para or meta positions reacted smoothly under the optimal conditions, delivering the corresponding products, 4r–4u, in excellent yields. Notably heteroaryl triflates containing a Lewis basic N(sp2) atom (4v and 4w), which are incompatible in many Pd-catalyzed alkene functionalizations, are tolerated in this reaction, though slightly lower yields were obtained. Gratifyingly, when we used alkenyl triflates 2h and 2i as carbon electrophiles, 1,2-alkenylboration could be achieved. Both 3-butenoic acid and 4-pentenoic acid derivatives underwent alkenylboration in good to excellent yields (4x–4z).
Table 2.
Electrophile Scope.[a]
|
Unless otherwise specified, reaction conditions were as in Table 1. dr values were found to be >30:1 in all cases. [b] 2d (2 equiv), 3a (3 equiv). [c] The final product was oxidized to the corresponding alcohol with NaBO3•4H2O (5 equiv) for ease of isolation.
In order to demonstrate the method’s operational simplicity and practicality, we scaled up the alkene arylboration reaction using 1c as a model substrate (Scheme 2). Under standard conditions, boronate 4c was isolated in 91% yield (1.2 g).
Scheme 2.
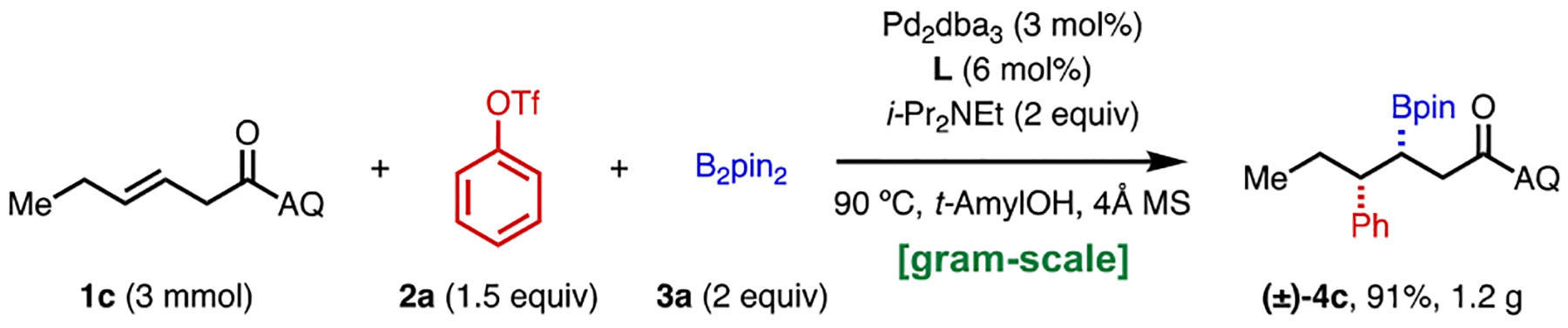
Gram-Scale 1,2-Arylboration.
Considering the relatively expensive nature of aryl triflates, we also investigated other aryl electrophiles as triflate surrogates. Aryl fluorosulfates,[15] which can be readily prepared from phenols and sulfurylfluoride (SO2F2), are a class of inexpensive aryl electrophiles that are used on industrial scale, making them attractive yet underutilized coupling partners in alkene functionalization. To our delight, phenyl fluorosulfate (2j) was found to be a highly effective electrophile in this 1,2-arylboration reaction, providing nearly quantitative yield of 4c (Scheme 3).
Scheme 3.
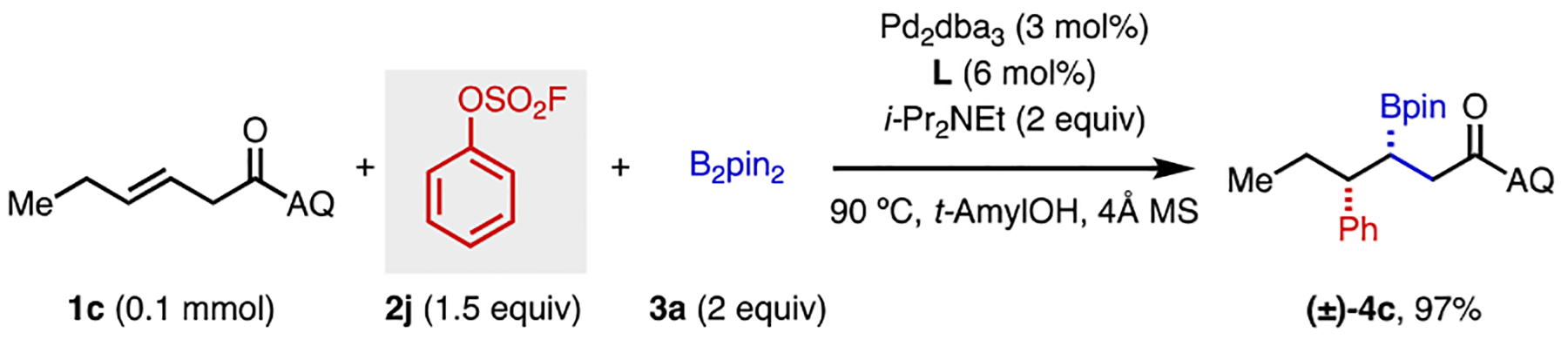
Arylboration using Phenyl Fluorosulfate as the Electrophile.
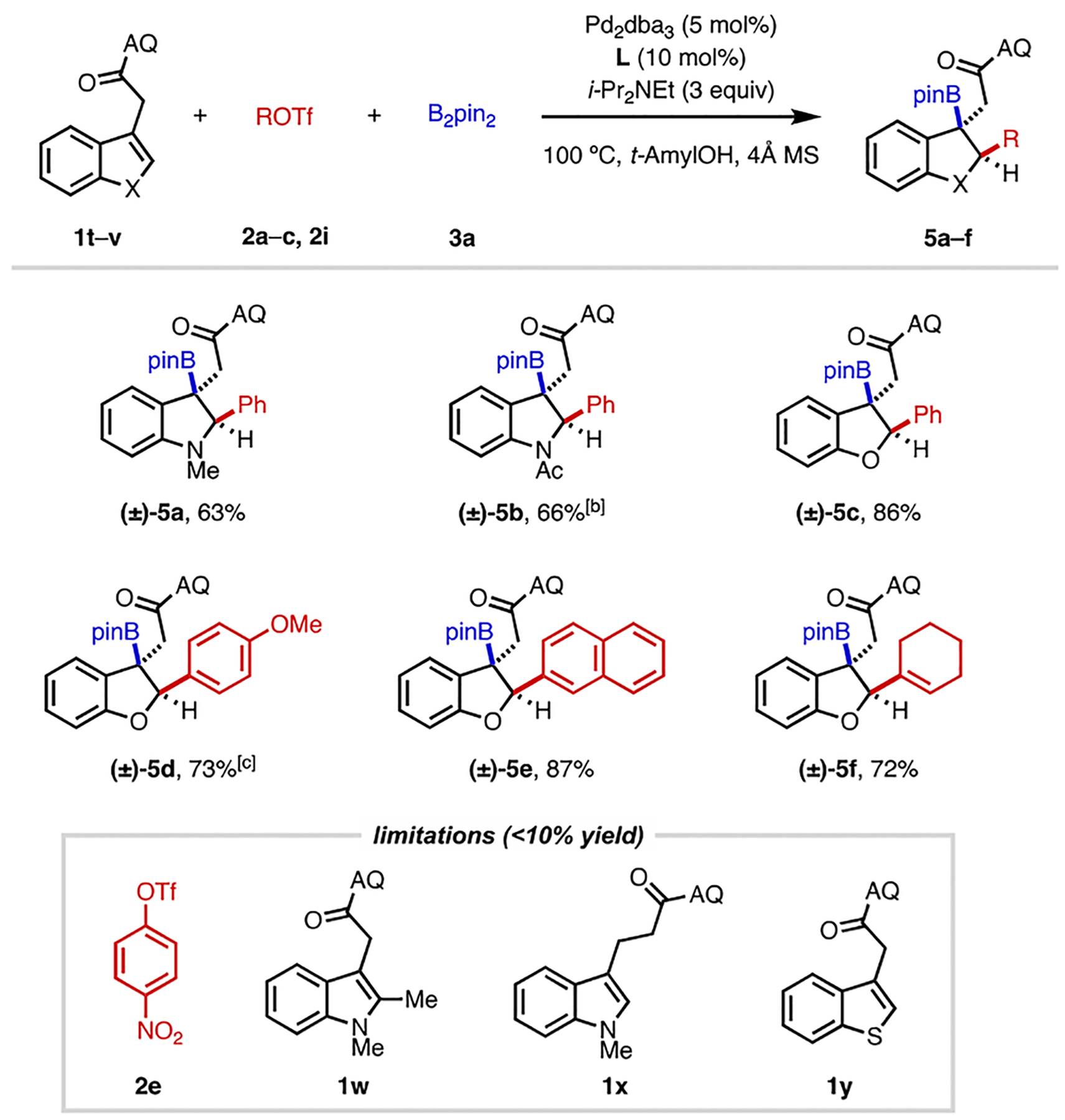
Given the broad scope of this reaction and its insensitivity to alkene substitution patterns, we next questioned whether it was possible to carry out directed dearomative 1,2-difunctionalization[16] of heteroarenes (Table 3). The three-component 1,2-carboboration of heteroarenes would represent a powerful way to prepare highly functionalized heterocycles. However, this type of reaction has not been previously reported in the literature to the best of our knowledge. The envisioned transformation is challenging for several reasons, including the potential for competitive two-component Miyaura borylation, the relatively large energy barrier associated with breaking aromaticity, and the possibility of undergoing rearomatization. Taking inspiration from an elegant recent report by Jia, Lautens and coworkers on Pd(0)-catalyzed intramolecular dearomative arylboration of indoles,[17] we reasoned that the aforementioned issues could be overcome through AQ-directed Heck-type migratory insertion.
Table 3.
Dearomative Aryl/Alkenylboration of Heterocycles.[a]
|
Reaction conditions: 1t–v (0.05 or 0.1 mmol), 2a–c, or 2i (3 equiv), 3a (3 equiv), Pd2dba3 (5 mol%), L (10 mol%), i-Pr2NEt (3 equiv), 4Å MS (20–30 mg), 100 °C, N2, 38–44 h. Percentages refer to isolated yields. The diastereomeric ratio (dr) was found to be >30:1 in all cases. [b] Reaction time of 96 h. [c] 1v (0.1 mmol), 2c (2 equiv), Pd2dba3 (4.5 mol%), L (9 mol%).
Gratifyingly, using this substrate directivity strategy, we were able to effect 1,2-carboboration of indoles and benzofurans in good yields and with excellent diastereoselectivity (5a–5f). Several benzo-fused heterocyclic products containing a tertiary boronate group could thus be prepared in a succinct manner. This dearomative transformation is not without its limitations. Aryl triflates bearing strong electron-withdrawing groups are incompatible in this reaction. With indole substrates bearing a 2-substituent (1w) or one extra methylene spacer (1x), similarly no reaction was observed. A benzothiophene derivative 1y was also unreactive in this transformation.
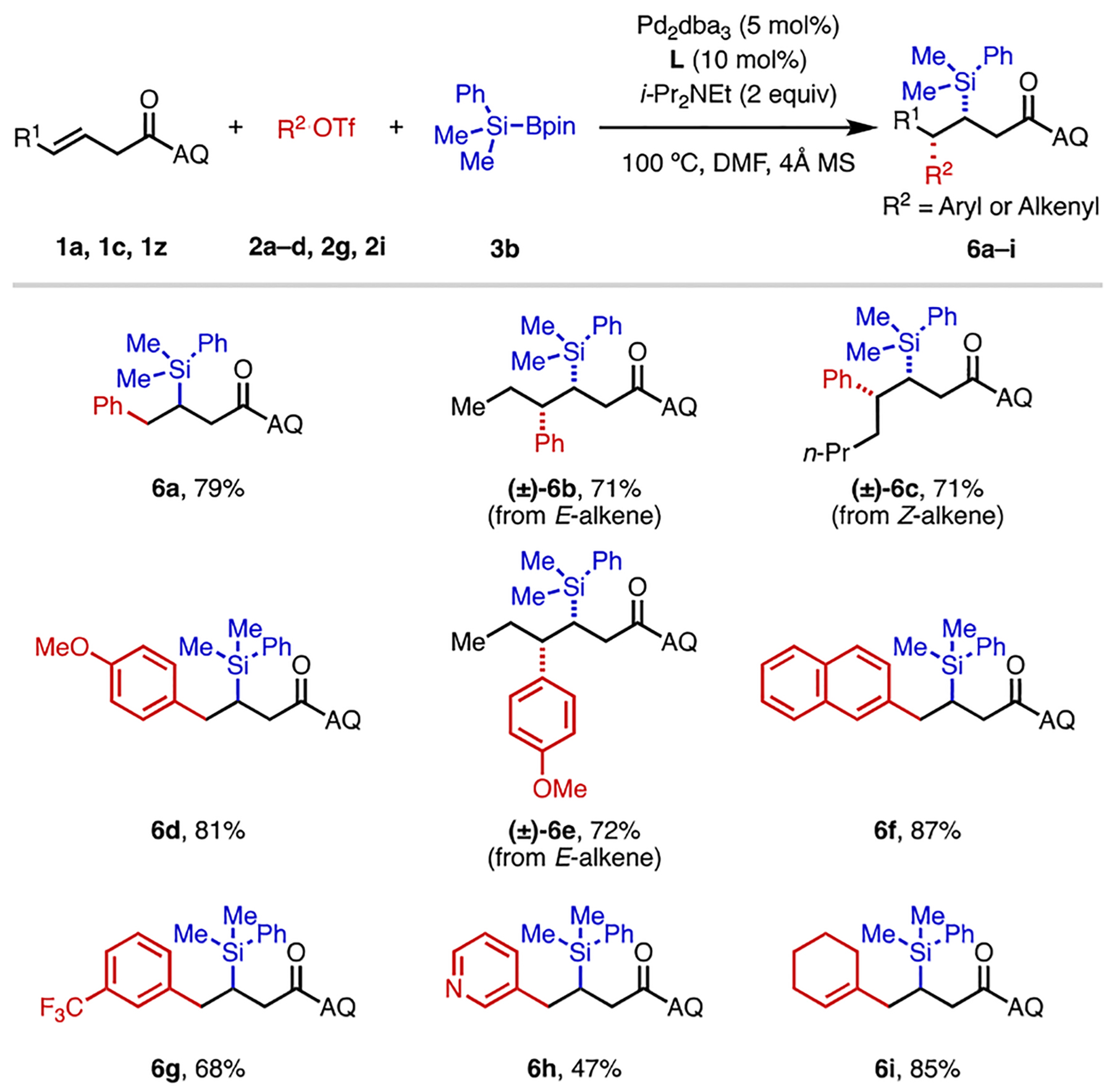
To demonstrate the versatility of directed Heck-type 1,2-difunctionalization chemistry, we next sought to develop a method for three-component alkene 1,2-carbosilyation, a transformation that would be synthetically enabling yet remains underdeveloped in the literature.[18,19] To this end, we tested Suginome’s PhMe2Si–Bpin reagent[12] (3b) in place of B2pin2 (3a). Gratifyingly, after brief optimization we identified conditions for alkene 1,2-carbosilylation (Table 4).[20] Although silyl group transfer to the Pd(II) center was generally favored over boryl group transfer, small amounts of the 1,2-carboborylated byproduct could nevertheless be detected. This competitive pathway accounts for the slightly lower product yields in this transformation. Representative examples of alkene substrates and electrophiles were examined, and yields were consistently in the moderate to good range (6a–6i). 4-Pentenoic acid derivatives (such as 1l), however, were incompatible in this transformation. In this case, only a small amount of arylsilylated product was generated, together with alkene arylboration and hydroarylation byproducts.
Table 4.
Scope of Alkene Aryl/Alkenylsilylation.[a]
|
Reaction conditions: 1a, 1c, 1z (0.1 mmol), 2a–d, 2g, 2i (1.5 equiv), 3b (3 equiv), Pd2dba3 (5 mol%), L (10 mol%), i-Pr2NEt (2 equiv), 4Å MS (30 mg), 100 °C, N2, 38–44 h. All the yields refer to the isolated yields.
To illustrate the synthetic utility of this 1,2-carboboration reaction, we performed a series of transformations on representative 1,2-carboborated products (Scheme 4). First, we carried out a 2-step transamination sequence to remove the AQ directing group, following Verho’s procedure.[21] Compounds 4c, 4j, and 4l were successfully transformed to the corresponding pyrrolidine amides in excellent yields. In addition, with Ni(tmhd)2 as a mediator, methanolysis of 4c was achieved in 78% yield.[22] Finally, the boronate group of 8c was transformed to a vinyl and fluoro group, respectively (10 and 11), using literature methods.[23,24]
Scheme 4.
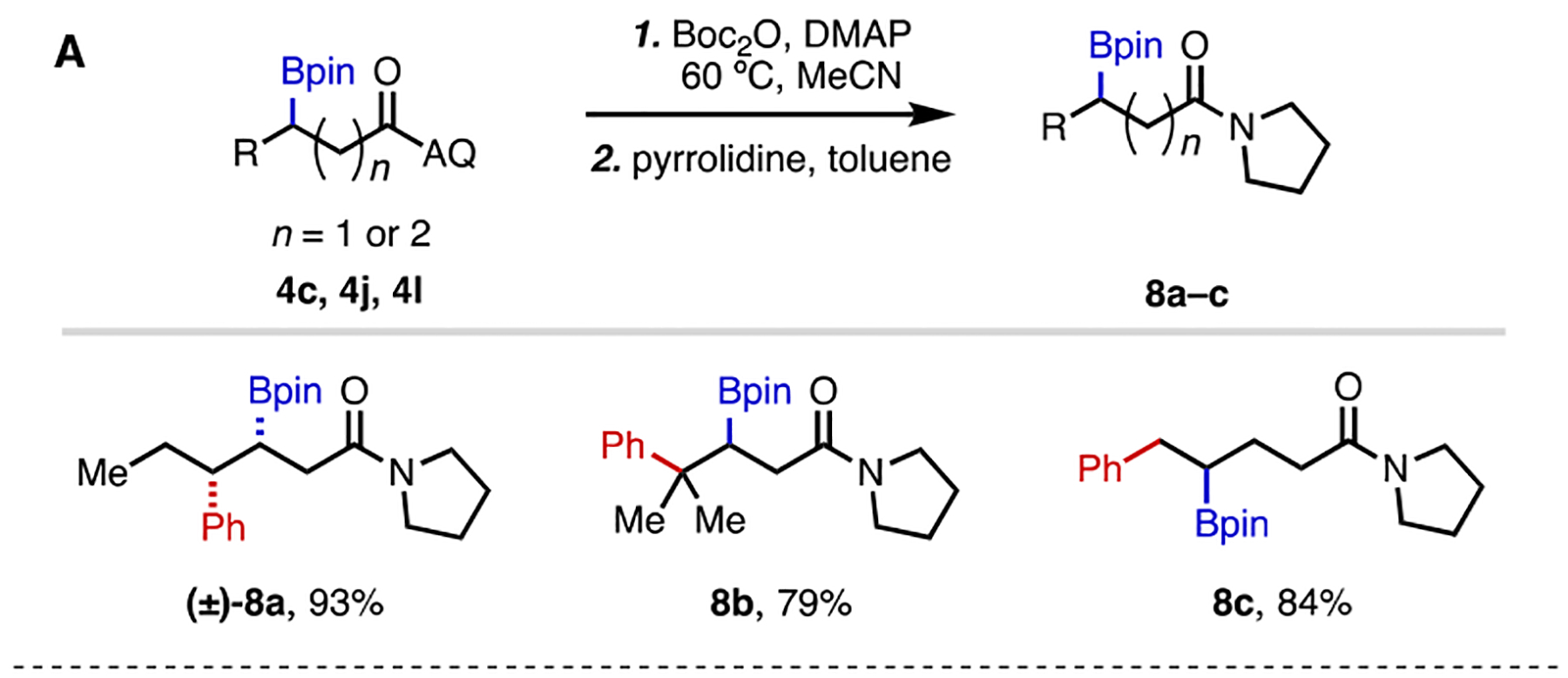
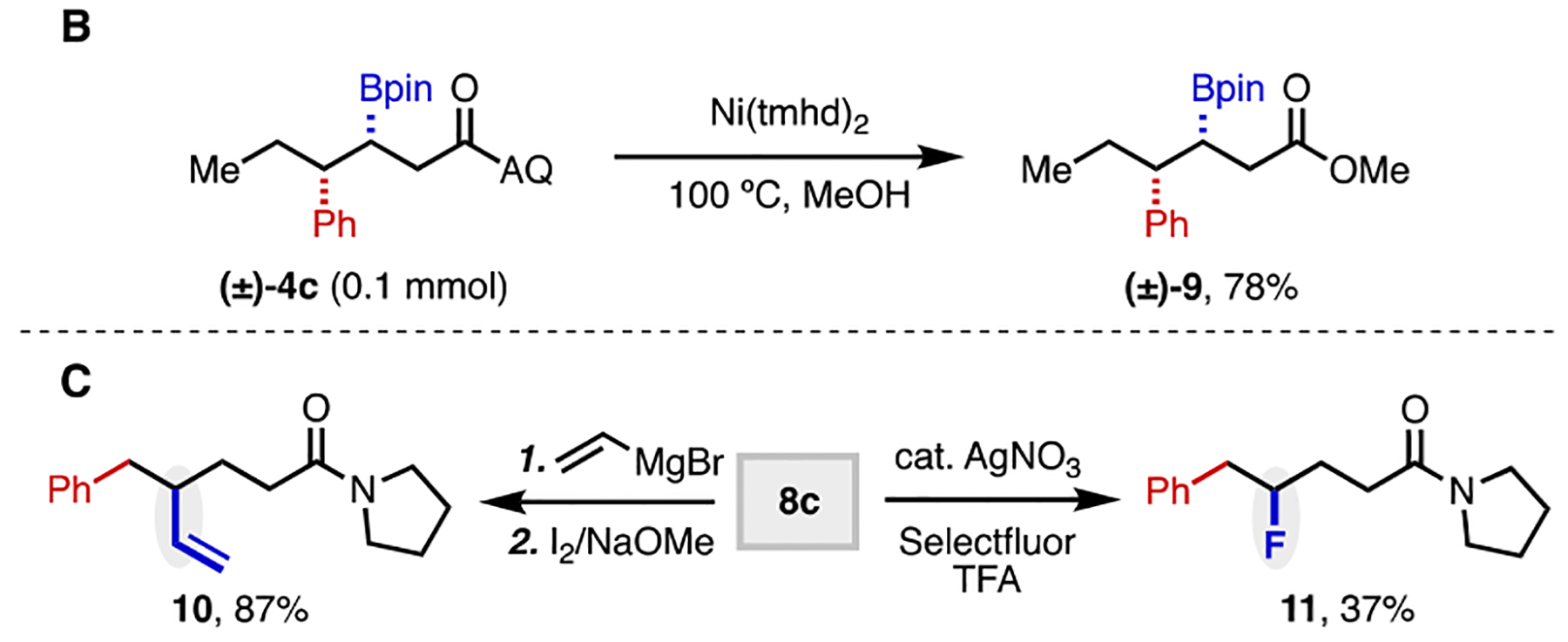
Diversification of Arylborylated Products.
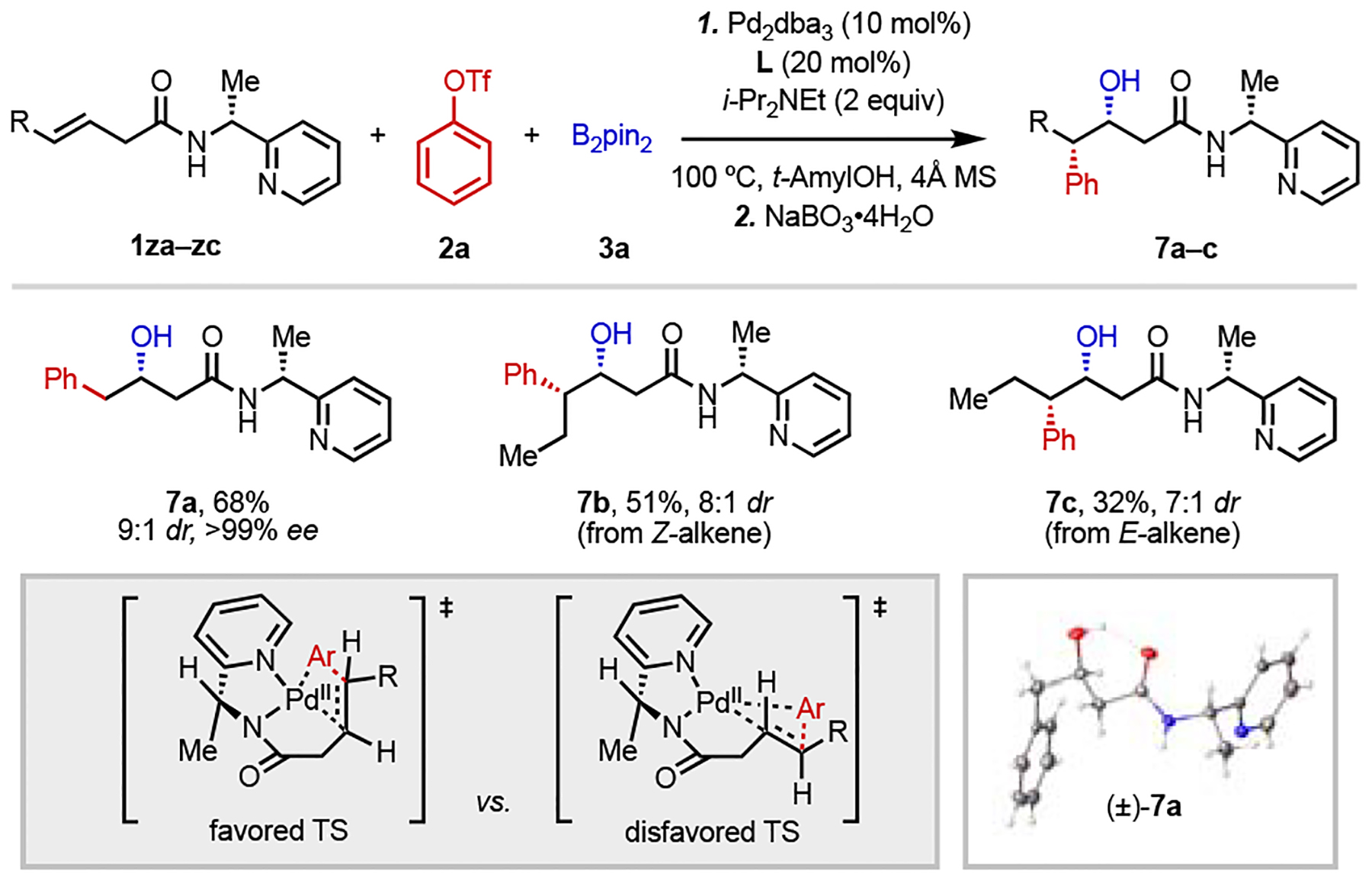
To conclude this study, we aimed to demonstrate the viability of establishing the absolute configuration of the newly installed stereocenters through use of a chiral bidentate directing group to facilitate the migratory insertion step.[25,26] After some experimentation, we eventually identified 1-(pyridin-2-yl)ethan-1-amine (as shown in substrates 1za–1zc, Table 5),[27] which is commercially available in enantiopure form, as a suitable chiral directing group. Under modified conditions using increased catalyst and ligand loading, 1,2-arylboration proceeded smoothly with terminal alkene substrate 1za, forming product 7a in good yield with 9:1 dr and no epimerization of the chiral auxiliary. With internal alkene substrates 1zb and 1zc, this strategy could be used to establish two contiguous stereogenic centers (7b and 7c). While both reactions provided similar diastereoselectivity to the terminal case, the yields were diminished (51% and 32% for the Z- and E- alkene isomers, respectively) with unreatced starting material in both cases, reflecting the sensitivity of the reaction to steric hindrance. Based on an X-ray crystal structure of the racemic form of product 7a, we were able to assign the relative stereochemistry of the major diastereomer and hence establish the absolute configuration of the newly formed C(sp3)–O bond. The stereochemistry of 7b and 7c was assigned by analogy assuming a syn-migratory insertion event. One plausible stereoinduction model is shown in Table 5 (bottom left). In this model, the directing group binds in a bidentate fashion, displacing phosphine. The bottom-facing methyl group then affects the preferred coordination mode of the alkene to the palladium center during the migratory insertion step through steric clashing. Alternatively, the directing group could be bound in a monodentate fashion, with the phosphine still coordinated (see SI for additional discussion).
Table 5.
Diastereoselective Arylboration Using a Removable Chiral Directing Group.[a]
|
Reaction conditions: 1za–zc (0.05 mmol), 2a (1.5 equiv), 3a (2 equiv), Pd2dba3 (10 mol%), L (20 mol%), i-Pr2NEt (2 equiv), 4Å MS (20 mg), 100 °C, N2, 44–48 h. The final products were oxidized to the corresponding alcohol with NaBO3•4H2O (5 equiv) for ease of isolation. All the yields refer to the isolated yields.
Conclusion
In conclusion, we have developed highly regio- and diastereoselective carboboration and carbosilylation reactions of alkenyl carbonyl compounds using a substrate directivity strategy. Compatible substrates include alkenes with various substitution patterns and benzo-fused heteroaromatics that react through five- and six-membered palladacycle intermediates. These methods allow direct access to carbonyl products containing boron and silicon groups at the β and γ positions. Furthermore, an array of aryl and alkenyl triflates are suitable carbon electrophiles. The reactions are scalable and operationally simple. Diversification of arylborylated products was also demonstrated, showcasing the synthetic versatility of the products. Finally, an example of diastereoselective 1,2-arylboration using a chiral directing group was shown.
Experimental Section
General procedure for aryl- and alkenylboration of alkenes.
To a 4-mL scintillation vial equipped with a Teflon-coated magnetic stir bar were added the alkene substrate (0.1 mmol), bis(pinacolato)diboron (50.8 mg, 0.2 mmol), Pd2(dba)3 (2.8 mg, 3 mol%), Cy-JohnPhos (2.2 mg, 6 mol%), and 4Å molecular sieves (~28 mg). The vial was sealed with a screw-top septum cap and was then evacuated and backfilled with N2 (×3). Under positive N2 pressure, aryl or alkenyl triflate (0.15 mmol), i-Pr2NEt (34.8 μL, 0.2 mmol), and t-AmylOH (0.2 mL) were added. All needle inlets/outlets were removed, and the reaction was placed in a heating block that was pre-heated to 90 °C. After 40–44 h, the black reaction mixture was allowed to cool to room temperature and filtered through a short plug of celite. Upon purification by column chromatography with 5:1 to 3:1 hexanes:EtOAc as the eluent, the pure product was obtained.
Supplementary Material
Acknowledgements
This work was financially supported by Scripps Research, Bristol-Myers Squibb (Unrestricted Grant), and the National Institutes of Health (5R35GM125052-02). Additionally, we gratefully acknowledge Bristol-Myers Squibb for a Graduate Fellowship (Z.L.), the A. P. Sloan Foundation and Dreyfus Foundation for young faculty fellowships (K.M.E.), and Nankai University College of Chemistry for an International Research Schlorship (X.L.). We thank Joseph Derosa, Xin Wang and Mingyu Liu for donation of alkene substrates. Dr. Jason Chen and Brittany Sanchez (Scripps Research Automated Synthesis Facility) are acknowledged for SFC and HRMS analysis. We further thank Dr. Milan Gembicky and Jake B. Bailey (UCSD) for X-ray crystallographic analysis.
References
- [1].For reviews, see:; a) Suginome M, Chem. Rec 2010, 10, 348–358; [DOI] [PubMed] [Google Scholar]; b) Liu Z, Gao Y, Zeng T, Engle KM, Isr. J. Chem 2019, DOI: 10.1002/ijch.201900087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].For examples of copper-catalyzed intermolecular 1,2-carboboration, see:; a) Yoshida H, Kageyuki I, Takaki K, Org. Lett 2013, 15, 952–955; [DOI] [PubMed] [Google Scholar]; b) Meng F, Haeffner F, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 11304–11307; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Su W, Gong T-J, Lu X, Xu M-Y, Yu C-G, Xu Z-Y, Yu H-Z, Xiao B, Fu Y, Angew. Chem 2015, 127, 13149–13153; [Google Scholar]; Su W, Gong T-J, Lu X, Xu M-Y, Yu C-G, Xu Z-Y, Yu H-Z, Xiao B, Fu Y, Angew. Chem. Int. Ed 2015, 54, 12957–12961; [DOI] [PubMed] [Google Scholar]; d) Parra A, López A, Díaz-Tendero S, Amenós L, Ruano JLG, Tortosa M, Synlett 2015, 26, 494–500; [Google Scholar]; e) Huang Y, Smith KB, Brown MK, Angew. Chem 2017, 129, 13499–13503; [Google Scholar]; Huang Y, Smith KB, Brown MK, Angew. Chem. Int. Ed 2017, 56, 13314–13318; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kim N, Han JT, Ryu DH, Yun J, Org. Lett 2017, 19, 6144–6147. [DOI] [PubMed] [Google Scholar]
- [3].For examples of palladium-catalyzed intermolecular 1,2-carboboration, see:; a) Yang K, Song Q, Org. Lett 2016, 18, 5460–5463; [DOI] [PubMed] [Google Scholar]; b) Yang K, Song Q, J. Org. Chem 2016, 81, 1000–1005; [DOI] [PubMed] [Google Scholar]; c) Liu Z, Ni H-Q, Zeng T, Engle KM, J. Am. Chem. Soc 2018, 140, 3223–3227; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu Z, Li X, Zeng T, Engle KM, ACS Catal. 2019, 9, 3260–3265; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Bai Z, Zheng S, Bai Z, Song F, Wang H, Peng Q, Chen G, He G, ACS Catal. 2019, 9, 6502–6509. [Google Scholar]
- [4].For examples of nickel-catalyzed intermolecular alkene 1,2-carboboration, see:; a) Logan KM, Sardini SR, White SD, Brown MK, J. Am. Chem. Soc 2018, 140, 159–162; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sardini SR, Lambright AL, Trammel GL, Omer HM, Liu P, Brown MK, J. Am. Chem. Soc 2019, 141, 9391–9400; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang W, Ding C, Pang H, Yin G, Org. Lett 2019, 21, 3968–3971; [DOI] [PubMed] [Google Scholar]; d) Chen L-A, Lear AR, Gao P, Brown MK, Angew. Chem 2019, 131, 11072–11076; [DOI] [PMC free article] [PubMed] [Google Scholar]; Chen L-A, Lear AR, Gao P, Brown MK, Angew. Chem. Int. Ed 2019, 58, 10956–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For 1,2-carboboration using cooperative catalysis, see:; a) Semba K, Nakao Y, J. Am. Chem. Soc 2014, 136, 7567–7570; [DOI] [PubMed] [Google Scholar]; b) Smith KB, Logan KM, You W, Brown MK, Chem. Eur. J 2014, 20, 12032–12036; [DOI] [PubMed] [Google Scholar]; c) Jia T, Cao P, Wang B, Lou Y, Yin X, Wang M, Liao J, J. Am. Chem. Soc 2015, 137, 13760–13763; [DOI] [PubMed] [Google Scholar]; d) Semba K, Ohtagaki Y, Nakao Y, Org. Lett 2016, 18, 3956–3959. [DOI] [PubMed] [Google Scholar]
- [6].Xu H, White PB, Hu C, Diao T, Angew. Chem 2017, 129, 1557–1560; [DOI] [PubMed] [Google Scholar]; Xu H, White PB, Hu C, Diao T, Angew. Chem. Int. Ed 2017, 56, 1535–1538. [DOI] [PubMed] [Google Scholar]
- [7].For an example of Ni(II)-catalyzed chain-walking arylboration, see:; a) Wang W, Ding C, Li Y, Li Z, Li Y, Peng L, Yin G, Angew. Chem 2019, 131, 4660–4664; [DOI] [PubMed] [Google Scholar]; Wang W, Ding C, Li Y, Li Z, Li Y, Peng L, Yin G, Angew. Chem. Ind. Ed 2019, 58, 4612–4616. [DOI] [PubMed] [Google Scholar]; For an example of Pd(0)-catalyzed 1,1-arylboration, see:; b) Nelson HM, Williams BD, Miró J, Toste FD, J. Am. Chem. Soc 2015, 137, 3213–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huffman TR, Wu Y, Emmerich A, Shenvi RA, Angew. Chem 2019, 131, 2393–2398; [DOI] [PMC free article] [PubMed] [Google Scholar]; Huffman TR, Wu Y, Emmerich A, Shenvi RA, Angew. Chem. Ind. Ed 2019, 58, 2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Gurak JA Jr., Yang KS, Liu Z, Engle KM, J. Am. Chem. Soc 2016, 138, 5805–5808; [DOI] [PubMed] [Google Scholar]; b) Liu Z, Zeng T, Yang KS, Engle KM, J. Am. Chem. Soc 2016, 138, 15122–15125; [DOI] [PubMed] [Google Scholar]; c) Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM, J. Am. Chem. Soc 2017, 139, 11261–11270; [DOI] [PubMed] [Google Scholar]; d) Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM, J. Am. Chem. Soc 2017, 139, 10657–10660; [DOI] [PubMed] [Google Scholar]; e) Zeng T, Liu Z, Schmidt MA, Eastgate MD, Engle KM, Org. Lett 2018, 20, 3853–3857; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Matsuura R, Jankins TC, Hill DE, Yang KS, Gallego GM, Yang S, He M, Wang F, Marsters RP, McAlpine I, Engle KM, Chem. Sci 2018, 9, 8363–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For selected examples, see:; a) Wang H, Bai Z, Jiao T, Deng Z, Tong H, He G, Peng Q, Chen G, J. Am. Chem. Soc 2018, 140, 3542–3546; [DOI] [PubMed] [Google Scholar]; b) Wang C, Xiao G, Guo T, Ding Y, Wu X, Loh T-P, J. Am. Chem. Soc 2018, 140, 9332–9336; [DOI] [PubMed] [Google Scholar]; c) Lv H, Xiao L-J, Zhao D, Zhou Q-L, Chem. Sci 2018, 9, 6839–6843; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shen H-C, Zhang L, Chen S-S, Feng J, Zhang B-W, Zhang Y, Zhang X, Wu Y-D, Gong L-Z, ACS Catal. 2019, 9, 791–797; [Google Scholar]; e) Zhang Y, Chen G, Zhao D, Chem. Sci 2019, DOI: 10.1039/C9SC02182E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For a representative review, see:; Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Suginome M, Matsuda T, Ito Y, J. Am. Chem. Soc 2000, 122, 11015–11016. [Google Scholar]
- [13].Martin R, Buchwald SL, Acc. Chem. Res 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Singh R, Vince R, Chem. Rev 2012, 112, 4642–4686. [DOI] [PubMed] [Google Scholar]
- [15].a) Dong J, Krasnova L, Finn MG, Sharpless KB, Angew. Chem 2014, 126, 9584–9603; [DOI] [PubMed] [Google Scholar]; Dong J, Krasnova L, Finn MG, Sharpless KB, Angew. Chem. Int. Ed 2014, 53, 9430–9448; [DOI] [PubMed] [Google Scholar]; b) Liang Q, Xing P, Huang Z, Dong J, Sharpless KB, Li X, Jiang B, Org. Lett 2015, 17, 1942–1945. [DOI] [PubMed] [Google Scholar]
- [16].Wertjes WC, Southgate EH, Sarlah D, Chem. Soc. Rev 2018, 47, 7996–8017. [DOI] [PubMed] [Google Scholar]
- [17].Shen C, Zeidan N, Wu Q, Breuers CBJ, Liu R-R, Jia Y-X, Lautens M, Chem. Sci 2019, 10, 3118–3122. [Google Scholar]
- [18].For examples of palladium-catalyzed carbosilylation of different π-systems, see:; a) Obora Y, Tsuji Y, Kawamura T, J. Am. Chem. Soc 1995, 117, 9814–9821; [Google Scholar]; b) Wu M-Y, Yang F-Y, Cheng C-H, J. Org. Chem 1999, 64, 2471–2474; [Google Scholar]; c) Hande SM, Nakajima M, Kamisaki H, Tsukano C, Takemoto Y, Org. Lett 2011, 13, 1828.–; [DOI] [PubMed] [Google Scholar]; d) Shintani R, Kurata H, Nozaki K, J. Org. Chem 2016, 81, 3065–3069. [DOI] [PubMed] [Google Scholar]
- [19].For selected examples of conceptually distinct approaches to catalytic alkene 1,2-carbosilylation, see:; a) Nii S, Terao J, Kambe N, J. Org. Chem 2000, 65, 5291–5297; [DOI] [PubMed] [Google Scholar]; b) Liepins V, Bäckvall J-E, Chem. Commun 2001, 265–266; [Google Scholar]; c) Nakamura S, Uchiyama M, J. Am. Chem. Soc 2007, 129, 28–29; [DOI] [PubMed] [Google Scholar]; d) Yang Y, Song R-J, Ouyang X-H, Wang C-Y, Li J-H, Luo S, Angew. Chem 2017, 129, 8024–8027; [Google Scholar]; Yang Y, Song R-J, Ouyang X-H, Wang C-Y, Li J-H, Luo S, Angew. Chem. Int. Ed 2017, 56, 7916–7919. [DOI] [PubMed] [Google Scholar]
- [20].For selected examples of Pd-catalyzed C(sp3)–H silylation enabled by bidentate directing groups, see:; a) Kanyiva KS, Kuninobu Y, Kanai M, Org. Lett 2014, 16, 1968–1971; [DOI] [PubMed] [Google Scholar]; b) Liu Y-J, Liu Y-H, Zhang Z-Z, Yan S-Y, Chen K, Shi B-F, Angew. Chem 2016, 128, 14063–14066; [Google Scholar]; Liu Y-J, Liu Y-H, Zhang Z-Z, Yan S-Y, Chen K, Shi B-F, Angew. Chem. Int. Ed 2016, 55, 13859–13862; [DOI] [PubMed] [Google Scholar]; c) Deb A, Singh S, Seth K, Pimparkar S, Bhaskararao B, Guin S, Sunoj RB, Maiti D, ACS Catal. 2017, 7, 8171–8175; [Google Scholar]; d) Zhan B-B, Fan J, Jin L, Shi B-F, ACS Catal. 2019, 9, 3298–3303. [Google Scholar]
- [21].Verho O, Lati MP, Oshmann M, J. Org. Chem 2018, 83, 4464–4476. [DOI] [PubMed] [Google Scholar]
- [22].Deguchi T, Xin H-L, Morimoto H, Ohshima T, ACS Catal. 2017, 7, 3157–3161. [Google Scholar]
- [23].a) Sonawane RP, Jheengut V, Rabalakos C, Larouche-Gauthier R, Scott HK, Aggarwal VK, Angew. Chem 2011, 123, 3844–3847; [DOI] [PubMed] [Google Scholar]; Sonawane RP, Jheengut V, Rabalakos C, Larouche-Gauthier R, Scott HK, Aggarwal VK, Angew. Chem. Int. Ed 2011, 50, 3760–3763; [DOI] [PubMed] [Google Scholar]; b) Hoang GL, Takacs JM, Chem. Sci 2017, 8, 4511–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li Z, Wang Z, Zhu L, Tan X, Li C, J. Am. Chem. Soc 2014, 136, 16439–16443. [DOI] [PubMed] [Google Scholar]
- [25].For related examples of Heck-type processes employing chiral directing groups, see:; a) Buezo ND, Mancheño OG, Carretero JC, Org. Lett 2000, 2, 1451–1454. [DOI] [PubMed] [Google Scholar]; (b) Nilsson P, Larhed M, Hallberg A, J. Am. Chem. Soc 2003, 125, 3430–3431. [DOI] [PubMed] [Google Scholar]; For a review, see:; c) Oestreich M, Eur. J. Org. Chem 2005, 783–792. [Google Scholar]
- [26].For use of a chiral monodentate directing group in Pd(II)-catalyzed alkene dioxygenation, see:; Neufeldt SR, Sanford MS, Org. Lett 2013, 15, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim Y, Kim S-T, Kang D, Sohn T.-i., Jang E, Baik M-H, Hong S, Chem. Sci 2018, 9, 1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.