

Differential analysis (DA) on a label-free mass-spectrometry benchmark study shows that leading summarization strategies often fail compared with the peptide-based tool MSqRob, and, we provide deep insights in performance gaps of the former methods.
MSqRob, however, is computationally expensive and does not provide protein-summaries for downstream analysis and visualization.
Therefore, we propose MSqRobSum, which fits MSqRob's model in a two-stage approach, providing a fast modular framework for robust protein summarization and inference that outperforms leading summarization-based methods for DA.
Keywords: Biostatistics, label-free quantification, bioinformatics, mass spectrometry, bioinformatics software, differential expression, ridge regression, shotgun proteomics, summarization
Graphical Abstract

Highlights
In depth performance assessment of leading tools for differential protein abundance.
Novel fast modular framework MSqRobSum for robust protein summarization and inference.
MsqRobSum outperforms leading protein summarization-based tools.
MSqRobSum is on par with top-performing peptide based tool MSqRob.
Abstract
Label-Free Quantitative mass spectrometry based workflows for differential expression (DE) analysis of proteins impose important challenges on the data analysis because of peptide-specific effects and context dependent missingness of peptide intensities. Peptide-based workflows, like MSqRob, test for DE directly from peptide intensities and outperform summarization methods which first aggregate MS1 peptide intensities to protein intensities before DE analysis. However, these methods are computationally expensive, often hard to understand for the non-specialized end-user, and do not provide protein summaries, which are important for visualization or downstream processing. In this work, we therefore evaluate state-of-the-art summarization strategies using a benchmark spike-in dataset and discuss why and when these fail compared with the state-of-the-art peptide based model, MSqRob. Based on this evaluation, we propose a novel summarization strategy, MSqRobSum, which estimates MSqRob's model parameters in a two-stage procedure circumventing the drawbacks of peptide-based workflows. MSqRobSum maintains MSqRob's superior performance, while providing useful protein expression summaries for plotting and downstream analysis. Summarizing peptide to protein intensities considerably reduces the computational complexity, the memory footprint and the model complexity, and makes it easier to disseminate DE inferred on protein summaries. Moreover, MSqRobSum provides a highly modular analysis framework, which provides researchers with full flexibility to develop data analysis workflows tailored toward their specific applications.
Label-free quantitation (LFQ) mass spectrometry (MS) based workflows have become standard practice in quantitative proteomics (e.g. (1, 2)). This technology typically starts with protein extraction followed by an enzyme digestion step to produce peptides of a convenient length. The thus obtained peptide mixture is then analyzed in a mass spectrometer where intact peptide masses and their intensities are measured, resulting in an MS1 spectrum. In typical LFQ, the intensities of the thus recorded peaks are taken as proxies for peptide abundance. To identify the peaks observed in the MS1 spectrum, these peaks are first isolated in the instrument, and then subjected to fragmentation. Each of the resulting fragmentation (MS2) spectra is then used for peptide identification. In LFQ, each sample is separately analyzed on the mass spectrometer, and differential expression is obtained by comparing relative intensities between runs for the same identified peptide (1).
However, this workflow also induces challenging data analysis problems. First, different peptides from the same protein often have very distinct physio-chemical properties, leading to large differences in their MS1 intensities even though these peptides are of similar abundance (supplemental Fig. S1A1). Second, because of technological constraints not all peptides can be subjected to fragmentation. Indeed, only those peptides with the highest MS1 intensities within a certain retention window are typically selected for fragmentation (3). As a result, the identification in any given run depends not only on the abundance of that peptide, but also on the abundances of any co-eluting peptides. There can thus be context-depending missingness in a given run. Moreover, there are many other potential sources of (random or non-random) missingness, including peptide misidentification, ambiguous matching of MS1 peaks, and poor quality MS2 spectra (4). Hence, there is considerable variation in terms of the peptides that are identified in each of the different MS runs in an experiment. Taken together, the identification issue and the peptide specific effects on quantification have a severe impact on the downstream summarization of peptide intensities toward protein abundances (5).
Indeed, because of these issues, simple summarization methods such as the mean or median peptide intensity are known to give unreliable protein abundance estimates (5) and more advanced summarization strategies have therefore been proposed for LFQ data in the literature (6, 7, 8, 9). In Fig. 1A we show the performance of these different data analysis strategies on a benchmark dataset. Notably, we observe huge differences in performance between the different summarization strategies, which are driven by the absolute abundance, and any differences in this abundance, of a protein between conditions. Moreover, none of the summarization strategies outperforms the others across all conditions.
Fig. 1.

Comparison of current state-of-the-art tools for DE analysis of proteins. We compare one peptide based tool, MSqRob and four summarization based tools. Of these, Perseus and Differential Enrichment analysis of Proteomics data (DEP) with mixed imputation are both based on maxLFQ protein intensities. MSstats uses median polish summarized protein intensities, whereas Proteus uses high-flyers summarization. The data consists of E. Coli proteins spiked at four different concentrations (a, b, c, and d) in a human proteome. The plot in Panel A shows the performance of each method for the pairwise comparisons b-a, c-b, and d-c (True Positive Rate = E. Coli/(Total E. Coli); False Discovery Proportion = Human/(Human + E. Coli)). MSstats outperforms Proteus, DEP, and Perseus at higher fold changes, but drops in performance down to Perseus levels at the lowest fold change. Proteus outperforms Perseus at higher fold changes but is less performant at the lowest fold change. MSqRob always outperforms the other methods. The boxplots in panel B show estimated log2 fold changes of differentially (E. Coli) and non-differentially (human) expressed proteins in the a versus b comparison. The thick gray line indicates the real log2 fold change for the E. Coli proteins. Perseus has biased fold changes for the E. Coli proteins, but has more precise fold changes for human proteins than DEP and MSstats. MSqRob has more precise and more accurate fold changes than any other method.
To avoid these summarization issues, peptide-based models, such as MSqRob (10), allow testing for differentially expression (DE) of proteins directly from the observed peptide intensities. The result is that these methods uniformly outperform summarization-based methods ((10) and Fig. 1A). Indeed, by modeling peptide intensities directly, MSqRob naturally accounts for differences in peptide characteristics, and for differences in the number of identified peptides for a given protein in each sample, resulting in a bias reduction and a better uncertainty estimation on the fold change estimates. However, the MSqRob method also suffers from some drawbacks compared with summarization methods. MSqRob must introduce random sample effects to account for correlation between the peptide intensities for a given protein in the same sample. This makes data analysis computationally more demanding, renders appropriate degrees of freedom of the test statistics unavailable, and even approximating these is impossible because of imbalances in the peptides across samples. The use of random effects also makes it difficult to disseminate the method toward non-specialized end-users as the interpretation of the result becomes correspondingly more complex. Moreover, MSqRob does not readily provide protein summaries for each sample, which are important for end-users to explore and visualize the data, and for further processing in downstream applications.
We therefore here introduce a novel estimation strategy for MSqRob using a two-stage approach, which we call MSqRobSum. MSqRobSum provides robust protein level summaries that account for peptide specific effects, which are then further processed using robust ridge regression. Hence, MSqRobSum combines the advantage of MSqRob's robust inference framework with the benefits of summarization, which allows fast and modular data analysis workflows. In addition, these workflows benefit from the straightforward visualization and interpretation of results at the protein level that is offered by MSqRobSum. We illustrate the high performance of MSqRobSum on a spike-in dataset, explain why it surpasses existing state-of-the-art summarization-based tools for DE in LFQ MS-based quantitative proteomics, and apply it on two biological case studies.
EXPERIMENTAL PROCEDURES
We performed a comparison of current state-of-the-art software tools for DE analysis of proteins on a benchmark spike-in dataset. We compared one peptide-based tool, MSqRob and four summarization-based tools: Proteus, Perseus, MSstats, and Differential Enrichment analysis of Proteomics data (DEP). For all tools we aimed to use the default workflow as suggested by the respective documentation. We also introduce our own novel summarization strategy for DE analysis, MSqRobSum, which aims to maintain MSqRob's superior performance while also providing useful protein expression summaries.
Spike-in Data Set
The performance of MSqRobSum and other state-of-the-art software tools for differential expression analysis were benchmarked using a publicly available dataset (PRIDE identifier: PXD003881 (11)). E. Coli lysates were spiked at five different concentrations (3%, 4.5%, 6%, 7.5, and 9% wt/wt) in a stable human background (four replicates per treatment). The twenty resulting samples were run on an Orbitrap Fusion mass spectrometer. Raw data files were processed with MaxQuant (version 1.6.1.0, (12)) using default search settings unless otherwise noted. Spectra were searched against the UniProtKB/SwissProt human and E. Coli reference proteome databases (07/06/2018), concatenated with the default MaxQuant contaminant database. Carbamidomethylation of cysteine was set as a fixed modification, and oxidation of Methionine and acetylation of the protein N terminus were allowed as variable modifications. In silico cleavage was set to use trypsin/P, allowing two missed cleavages. Match between runs was also enabled using default settings. The resulting peptide-to-spectrum matches (PSMs) were filtered by MaxQuant at 1% False Discovery Rate (FDR). In all analyses, E. coli proteins are labeled as DE (true positives), and all human proteins as equally expressed (true negatives).
To benchmark performance and FDR control of these different quantification strategies, the False Discovery Proportion (FDP) and True Positive Rate (TPR) of a set of proteins returned by the method were calculated, with
and,
We define a set of significant DE proteins as the proteins with a p value lower then a certain threshold. The FDP is then the fraction of human proteins in the set of human and E. Coli proteins recovered, whereas the TPR is the fraction of all E. Coli proteins recovered.
Biological Data Sets
We also illustrate MSqRobSum on two biological case studies.
In the first experiment, tissue samples were collected from patients undergoing transurethral resection of bladder cancer. The dataset consists of LFQ MS data from four non-invasive (pTa stage) and four invasive tumor tissue samples (pT2+ stage) (ProteomeXchange identifier: PXD002170) and is henceforth referred to as the Latosinska dataset (13). The raw data files were processed with MaxQuant by The et al. (2019) (14).
In a second experiment, Ramond et al. (15) knocked out the arginine transporter gene, ArgP, in the pathogenic coccobacillus Francisella tularensis and they assessed how this affects the proteome. To this end, both wild-type and ArgP knockout mutants were grown in biological triplicate and each replicate was analyzed in technical triplicate with LFQ MS. The raw data files were processed with MaxQuant (ProteomeXchange identifier: PXD001584) (15). This dataset is henceforth referred to as the Francisella dataset.
Proteus Analysis
We performed the default workflow in the R package Proteus (0.2.9) starting from the PSM values as reported in the evidence.txt file in MaxQuant's output (16). Proteins that are only identified as contaminants or reversed sequences are removed from the data set. The intensities of PSMs in a given sample that can be assigned to the same peptide sequence are summed. Peptide intensities are summarized to protein intensities using the high-flyer method (6). Peptides were assigned to their leading razor protein. Protein intensities are normalized to the median, and median intensities in each sample are equal. Protein intensities are log2 transformed.
DE of proteins is analyzed in Proteus with empirical Bayes moderated t-tests using the bioconductor limma package (17). Note, that we surpress an index for protein in all our model specifications for notational convenience.
In the limma analysis the following protein-wise linear models are considered.
with yst the normalized log2-transformed protein intensity in sample s of treatment t, β0 the intercept, βttreatment the effect of spike-in condition t, and, ϵst the protein-wise random error terms, which are assumed to be normally distributed with mean 0 and variance σ2. The variances σ2 are estimated with empirical Bayes, which stabilizes the estimates by borrowing strength across proteins. Proteus corrects for multiple testing using the Benjamini-Hochberg FDR procedure.
Perseus Analysis
We performed a standard Perseus workflow starting from the MaxLFQ protein summaries calculated by MaxQuant. MaxLFQ protein summaries are normalized and summarized intensity values for each protein in each sample. We can summarize the maxLFQ method as follows. The median ratio of the common peptides from a protein in all pairwise sample comparisons is calculated. Non-linear least-squares regression on these ratios is used to define an optimal protein expression profile across samples. This profile is rescaled to match the total summed peptide intensities from this protein in all samples (7). MaxLFQ protein summaries, as reported in MaxQuant's proteinGroups.txt file were further assessed in Perseus version 1.6.0.7. Proteins that are only identified by a modification site, contaminants, and reversed sequences are removed from the data set. Protein-wise two-sample t-tests on the log2 transformed maxLFQ values are performed for all pairwise treatment combinations. Perseus corrects for multiple testing using the Benjamini-Hochberg FDR procedure.
MSstats Analysis
A standard MSstats (version 3.12 (9)) workflow starts from the peptide intensities reported in MaxQuant's evidence.txt file. Peptides with only one or two measurements across all samples, and peptides that occur in more than one protein are filtered out. When a peptide is measured multiple times in a sample, only the maximum intensity is kept. The log2 peptide intensities are median normalized and missing values are imputed using an Accelerated Failure Model (AFM). Peptide intensities are summarized to protein intensities using Tuckey's median polish algorithm (18). MSstats builds protein-wise linear models based on these protein summaries.
with yst the normalized log2-transformed protein intensity in sample s of treatment t, β0 the intercept, βttreatment the effect of spike-in condition t, and, ϵst the protein-wise random error terms, which are assumed to be normally distributed with mean 0 and variance σ2. The multiple testing problem is corrected using the Benjamini-Hochberg FDR procedure.
Differential Enrichment Analysis of Proteomics Data (DEP)
MaxLFQ values are analyzed with the standard workflow in the Bioconductor software package DEP version 1.2.0 (8). Proteins that are contaminants or that originate from reversed sequences are removed from the data set. Only proteins with no missing values in at least one treatment group are kept. The data are normalized using Variance Stabilizing Normalization (VSN) (19).
Missing values are imputed differently for proteins that are missing completely at random (MCAR), and proteins that are missing not at random (MNAR) (4). MCAR proteins are defined as proteins observed in at least one replicate for every condition, and these are imputed with k-nearest neighbors averaging. MNAR proteins are assumed to be missing under low abundance and are thus considered left-censored data. Proteins are labeled MNAR when completely missing in at least one condition and are imputed with a stochastic minimal value approach. In short, a value is drawn from a normal distribution centered around the first percentile of all observed protein expressions in the sample, and with a standard deviation estimated as the median protein-wise standard deviation.
DE of proteins is analyzed in DEP with empirical Bayes moderated t-tests using the bioconductor limma package (17), like the Proteus workflow. Multiple testing is corrected using an empirical FDR estimation approach as implemented in the R package fdrtool.
MSqRob Analysis
The data is preprocessed using the MSnBase R/Bioconductor package version 2.6.2 (20). The analysis is done using the summarized peptide intensities as reported in the peptides.txt file in MaxQuant's output. The spike-in data and the Latosinka data are normalized using VSN, as in the default DEP workflow. The Francisella data, however, are normalized using quantile normalization (21), like the MSqRob analysis described in the paper of Goeminne et al. (2016) (10). Proteins that are only identified by a modification site, contaminants, and reversed sequences are removed from the data set. To avoid ambiguity, peptide sequences attributed to both E. coli and human proteins are removed. Peptides that are only observed once across all samples are also removed. Finally, treatments in which a protein is only observed in one replicate are still included in the DE analysis for this protein.
MSqRob is a linear regression peptide-based mixed model.
We consider the protein-wise models.
with ytsp the normalized log2-transformed intensity of peptide p in sample s with treatment t, β0 the intercept, βttreatment the effect of spike-in condition t, βssample a random effect that corrects for the correlation in measured expression levels between the peptides from the same protein in samples (pseudo replication on the sample level), and, βppeptide the effect of peptide p. Again the error term ϵtsp is assumed to be normally distributed with mean 0 and variance σ2.
When only one peptide is measured for a protein in all samples, the model reduces to.
The parameters for treatment and peptide are tuned using penalised estimation by exploiting the link between random effects and ridge regression.
Variability in the parameter estimators is reduced by shrinkage toward zero when there are only few observations. This protects against overfitting and makes the estimators more stable and accurate. The influence of outliers is weighed down by M-estimation using Huber weights. The variance of the protein-wise random error terms ϵtsp are again estimated with limma's empirical Bayes variance estimator.
Multiple testing is corrected using the Benjamini-Hochberg FDR procedure.
MSqRobSum Analysis
MSqRob's mixed model can also be estimated through a two-stage regression analysis (22). Here we first summarize peptide intensities to the protein level and subsequently test for DE on these protein summaries.
The same preprocessing is used as for the MSqRob analysis described in section 2.6. In the first stage we aggregate all normalized peptide intensities of a protein using robust regression with M-estimation using Huber weights. We consider the protein-wise linear model:
With ysp the normalized log2-transformed intensity of peptide p in sample s and βppeptide the effect of peptide p. By encoding the peptide effect as a sum contrast, βssample can be interpreted as the mean intensity in sample s for this protein. The error term ϵsp is assumed to be normally distributed with mean 0 and variance σpeptide2.
In the second stage, we perform an MSqRob analysis on protein intensities with the reduced model.
With yts the summarized log2-transformed protein intensity in sample s of treatment t, β0 the intercept, and, βttreatment the effect of spike-in condition t. Again, the error term ϵts is assumed to be normally distributed with mean 0 and variance σ2. We correct for multiple testing using the Benjamini-Hochberg FDR procedure.
We can expect a drop in performance in MSqRobSum compared with MSqRob because we lose information on the measured intensities and introduce some random variation during summarization. We also do not take into account that the covariance matrix of the estimated sample estimates is highly dependent on the number of measured peptide intensities in the sample (22). However, we expect that the resulting impact on performance is minimal in practice.
Software
Data preprocessing, statistical analysis and figures were done using the R programming language version 3.5.1. All R code is open sourced for reproducibility (https://github.com/statOmics/MSqRobSumPaper). The MSqRob algorithm has been implemented in R previously (10). However, we re-implemented MSqRob in R, and extended it to also allow for our proposed two-stage parameter estimation strategy, MSqRobSum. Because we fit a mixed model for each protein separately, we could easily parallelize the computations, which greatly speeds up the MSqRob and MSqRobSum analysis. In a full MSqRob peptide-level analysis, we typically allow for twenty iterations in the M-estimation using Huber weights for robust estimation of the model parameters. However, the MSqRob protein-level analysis in MSqRobSum only does one iteration by default. This sufficiently robustifies against outliers while maintaining proper FDR-control. The robust summarization, MSqRob and MSqrobSum algorithms are implemented as a open source R package MSqRobSum (https://github.com/statOmics/MSqRobSum). The robust summarization algorithm is also ported to the combineFeatures function for summarization in the R bioconductor package MSnbase (20).
RESULTS
State-of-the-art methods and our novel MSqRob approach are all benchmarked using a dataset where an E. Coli proteome was spiked at five different concentrations in a human background. We first compare existing tools for DE analysis of LFQ based quantitative proteomics, and critically assess why the performance of summarization-based approaches breaks down. Next, we show that our novel summarization-based method, MSqRobSum, maintains the high performance of the peptide-level based approach MSqRob. We further illustrate that MSqRobSum unlocks MSqRob toward modular data analysis workflows and we explain how and why MSqRobSum improves upon competitive summarization-based approaches. We conclude this section by assessing MSqRobSum in two biological case studies.
Comparison Between Methods
In this section we compare four summarization-based methods (Proteus, MaxQuant-Perseus, DEP, and MSstats), and one peptide-based model (MSqRob).
The Proteus workflow corrects for missingness of peptides under low abundance by summarizing using the high-flyer method, which provide protein-level intensities by taking the mean intensity of the three most intense peptides (6, 16).
However, this method does not correct for peptide specific effects and removes information by only using the top three peptide intensities. This introduces variability and bias in the estimated protein summaries (supplemental Fig. S1B2). Indeed, the most abundant peptides typically differ between samples, leading to a low performance compared with all other methods (Fig. 1A).
The popular MaxQuant-Perseus workflow is based on MaxQuant's MaxLFQ summarization and subsequent statistical analysis with Perseus using t-tests (7). MaxLFQ corrects for peptide specific effects by looking at pair-wise abundance ratios of shared peptides between samples. However, the heuristics in MaxLFQ often removes considerable information, which leads to increased missingness and imprecise summaries (supplemental Fig. 1C3). Comparisons that involve low spike-in concentrations often have too few shared peptides between samples, i.e. less than two, and these ratios are unreliable for summarization. Even though MaxLFQ corrects for peptide species by calculating ratios for shared peptides, it still appears to produces biased fold change estimates. The use of t-tests also results in suboptimal analysis as their variance estimator only includes the information of the data for the samples that are involved in the comparison. The summarization method combined with the less efficient downstream analysis often results in a low performance compared with the other methods (Fig. 1).
The recent Differential Enrichment analysis of Proteomics data (DEP) software package greatly improved MaxLFQ based analysis by adopting a mixed imputation strategy for missing protein intensities that infers whether random missingness or missingness because of low abundance occurs (8). It also provides a more robust downstream DE analysis using protein-wise linear models combined with empirical Bayes statistics (through the limma package (17)). Hence, DEP produces both more accurate as well as more precise fold change estimates and vastly outperforms the Perseus analysis (Fig. 1).
MSstats (9) is another popular software suite for proteomics data analysis. Initially, MSstats performed peptide-based modeling using linear mixed models. However, recent releases adopt summarization-based workflows in which peptide intensities are first summarized to protein intensities, and linear modeling is then performed on the protein level. The default choice of summarization in MSstats is median polish, which corrects for peptide specific effects and is robust against outliers. Median polish is, however, unstable in the presence of too much missing data but this is alleviated in MSstats by imputing missing peptide intensities by default using an Accelerated Failure Model (AFM). However, unlike DEP's mixed imputation strategy, AFM assumes that all intensities are missing because of low abundance, thus neglecting to consider other sources of missingness. Indeed, it turns out that MSstats' performance increases when the imputation step is omitted, especially in comparisons with high spike-in concentrations (supplemental Fig. S2), indicating that AFM's assumptions are insufficient.
The median polish summarization in MSstats produces more accurate fold change estimates compared with MaxLFQ (Fig. 1B). However, whereas MSstats outperforms MaxLFQ based workflows at high fold changes (Fig. 1A, comparison b-a and supplemental Fig. S3A, comparisons c-a and d-a), its performance becomes increasingly worse in comparisons with low fold changes (Fig. 1A, comparisons c-b and d-c). This happens because the high fold change comparisons are achieved by a low concentration of spike-in proteins, with missingness predominantly caused by low abundance, whereas the low fold change comparisons contain a high concentration of spike-in proteins, with missingness originating from other sources. The missingness by low abundance assumption of MSstats is therefore much more likely to be violated for low fold change cases, leading to a suboptimal ranking and a breakdown of MSstats for these comparisons. In contrast, DEP, which also accounts for random missingness, does not breakdown for these comparisons.
It should be noted that DEP's default preprocessing includes more stringent filtering for dubious proteins and thus returns less proteins overall than MSstats, which renders the better performance of MSstats in comparisons involving concentration a (b-a, c-a, d-a) superficial. Indeed, when only considering common proteins, DEP shows higher sensitivity then MSstats (supplemental Fig. S3B).
MSqRob, finally, uses a peptide-based approach that provides robustness against outliers and overfitting by adopting M-estimation, ridge regression and a limma style empirical Bayes procedure for variance estimation (10). MSqRob thus derives unbiased fold change estimates with high precision and outperforms all summarization-based models (Fig. 1A). The increase in performance is even more apparent at low fold changes (Fig. 1A comparison c-b and d-c).
MSqRobSum Has Similar Overall Performance to MSqRob
In this section, we show that we can fit the MSqRob model in a two-stage approach, with minimal impact on performance.
In the first stage of MSqRobSum we summarize peptide intensities in a sample to protein intensities using robust regression. This summarization is precise, and more robust than both high-flyer and maxLFQ summarization (supplemental Fig. S1). In the second stage, MSqRobSum provides precise and unbiased fold change estimates, comparable to MSqRob (supplemental Fig. S4).
MSqRobSum has a similar performance to MSqRob for medium to highly differentially expressed proteins (Fig. 2, comparison b-a and supplemental Fig. S5, comparisons c-a, d-a, and d-b). Although the performance of MSqRobSum is lower than that of MSqRob for increasingly lower fold changes (Fig. 2 comparison c-b and d-c), it should be noted that all summarization methods suffer from a drop in performance at lower fold changes (Fig. 1).
Fig. 2.

Comparison of performance of MSqRob and MSqRobSum. We compare the performance of MSqRob and MSqRobSum. The data consists of E. Coli proteins spiked at four different concentrations (a, b, c, and d) in a human proteome. The plot shows the performance of each method for the pairwise comparisons b-a, c-b, and d-c (True Positive Rate = E. Coli/(Total E. Coli); False Discovery Proportion = Human/(Human + E. Coli)). The estimated 1% (circle) and 5% (triangle) FDR is controlled if it remains below 1 and 5% FDP, respectively (indicated by vertical gray lines). Performance of MSqRobSum is close to MSqRob in all comparisons, and MSqRobSum even outperforms MSqRob in the b-a comparison. The performance of MSqRobSum does decline compared with MSqRob at decreasing fold changes between treatments (e.g. c-b and d-c), but the FDR is controlled in all comparisons.
A major contributor to the performance drop of MSqRobSum is human protein Q9BZJ0, which has relatively low protein summaries for the samples in condition c because of outlying intensities of one peptide in all samples of condition c (supplemental Fig. S6). As a result, this protein receives a very low p value from the MSqRobSum analysis for comparison d-c and is thus returned as a false positive at 1% FDR (supplemental Fig. S7). The MSqRob analysis, however, explicitly models the variance at the peptide level and the between sample variability and correctly rejects this protein.
The FDR is controlled at the 1 and 5% level for both MSqRob and MSqRobSum across almost the whole range of fold changes in differential expression (Fig. 2), except in comparison c-a and d-a (supplemental Fig. S5). The loss of FDR control in the latter comparison occurs because overspiking (high spike-in concentrations) causes increased ion competition between the peptide molecules in the sample (23, 5). This in turn causes peptides with equal abundance in two samples to be less ionized in the sample with the higher total protein concentration, resulting in a lower measured intensity for those peptides in that sample.
This effect is clearly visible as the average estimated fold changes of the human proteins steadily decreases as the spiked-in E. Coli concentration increases (supplemental Fig. S8A). At higher spiked-in E. coli concentrations, more human proteins thus appear to be differentially downregulated, and these additional false positives artificially inflate the estimated FDR (supplemental Fig. S8B).
The rationale for switching to MSqRobSum instead of MSqRob is based on two issues with MSqRob. The first issue is that it is unclear which degrees of freedom should be used for the test with MSqRob. MSqRob uses the degrees of freedom of the variance at the peptide level (within sample variance), but these do not correspond to the degrees of freedom of the standard errors on the fold change estimates. Indeed, these standard errors include both the within sample variance and the between sample variance, and the correct degrees of freedom therefore vary between those of the within sample variance, and those that would be obtained for a tool that models the data at protein level. For unbalanced data, the correct degrees of freedom cannot be approximated and the results of MSqRob are thus bound to be too liberal.
The second issue is speed, as fitting the large mixed models in a peptide-level MSqRob workflow is computationally quite expensive. In contrast, the robust summarization in the first stage of MSqRobSum is a relatively cheap operation computationally. By switching to the two-stage approach in MSqRobSum, analysis time is reduced to less than a third of the MSqRob computation time (∼10 min versus ∼3 min). Parallelization of both methods maintains this speed difference, while decreasing processing time even further (∼3 min versus ∼1 min, supplemental Fig. S9).
Moreover, MSqRobSum also returns protein summaries that are useful for visualization and for downstream analyses, which are not available in MSqRob as it models the peptide intensities directly.
MSqRobSum Allows for a Modular Data Analysis Workflow
The MSqRobSum workflow consists of three steps: preprocessing, summarization with robust regression, and DE analysis with robust ridge regression. Because each step can have an important impact on the performance of the entire data analysis workflow, the decoupling of summarization and inference provides optimal flexibility to combine each of the MSqRobSum steps with other tools in modular workflows. To illustrate the usefulness of such modular workflows, we will start from the default Perseus workflow and we will show how each step in the MSqRobSum workflow ramps up the performance.
The default Perseus workflow consists of maxLFQ summarization combined with t-tests for statistical inference. Its performance is relatively low and the FDR is not controlled at either the 1% or the 5% level (Fig. 3). However, exploratory data analysis revealed a strong batch effect across the samples which is undocumented in the experimental design. Batch effects should be corrected for during the statistical analysis but if undocumented, it is not always obvious if and how samples are organized in batches (supplemental Fig. S10). Often, normalization strategies already sufficiently correct for these sample effects. We can thus improve the performance and FDR control of the Perseus analysis by preprocessing the maxLFQ summarized intensities with VSN (Fig. 3).
Fig. 3.

Improvements of DE analysis using a modular data analysis workflow. We show incremental improvements in DE analysis by incrementally changing components in the workflow. The data consists of E. Coli proteins spiked at four different concentrations (a, b, c, and d) in a human proteome. The plot shows the performance of each method for the pairwise comparisons b-a, c-b, and d-c (True Positive Rate = E. Coli/(Total E. Coli); False Discovery Proportion = Human/(Human + E. Coli)). The circle and triangle are at 1 and 5% FDR, respectively, as estimated by the method. Perseus default performs t-tests on maxLFQ protein summaries for DE analysis. However, its performance is low and FDR is not controlled. Adding VSN normalization to the protein summaries boosts the performance of the DE analysis (perseus vsn). This workflow is further improved by replacing conventional t-tests by MSqRobSum's inference step (MSqRobSum maxLFQ). Adopting DEP's mixed imputation scheme results in an additional gain in performance (MSqRobSum DEP), whereas the best results are obtained by replacing maxLFQ and mixed imputation with our robust summarization (MSqRobSum default).
The statistical inference in Perseus is based on t-tests, which are underpowered when dealing with more than two conditions and other more complex study designs. We can therefore further improve performance by modeling intensities with MSqRob's robust ridge regression approach, which allows for higher performance and good FDR control. Note, however, that FDR is not controlled in conditions c-a and d-a because of ion competition, as also highlighted above (supplemental Fig. S11).
MaxLFQ's summarization strategy, based on pairwise ratios between samples, is inefficient for samples with low concentrations, which leads to unstable summaries and/or missingness. DEP dealt with this through a context-dependent imputation strategy, which increases the power of the subsequent statistical inference (Fig. 3). At high protein concentrations, there is low missingness and the effect of imputation will be small (Fig. 3 comparison d-c).
With MSqRobSum, we correct for peptide-specific effects through a model-based robust summarization strategy which models the log-transformed peptide intensities directly through robust regression. This robust regression efficiently uses all available protein intensities and imputation such as used in MaxLFQ is therefore not required (supplemental Fig. S12). The full MSqRobSum workflow thus further boosts performance while maintaining good FDR control (Fig. 3). Moreover, this MsqRobSum workflow uniformly outperforms all other modular approaches.
Performance of MSqRobSum in Biological Data Sets
In this section we adopt MSqRobSum on two case studies. The Latosinka study that compares the proteome of non-invasive and invasive human bladder cancer tumors, and the Franciscella study that assesses the impact of knocking out the arginine transporter gene on the proteome of the pathogenic coccobacillus Francisella tularensis.
Fig. 4 compares the performance of MSqRob, MSqRobSum, MSstats and DEP in the Latosinka study. It illustrates that MSqRob is much more liberal and returns many more proteins at a fixed FDR level than the summarization based approaches. At 5% FDR MSqRob returns 45 DE proteins, whereas MSqRobSum and DEP return 4, and MSstats returns none. At 1% FDR, DEP returns 2 DE proteins and MSqRobSum returns none. However, MSqRobSum takes over at more liberal FDR levels. Also note that DEP performs a more stringent filtering, which results in a lower multiple testing burden. When only considering proteins that were analyzed with both tools, MSqRobSum returns more proteins over the entire FDR range (supplemental Fig. S13). This shows that the difference in DE proteins between MSqRobSum and DEP at low FDR levels is induced by the filtering strategy, and that MSqRobSum is the most liberal method among the summarization-based approaches.
Fig. 4.
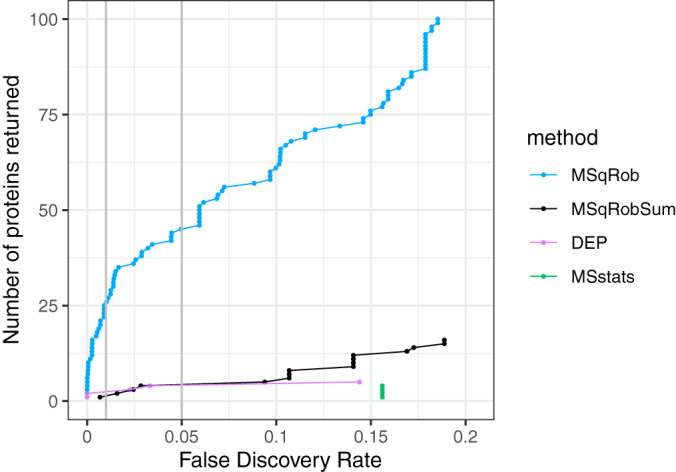
Comparison of different tools for DE analysis of proteins on the Latosinka dataset. We compare MSqRob, MSqRobSum, Differential Enrichment analysis of Proteomics data (DEP), and MSstats. The plot shows the number of proteins that are returned by each method at a certain FDR level. The two vertical gray lines indicate the 1 and 5% FDR level. MSqRob is the methods that the largest number of proteins as DE. The DEP analysis returns more proteins than MSqRobSum at 1% FDR, and an equal number of proteins at 5% FDR; but MSqRobSum always returns proteins at higher FDR levels. MSstats has the lowest sensitivity.
When we further dissect the differences between MSqRob and MSqRobSum (supplemental Fig. S14), both methods show similar fold change (FC) estimates (gray dots panel A), similar t test statistics (gray dots panel B), and similar standard errors on the fold change estimates (gray dots panel D) for the bulk of the proteins. For a small set of proteins, indicated by dots close to the x or y axis, very distinct estimates are obtained. The degrees of freedom, however, are generally much larger for MSqRob than for MSqRobSum for many proteins (gray dots in panel C). When we focus on the 45 DE proteins flagged by MSqRob, we can identify several reasons why more DE proteins are reported than with MSqRobSum. (1) The t test of MSqRob cannot correctly account for the degrees of freedom (DF) of the standard error on the FC estimate, which consists of the within and between sample error. Its DF is therefore solely based on the DF of the within sample variance estimator, which is vastly overestimated for proteins with many peptides. When the MSqRobSum analysis is run with the MSqRob DF, the number of DE proteins rises from 4 to 15 at 5% FDR (full red and blue dots in supplemental Fig. S14). (2) The t test result is larger for those proteins that are only significant for MSqRob. This is mainly because of a lower standard error for MSqRob (empty dots in supplemental Fig. S14B and S14D). Note that the estimator for the between sample variance can be expected to have a large uncertainty in experiments with few samples and that it will therefore be often underestimated by random chance (blue dots in supplemental Fig. S14B and S14D). For such proteins, the statistical test of MSqRob almost acts as if all observed peptide-level data are independent, which might lead to an underestimation of the standard error on the estimated FC estimator. (3) For a few proteins, the shrinkage of the FC estimator is also higher in MSqRobSum, leading to a more conservative fold change estimate (supplemental Fig. S14A). Issues (1) and (2) thus indicate that MSqRob can be expected to produce too liberal protein lists in experiments with small sample sizes.
Like the paper of Goeminne et al. (2016) (10), we also evaluate the performance of the method in the Francisella dataset of Ramond et al. (2015) (15). In Fig. 5, we observe that the Ramond et al. analysis flags many DE proteins. Most of these DE proteins, however, have fold changes close to zero and are downregulated in the wild type as compared with the knockout (supplemental Fig. S15). This is counter intuitive because the knockout induces arginine deficiency, which can be expected to affect many proteins negatively rather than positively. Goeminne et al. (2016) argue that the Ramond et al. (2015) analysis treats all technical repeats as biological repeats because Perseus cannot account for technical replicates. Hence, the Perseus analysis can be expected to produce too liberal results and reports many DE proteins with near-zero FC estimates. Goeminne et al. (2016) also showed that the proteins that were discovered by Ramond et al. (2015) and not by MSqRob did not bear strong evidence for differential expression between WT and mutant. The MSqRob and the MSqRobSum analysis report fewer DE proteins, and, as expected from biology, most of these DE proteins are up-regulated in wild type as compared with knockout (supplemental Fig. S15). Here again, MSqRobSum reports less proteins that MSqRob. However, in this experiment with more MS runs, we can observe that the major difference between both tools is driven by the difference in degrees of freedom. Indeed, the performance of MSqRob and MSqRobSum is much closer when they are both based on the asymptotic z-test (supplemental Fig. S16).
Fig. 5.
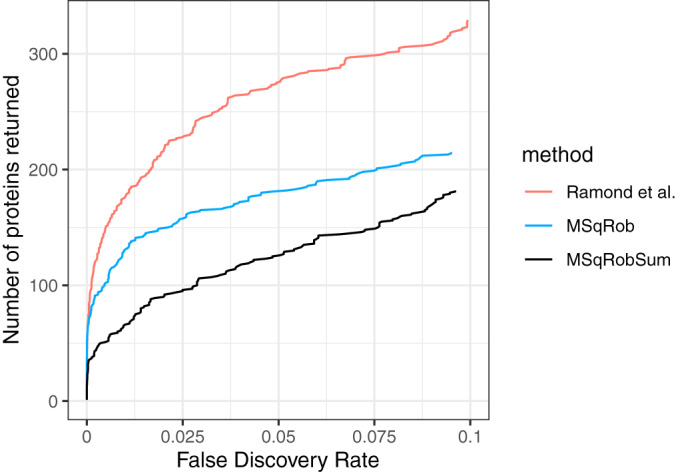
Comparison of MSqRob and MSqRobSum for DE analysis of proteins on the Francisella dataset. We compare the results of the original analysis by Ramond et al. (2015), MSqRob and MSqRobSum. Ramond et al. return the largest number of proteins as DE and MSqRobSum returns the lowest number of DE proteins.
DISCUSSION
In this work, we introduced MSqRobSum, a novel summarization-based method for LFQ which offers stable protein intensity estimation and high-performance protein DE analysis. We performed a benchmark study of different existing software implementations for summarization based LFQ methods and the state-of-the-art peptide-based model, MSqRob. MSqRob uses the information on all peptides during statistical inference and outperforms all summarization-based methods, which can only carry out inference on the protein summaries. However, MSqRob models are computationally quite expensive, can be hard to understand by experimentalists, include tests with unspecified degrees of freedom, and do not provide protein summaries for visualization and downstream processing. These MSqRob drawbacks are not present in summarization-based methods. Indeed, summarization is usually a relatively cheap operation and reduces the number of data points, whereas the obtained protein summaries allow easy visual inspection of the data. The use of protein summaries also reduces model complexity and enables statistical inference with t-statistics that have well-defined degrees of freedom. However, many existing summarization-based methods suffer a considerable drop in performance compared with MSqRob (Fig. 1). Our analysis shows that this drop in performance is dependent on issues with the summarization method used. Methods that do not take into account peptide specific effects, such as the high-flyer method in Proteus, show a clear drop in performance, whereas a method like MaxLFQ does consider peptide specific effects, but is based on heuristics and is not very data efficient. With MSqRobSum, we instead rely on robust regression for summarization, which allows correction for peptide-specific effects, effectively exploits all data in its model based summarization, and is robust against outliers. Taken together, the result is a considerable boost in performance in the DE analysis when compared with MaxLFQ.
We also show that preprocessing is crucial for the performance of a DE workflow. The first type of such preprocessing is normalization, which can have a large impact on DE analysis (Fig. 3). The second type of preprocessing is imputation of missing values, and this too can be beneficial (Fig. 3). However, because several different imputation methods exist, and because each of these applies to different sources of missingness, the best results are typically achieved when using a mixed imputation, where randomly missing values and values missing under low abundance are imputed differently (8). It should be noted, however, that the robust modeling in MSqRobSum can safely omit imputation altogether (supplemental Fig. S12).
Another crucial component in LFQ is the statistical model for discovering DE proteins. Perseus utilizes standard t-tests, but these are vastly underpowered compared with linear regression based models in more complex experimental designs. Moreover, MSqRob extends the linear model to robustify it against outliers and to improve uncertainty estimation (5). In the MSqRobSum workflow, we therefore use MSqRob's robust linear model approach instead of t-tests on the protein summaries. This considerably improves performance of the DE analysis, reaching a level comparable to MSqRob for a wide range of DE proteins (Fig. 3). And although MSqRob does show lower performance for increasingly lower fold changes in DE (Fig. 2), all summarization methods suffer from a drop in performance in these cases, often more severe than that of MSqRobSum (Fig. 1 and 2 comparisons c-b and d-c).
Moreover, we show in the analysis of the two biological datasets that MSqRob can be too liberal in datasets with few samples because of overestimation of the degrees of freedom and an underestimation of the between sample error.
Lastly, the robust summarization approach has the merit that the entire analysis workflow has become modular: the provided robust protein abundance estimates can be used for visualization and integration in other tools for DE, whereas MSqRob can now also start from protein summaries provided by other tools. This gives users considerable additional flexibility to develop modular workflows that are tailored toward their specific applications and renders MSqRob future proof when novel and more performant summarization procedures become available.
Supplementary Material
Acknowledgments
We thank the students of the Statistical Genomics course 2017–2018, Ghent University, who assisted us in assessing the initial implementation of MsqRobSum.
This article contains supplemental data.
Funding and additional information—L.M. is supported by European Union's Horizon 2020 Programme under Grant Agreement 823839 [H2020-INFRAIA-2018-1] and Research Foundation Flanders (FWO) [grant number G042518N].
Author contributions—A.S. and L.C. designed research; A.S. and L.C. performed research; A.S. and L.G. analyzed data; A.S., L.M., and L.C. wrote the paper.
Conflict of interest—Authors declare no competing interests.
Abbreviations—The abbreviations used are:
- DE
- differential expression
- LFQ
- label-free quantitation
- MS
- mass spectrometry
- DEP
- differential enrichment analysis of proteomics data
- PSM
- peptide-to-spectrum match
- FDR
- false discovery rate
- FDP
- false discovery proportion
- TPR
- true positive rate
- AFM
- accelerated failure model
- VSN
- variance stabilizing normalization
- MCAR
- missing completely at random
- MNAR
- missing not at random
- FC
- fold change
- DF
- degrees of freedom.
REFERENCES
- 1. Goeminne L. J. E., Gevaert K., and Clement L. (2018) Experimental design and data-analysis in label-free quantitative LC/MS proteomics: A tutorial with MSqRob. J. Proteomics 171, 23–36 [DOI] [PubMed] [Google Scholar]
- 2. Tebbe A., Klammer M., Sighart S., Schaab C., and Daub H. (2015) Systematic evaluation of label-free and super-SILAC quantification for proteome expression analysis. Rapid Commun. Mass Spectrom. 29, 795–801 [DOI] [PubMed] [Google Scholar]
- 3. Tu C., Li J., Sheng Q., Zhang M., and Qu J. (2014) Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data. J. Proteome Res. 13, 2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lazar C., Gatto L., Ferro M., Bruley C., and Burger T. (2016) Accounting for the multiple natures of missing values in label-free quantitative proteomics data sets to compare imputation strategies. J. Proteome Res. 15, 1116–1125 [DOI] [PubMed] [Google Scholar]
- 5. Goeminne L.J.E., Argentini A., Martens L., and Clement L. (2015) Summarization vs peptide-based models in label-free quantitative proteomics: performance, pitfalls, and data analysis guidelines. J. Proteome Res. 14, 2457–2465 [DOI] [PubMed] [Google Scholar]
- 6. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P. C., Geromanos S. J. (2005) Absolute quantification of proteins by Lcmse. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 7. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed Maxlfq. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X., Smits A. H., van Tilburg G. B., Ovaa H., Huber W., and Vermeulen M. (2018) Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protocols 13, 530–550 [DOI] [PubMed] [Google Scholar]
- 9. Choi M., Chang C.Y., Clough T., Broudy D., Killeen T., MacLean B., and Vitek O. (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 [DOI] [PubMed] [Google Scholar]
- 10. Goeminne L. J. E., Gevaert K., and Clement L. (2016) Peptide-level robust ridge regression improves estimation, sensitivity, and specificity in data-dependent quantitative label-free shotgun proteomics. Mol. Cell. Proteomics 15, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen X., Shen S., Li J., Hu Q., Nie L., Tu C., et al. (2018) Ionstar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. U.S.A. 115, E4767–E4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 13. Latosinska A., Vougas K., Makridakis M., Klein J., Mullen W., Abbas M., et al. (2015) Comparative analysis of label-free and 8-Plex iTRAQ approach for quantitative tissue proteomic analysis. PLOS ONE 10, e0137048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. The M., and Käll L. (2018) Focus on the spectra that matter by clustering of quantification data in shotgun proteomics. bioRxiv 488015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramond E., Gesbert G., Guerrera I.C., Chhuon C., Dupuis M., Rigard M., et al. (2015) Importance of host cell arginine uptake in Francisella phagosomal escape and ribosomal protein amounts. Mol. Cell. Proteomics 14, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gierlinski M., Gastaldello F., Cole C. Barton G. J. (2018) Proteus: an R package for downstream analysis of MaxQuant output. bioRxiv 41651 [Google Scholar]
- 17. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., et al. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holder R. L., Mosteller F., and Tukey J. W. (1979) Data analysis and regression. Appl. Statistics 28, 177 [Google Scholar]
- 19. von Heydebreck A., Huber W., Poustka A., Vingron M. (2002) Variance Stabilization and Robust Normalization for Microarray Gene Expression Data. In: Compstat (Härdle W., Rönz B., eds), pp. 623–628, Physica, Heidelberg [Google Scholar]
- 20. Gatto L., and Lilley K. S. (2011) MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28, 288–289 [DOI] [PubMed] [Google Scholar]
- 21. Bolstad B. M., Irizarry R. A., Astrand M., and Speed T. P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 [DOI] [PubMed] [Google Scholar]
- 22. Molenberghs G., and Verbeke G. (2000) Linear mixed models for longitudinal data. Springer Series in Statistics. Springer, New York [Google Scholar]
- 23. Milac T. I., Randolph T. W., Wang P. (2012) Analyzing LC-MS/MS data by spectral count and ion abundance: two case studies. Statistics and Its Interface 5, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.