

Abstract
One-carbon metabolism is a collection of metabolic cycles that supports methylation and provides one-carbon bound folates for the de novo synthesis purine and thymidine nucleotides. The methylation of phosphatidylethanolamine to form choline has been extensively studied in the context of fatty liver disease. However, the role of one-carbon metabolism in supporting nucleotide synthesis during liver damage has not been addressed. The objective of this study is to determine how the disruption of one-carbon metabolism influences nucleotide metabolism in the liver after dietary methionine and choline restriction. Mice (n=8) were fed a methionine–choline-deficient or control diet for 3 weeks. We treated mice with the compound alloxazine (0.5 mg/kg), a known adenosine receptor antagonist, every second day during the final week of feeding to probe the function of adenosine signaling during liver damage. We found that concentrations of several hepatic nucleotides were significantly lower in methionine-and choline-deficient mice vs. controls (adenine: 13.9±0.7 vs. 10.1±0.6, guanine: 1.8±0.1 vs. 1.4±0.1, thymidine: 0.0122±0.0027 vs. 0.0059±0.0027 nmol/mg dry tissue). Treatment of alloxazine caused a specific decrease thymidine nucleotides, decrease in mitochondrial content in the liver and exacerbation of steatohepatitis as shown by the increased hepatic lipid content and altered macrophage morphology. This study demonstrates a role for one-carbon metabolism in supporting de novo nucleotide synthesis and mitochondrial function during liver damage.
Keywords: Nucleotides, Inflammation, Amino acids, Macrophage, Steatohepatitis
Graphical Abstract
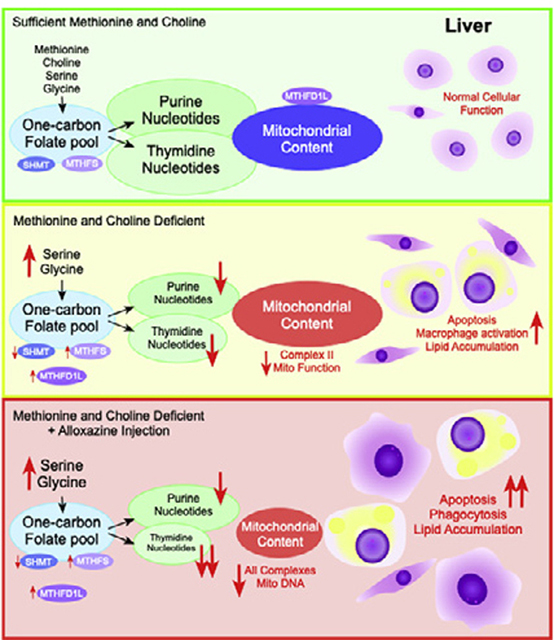
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a growing health epidemic with an estimated prevalence of one in three healthy adults in the United States; it is estimated that up to 80% of the type 2 diabetic and obese population have NAFLD [1]. Approximately 15% of patients with NAFLD progress to nonalcoholic steatohepatitis (NASH) with a potential prevalence of 12% of the U.S. population [2]. Chronic damage coupled with immune-directed tissue turnover is at the center of progression of fibrosis that eventually leads to cirrhosis and liver failure and greatly enhances the risk of developing hepatocellular carcinoma [3,4]. Two common observations in liver damage are dysfunction of immune cells and impaired mitochondrial function, but the metabolic interactions between energy metabolism and immune signaling are not fully known. Understanding the molecular mechanisms that contribute to initiation, progression and resolution of liver damage is of the utmost importance and will allow for the development of more effective treatments and management of this disease. The goal of this study is to examine the impact of impaired one-carbon metabolism on macrophage function and its role in the progression of liver damage.
One-carbon metabolism (Supplemental Fig. S1) plays a crucial role in liver function and has been shown to have a broad and robust association with NAFLD and NASH [5,6]. Pogribny et al. have demonstrated that mice with reduced expression of enzymes involved in the regulation of one-carbon metabolism including methionine adenosyltransferase 1a and S-adenosylhomocysteine hydrolase have increased susceptibility to developing fatty liver [7]. Furthermore, humans with cirrhotic livers were shown to have reduced capacity for synthesis and utilization of S-adenosylmethionine (AdoMet), and several rodent models with genetic perturbations of one-carbon metabolism exhibit liver damage [8–10]. AdoMet is a substrate for many methyltransferase enzymes including glycine-N-methyltransferase and phosphatidylethanolamine-N-methyltransferase which have been implicated in NAFLD and NASH using genetic models [11,12]. Flux through methyltransferase enzymes is partially dictated by the concentration of AdoMet and end product inhibition by S-adenosylhomocysteine (AdoHcy) [13]. The observation of inflammation under conditions where one-carbon metabolism is impaired suggests that this pathway plays a role in the regulation of immune cell function. One-carbon metabolism is crucial for de novo synthesis of purine and thymidine nucleotides that are required for cell proliferation [14]. However, the influence of impaired one-carbon metabolic system on de novo purine and pyrimidine synthesis has not been well studied [15].
Purine and pyrimidine metabolites are known to influence immune signaling and function of macrophages [16]. De novo thymidine synthesis requires one-carbon bound folate as a substrate and is well known to influence cell cycle activity and proliferation [17], but the effects of purines are less well described. The adenosine receptors (A1, A2A, A2B or A3) are expressed on many types of immune cells and are known to play diverse roles in immune function [18]. Deletion of the A2B adenosine receptor was shown to up-regulate inflammatory cytokine production from adipose tissue macrophages [19,20]. A2B receptor knockout mice also had poor glucose tolerance and increased liver inflammation and macrophage markers after high-fat diet feeding [21]. Moreover, cultured RAW 264.7 mouse macrophages stimulated with lipopolysaccharides showed increased production of the anti-inflammatory cytokine interleukin-10 (IL-10) through stimulation of adenosine receptors [22]. Given the key roles that macrophages play in tissue repair, we investigate one-carbon purine and pyrimidine metabolism and their potential for influencing macrophage function during liver damage. Therefore, we employed the methionine-and choline-deficient diet (MCD) to induce liver damage and used the selective A2B receptor antagonist [23,24] alloxazine in an attempt to probe the role of adenosine signaling through the A2B receptor during liver damage. Our hypothesis was that blockade of the A2B receptor would exacerbate inflammation in the livers of MCD mice. We find that MCD feeding resulted in significant reduction in hepatic purine and thymidine content. Alloxazine disrupted folate and one-carbon metabolism and further decreased hepatic thymidine nucleotide content and mitochondrial respiratory complexes in mice fed the MCD diet. These metabolic changes were associated with an exacerbated NASH phenotype characterized by enhanced triglyceride accumulation, apoptosis and macrophage activation in the liver.
2. Materials and methods
2.1. Animals
C57BL/6J male mice were housed in ventilated cages with corn cob bedding and maintained on a 14:10-h light/dark cycle with ad libitum access to standard nonpurified diet (Envigo #2918). At 10 weeks of age, mice were randomly divided into two groups. One group was fed an MCD diet (Envigo #TD.90262), and a control group was fed a nutrient-matched control diet (CTR) (Envigo #TD.94149) containing sufficient methionine and choline. Mice were fed the diets ad libitum for 3 weeks. During the final week of the study, each diet group was divided into two subgroups (n=8 per group) that received intraperitoneal (IP) injections every other day (four total) of the following solutions: vehicle [1% dimethylsulfoxide (DMSO) in saline: Veh] or alloxazine (Alx) at a dose of 0.5 mg/kg body weight dissolved in vehicle. All study protocols were approved by the Institutional Animal Care and Use Committee of the University of Florida and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication 86–23 revised 1985).
2.2. Tissue collection
Mice were anesthetized using ketamine/xylazine prior to tissue collection on the day after the final IP injection. Whole blood was collected via heart puncture into EDTA-treated tubes and centrifuged at 3000g for 10 min at 4°C. Plasma was separated and stored at −80°C until further analysis. The whole liver was excised and then subdivided for immediate snap-freezing and storage at −80°C for metabolite analysis or fixation with 10% phosphate-buffered formalin for histological staining. For mitochondrial respiration studies, mitochondria were freshly isolated from livers of C57BL/6J mice by differential centrifugation protocol, and respiration was measured as previously described [25].
2.3. Histological analysis
Liver tissues were embedded in paraffin, sliced and stained with hematoxylin & eosin (H&E) or picrosirius red at the University of Florida Molecular Pathology Core Facility. Liver sections were scored for NASH in the Department of Pathology at the University of Florida.
2.4. Immunofluorescence
Immunofluorescent staining was performed on deparaffinized mouse liver sections obtained from the University of Florida Molecular Pathology Core Facility. Slides were blocked in 10% normal goat serum for 1 h and then probed with rabbit anti-α-smooth muscle actin, 1:250 (Cell Signaling Technologies, Danvers, MA,USA); rat anti-F4/80, 1:50 (Fisher); or rabbit anti-cleaved caspase-3, 1:400 (Cell Signaling Technologies) overnight at 4°C in a humidified chamber. Slides were then washed with Tris-buffered saline with 1% Tween-20 and probed with secondary antibodies goat anti-rabbit Dylight 488 (1:500; Fisher, Hampton, NH, USA) or goat anti-rat Dylight 633 (1:500; Fisher) for 30 min at room temperature in the dark. These procedures were followed by incubation of the slides with Hoechst 33342 for 10 min at a concentration of 1:4000 to visualize nuclei. Fluorescent images were visualized using a Nikon Eclipse Ti2 fluorescent microscope and quantified using Nikon Elements version 4.6 (Nikon, Melville, NY, USA).
2.5. Western blotting
Mouse liver homogenates were diluted in Laemmli loading buffer and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (either 7.5% or 10% acrylamide) before being transferred to a polyvinylidene difluoride membrane. Antibodies (Supplementary Table S3) were obtained from Cell Signaling Technologies with the following exceptions: adenosine A1 receptor (ADORA1) (Proteintech), adenosine A2B receptor (ADORA2B) (Millipore), OXPHOS Total Complex (Abcam, UK), adenosine A2A receptor (ADORA2A), adenosine A3 receptor (ADORA3), serine hydroxymethyltransferase 1 (SHMT1), serine hydroxymethyltransferase 2 (SHMT2), and alpha tubulin, Vinculin (Fisher) and StarBright Blue 700 Goat Anti-Rabbit IgG (BioRad, Hercules, CA, USA). Blots were visualized using the Chemidoc MP imaging system (BioRad, CA, USA). Densities were calculated and normalized to the density of a loading control protein and to the density of the total lane protein.
2.6. Metabolite and biomarker analysis
Frozen liver samples were shipped on dry ice to the Southeast Center for Integrated Metabolomics (Florida, USA) for analysis of nucleotide metabolites by liquid chromatography–tandem mass spectrometry (MS) with isotope dilution. AdoMet and AdoHcy were determined using ultra-performance liquid chromatography (UPLC) as previously described [26]. Plasma total homocysteine was determined by derivatization with 7-fluorobenzo-2,1,3-oxadiazole-4-sulfonic acid before separation using UPLC as previously described [27]. Amino acids were measured by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidylcarbamate followed by UPLC separation and fluorescence detection using a published method [28]. Folates were analyzed in mouse liver tissue by the method adapted from Bagley and Selhub [29]. Briefly, folates were extracted from mouse liver tissue by sonicating in a hypotonic buffer and then incubating with recombinant conjugase from Arabidopsis. Unconjugated folates were loaded onto a folate binding protein-affinity chromatography column, extensively washed to remove contaminants and then eluted by addition of concentrated phosphoric acid. Isolated folates were then separated on a Luna C18 column and quantified using an electrochemical detection method. Total formate in liver was measured using isotope-dilution gas chromatography–MS after derivatization with 2,3,4,5,6-pentaflurobenzyl bromide as described [30]. Plasma alanine aminotransferase (ALT) activity was determined kinetically using a commercial kit (Biovision, Milpitas, CA, USA). Triglycerides were extracted using a modified Folch extraction as previously described using a colorimetric kit (Sekiusi, Secaucus, NJ, USA) [31]. Absorbance was read using a Spectramax 340 PC microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Since alloxazine structurally mimics flavin molecules, the erythrocyte glutathione reductase activity coefficient was determined as an indicator of riboflavin status using the method as previously described [32].
2.7. Gene expression and mitochondrial DNA abundance
Real-time quantitative polymerase chain reaction (PCR) was used to determine gene expression of Shmt1, Shmt2 and Pcna in mouse liver. Total RNA was extracted using the Qiagen RNeasy Plus Mini Kit (Qiagen, Germany). cDNA was synthesized using the Applied Biosystems High-Capacity Reverse Transcription Kit (Applied Biosystems, MA, USA). DNA was isolated from liver using the DNeasy Mini Kit (Qiagen). Concentration and purity of samples were assessed using a nanophotometer (Implen, Germany). Reverse-transcription quantitative PCR (RT-qPCR) was performed using the PowerUp SYBR Green master mix (Applied Biosystems). Gene expression was normalized to the housekeeping gene Rpl13a, and results were quantified using the ΔΔCT method. Mitochondrial DNA abundance was measured by determining the relative abundance of mitochondrially encoded NADH dehydrogenase 2 (ND2) compared to genomic phosphoglycerate kinase.
2.8. Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad, San Diego, CA, USA). A two-way analysis of variance (MCD diet vs. alloxazine) was used in combination with Tukey’s multiple comparison test. A P value of less than .05 was considered significant. n=8 mice per treatment group unless otherwise specified. All data are presented as mean ± standard deviation.
3. Results
3.1. Alloxazine treatment increased inflammation and lipid accumulation in MCD mice
Mice fed the MCD diet lost a significant amount of body weight over 3 weeks compared to the CTR mice (Fig. 1A). Gross liver morphology was determined by H&E staining (Fig. 1B) and showed increased lipid accumulation and lobular inflammation in MCD mice compared to CTR. MCD feeding induced significant liver damage and lobular inflammation that was exacerbated by alloxazine (Table 1 and Fig. 1C). Lipid accumulation was confirmed by TG analysis (Fig. 1D). MCD feeding yielded a greater than tenfold increase in plasma ALT (Fig. 1E) that was not further enhanced by alloxazine.
Fig. 1.
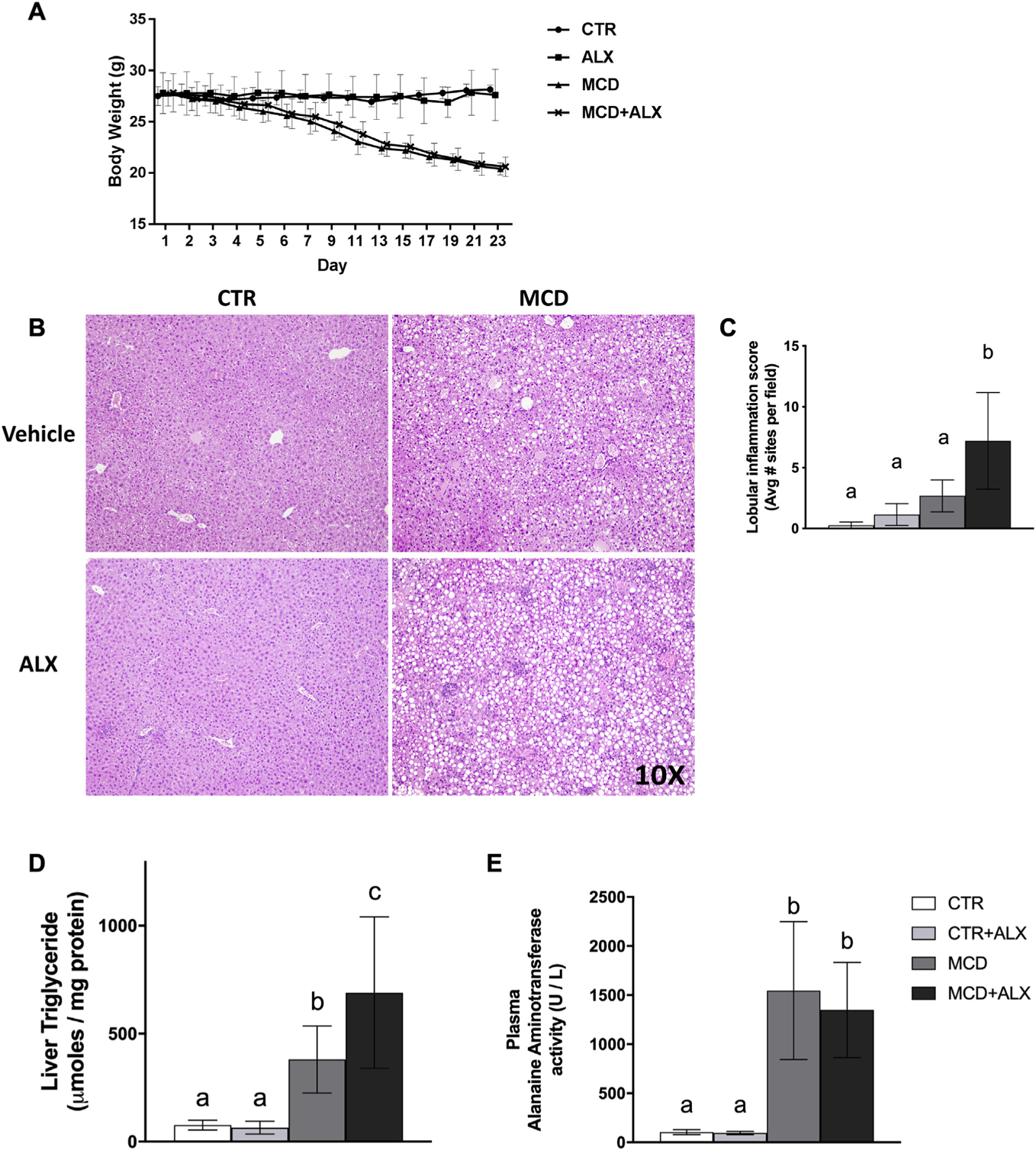
MCD diet and alloxazine treatment induced liver damage.Body weight in grams was recorded over the time course of feeding (A), black circles; CTR, black squares; CTR+ALX, black triangles; MCD, black x; MCD+ALX. H&E staining of paraffin-embedded liver sections from a representative mouse from each group at 10× magnification (B). Lobular inflammation score (C). Liver triglyceride concentrations (D). Plasma alanine aminotransferase activity (E). Different lowercase letters denote a significant difference (P<.05, n=4–8). All data are presented as mean ± standard deviation.
Table 1.
NASH scoring
| Measurement | CTR | CTR+ALX | MCD | MCD+ALX |
|---|---|---|---|---|
| H | 0.9±1.8a | 2.9±7.6a | 43.1±19.6b | 60.0±21.6b |
| Lobular inflammation | 0.3±0.3a | 0.8±0.9a | 3.1±1.7a | 7.2±4.0b |
| Ballooning score (0–2) | 0±0 | 0±0 | 1.0±1.1a | 1.3±0.9a |
| Portal inflammation score (0–1) | 0.1±0.4 | 0.1±0.4 | 0.5±0.5 | 0.5±0.5 |
| NASH score | 0.5±0.5 | 1.1±0.9 | 4.9±1.4a | 6.5±1.7a |
NASH scoring was determined in a blinded manner. Different lowercase letters denote a significant difference (P<.05, n=8). All values presented as mean ± standard deviation.
Adenosine receptors were probed in livers of mice treated with MCD and alloxazine (Fig. 2A–E). The MCD diet decreased the relative expression of the A1 and A2B receptors. Alloxazine also significantly decreased A1 receptor expression in control mice. The treatments did not significantly influence the expression of any of the other adenosine receptors.
Fig. 2.
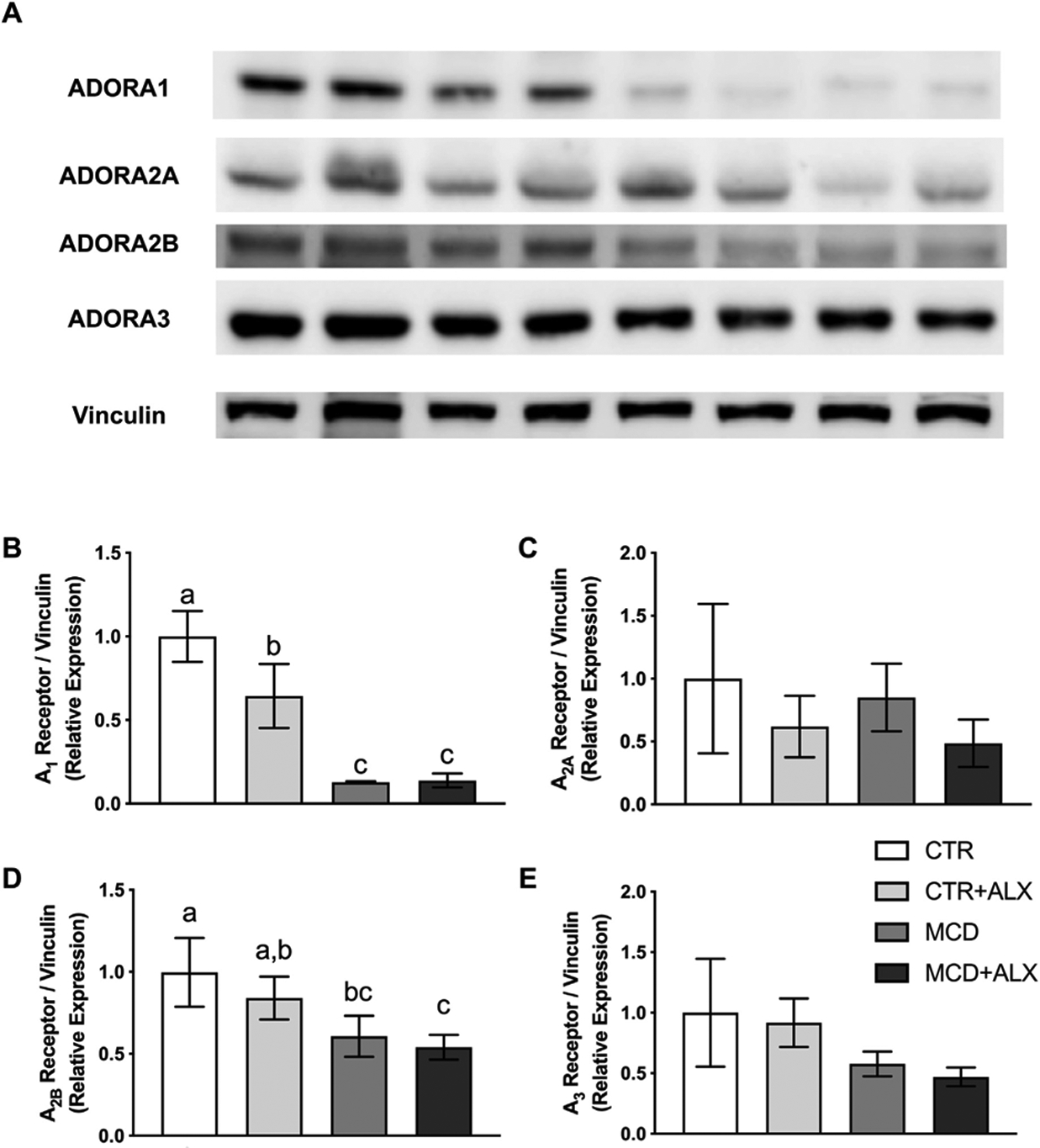
Adenosine receptor expression in livers of MCD and alloxazine-treated mice.Western blot images of ADORA1, ADORA2A, ADORA2B and ADORA3 in mouse liver (A). Relative expression of proteins measured by western blotting for ADORA1 (B), ADORA2A (C), ADORA2B (D) and ADORA3 (E). White bars represent CTR+Veh, light gray represent CTR+ALX, dark gray bars represent MCD+Veh, and dark bars represent MCD+ALX. Different lowercase letters denote a significant difference (P<.05, n=3). All data are presented as mean ± standard deviation.
3.2. Alloxazine treatment increased the size and reduced total number of hepatic macrophages
Our histological observation of enhanced immune cell activity and apoptosis in MCD mice was also corroborated by immunohistochemical detection of the macrophage marker F4/80 and the apoptotic marker cleaved caspase-3 (Fig. 3A). Analysis revealed increased intensity of F4/80 staining in the livers of MCD mice that was further increased by alloxazine injection (Fig. 3B). Cleaved caspase-3 mirrored the expression of F4/80, indicating that apoptosis was driving this effect (Fig. 3C). The overlay of cleaved caspase-3 and F4/80 staining was significantly higher in MCD mice treated with alloxazine, indicating more macrophages at apoptotic foci (Fig. 3D). In addition, macrophages in alloxazine-treated MCD livers were fewer in number but larger, as indicated by average macrophage area (Fig. 3E). Visually, macrophages in the alloxazine-treated mouse liver show a distinct bulbous morphology with a honeycomb-like staining pattern with several large intracellular vacuoles, resembling a foam-cell phenotype [33,34] (Fig. 3A magnified section).
Fig. 3.
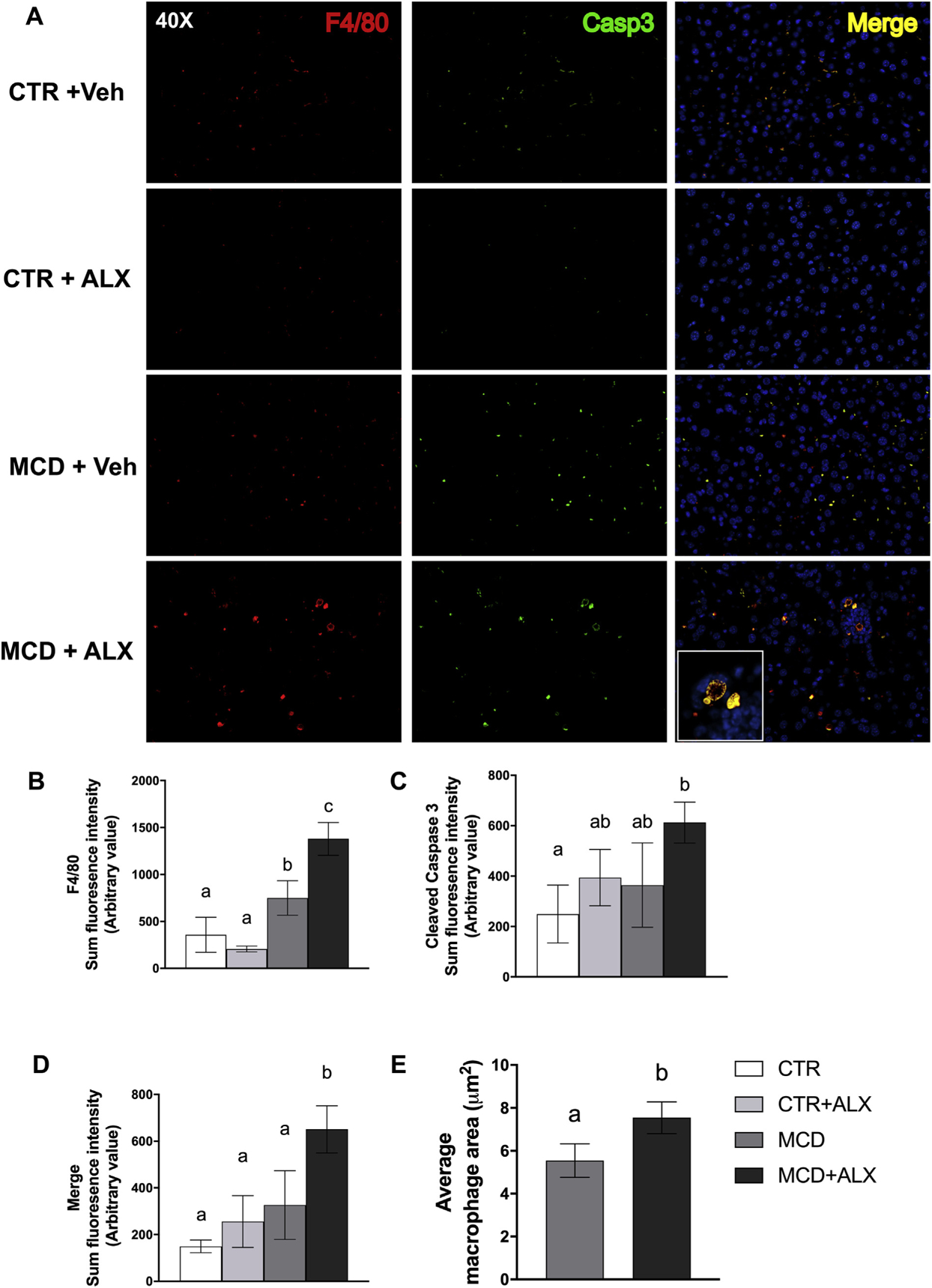
Alloxazine alters macrophage function in livers of MCD mice.Liver sections stained for F4/80 and cleaved caspase-3, 40× magnification (A). Sum fluorescent intensity of F4/80 (B), cleaved caspase-3 (C), merged fluorescence of F4/80 and cleaved caspase-3 (MERGE) (D) staining and average macrophage area (E). White bars represent CTR+Veh, light gray bars represent CTR+ALX, dark gray bars represent MCD+Veh, and dark bars represent MCD+ALX. Different lowercase letters denote a significant difference (P<.05, n=3–8). All data are presented as mean ± standard deviation.
3.3. MCD feeding alters folate metabolism and reduces hepatic purine and pyrimidine nucleotide content
MCD diet feeding resulted in decreased liver AdoMet concentrations, a decreased ratio of AdoMet to AdoHcy and significantly elevated plasma homocysteine. Notably, these metabolites were not altered by alloxazine treatment in either diet (Table 2). MCD feeding did not significantly alter the hepatic folate species measured (Table 2). However, hepatic folate species in both CTR and MCD mice treated with alloxazine were significantly altered, indicating that alloxazine disrupted hepatic folate metabolism. CTR mice treated with alloxazine showed increased hepatic concentrations of several folate species, and total hepatic folates increased threefold. We also observed significantly reduced formate concentrations in livers of MCD mice treated with alloxazine (Table 2). Plasma serine and glycine concentrations were significantly elevated in MCD mice, increasing from 97.4±24.4 to 282±149 μM and 191±442 to 260±231 μM, respectively. Hepatic glycine was significantly higher in livers of alloxazine-treated MCD mice and trended higher in other groups compared to CTR mice. Hepatic glycine was significantly higher in livers of alloxazine-treated MCD mice and trended higher in other groups compared to CTR mice. When we consider this finding along with reduced hepatic nucleotide and formate concentrations, the data suggest that mitochondrial catabolism of glycine by the liver is insufficient to support one-carbon needs during MCD feeding and is further impaired by alloxazine treatment (Table 2). MCD mice treated with alloxazine did not accumulate folates in the liver to the degree observed in CTR mice, but dihydrofolate (DHF) and 5-formyl-tetrahydrofolate (THF) were significantly elevated.
Table 2.
Liver and plasma amino acid, folate and one-carbon metabolites
| Metabolite | CTR | CTR+ALX | MCD | MCD+ALX |
|---|---|---|---|---|
| Liver | ||||
| AdoMet | 88.6±11.5a | 83.5±13.7a | 44.6±8.12b | 28.2±11.8b |
| AdoHcy | 13.8±2.04ab | 11.3±10.7b | 17.5±5.69ab | 19.59±6.85a |
| AdoMet: AdoHcy | 6.52±0.99a | 7.46±1.11a | 2.71±1.09b | 2.06±0.56b |
| Methionine | 83.0±60.1 | 85.3±48.2 | 77.4±33.1 | 84.7±48.6 |
| Serine | 799±253 | 785±294 | 933±253 | 1007±218 |
| Glycine | 242±114 | 365±114 | 403±170 | 416±159 |
| Alanine | 2550±863 | 1890±411 | 2510±1626 | 2160±2098 |
| Ornithine | 298±118a | 256±135a | 417±83.0ab | 500±68.0b |
| Total folates | 11.94±5.55a | 36.76±4.87b | 11.8±3.87a | 18.0±4.67a |
| THF | 5.63±2.70a | 21.9±2.90b | 5.05±1.30a | 9.33±3.74a |
| DHF | 0.54±0.36a | 1.51±0.45b | 0.48±0.15a | 1.55±0.36b |
| 5-Methyl-THF | 0.85±0.51a | 2.74±0.75b | 0.89±0.38a | 1.68±0.66ab |
| 5,10-Methenyl-THF * | 4.37±2.12 | 9.11±3.15 | 5.43±2.36 | 6.20±1.31 |
| 5-Formyl-THF | 0.72±0.49ab | 1.36±0.84ab | 0.28±0.21a | 2.04±0.80b |
| Formate | 0.20±0.04a | 0.18±0.02a | 0.18±0.03a | 0.11±0.01b |
| Plasma (μM) | ||||
| Methionine | 28.4±5.71a | 38.0±4.40b | 43.2±7.49b | 43.8±5.70b |
| Serine | 97.4±24.4a | 143±14.8ab | 282±149b | 301±151b |
| Glycine | 191±46.1a | 260±52.6a | 442±231b | 492±182b |
| Homocysteine | 11.5±4.25a | 14.5±5.75a | 39.3±2.74b | 43.73±14.5b |
Concentrations of amino acids and one-carbon metabolites in liver and plasma. Liver one-carbon, amino acid metabolites and formate are reported as nmol/mg wet liver weight. Folate metabolites are reported as pmole/mg wet liver weight. Plasma metabolites are reported as μM. Concentrations of folate metabolites in liver tissue are reported as μM/mg wet liver weight. Measurements were obtained by HPLC. Different lowercase letters denote a significant difference (P<.05, n=4–8). All values presented as mean ± standard deviation.
Represents both 10-formyl-THF and 5,10-methenyl-THF.
Almost all the amino acids were increased in the plasma of MCD mice, and alloxazine treatment significantly increased plasma concentration of several amino acids in CTR mice including glutamate, arginine, methionine, phenylalanine, tyrosine and tryptophan (Supplementary Table S1).
We observed a significant increase in mitochondrial methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1 like (MTHFD1L) expression in the MCD mice. Methylenetetrahydrofolate synthetase (MTHFS) was significantly increased in MCD mice (Fig. 4 A, B). Protein levels of SHMT1 and SHMT2 were reduced by MCD feeding and were accompanied by a decrease in glycine decarboxylase (GLDC) expression (Fig. 4A–B). Alloxazine did not influence expression of SHMT1, SHMT2 or GLDC but did significantly decrease mRNA abundance of shmt1 and shmt2 (Fig. 4C). MCD mice showed significantly higher thymidylate synthase (TYMS) protein in the liver (Fig. 4A–B).
Fig. 4.
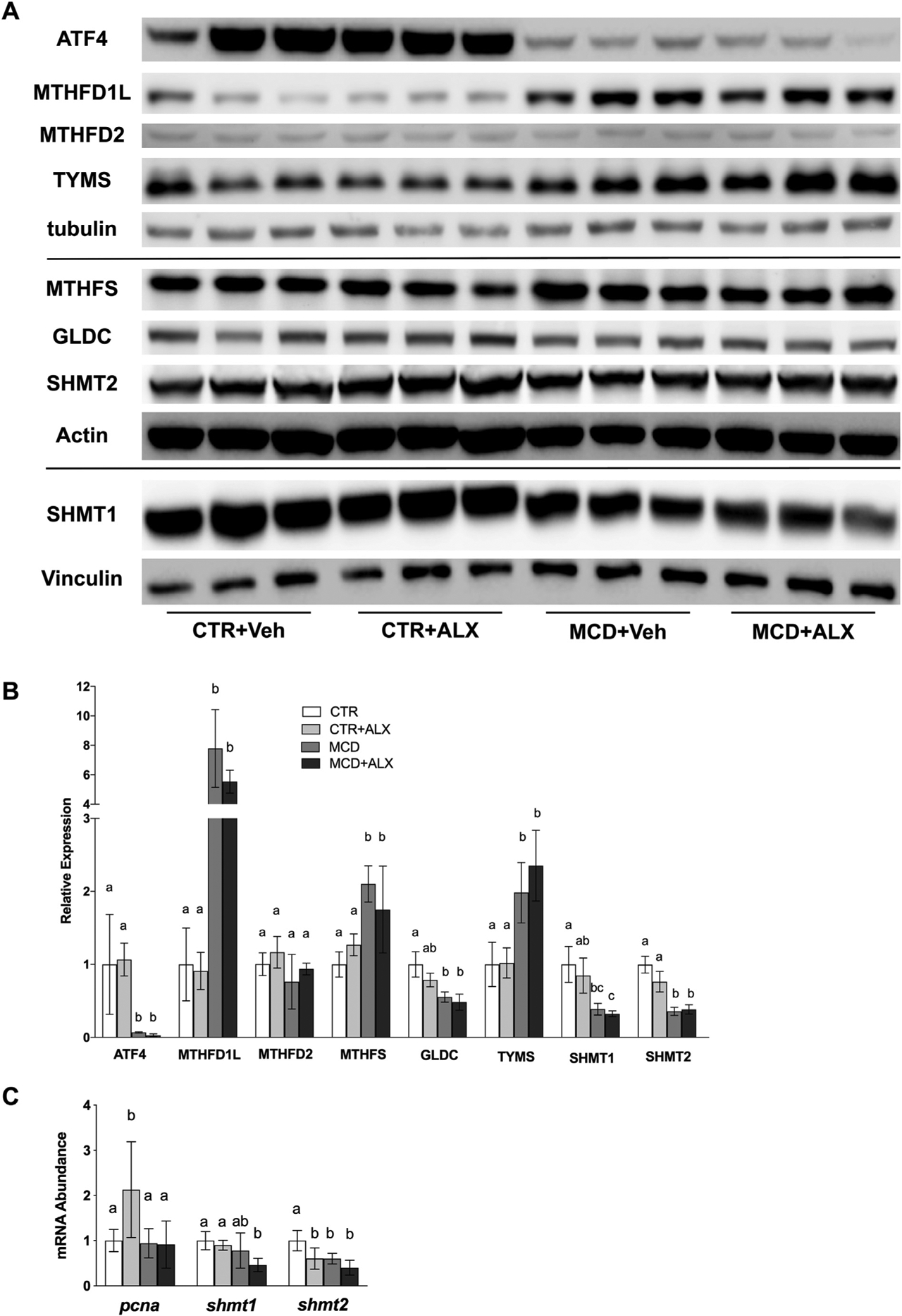
MCD diet and alloxazine treatment both disrupt hepatic one-carbon metabolism.Western blots of ATF4, MTHFD1L, MTHFD2, TYMS, MTHFS, GLDC, SHMT1, and SHMT2 in mouse liver (A). Relative expression of proteins measured by western blotting (B). mRNA abundance of pcna, shmt1 and shmt2 was measured in mouse liver using RT-qPCR (C). White bars represent CTR+Veh, white bars with pattern represent CTR+ALX, dark bars represent MCD+Veh, and dark bars with pattern represent MCD+ALX. Different lowercase letters denote a significant difference (P<.05, n=3). PCNA, proliferating cell nuclear antigen. All data are presented as mean ± standard deviation.
Activating transcription factor 4 (ATF4) has previously been shown to up-regulate flux through mitochondrial folate metabolism and is also an important transcription factor in the unfolded protein response [35]. However, ATF4 expression was reduced so much that it was almost undetectable in the livers of MCD mice (Fig. 4A–B). Alloxazine treatment up-regulated mRNA abundance of pcna in CTR mice but not in MCD mice (Fig. 4C).
Total adenine, guanine and thymidine nucleotides were significantly reduced by MCD feeding (Fig. 5A–E). Individual nucleotide measurements are presented in Supplemental Table 2. Both CTR and MCD mice injected with alloxazine showed significant reduction in total thymidine nucleotide concentrations (Fig. 5E). Mice fed the CTR diet injected with alloxazine also showed a significant reduction in total hepatic thymidine nucleotide content, suggesting that alloxazine may inhibit TYMS. This is further supported by greater DHF observed in CTR and MCD mice injected with alloxazine (Table 2).
Fig. 5.
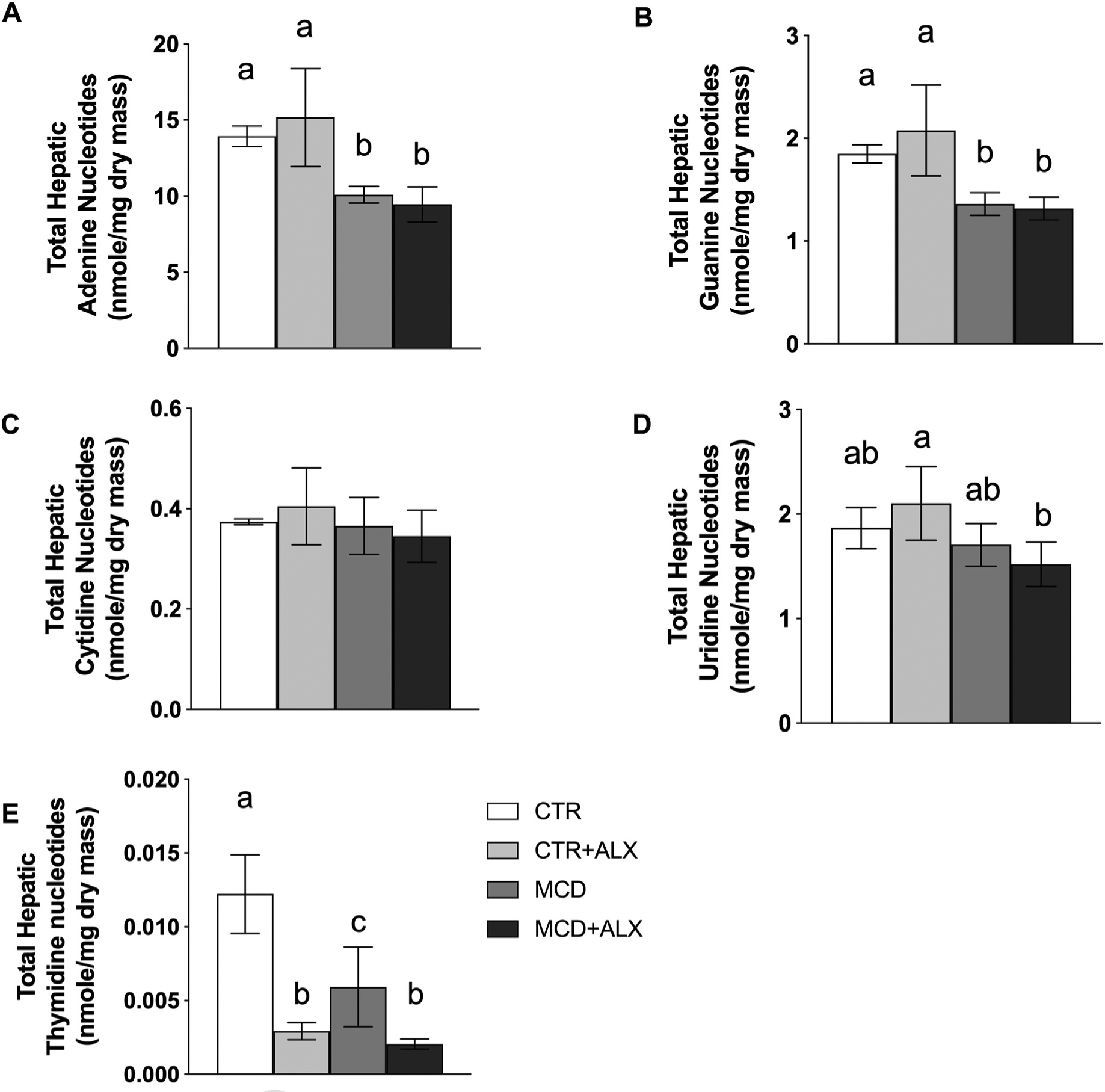
Alloxazine depletes hepatic thymidine nucleotide content.Total hepatic adenine nucleotides (A), guanine nucleotides (B), cytidine nucleotides (C), uridine nucleotides (D), and thymidine nucleotides (E). White bars represent CTR+Veh, light gray bars represent CTR+ALX, dark gray bars represent MCD+Veh, and dark bars represent MCD+ALX. Different lowercase letters denote a significant difference (P<.05, n=4). All data are presented as mean ± standard deviation.
3.4. Disruption of one-carbon and nucleotide metabolism exacerbates mitochondrial dysfunction in MCD mice
Analysis of respiratory complexes showed a trend towards lower expression of complex I, III, IV and V and a significant reduction in complex II expression in liver of MCD mice (Fig. 6A). MCD mice treated with alloxazine showed significant reductions in all mitochondrial complexes and a corresponding decrease in mitochondrial DNA abundance (Fig. 6B).
Fig. 6.
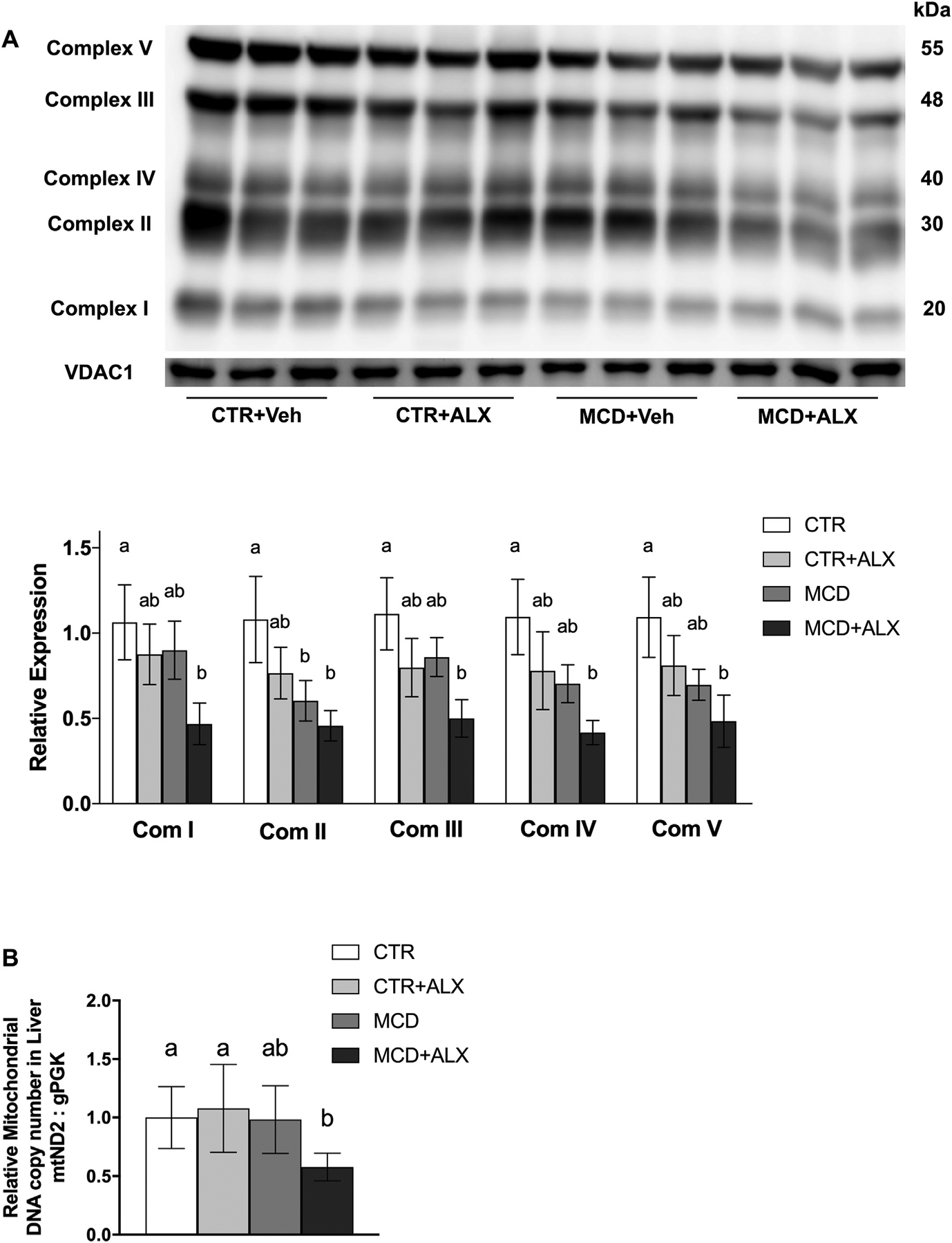
Alloxazine exacerbates mitochondrial dysfunction in MCD-diet-fed mice.Western blot of mitochondrial oxidative phosphorylation complexes I, II, III, IV, V, and VDAC1 measured in liver. Complexes were normalized to VDAC1, and VDAC1 was normalized to total lane protein (A). Relative hepatic mitochondrial DNA copy number determined by ND2 relative abundance (B). White bars represent CTR+Veh, light gray bars CTR+ALX, dark gray bars represent MCD+Veh, and dark bars with pattern represent MCD+ALX. Different lowercase letters denote a significant difference (P<.05, n=3). mND2, mitochondrially encoded NADH dehydrogenase 2, nPGK, nuclear encoded phosphoglycerate kinase, VDAC1, voltage-dependent anion channel 1. All data are presented as mean ± standard deviation.
4. Discussion
The MCD diet has been used for decades as a model of hepatic fibrosis and hepatocellular carcinoma [36]. However, the precise etiology of these conditions is not known. We recognized a deficit in the literature regarding the impact of MCD feeding on hepatic nucleotide metabolism. We hypothesized that impaired one-carbon metabolism would reduce hepatic nucleotides that depend on one-carbon metabolism for de novo synthesis. Indeed, eliminating the dietary methyl-donors methionine and choline reduced hepatic purine and thymidine content despite compensatory up-regulation of MTHFD1L, MTHFS and TYMS enzymes, which direct flux of folate-conjugated one-carbon units towards nucleotide biosynthesis. These findings indicate that de novo purine and thymidine synthesis and the respective salvage pathways are unable to maintain the tissue content of these metabolites. Purine and thymidine nucleotides are crucial in modulating the inflammatory response, both for proliferation and signaling, to promote tissue repair during tissue damage [16,17]. All of the adenosine receptors were expressed in the liver, and the MCD diet reduced expression of the A1 and A2B receptors, indicating that expression of these receptors is susceptible to changes when one-carbon metabolism in disrupted. To our knowledge, this is the first study that has attempted to probe purine signaling in the context of MCD-diet-induced liver damage.
A great deal of research has contributed to the understanding of the role of folate-mediated one-carbon metabolism for regulating the cell cycle with implications for mitochondrial function, cell survival, apoptosis and cancer [37–40]. It was not expected that alloxazine would disrupt folate metabolism and reduce formate content in MCD-fed mice, and we are unable to fully explain the increase in total hepatic folates. However, we have ruled out respective changes in food intake (Supplementary Fig. S2) and body weight gain since neither was significantly different. Structurally, alloxazine is very similar to the isoalloxazine portion of flavin molecules and could feasibly interact with flavin binding sites in cells. An additional mechanism by which alloxazine may interfere with mitochondrial folate metabolism is through SLC25A32 (mitochondrial folate transporter) that is responsible for the import of both folate and flavin adenine dinucleotide into the mitochondria [41]. Impairment of SLC25A32 by alloxazine could also explain the changes we observed in hepatic folate metabolites in alloxazine-treated mice. We did not find any evidence that alloxazine interferes with flavoproteins by showing that alloxazine did not interfere with the erythrocyte glutathione reductase activation coefficient assay in mice (Supplementary Fig. S3). Moreover, we did not observe any influence of alloxazine on mitochondrial respiration in isolated mitochondria; it is unlikely that this mechanism is responsible for alterations in hepatic folate content in our mice (Supplementary Fig. S4). Since alloxazine has previously been demonstrated to be A2B receptor antagonist, the possibility that the A2B receptor influences folate metabolism exists and bears further examination.
Impaired respiration has been previously reported in MCD mice [42], and together, elevations in hepatic triglycerides, circulating amino acids, decreased ATP concentrations (Supplemental Table 1) and reduced mitochondrial complexes suggested compromised mitochondrial function. More recently, it has been shown that mitochondrial respiratory chain expression and function are dependent on SHMT2 activity [43]. Hepatic ATP content was also reduced in MCD mice when compared to CTR mice, which corroborates with reduced mitochondrial activity but also supports reduced capacity for de novo nucleotide synthesis. A decrease in cellular mitochondrial content would compromise the generation of one-carbon units from this organelle and lower nutrient oxidation capacity. Moreover, SHMT2 has recently been shown to have a critical role in oxidative phosphorylation and translation of mitochondrial complexes. Loss of SHMT2 in HEK293T cells was shown to impair translation and functional activity of mitochondrial complexes by inhibiting tRNA methylation by 5,10-methylene-THF [43]. Indeed, knocking out SHMT2 in mice is embryonically lethal due to defects in mitochondrial respiration [44]. Notably, the antifolate drugs 5-fluoruracil and methotrexate have been reported to cause hepatic lipid accumulation, but the mechanism has not been elucidated [45–47]. Our current results demonstrate that alloxazine treatment in the MCD model disrupts folate metabolites, significantly decreases hepatic mitochondrial content, increases lipid content and enhances inflammation. Taken together with the recent literature describing the role of one-carbon metabolism in mitochondrial biogenesis and the effect of antifolates on liver lipid accumulation, our results support the role of one-carbon metabolism influencing mitochondrial biogenesis in the pathogenesis of liver inflammation.
Purine receptors and the regulation of purinergic signaling are very complex and not well understood [48], no doubt due to the challenge of differentiating between intracellular vs. extracellular metabolite abundance and the large number of structurally analogous molecules. However, the balance of extracellular purine metabolites modulates immune function [49]. Recently, ATP has been shown to play a role as an extracellular signaling molecule, and the P2X4 receptor that responds to ATP has been implicated in hepatic fibrogenesis [50]. On the other hand, adenosine has been shown to have an immunosuppressive property through adenosine receptor signaling and subsequent IL-10 secretion [51]. The reduced hepatic ATP concentration in the MCD mice may be protective against fibrosis but likely contributes to apoptosis. After tissue damage, adenosine produced from degraded ATP or from the intracellular adenosine generated from one-carbon metabolism signals through adenosine receptors to reduce inflammation and promote tissue repair. In the MCD diet, there are increased demand for purine salvage and reduced capacity for de novo purine synthesis, and thus, purine signaling is impaired in this model and contributes to the enhanced liver damage observed.
Significant fibrosis was not observed in this study, most likely due to the relatively short feeding duration. However, we did observe significant lobular inflammation concurrently with a disruption in folate metabolism in the alloxazine-injected MCD mice. During high apoptotic burden, the response of overwhelmed macrophages and Kupffer cells is to signal immune cells from circulation to infiltrate the liver. In vivo, alloxazine greatly increased apoptosis and hepatic macrophage staining. Macrophages in MCD mice treated with alloxazine were directed to apoptotic foci and were larger in size, which suggest that these macrophages were more actively phagocytosing apoptotic cell remnants. We propose that impaired mitochondrial capacity increases apoptosis, which in turn overwhelms similarly compromised hepatic macrophages, and thus, peripheral immune cells are recruited to the liver.
In summary, disruption of one-carbon metabolism resulted in reduced hepatic purine and thymidine content. These changes induced hepatic inflammation and were accompanied by increased hepatic macrophage activation. The role of one-carbon metabolism in liver damage has not previously explored the impact of alterations in de novo nucleotide synthesis and has primarily focused on disruption of phospholipid metabolism. This study provides an alternative mechanism by which impaired one-carbon metabolism can influence mitochondrial function, hepatic TG content and liver damage. We also show that the previously described A2B receptor antagonist alloxazine turned out to disrupt hepatic folate metabolism, reduce thymidine content, decrease mitochondrial content, and increase apoptosis and macrophage phagocytic activity. Further research to elucidate the mechanisms that are responsible for the current observations is warranted.
Supplementary Material
Supplementary Figure S2 Food intake over time. Open triangles represent CTR+Veh, black squares represent CTR+ALX, open squares represent MCD+Veh, and black diamonds represent MCD+ALX. Different lowercase letters denote a significant difference (P <.05, n = 8). All values presented as mean ± standard deviation.
Supplementary Figure S3 Alloxazine does not affect flavin-dependent enzyme activity. Erythrocyte glutathione reductase activity (A). White bars represent CTR+Veh, white bars with pattern represent CTR+ALX, dark bars represent MCD+Veh, and dark bars with pattern represent MCD+ALX. Different lowercase letters denote a significant difference (P b .05, n = 3). All values presented as mean ± standard deviation.
Supplementary Figure S4 Alloxazine does not acutely inhibit respiration in isolated mouse mitochondria in vitro. (A and B) Respiration rates using substrates for complex I (glutamate and malate) and complex II (succinate). Respiration rates were reported as oxygen concentration per second, normalized to total mitochondrial protein. (B) Calculated respiratory control ratios for complex I and II respiration. All values presented as mean ± standard deviation.
Supplementary Figure S1 One-carbon metabolism. Ado, adenosine; ALDH1L2, aldehyde dehydrogenase 1L2; BHMT, betaine; homocysteine methyltransferase; DAG, diacylglycerol; DMG, dimethylglycine; DMGDH, dimethylglycine dehydrogenase; GCC, glycine cleavage complex; GNMT, glycine-N-methyltransferase; MATI/III, methionine adenosyltransferase; MS, methionine synthase; MTHFR, methyltetrahydrofolate reductase; 5-CH3THF, 5-methyltetrahydrofolate; 5,10-CH2THF, 5,10-methylenetetrahydrofolate; 10-CHO-THF, 10-formyltetrahydrofolate; PC, phosphatidylcholine; PEMT, phosphatidylethanolamine-N-methyltransferase; SAHH, S-adenosylhomocysteine hydrolase; SDH, sarcosine dehydrogenase.
Acknowledgments
The authors would like to thank Dr. Jesse Gregory III and Ms. Maria Ralat for measuring folates and Dr. Martha Field for kindly providing the antibody for methylenetetrahydrofolate synthetase. We would also like to thank Dr. John Brosnan, Dr. Margaret Brosnan and Theerawat Pongnopparat for measuring formate.
Financial support: University of Florida Institute of Food and Agricultural Sciences and the Department of Food Sciences and Human Nutrition and R01 DK112865 (C.E.Mat.).
Abbreviations:
- ATF4
activating transcription factor 4
- ALX
alloxazine
- CTR
control
- IL-10
interleukin-10
- MCD
methionine and choline deficient
- MTHFD1L
methylenetetrahydrofolate dehydrogenase 1 like
- MTHFD2
methylenetetrahydrofolate dehydrogenase 2
- NAFLD
nonalcoholic fatty liver disease
- NAD
nicotinamide adenine dinucleotide
- NASH
nonalcoholic steatohepatitis
- AdoHcy
S-adenosylhomocysteine
- AdoMet
S-adenosylmethionine
- RANKL
receptor activator of nuclear factor kappa-B ligand
- SHMT
serine hydroxymethyltransferase
- THF
tetrahydrofolate
- TYMS
thymidylate synthase
- Veh
vehicle
Footnotes
Disclosures: The authors have no conflict of interest to declare.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jnutbio.2020.108381.
References
- [1].Bellentani S The epidemiology of non-alcoholic fatty liver disease. Liver Int 2017; 37(Suppl. 1):81–4. [DOI] [PubMed] [Google Scholar]
- [2].Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 2011;140:124–31. [DOI] [PubMed] [Google Scholar]
- [3].Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol 2018;68:238–50. [DOI] [PubMed] [Google Scholar]
- [4].Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155:1828–37 [e2]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Walker AK. 1-Carbon cycle metabolites methylate their way to fatty liver. Trends in endocrinology and metabolism: TEM 2017;28:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mato JM, Alonso C, Noureddin M, Lu SC. Biomarkers and subtypes of deranged lipid metabolism in non-alcoholic fatty liver disease. World J Gastroenterol 2019; 25:3009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pogribny IP, James SJ, Beland FA. Molecular alterations in hepatocarcinogenesis induced by dietary methyl deficiency. Mol Nutr Food Res 2012;56:116–25. [DOI] [PubMed] [Google Scholar]
- [8].Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2011;8:35–44. [DOI] [PubMed] [Google Scholar]
- [9].Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol 2013;12:183–9. [PMC free article] [PubMed] [Google Scholar]
- [10].Cabrero C, Duce AM, Ortiz P, Alemany S, Mato JM. Specific loss of the high-molecular-weight form of S-adenosyl-L-methionine synthetase in human liver cirrhosis. Hepatology (Baltimore, Md) 1988;8:1530–4. [DOI] [PubMed] [Google Scholar]
- [11].Watkins SM, Zhu X, Zeisel SH. Phosphatidylethanolamine-N-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J Nutr 2003;133:3386–91. [DOI] [PubMed] [Google Scholar]
- [12].Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology (Baltimore, Md) 2008;47: 1191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hoffman DR, Cornatzer WE, Duerre JA. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can J Biochem 1979;57:56–65. [DOI] [PubMed] [Google Scholar]
- [14].Tibbetts AS, Appling DR. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr 2010;30:57–81. [DOI] [PubMed] [Google Scholar]
- [15].Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ohradanova-Repic A, Machacek C, Charvet C, Lager F, Le Roux D, Platzer R, et al. Extracellular purine metabolism is the switchboard of immunosuppressive macrophages and a novel target to treat diseases with macrophage imbalances. Front Immunol 2018;9:852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ. New insights into the metabolic and nutritional determinants of severe combined immunodeficiency. Rare Dis 2015;3:e1112479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J 2013;21:245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Csoka B, Koscso B, Toro G, Kokai E, Virag L, Nemeth ZH, et al. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 2014;63:850–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hasko G, Csoka B, Nemeth ZH, Vizi ES, Pacher P. A(2B) adenosine receptors in immunity and inflammation. Trends Immunol 2009;30:263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Johnston-Cox H, Eisenstein AS, Koupenova M, Carroll S, Ravid K. The macrophage A2B adenosine receptor regulates tissue insulin sensitivity. PloS one 2014;9: e98775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Song Z, Uriarte S, Sahoo R, Chen T, Barve S, Hill D, et al. S-adenosylmethionine (SAMe) modulates interleukin-10 and interleukin-6, but not TNF, production via the adenosine (A2) receptor. Biochim Biophys Acta 1743;2005:205–13. [DOI] [PubMed] [Google Scholar]
- [23].Ji XD, Jacobson KA. Use of the triazolotriazine [3H]ZM 241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Des Discov 1999;16:217–26. [PMC free article] [PubMed] [Google Scholar]
- [24].Brackett LE, Daly JW. Functional characterization of the A2b adenosine receptor in NIH 3T3 fibroblasts. Biochem Pharmacol 1994;47:801–14. [DOI] [PubMed] [Google Scholar]
- [25].Gusdon AM, Fernandez-Bueno GA, Wohlgemuth S, Fernandez J, Chen J, Mathews CE. Respiration and substrate transport rates as well as reactive oxygen species production distinguish mitochondria from brain and liver. BMC Biochem 2015;16: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jacobs RL, Stead LM, Devlin C, Tabas I, Brosnan ME, Brosnan JT, et al. Physiological regulation of phospholipid methylation alters plasma homocysteine in mice. J Biol Chem 2005;280:28299–305. [DOI] [PubMed] [Google Scholar]
- [27].Vester B, Rasmussen K. High performance liquid chromatography method for rapid and accurate determination of homocysteine in plasma and serum. Eur J Clin Chem Clin Biochem 1991;29:549–54. [DOI] [PubMed] [Google Scholar]
- [28].Fiechter G, Mayer HK. Characterization of amino acid profiles of culture media via pre-column 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate derivatization and ultra performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2011;879:1353–60. [DOI] [PubMed] [Google Scholar]
- [29].Bagley PJ, Selhub J. Analysis of folate form distribution by affinity followed by reversed-phase chromatography with electrical detection. Clin Chem 2000;46: 404–11. [PubMed] [Google Scholar]
- [30].Lamarre SG, MacMillan L, Morrow GP, Randell E, Pongnopparat T, Brosnan ME, et al. An isotope-dilution, GC-MS assay for formate and its application to human and animal metabolism. Amino Acids 2014;46:1885–91. [DOI] [PubMed] [Google Scholar]
- [31].da Silva RP, Leonard KA, Jacobs RL. Dietary creatine supplementation lowers hepatic triacylglycerol by increasing lipoprotein secretion in rats fed high-fat diet. J Nutr Biochem 2017;50:46–53. [DOI] [PubMed] [Google Scholar]
- [32].Tillotson JA, Baker EM. An enzymatic measurement of the riboflavin status in man. Am J Clin Nutr 1972;25:425–31. [DOI] [PubMed] [Google Scholar]
- [33].Lin R, Zhang J, Zhou L, Wang B. Altered function of monocytes/macrophages in patients with autoimmune hepatitis. Mol Med Rep 2016; 13:3874–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zheng H, Fu Y, Huang Y, Zheng X, Yu W, Wang W. mTOR signaling promotes foam cell formation and inhibits foam cell egress through suppressing the SIRT1 signaling pathway. Mol Med Rep 2017;16:3315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Celardo I, Lehmann S, Costa AC, Loh SH, Miguel Martins L. dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ 2017;24:638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stephenson K, Kennedy L, Hargrove L, Demieville J, Thomson J, Alpini G, et al. Updates on dietary models of nonalcoholic fatty liver disease: current studies and insights. Gene Expr 2018;18:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci 2014;39:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ormazabal A, Casado M, Molero-Luis M, Montoya J, Rahman S, Aylett SB, et al. Can folic acid have a role in mitochondrial disorders? Drug Discov Today 2015;20: 1349–54. [DOI] [PubMed] [Google Scholar]
- [39].Newman AC, Maddocks ODK. Serine and functional metabolites in cancer. Trends Cell Biol 2017;27:645–57. [DOI] [PubMed] [Google Scholar]
- [40].Newman AC, Maddocks ODK. One-carbon metabolism in cancer. Br J Cancer 2017; 116:1499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Spaan AN, Ijlst L, van Roermund CW, Wijburg FA, Wanders RJ, Waterham HR. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2005; 86:441–7. [DOI] [PubMed] [Google Scholar]
- [42].Romestaing C, Piquet Ma Fau-Letexier D, Letexier D Fau-Rey B, Rey B Fau-Mourier A, Mourier A Fau-Servais S, Servais S Fau-Belouze M, et al. Mitochondrial adaptations to steatohepatitis induced by a methionine- and choline-deficient diet [DOI] [PubMed] [Google Scholar]
- [43].Morscher RJ, Ducker GS, Li SH, Mayer JA, Gitai Z, Sperl W, et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018;554: 128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tani H, Ohnishi S, Shitara H, Mito T, Yamaguchi M, Yonekawa H, et al. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci Rep 2018;8:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sakthiswary R, Chan GYL, Koh ET, Leong KP, Thong BYH. Methotrexate-associated nonalcoholic fatty liver disease with transaminitis in rheumatoid arthritis. Scientific World Journal 2014;2014:823763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stankov MV, Panayotova-Dimitrova D, Leverkus M, Vondran FW, Bauerfeind R, Binz A, et al. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS 2012;26:1995–2006. [DOI] [PubMed] [Google Scholar]
- [47].da Silva RP, Kelly KB, Al Rajabi A, Jacobs RL. Novel insights on interactions between folate and lipid metabolism. Biofactors 2014;40:277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Burnstock G, Vaughn B, Robson SC. Purinergic signalling in the liver in health and disease. Purinergic Signal 2014;10:51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Faas MM, Saez T, de Vos P. Extracellular ATP and adenosine: the yin and yang in immune responses? Mol Aspects Med 2017;55:9–19. [DOI] [PubMed] [Google Scholar]
- [50].Le Guilcher C, Garcin I, Dellis O, Cauchois F, Tebbi A, Doignon I, et al. The P2X4 purinergic receptor regulates hepatic myofibroblast activation during liver fibrogenesis. J Hepatol 2018;69:644–53. [DOI] [PubMed] [Google Scholar]
- [51].Nemeth ZH, Lutz CS, Csoka B, Deitch EA, Leibovich SJ, Gause WC, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol 2005;175:8260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S2 Food intake over time. Open triangles represent CTR+Veh, black squares represent CTR+ALX, open squares represent MCD+Veh, and black diamonds represent MCD+ALX. Different lowercase letters denote a significant difference (P <.05, n = 8). All values presented as mean ± standard deviation.
Supplementary Figure S3 Alloxazine does not affect flavin-dependent enzyme activity. Erythrocyte glutathione reductase activity (A). White bars represent CTR+Veh, white bars with pattern represent CTR+ALX, dark bars represent MCD+Veh, and dark bars with pattern represent MCD+ALX. Different lowercase letters denote a significant difference (P b .05, n = 3). All values presented as mean ± standard deviation.
Supplementary Figure S4 Alloxazine does not acutely inhibit respiration in isolated mouse mitochondria in vitro. (A and B) Respiration rates using substrates for complex I (glutamate and malate) and complex II (succinate). Respiration rates were reported as oxygen concentration per second, normalized to total mitochondrial protein. (B) Calculated respiratory control ratios for complex I and II respiration. All values presented as mean ± standard deviation.
Supplementary Figure S1 One-carbon metabolism. Ado, adenosine; ALDH1L2, aldehyde dehydrogenase 1L2; BHMT, betaine; homocysteine methyltransferase; DAG, diacylglycerol; DMG, dimethylglycine; DMGDH, dimethylglycine dehydrogenase; GCC, glycine cleavage complex; GNMT, glycine-N-methyltransferase; MATI/III, methionine adenosyltransferase; MS, methionine synthase; MTHFR, methyltetrahydrofolate reductase; 5-CH3THF, 5-methyltetrahydrofolate; 5,10-CH2THF, 5,10-methylenetetrahydrofolate; 10-CHO-THF, 10-formyltetrahydrofolate; PC, phosphatidylcholine; PEMT, phosphatidylethanolamine-N-methyltransferase; SAHH, S-adenosylhomocysteine hydrolase; SDH, sarcosine dehydrogenase.