

Data independent acquisition (DIA) is an attractive method for quantitative proteomics. However, most DIA methods require collecting exhaustive, sample-specific spectrum libraries with data dependent acquisition (DDA) to detect and quantify peptides. Studies of non-human organisms, splice junctions, sequence variants, or simply working with small sample yields can make developing spectrum libraries impractical. Here we illustrate a method to efficiently generate DIA-only chromatogram libraries and demonstrate best practices for how to acquire, queue, and validate DIA data without spectrum libraries.
Keywords: Data evaluation, label-free quantification, mass spectrometry, protein identification, quantification, data independent acquisition, DIA
Graphical Abstract

Highlights
Rapid DIA-only library building with gas-phase fractionation.
Recommended DIA acquisition strategies with staggered windows and forbidden zones.
Optimized DIA instrument settings for several Thermo Orbitrap instruments.
Data analysis tutorial using open source DIA software.
Abstract
Data independent acquisition (DIA) is an attractive alternative to standard shotgun proteomics methods for quantitative experiments. However, most DIA methods require collecting exhaustive, sample-specific spectrum libraries with data dependent acquisition (DDA) to detect and quantify peptides. In addition to working with non-human samples, studies of splice junctions, sequence variants, or simply working with small sample yields can make developing DDA-based spectrum libraries impractical. Here we illustrate how to acquire, queue, and validate DIA data without spectrum libraries, and provide a workflow to efficiently generate DIA-only chromatogram libraries using gas-phase fractionation (GPF). We present best-practice methods for collecting DIA data using Orbitrap-based instruments and develop an understanding for why DIA using an Orbitrap mass spectrometer should be approached differently than when using time-of-flight instruments. Finally, we discuss several methods for analyzing DIA data without libraries.
Shotgun proteomics (1) using liquid chromatography (LC) inline with tandem mass spectrometry (MS/MS) is enabling a revolution in the study of large-scale systems biology (2). Although the most common approach to large-scale proteomics relies on data-dependent acquisition (DDA) (3), data-independent acquisition (DIA) (4, 5) is emerging as a powerful tool for studying the proteome (6). DIA workflows attempt to acquire the same precursor isolation windows repeatedly across the elution profile of a peptide. Unlike parallel reaction monitoring (PRM) (7), which targets specific peptides, DIA targets wide, evenly spaced precursor isolation windows that are tiled across an m/z range of interest. Originally Venable et al. (4) envisioned DIA as a method to detect peptides without requiring a precursor signal. Consequently, the original methods focused on acquiring MS/MS with narrow precursor windows (10 m/z) at 3 Hz with an approximate 35 s cycle time. This approach allowed them to generally acquire at least one MS/MS spectrum within the elution profile of each peak, but quantitation could only be performed on precursor ions in interspersed MS spectra. Modern Orbitrap and ToF instruments can collect MS/MS at 10 Hz or faster and allow for PRM-like quantitation using MS/MS spectra.
DIA MS/MS differ from DDA MS/MS in several ways. First, DDA MS/MS are triggered by the presence of a Top-N intense MS1 ion, whereas DIA MS/MS are triggered systematically regardless of precursor ion intensities. This means that although many MS/MS contain either no peptides or signal that is so low that it is uninterpretable, DIA always contains at least one MS/MS near the apex of the peptide signal, and cannot suffer from the data-dependent ion selection problem of triggering a poor MS/MS from the shoulder of a precursor peak. Second, DDA fragmentation is performed with a charge-state optimized collision energy, whereas DIA fragmentation uses a fixed specified collision energy that may not fragment peptides the same way. Higher charged peptides typically fragment best with lower collision energies, there can be a tradeoff between over- and under-fragmenting classes of peptides. Because of this, it is important that DDA methods match DIA settings when building spectral libraries and use the same collision energy settings in both acquisition methods (8, 9). Third, DDA precursor selection and fragmentation theoretically contain only one precursor species per MS/MS, whereas DIA fragments all coeluting peptides within a specified precursor m/z range. Additionally, DDA analysis methods seldom consider multiple peptides per MS/MS, which commonly occur in congested regions of LC gradients (10) or with complicated or mixed proteomes such as ocean, soil, or gut metaproteomics studies (11).
Despite the wide appeal of DIA for quantitative proteomics, one drawback is that many approaches commonly require generating comprehensive DDA-based spectrum libraries (12) before interpreting any DIA data (9, 13, 14). Although this approach produces high-performance libraries with instrument-specific fragmentation and retention times (15), it does so at the expense of instrument time, sample, and significant effort offline fractionating that sample. However, several approaches have been developed to detect peptides directly from DIA experiments (16–18), and here we demonstrate how to use them to successfully acquire and analyze DIA experiments without spectrum libraries using a DIA-only workflow (19). This document is designed to build intuition to aid in decision making before starting a DIA experiment. Specifically, we focus on collecting DIA data, constructing DIA-only libraries, and analyzing DIA data with open-source software using Proteowizard (20), Skyline (21) and EncyclopeDIA (19). Although much of this intuition is transferable to ToF-based experiments, here we use DIA methods for Orbitraps as examples. Nevertheless, principles emphasized here, such as gas phase fractionation, are also valid on other instrumentations. Finally, it should be noted that other open-source (e.g. DIA-Umpire (17)) and commercial (e.g. ProteinLynx Global Server (16), Scaffold DIA or Spectronaut Pulsar) software for detecting peptides without spectrum libraries can also be used.
EXPERIMENTAL PROCEDURES
HeLa Cell Culture and Sample Preparation
We cultured HeLa S3 cervical cancer cells (ATCC, Manassas, Virginia) at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) with added l-glutamine, 10% fetal bovine serum (FBS), and 0.5% strep/penicillin. We washed cells three times with 4 °C phosphate-buffered saline (PBS) and frozen using liquid nitrogen. We lysed frozen cells in 9 m urea, 50 mm Tris (pH 8), 75 mm NaCl, and protease inhibitors (Roche, Basel, Switzerland, Complete-mini EDTA-free). After scraping, we probe sonicated cells for 2 × 15 s, then incubated them for 20 min on ice, followed by 10 min of centrifugation at 21,000 × g and 4 °C. We estimated supernatant protein content using bicinchoninic acid. We reduced proteins with 5 mm dithiothreitol for 30 min at 55 °C, alkylated with 10 mm iodoacetamide in the dark for 30 min at room temperature, and quenched with an additional 5 mm dithiothreitol for 15 min at room temperature. We diluted proteins to 1.8 m urea and digested with sequencing grade trypsin (Pierce, Waltham, Massachusetts) overnight at an estimated 1:50 enzyme to substrate ratio, before quenching with 10% trifluoroacetic acid to achieve approximately pH 2. We desalted the resulting peptides with 100 mg tC18 SepPak cartridges (Waters, Milford, Massachusetts) and dried them with vacuum centrifugation. We brought the peptides to 1 μg/3 μl in 0.1% formic acid and 2% acetonitrile prior to mass spectrometry acquisition.
Liquid Chromatography
We analyzed peptides with a Waters NanoAcquity UPLC coupled with a Thermo, Waltham, Massachusetts, Q-Exactive HF tandem mass spectrometer. We used an in-house pulled column created from 75 μm inner diameter fused silica capillary packed with 3 μm ReproSil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) to 300 mm, coupled with a Kasil fritted trap column created from 150 μm inner diameter that was packed with the same C18 beads to 25 mm. Solvent A was 0.1% formic acid in water, whereas solvent B was 0.1% formic acid in 98% acetonitrile. For each injection, we loaded ∼1 μg peptides and separated them using a 90-min gradient from 5 to 35% B, followed by a 40 min washing gradient.
Mass Spectrometry
We performed chromatogram library DIA experiments as described in Searle et al (19). Briefly, we acquired 8 chromatogram library acquisitions with 4 m/z DIA spectra (4 m/z precursor isolation windows at 30,000 resolution, AGC target 1e6, maximum inject time 55 ms, 27 NCE) using a staggered (also referred to as overlapping) window pattern from narrow mass ranges using window placements optimized by Skyline (i.e. 398.43–502.48, 498.48–602.52, 598.52–702.57, 698.57–802.61, 798.61–902.66, 898.6–1002.70 m/z, 998.61–1102.75, and 1098.75–1202.80 m/z). Two precursor spectra, a wide spectrum (400–1600 m/z at 60,000 resolution) and a narrow spectrum matching the range (i.e. 390–510, 490–610, 590–710, 690–810, 790–910, 890–1010, 990–1110, and 1090–1210 m/z) using an AGC target of 3e6 and a maximum inject time of 100 ms were interspersed every 25 MS/MS spectra. See supplementary Table S1 for the actual windowing schemes.
For single-injection runs, the Thermo Q-Exactive HF was configured to acquire either 25 × 16 m/z (covering 500–900 m/z), 25 × 20 m/z (400–900 m/z), 25 × 24 m/z (400–1000 m/z), 25 × 28 m/z (400–1100 m/z), or 25 × 32 m/z (400–1200 m/z) precursor isolation window DIA spectra (30,000 resolution, AGC target 1e6, maximum inject time 55 ms, 27 NCE) using a staggered window pattern using window placements optimized by Skyline. Precursor spectra (target range ± 15 m/z at 30,000 resolution, AGC target 3e6, maximum inject time 100 ms) were interspersed every 25 MS/MS spectra. Again, see supplementary Table S1 for the actual windowing scheme.
Experimental Design and Statistical Rationale
This study is focused on interpreting results from different DIA acquisition strategies, rather than any biological interpretation. As such, technical triplicate experiments of the single-injection runs were acquired from the same HeLa lysate using a block-based data acquisition strategy.
DIA Data Analysis
The library-free data analysis workflow used in this manuscript is fully described as a step-by-step walkthrough in supplementary Note 1. Briefly, we demultiplexed (22) staggered DIA data with 10 ppm accuracy in ProteoWizard (20) (version 3.0.1908). We used EncyclopeDIA (version 0.9.0) to search the resulting demultiplexed mzMLs. Walnut, a reimplementation of the PECAN FASTA search engine in EncyclopeDIA, was configured to search reviewed human proteins from Uniprot Swissprot downloaded December 13, 2019 (20379 total entries), with the default settings: fixed cysteine carbamidomethylation, 10 ppm precursor and fragment tolerances, using Y ions, and assuming full tryptic digestion with up to 1 missed cleavage. Library searching in EncyclopeDIA using both the Pan-Human Library (23) and chromatogram libraries was also configured with the same default settings, except both B and Y ions were used and library tolerances were also configured to 10 ppm. EncyclopeDIA search results were filtered to a 1% peptide-level using Percolator 3.1 and then filtered again to a 1% protein-level FDR assuming protein grouping parsimony.
RESULTS AND DISCUSSION
Designing A Balanced DIA Measurement Strategy
The intent of DIA is to measure as much of the proteome as possible, while still maintaining quantitative rigor. Balancing compromises between these two goals is critical for successful experiments. These compromises manifest as a result of three competing objectives: (a) maximize the total precursor range of targeted peptides, (b) maximize the number of points measured across every chromatographic peak, and (c) minimize the number of interfering peptides in each window.
The Precursor Range of Targeted Peptides
Although it is impractical to measure every possible tryptic peptide in a proteome, the total precursor range can be optimized to target most peptides. For example, although some peptides produce more intense signals below 400 m/z or above 900 m/z, we find that 93% of peptides in the Pan-Human library (23) can be observed within that range (Fig. 1). Assuming a fixed cycle time, narrowing our focus to this range allows us to collect narrower precursor isolation windows, and thus lowering signal interference for any given peptide. However, this same range only encapsulates 77% of the phosphopeptides in a human phosphopeptide library (24) of similar scope, suggesting that it is important to match the precursor range to the proteome of interest if specific peptides or modifications are targeted.
Fig. 1.
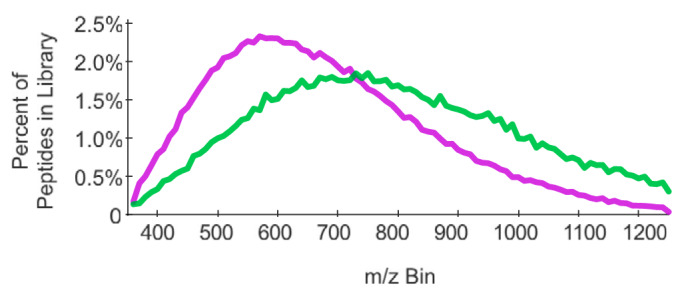
The distribution of peptide precursor m/z in the Pan-Human library (purple) and in the HeLa phosphopeptide library (green) differ significantly, indicating the importance of setting precursor isolation width ranges appropriately for each experiment. The bin size is 10 m/z.
The Number of Points Across Chromatographic Peaks
Because quantitative measurements are made at the fragment-level, it is imperative that there are sufficient fragment ion measurements for each precursor isolation window to appropriately represent the peptide peak shape. Following the conventional practice of quantitative mass spectrometry (25), most DIA tools use trapezoidal rule-based integration to measure peak area. Although generally robust, significant measurement errors can be caused simply by undersampling across the shape of the peak (Fig. 2A). Based on a model sampling at fixed intervals across Gaussian distributions (Fig. 2B), we estimate that restricting measurements to a minimum of nine points sufficiently limits bias caused by trapezoidal integration to below an average of 1% (Fig. 2C). In general, we recommend attempting to achieve a minimum average of 10 points to describe a peak to ensure that faster eluting peptides at the beginning and end of the chromatographic gradient are adequately measured.
Fig. 2.
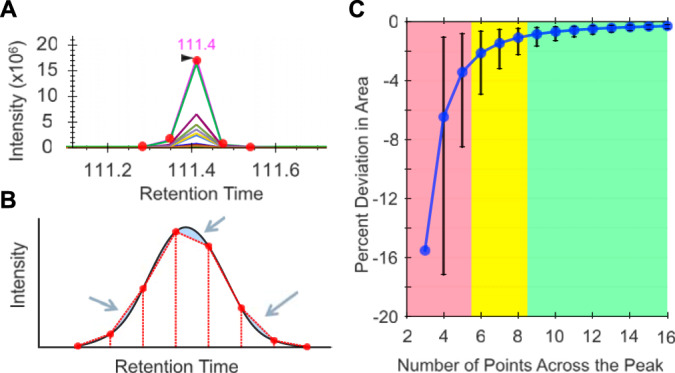
A, Trapezoidal quantitation can produce poor measurements with only five points (or fewer). B, Error (shaded in blue) in trapezoidal quantitation (red dashed lines) typically cancel out when measuring a Gaussian peak (black solid line) with eight to nine points across the peak. C, The average percent deviation with 95% error bars in actual/calculated area at different number of points across a Gaussian peak. The shaded regions indicate poorly quantified peptides with 5 or fewer transitions (red), moderately quantified peptides with between 6 and 8 transitions (yellow), and well quantified peptides with 9 or more transitions (green).
Several data acquisition variables can be adjusted to achieve this requirement: total precursor isolation range, scan rate, and peptide elution width. Average peak elution widths are dependent on the liquid chromatography setup and can be determined by looking at past runs (DIA or DDA) using a fixed gradient. The necessary cycle time is the ratio of the average peak width and the minimum number of points needed to describe a peak (typically 10):
The optimal scan rate is instrument specific. Combined with the estimated cycle time, it is possible to determine the relationship between total precursor range and the fixed precursor isolation width (or average width if using variable width windows):
This calculation assumes equal transmission of ions across the entire precursor isolation window, and no ions outside that window. It should be noted that no Q1 quadrupole produces a true square-wave transmission, and that some researchers increase the precursor isolation width to add small margins on either side that account for loss of sensitivity at each window edge. We find that this is typically not necessary for instruments that employ a hyperbolic segmented Q1 quadrupole (e.g. for Thermo instruments, the QE+, QE-HF, QE-HFX, and Fusion Lumos), as long as the window boundaries are placed in “forbidden zones” (discussed below and in supplementary Note 2). However, small margins may help with other Q1 designs.
The Cost of Interfering Peptides
At first it might appear logical to increase the total precursor range to be as large as possible to measure the greatest number of peptides. Although we previously observed that most tryptic peptides could be detected within the total precursor range of 400–900 m/z, there are some peptides for which the most intense charged ion falls outside that range, and others that are rarely (if ever) observed in that range. As the total precursor range increases, the precursor isolation width also increases, and with that the number of interfering peptides. We find that at some point, interference caused by wider precursor isolation widths outweighs any benefit gained from sampling more peptides. To demonstrate this, we analyzed the same tryptic peptide sample generated from a HeLa lysate using five different acquisition schemes that were scaled to keep cycle time constant (Fig. 3). We found that although the Pan-Human library (23) contains peptides from 400–1200 m/z, increasing the total precursor range from 500–900 m/z did not actually provide any meaningful increase in sensitivity when searching the resulting files with EncyclopeDIA, and indeed we found that the number of detected peptides starts to drop beyond 400–1000 m/z. However, when searching a sample-specific library, where retention times and fragmentation patterns are tuned specifically for the instrumentation and chromatographic setup used in this specific experiment, we found that we detected more peptides at wider windows, presumably because of increasing the precision for matching peptide signatures.
Fig. 3.
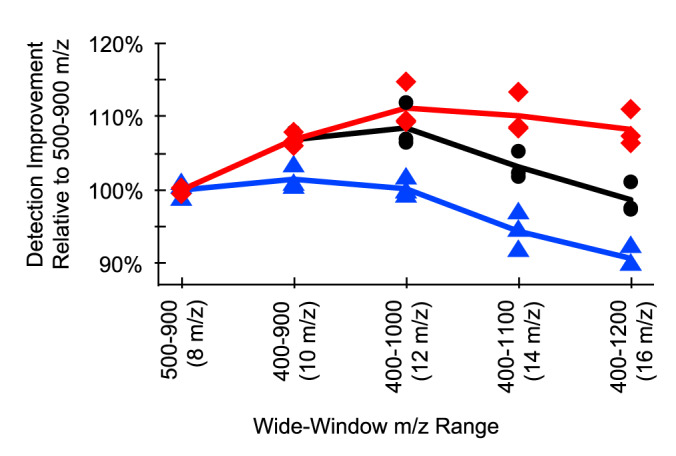
Although the percentage of new HeLa peptide detections increases with wider total precursor ranges, the number of detections in individual wide-window experiments is highest at 400–1000 m/z because of the tradeoff of increased overall m/z range with decreased window selectivity for any given window. This holds true for triplicate analyses searched with the Pan-Human library (blue triangle) and searched with two types of sample-specific libraries: a Pan-Human library-based chromatogram library (black circle), and a FASTA-based chromatogram library (red square) generated with Pecan.
Generating a Windowing Scheme
After deciding on a precursor range, the next step is to build a windowing scheme with an inclusion list. A more detailed review of various inclusion list approaches can be found elsewhere (9); here we will focus on general best practices for window width and placement. In addition to “normal” fixed-window DIA windowing schemes, there are several ways to improve peptide selectivity for the majority of peptides while maintaining a reasonable cycle time (Fig. 4). One approach is to use variable-width windows (26), which adjust the window-width based on the expected number of precursors in those windows. This approach has the advantage of producing narrow precursor isolation windows in regions of the mass range where there are the most peptide precursors. Conversely, windows in areas of the mass range with few precursors are often quite wide (as much as >50 m/z), and this can have a detrimental effect on the ability to differentiate coeluting precursors with similar peptide sequences.
Fig. 4.

Schematics for several types of windowing schemes, including (A) DDA, (B) PRM, (C) normal fixed-window DIA, (D) variable-window DIA, (E) staggered-window DIA, and (F) gas-phase fractionated DIA. Red bars indicate the isolation window for MS scans, whereas blue bars indicate the isolation window for MS/MS scans. Dashed lines indicate when an isolation window range is repeated.
Staggering precursor isolation windows from cycle to cycle (22) is a compressed sensing strategy (27) to achieve higher precursor specificity through undersampling (Fig. 5). Other applications of compressed sensing to achieve higher specificity include higher order multiplexing with MSX (28), and using a scanning quadrupole (29, 30). These windowing schemes take advantage of collecting peptide signals from multiple regions of the precursor space in the same MS/MS, and using other MS/MS nearby in time with different precursor space regions to computationally deconvolve signal specific to each individual region. Staggered window and MSX deconvolution are now a built-in feature in the freely available, open-source software tool, Proteowizard (20), while interpreting data from some scanning quadrupole windowing strategies is possible through Skyline (21).
Fig. 5.

Schematics for (A) staggered DIA windows, where (B) common peaks in the previous and next scan cycles can be used to computationally cut the precursor isolation window in half. Here the Time = T0 MS/MS scan acquired with a 600–624 m/z precursor isolation window is used as an example. Green ions, which correspond to peaks that can be found in the 588–612 m/z windows in the previous and next scans, can only come from peptides between 600–612 m/z. Similarly, blue ions can only come from peptides between 612–624 m/z. Red ions are found in both sets of previous and next scans and are fractionally allocated to both computationally demultiplexed MS/MS, whereas orange ions are not found in any neighboring scans and can be considered noise.
Most quadrupoles do not have perfectly rectangular isolation efficiency over the complete isolation window and DIA methods must account for the fact that transmission efficiency is lower at the window boundaries. The easiest way to do this is to add window margins (typically ± 0.5 m/z) to each precursor isolation window, and this works well for normal or variable width windows. However, these margins can significantly complicate the mathematics behind compressed sensing strategies, and coupling these approaches is not recommended.
Another strategy to account for non-rectangular quadrupole efficiency is to take advantage of peptide mass defects. Because peptides are all made of the same components (e.g. amino acids), they have an “avergine” elemental composition (31) and there are regions of m/z values (“forbidden zones”) where it is impossible to find a peptide precursor (32) (Fig. 6). Using an inclusion list with windows bordered by forbidden zones maximizes the transmission of precursor ions in the window range. By placing a window edge at one of these forbidden zones where no precursor can possibly exist, the quadrupole transmission edge effects are less pronounced. The forbidden zone width and sparsity changes across the m/z range, but is typically ± 0.1 m/z. This matches the isolation window transmission falloff on instruments that utilize a hyperbolic segmented Q1 quadrupole (33).
Fig. 6.

Frequency of precursor m/z in +2H and +3H peptides between 600 and 610 m/z in Uniprot Swissprot human tryptic peptides (blue) echoes the frequency of all precursors found in the Pan-Human library (red). Black circles indicate “forbidden zones” where no precursors are likely to exist, which make for excellent DIA precursor isolation window boundaries.
Practically, this entails shifting the width and edges of the windows off of the chosen integer value (for example, a 24 m/z window from 400 m/z to 424 m/z) so that the window boundaries where quadrupole ion transmission suffers fall in forbidden zones where no precursor can exist (e.g. 400.43 m/z to 424.44 m/z). Forbidden zones for +2H and +3H peptides can be expressed as:
where the optimal m/z increment is 1.00045475 and constant is 0.25 (34, 35). This formula is based on the same principles that motivate the 1.0005 m/z bin constant used in SEQUEST (36), and is common to Skyline and EncyclopeDIA. Both programs are capable of generating inclusion lists using these principles by using Skyline's Edit Isolation Scheme feature (21) and EncyclopeDIA's Window Scheme Wizard (19). It should be noted that the constant should be modified when analyzing samples enriched in PTMs, such as phosphorylation, because each phosphate introduces a different mass defect pattern (discussed in more detail in supplementary Note 2). In this case, phospho-enrichment shifts the constant to 0.18.
Constructing DIA-Only Libraries
Leveraging prior knowledge in the form of libraries is the most common approach for detecting peptides in DIA experiments (37–40). Most commonly, libraries are constructed from fractionated DDA data, at the cost of significant effort, sample quantity, and instrument time. Additionally, relying on DDA-based spectral libraries as prior knowledge for DIA-based experiments assumes that DDA MS/MS are reasonable representations of DIA MS/MS. Even if DDA-based spectral libraries are collected on the same instrumentation platform at the same time as the DIA measurements, that is not always the case. First, DIA co-fragments all peptides with the same collision energy, regardless of charge state. Typically, researchers must configure the instrument to fragment peptides assuming a specific charge state, and because DDA MS/MS are charge state optimized for collision energy, the fragmentation patterns may not match. Second, offline fractionation simplifies the matrix each individual peptide sees, relative to the unfractionated sample. Consequently, retention times can shift between the library and quantitative DIA injections because of missing on-column interactions between peptides (41). Finally, DDA spectral libraries from off-line fractionated samples do not result in MS/MS spectra that reflect possible co-fragmentation interferences that would occur in the original unfractionated sample, again because the matrix has been simplified. Regardless, this strategy of generating DDA-based libraries to accompany DIA acquisitions remains the most frequently used approach to analyzing DIA data.
Analyzing DIA Data Sets Without Spectrum Libraries
Circumstances often dictate if generating a new sample-specific DDA-based spectrum library is feasible for each experiment. Although human samples can be interpreted with the Pan-Human library (23), very few other species-specific public libraries exist that are designed for DIA analysis. Global libraries, such as NIST (42), MassIVE-KB (43), and PeptideAtlas (44) typically do not contain calibrated or indexed retention times (45) (iRTs) making reuse for DIA impractical. In addition to working with non-human samples, other research interests (such as the study of splice junctions, sequence variants, or simply working with small sample yields) can make developing DDA-based spectrum libraries impractical. An alternative strategy is to predict spectrum libraries from FASTA databases using machine learning (46, 47), although care must be taken to appropriately correct for false discoveries when searching such large libraries.
One way to address these disadvantages is to identify peptides directly from DIA experiments. Early on, DIA data was analyzed using typical database search engines (4, 48) such as SEQUEST (36) but new approaches take advantage of the repetitive MS/MS measurements in DIA. Two major classes of tools have emerged to detect and quantify peptides from DIA experiments. Spectrum-centric analysis tools attempt to demultiplex several peptide signals from the same MS/MS spectra (16–18), by time-aligning elution peaks for both fragment and precursor ions. Fragment ions that co-vary across retention time are likely to come from the same peptide, and matching precursor ions indicate the potential masses for that peptide. These time-aligned ions are converted into demultiplexed “pseudo” spectra that usually represent a single peptide and can be interpreted with any database searching engine. A powerful benefit for this approach is that it can leverage a wealth of downstream MS/MS software because the pseudo spectra effectively resemble DDA data. In contrast, peptide-centric analysis (49) looks for specific peptides across all spectra in a precursor isolation window. PECAN (18) (PEptide-Centric Analysis) is an implementation of this approach that attempts to detect peptide sequences in FASTA databases by scanning raw files across retention time for groups of fragment ions that could have resulted from those sequences. Rather than generating predicted fragmentation models (46, 47), PECAN scores target peptides by considering the frequency of observing sequence-specific fragment ions at random in a background FASTA database. Some regions of the retention time gradient produce more fragment ions than others, so PECAN performs the same task with a subset of decoys that represent how peptides might score by random chance, and subtracts this background score at each retention time point. A series of feature scores are calculated for high scoring peptide-retention time matches (in contrast to peptide-spectrum matches) and these features are aggregated and FDR corrected using Percolator (50). Walnut (19) is a reimplementation of PECAN that makes minor modifications to the original PECAN scoring algorithm to improve speed and memory consumption.
Building DIA-only Libraries With Gas-phase Fractionation
Another strategy is to use additional DIA injections to build DIA-only libraries. Libraries that are constructed with DIA data can act as better priors, particularly when the DIA data is collected on the exact experimental sample and on the exact instrument platform. Gas-phase fractionation (51) (GPF) was originally coupled with DIA in the PAcIFIC approach (48) to improve peptide detection in complex samples by injecting the same sample multiple times, where each injection focused on different precursor isolation ranges. When experiment-representative DIA-based libraries are generated from gas-phase fractionated, narrow-window DIA acquisitions of a pooled reference sample, they are called chromatogram libraries (19). We find that 6× fractions each covering 100 m/z windows provides parallel reaction monitoring (7) (PRM) quality data (2 m/z precursor isolation windows) for every peptide between 400–1000 m/z (Fig. 4F).
Preparing the chromatogram library sample requires skimming a small aliquot from a representative set of the experimental samples and pooling them together. Although spectrum libraries are typically generated by strong cation exchange or high-pH reverse-phase liquid chromatography coupled with DDA, a chromatogram library is generated by gas-phase fractionation coupled with DIA (see supplementary Note 2) (48). Separating the library sample by GPF rather than fractionation using chemical interactions preserves the sample matrix, granting more accurate library retention times and more representative fragmentation patterns. In addition, peptide fragmentation patterns are more similar when comparing DIA data to DIA libraries versus DDA libraries, due in part to differences in collision energy estimation assumptions made by the two approaches.
Tradeoffs of Chromatogram Libraries Over Spectral Libraries
GPF involves no additional sample preparation; instead, GPF injects the same sample multiple times, each time focusing the precursor isolation range on a fraction of the overall precursor range of interest. This approach has been shown to improve sensitivity in modern DIA workflows (18), but comes at the expense of requiring additional injections and sample. Any peptides present in only a few samples or present in low abundance in a subset of samples will be diluted below the level of detection, which may mean that the peptide will be missing in the chromatogram library. Although this may be, it is important to remember that most peptides of very low abundance in a pooled but narrow isolation window GP fractionated injection will not likely be detectable in a wide isolation window, unfractionated injection. Also, as not all peptides present in the pool are present in each individual sample, searches with chromatogram libraries must be rescored and FDR corrected as with DDA-based libraries.
For many quantitative experiments, adding an additional 6× GPF-DIA injections of a sample pool to a long queue of biological samples adds negligible work for substantial gains in the number of detected peptides. Additionally, because only 6 μg of pooled sample are required, it is possible to construct the pool by mixing the remaining volume left by the autosampler pickup after the single-injection DIA runs. Although this is not optimal in terms of run order (the GPF-DIA injections must be run after all of the single-injection DIA measurements are performed), it is extremely efficient at conserving sample material. However, in smaller experiments with few quantitative samples, these injections can be impractical. In these cases, we recommend using GPF-DIA to collect quantitative samples, similar to the experimental workflow presented in Ting et al. (18). For example, in an experiment with only 3 biological samples, it may make more sense to collect 2× GPF-DIA injections (e.g. 400–700 m/z and 700–1000 m/z) per sample (6 total runs), rather than 6× GPF-DIA injections of a pool plus single-injection DIA for each sample (9 total runs). Unlike with single-injection DDA, we find that there is little technical variation between single-injection DIA runs, and that these 2× GPF-DIA approaches can be preferable to performing technical replicates with DDA. In an extreme case for performing exhaustive detection work on a single biological sample (no quantitative comparisons), we find that PECAN analysis of 6 GPF-DIA injections (essentially constructing a chromatogram library for each sample) can perform comparably to 6× strong cation exchange or high-pH reverse-phase fractionated DDA injections with dramatically less sample material and preparation effort (supplementary Note 2) (41).
Recommended Data Acquisition Methods for Orbitrap Instruments
Rationale for Orbitrap Parameter Settings
Thermo Orbitrap instruments use parallel ion accumulation and ion analysis, where the Orbitrap analyzes ions for the current spectrum while the instrument accumulates ions for the next spectrum. Consequently, the time used for collecting the Orbitrap transient, which is dictated by the resolution setting, must be balanced with the time used for accumulating ions, which is dictated by the Automatic Gain Control (AGC) target and the maximum Ion Inject Time (max IIT). The AGC target setting describes the target ion population the instrument will accumulate before the MS/MS event. The max IIT setting is the maximum time the instrument will spend accumulating ions before the MS/MS event. These two parameters, AGC and max IIT, together define the time it takes to accumulate ions for each MS/MS event. Either ion accumulation stops because of hitting the AGC target, or ion accumulation stops because of reaching the max IIT.
With DIA MS/MS events, we want to optimize for collecting the greatest number of ions in the allotted time without encumbering space charge effects to increase the fragment-level quantification accuracy. Consequently, we set the AGC target higher than for typical DDA MS/MS events, which are optimized to just get enough peptide sequence ions to make a successful search engine match as fast as possible. As with DDA methods, we set the max IIT to roughly match the time it takes to collect an Orbitrap transient. Narrower precursor isolation windows (e.g. for building chromatogram libraries) require more time to collect enough ions for a successful MS/MS, and thus it makes sense to increase the resolution settings to match the required higher max IITs. In addition to increased mass accuracy, longer transients increase signal to noise, thus resulting in higher quality quantitative measurements at the cost of fewer MS/MS per duty cycle.
Adjusting these settings is important when using different precursor ranges and isolation window schemes, as these result in different populations of ions within the average isolation window. We typically recommend the 400–1000 m/z precursor range using Orbitrap mass spectrometers running at either 10 or 20 Hz as a good tradeoff between breadth and selectivity for most unenriched, tryptically digested proteomics experiments (Table I and II). Fig. 7 indicates where these specific settings are used for QE- and Fusion-class instruments method editors. Although QE-HFX and Fusion Lumos instruments can collect high-quality DIA MS/MS at 20 Hz with 8 m/z or larger windows, we find that these instruments are still benefit from acquiring at a slower 10 Hz with 4 m/z windows used by GPF-DIA because of the smaller population of ions in each scan (data not shown). Given these parameters, we provide examples of several general inclusion window strategies with which to cover a precursor range which we will refer to as normal, variable, and staggered (Table III). We provide more detailed windows for other window widths and GPF-DIA windows in supplementary Table S1, and other windowing schemes for experiments with PTMs such as phosphorylation can be created in EncyclopeDIA's Window Scheme Wizard.
Table I. Single injection DIA acquisition settings for quantitative proteomics with Thermo instruments.
| QE | QE+ | QE-HF | QE-HFX | Fusion | Lumos | |
|---|---|---|---|---|---|---|
| Est. Scan Rate | 10 Hz | 10 Hz | 10 Hz | 20 Hz | 10 Hz | 20 Hz |
| MS1 Scans | ||||||
| Range | 385–1015 m/z | 385–1015 m/z | 385–1015 m/z | 390–1010 m/z | 385–1015 m/z | 390–1010 m/z |
| Resolution | 35000 | 35000 | 60000 | 60000 | 60000 (Orbi) | 60000 (Orbi) |
| Max IIT (ms) | 60 | 60 | 60 | 60 | 60 | 60 |
| AGC Target | 1E+6 | 1E+6 | 1E+6 | 1E+6 | 4E+5 | 4E+5 |
| Data Type | Centroid | Centroid | Centroid | Centroid | Centroid | Centroid |
| MS2 Scans | ||||||
| Windowing Scheme | 24 m/z staggered | 24 m/z staggered | 24 m/z staggered | 8 m/z staggered | 24 m/z staggered | 8 m/z staggered |
| Resolution | 17500 | 17500 | 30000 | 15000 | 30000 (Orbi) | 15000 (Orbi) |
| Max IIT (ms) | 60 | 60 | 60 | 20 | 60 | 20 |
| AGC Target | 1e6 | 1e6 | 1e6 | 1e6 | 4e5 | 4e5 |
| Loop Count | 25 | 25 | 25 | 75 | 25 | 75 |
| Default Charge | 3 | 3 | 3 | 3 | 3 | 3 |
| (N)CE | 27 | 27 | 27 | 27 | 33 (HCD) | 33 (HCD) |
| Data Type | Centroid | Centroid | Centroid | Centroid | Centroid | Centroid |
Fig. 7.

Instrument settings from Tables I and II in two versions of the Thermo instrument method editor for both Q-Exactive class instruments (QE, QE+, QE-HF, and QE-HFX) and Fusion class instruments (Fusion, Fusion Lumos).
Table III. 400–1000 m/z precursor isolation window center m/zs for normal windows (a), variable width windows (b) and deltas (c), and overlapping windows (d, e).
| Scan | Normal Windows (A) | Variable Width Windows (B) | Variable Window Deltas (C) | Staggered Windows Cycle 1 (D) | Staggered Windows Cycle 2 (E) |
|---|---|---|---|---|---|
| 1 | 412.44 | 415.7 | 31.4 | 400.43 | 412.44 |
| 2 | 436.45 | 444.05 | 25.3 | 424.44 | 436.45 |
| 3 | 460.46 | 467.4 | 21.4 | 448.45 | 460.46 |
| 4 | 484.47 | 487.75 | 19.3 | 472.46 | 484.47 |
| 5 | 508.48 | 506.75 | 18.7 | 496.48 | 508.48 |
| 6 | 532.49 | 524.9 | 17.6 | 520.49 | 532.49 |
| 7 | 556.5 | 542.2 | 17 | 544.5 | 556.5 |
| 8 | 580.51 | 558.65 | 15.9 | 568.51 | 580.51 |
| 9 | 604.52 | 574.6 | 16 | 592.52 | 604.52 |
| 10 | 628.54 | 590.3 | 15.4 | 616.53 | 628.54 |
| 11 | 652.55 | 605.7 | 15.4 | 640.54 | 652.55 |
| 12 | 676.56 | 621.65 | 16.5 | 664.55 | 676.56 |
| 13 | 700.57 | 638.15 | 16.5 | 688.56 | 700.57 |
| 14 | 724.58 | 655.45 | 18.1 | 712.57 | 724.58 |
| 15 | 748.59 | 673.85 | 18.7 | 736.58 | 748.59 |
| 16 | 772.6 | 693.1 | 19.8 | 760.6 | 772.6 |
| 17 | 796.61 | 713.2 | 20.4 | 784.61 | 796.61 |
| 18 | 820.62 | 734.1 | 21.4 | 808.62 | 820.62 |
| 19 | 844.63 | 756.65 | 23.7 | 832.63 | 844.63 |
| 20 | 868.64 | 780.85 | 24.7 | 856.64 | 868.64 |
| 21 | 892.66 | 807.25 | 28.1 | 880.65 | 892.66 |
| 22 | 916.67 | 836.7 | 30.8 | 904.66 | 916.67 |
| 23 | 940.68 | 870.25 | 36.3 | 928.67 | 940.68 |
| 24 | 964.69 | 911.2 | 45.6 | 952.68 | 964.69 |
| 25 | 988.7 | 967 | 66 | 976.69 | 988.7 |
| 26 | 1000.7 |
Table II. 6× GPF-DIA acquisition settings for DIA-only library generation with Thermo instruments.
| QE | QE+ | QE-HF | QE-HFX | Fusion | Lumos | |
|---|---|---|---|---|---|---|
| Est. Scan Rate | 10 Hz | 10 Hz | 10 Hz | 10 Hz | 10 Hz | 10 Hz |
| MS1 Scans | ||||||
| Range | 395–505 m/z, 495–605 m/z, etc… | 395–505 m/z, 495–605 m/z, etc… | 395–505 m/z, 495–605 m/z, etc… | 395–505 m/z, 495–605 m/z, etc… | 395–505 m/z, 495–605 m/z, etc… | 395–505 m/z, 495–605 m/z, etc… |
| Resolution | 35000 | 35000 | 60000 | 60000 | 60000 (Orbi) | 60000 (Orbi) |
| Max IIT (ms) | 60 | 60 | 60 | 60 | 60 | 60 |
| AGC Target | 1E+6 | 1E+6 | 1E+6 | 1E+6 | 4E+5 | 4E+5 |
| Data Type | Centroid | Centroid | Centroid | Centroid | Centroid | Centroid |
| MS2 Scans | ||||||
| Windowing Scheme | 6x GPF-DIA w/4 m/z staggered | 6x GPF-DIA w/4 m/z staggered | 6x GPF-DIA w/4 m/z staggered | 6x GPF-DIA w/4 m/z staggered | 6x GPF-DIA w/4 m/z staggered | 6x GPF-DIA w/4 m/z staggered |
| Resolution | 17500 | 17500 | 30000 | 30000 | 30000 (Orbi) | 30000 (Orbi) |
| Max IIT (ms) | 60 | 60 | 60 | 60 | 60 | 60 |
| AGC Target | 1e6 | 1e6 | 1e6 | 1e6 | 4e5 | 4e5 |
| Loop Count | 25 | 25 | 25 | 25 | 25 | 25 |
| Default Charge | 3 | 3 | 3 | 3 | 3 | 3 |
| (N)CE | 27 | 27 | 27 | 27 | 33 (HCD) | 33 (HCD) |
| Data Type | Centroid | Centroid | Centroid | Centroid | Centroid | Centroid |
We find that because of the relatively long scan time required by Orbitraps, staggered windows produce the highest quality results in our hands. Because of filtering caused by the Fourier Transform analysis, Orbitraps usually produce MS/MS with relatively low noise, which makes staggered-window deconvolution more accurate. However, staggered windows may not work as well for time-of-flight (ToF) instruments because of their poor noise rejection. Instead, we recommend variable width windows with small margins as specified by Zhang et al. (26), to take advantage of the high scan speed ToF mass spectrometers typically provide. Either way, because of software limitations, it is not advisable to use an isolation scheme with both staggered windows and margins.
Precursor spectra are required by DDA to trigger MS/MS. In contrast, with DIA it is possible to detect high confidence peptides without either seeing any precursor signal (18), or even acquiring MS1 precursor spectra at all (37). However, with Orbitrap instruments, we recommend collecting periodic precursor spectra because they are collected by the instrument to aid in the AGC calculation, regardless of whether they are saved with the raw file. In addition, MS1 precursor spectra improve detection rates with EncyclopeDIA, which does not require seeing precursor signals for each peptide but can use those signals when present to calculate several scores including fitting the isotopic distribution. We recommend acquiring precursor spectra for only the range of the MS/MS precursor isolation windows, for gas-phase fractions. We find that by focusing on regions of the precursor space, we observe more sensitive precursor signals for the peptides of interest, similar to that found in windowed accurate mass and tag approaches such as BoxCar (52).
Queueing
Accurate measured retention times contribute powerful prior knowledge to chromatogram libraries, so it is crucial that the retention times measured in the chromatogram library can be precisely aligned to the wide-window, single-shot quantitative samples. As gas-phase fractions typically do not include the same peptides, unstable retention times between these injections complicate retention time alignment between the fractions in a way that cannot be corrected with the addition of iRT (45) peptides. Because gas-phase fractions subset peptides by mass-to-charge, the only peptides that are present in multiple fractions are different charge states of the same peptide (supplementary Note 2). Without the same peptides detected across gas-phase fractions, retention time alignment cannot easily be performed between them. Therefore, it is important with GPF that retention times between each fraction are as stable as possible. There are two strategies to ensure stable chromatographic retention times: column conditioning and library sandwich queueing.
Column conditioning simply means running several samples after equilibrating a new column. Retention time is sensitive to changes in column age, so it is best to first condition the column to a sample with similar matrix complexity as the experimental samples. As the column ages and is exposed to the sample matrix, retention times should stabilize. In practice, tracking retention time stabilization can be done by choosing a handful of endogenous peptides or spiking a synthetic set of peptides into the conditioning sample and tracking the retention times and peak shapes over each injection before queueing any experimental samples.
Second, the order in which the library and the quantitative samples are acquired can help to capture the average retention time in the GPF library. It is recommended that the chromatogram library pool should be run in the middle of the experimental sample queue, “sandwiched” on either side by the unpooled, single-shot quantitative samples (Fig. 8). Because the pooled sample is an empirical average of the unpooled, single-shot quantitative samples, running at least 6 of the single-shot quantitative samples before running the pooled library sample gives the most stable retention times. Although retention times may shift between the first acquisition and the last, the order in which peptides elute typically does not. Because retention time ordering stays constant with the same column, it is easy to precisely retention time align peptides between two runs on the same column with EncyclopeDIA, including samples that are acquired early in the column's lifetime. However, this process assumes that there are no retention time deviations between GPF-DIA library-building injections, which cannot easily be aligned together (supplementary Note 2). Consequently, we find it better to run the GPF-DIA injections in the middle of the acquisition queue after the column retention properties have stabilized.
Fig. 8.
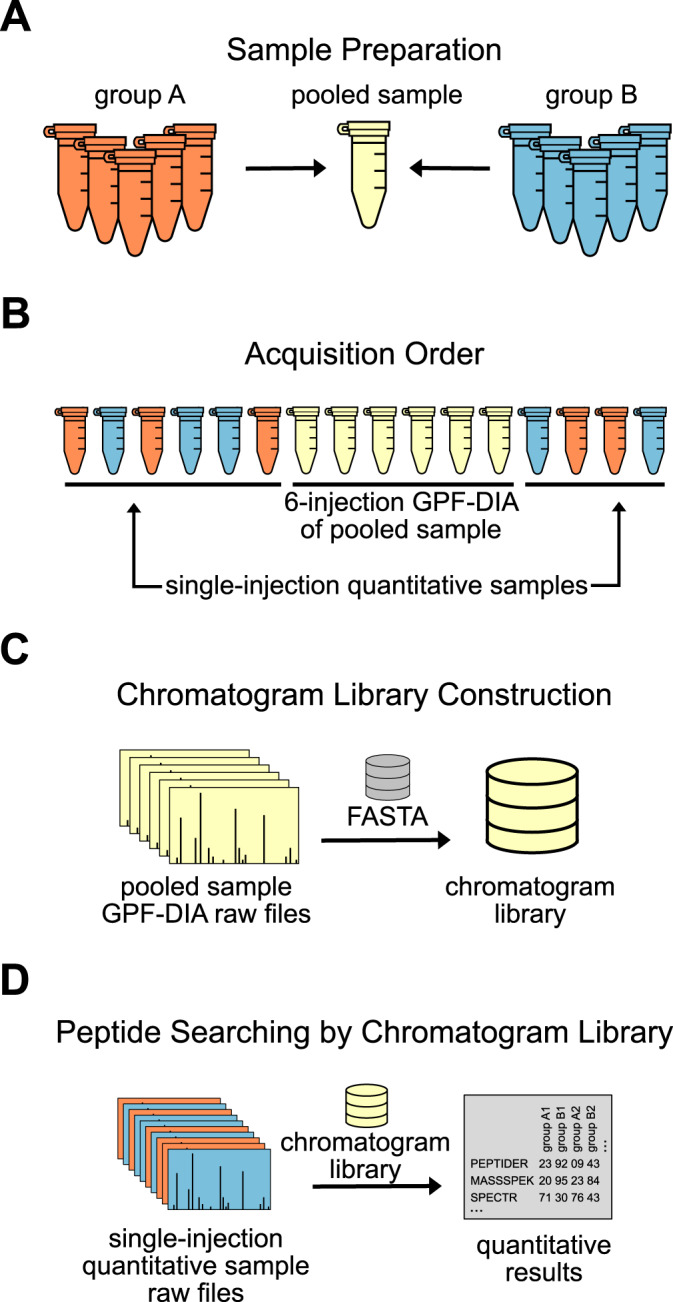
Recommended acquisition strategy for chromatogram library data collection. A, Orange and blue groups of biological samples are pooled, and B, run using GPF-DIA after at least 6 injections of the same matrix to ensure chromatographic consistency within the GPF runs. C, A chromatogram library is created from the pooled samples and D, used to interpret results from the single-shot DIA injections for each biological replicate.
Verifying Raw Data Quality
The raw data quality of DIA data can be assessed either qualitatively like shotgun or quantitatively like targeted proteomics before any peptide detection or quantification is performed. There are three ways to quickly assess data quality: comparing file size, observing total ion current (TIC) trace shape, and evaluating maximum ion inject time (IIT). The most basic assessment is noting file size across the runs. Quantitative single-shot samples should all be similarly sized files; likewise, each gas phase fraction should be roughly similar in file size. If any file is substantially lower in size, this may indicate a sample or acquisition issue that should be investigated further. Next, the TIC profile ideally makes a right-angled plateau with no obvious spikes in the gradient. Visual evaluation of the TIC can be performed by opening the acquisition file using vendor-specific software like XCalibur (Thermo) and Analyst (SCIEX), or with tools like SeeMS (ProteoWizard) and RawMeat (Vast Scientific). Finally, the IIT across a DIA experiment ideally is not affected by the precursor isolation windows. Plots of the IIT across retention time and isolation windows can be constructed with RawDiag (53) or EncyclopeDIA's built-in Raw File Browser function (supplementary Note 1).
Although IIT should ideally remain unchanged across retention time, it is common to observe that the average IIT across RT forms an upside-down U shape, where the maximum IIT is reached at the beginning and ends of a chromatographic gradient because less ions are eluting at those times. This is because more ions are eluting in the middle of the gradient, requiring less time to collect enough ions to trigger the MS/MS. However, at the beginning and ends of the gradient, there are fewer ions, and with fewer ions to fill the ion trap, the instrument spends more time accumulating ions and more likely triggers once reaching the maximum IIT.
Interpreting DIA Data
Data File Preparation
Acquisition files from the mass spectrometer are first converted using MSConvert (supplementary Note 1). For data acquired using the staggered window scheme discussed above, files require a computational demultiplexing step before they can be processed by data analysis softwares. Demultiplexing the files separates the staggered precursor isolation windows into their effective parts, for example the first few cycles in the staggered scheme described in Table III (D, E) are computationally demultiplexed into 12 m/z effective windows such that the converted output file contains isolation windows 400–412 m/z, 412–424 m/z, 424–436 m/z, etc. This requires only one additional parameter flag during the MSconvert step.
Constructing a Chromatogram Library
A chromatogram library is generated from the gas-phase fractionated, narrow-window acquisitions of the pooled reference sample. Peptide detections made in the GPF, narrow-window acquisitions of a pooled library sample are then the set of all possible peptide detections that can be made in the wide-window acquisitions. Although these acquisitions can be analyzed by searching against a spectral library, here we describe searching against a FASTA proteome with Walnut, a performance optimized implementation of the PECAN algorithm (supplementary Note 1).
Walnut detects peptides in the pooled reference sample acquisitions by searching a “target” proteome fasta in the context of a “background” proteome fasta. In global proteome experiments, the target and background proteome will be the same. In enriched or targeted proteome experiments, the target proteome should contain only proteins the researcher is interested in, whereas the background proteome should be all possible proteins in the sample. For example, an experiment investigating global protein abundances in yeast should use the yeast reference proteome as both the target and background; for an experiment focused on changes in mitochondrial respiration in yeast, the reference proteome would be yeast mitochondrial proteins whereas the background is the reference yeast proteome.
When searching for all peptides in a FASTA database, we suggest limiting searches narrowly to only peptides and proteins that are likely to be present in samples. Unlike with DDA where the search space is proportional to the number of acquired MS/MS, with peptide-centric DIA the search space is proportional to the number of peptides considered. Consequently, we do not recommend searching for PTMs, such as oxidation or phosphorylation, unless the experiment is likely enriched for oxidized or phosphorylated peptides. Similarly, we recommend only searching for ions that are likely to be present, either by limiting Walnut searches to Y-ions only when using beam-type fragmentation, or by limiting spectral predictions to only ions predicted to be above an appropriate intensity threshold.
FDR Control
When using Walnut to generate a chromatogram library, the gas phase fractions are searched against the target fasta and an equal number of decoys. The 1% peptide-level FDR-thresholded detections in each gas phase fraction are combined into a single chromatogram library, which is additionally controlled to a global 1% peptide-level FDR.
In a chromatogram library workflow as described here, the size of the library is sample-specific, as opposed to using repository-based libraries which may include proteins and peptides not expressed in the current experiment. Using a sample-specific library such as this decreases the statistical burden of false discovery rate (FDR) control by reducing the size, comprehensiveness, and heterogeneity of peptides and proteins represented in the library. However, even with the smaller statistical burden of a sample-specific chromatogram library, it is important to control for the various FDR control levels represented in a quantitative DIA experiment (54). Not only is the library FDR controlled, in this workflow each quantitative single-injection DIA file searched is itself additionally FDR controlled and each quantitative experiment is also globally FDR controlled at the 1% peptide- and the 1% protein-level.
Library searching assumes that all library entries are correct, and incorrect entries can propagate as true positives in target/decoy analysis (55). This concern is partially mitigated by multiple levels of filtering when building the library, and may benefit from additional filtering at a protein-level FDR. Further work is necessary to validate and improve FDR estimates for library searching in DIA experiments.
Refining Transitions for Quantification
EncyclopeDIA calculates a global interference score for transitions across all wide-window samples in the experiment and only uses the set of best scoring, interference-free transitions to integrate and sum for peptide quantification. Using this method for automated transition refinement, we see that as more interference-free fragments are required for quantification, the reproducibility of peptide quantification improves. We find that the peak area quantifications for peptides with just one or two interference-free transitions are extremely variable, with median coefficient of variations (CV) greater than 50% (Fig. 9). When we increase the number of interference-free transitions to a required three, the median peak area CV improves to 20%. Requiring five or more interference-free transitions improves the upper quartile peak area CV is also below 20%.
Fig. 9.
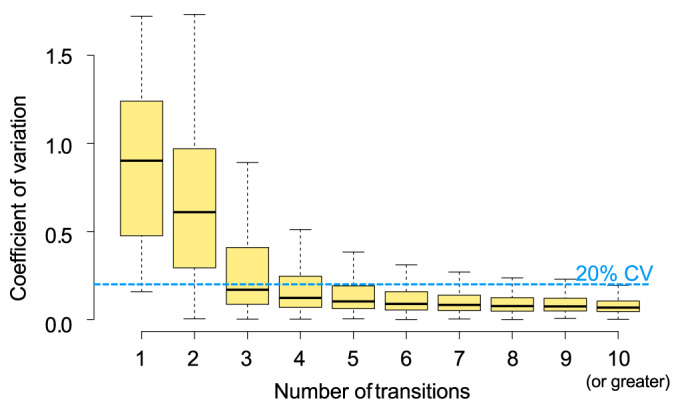
Coefficient of variation (CV) in TIC normalized peptide intensity based on the number of quantitative transitions used in technical triplicate 400–1000 m/z single-injection DIA experiments when searched with the PECAN-based chromatogram library. Boxes indicate the interquartile range around the median bold line with Tukey-style whiskers.
As with other targeted proteomics approaches, the chromatogram library approach integrates signal over the chromatographic area where a peptide is detected by placing integration boundaries on either side of a peak group. Background is subtracted from each peak group's signal area, and finally the background-subtracted peak areas are TIC normalized. For peptides, whose signal does not exceed the background noise, the peak area is reported as zero, as opposed to “NaN”. Use of zero in quantification as opposed to NaN reflects the systematic sampling of DIA; peptides that are not detected in every sample of a DIA experiment are not missing at random, but rather are not detected in every sample because they are either present but below the limit of quantification, present but below the limit of detection, or truly not present in the sample. Without further validation experiments (56), it is not possible to discern which scenario describes the failure to detect a peptide in a given sample.
Current Limitations
The Tradeoff of Mass and Temporal Separation
Many peptides generate the same fragment ions, either because of sequence similarity, or post-translational modifications (PTMs). With DDA, whereas it is rare that peptides share both fragment ions and the same precursor mass, certain circumstances, such as PTM-site localization require additional software and statistics for reliable peptide detection. With DIA this problem is both better and worse. One major advantage of DIA is that because of the regular fragment ion measurements, is possible to retention time-align fragment ions to temporally separate similar peptide species (57–59). However, unlike DDA, DIA lacks precursor selectivity and similar peptide sequences can fall in the same precursor isolation window: for example triply charged HSASQDGQDTIR (438.87 m/z) HSASQEGQDTIR (443.54 m/z) are both from Human Filaggrin and fall within the same 12 m/z precursor isolation window (436.45 m/z to 448.45 m/z). Here shared fragment ions may indicate the presence of either one or both peptides, and retention time or higher precursor isolation are the only methods to differentiate those outcomes. Although the narrow windows in GPF-DIA can somewhat mitigate the effect of shared fragment ions, care must be taken when reporting peptides with similar sequences to avoid double counting the same fragment ion evidence.
Longitudinal Studies and Batch Effects
Although a single pooled library sample is appropriate for most basic research purposes, there are some experimental scenarios where acquiring multiple chromatogram libraries may be best practice. For example, experimental designs with 3+ sample groups may benefit from a multiple chromatogram library strategy, in which each sample group is pooled for a sample type-specific chromatogram library. Large experiments spanning multiple columns, whether because of planned instrument maintenance or unplanned column changes because of column clogs, may benefit from treating each column as a separate set of experiments and calibrating to normalize between them (60). To note, a chromatogram library should be collected on each column used in an experiment. This requirement can be satisfied either by pooling the representative sample with enough volume to acquire chromatogram libraries on the same sample on multiple columns, or the experiment acquisition queue can be designed such that the chromatogram library for each column reflects the sample replicate blocks acquired on that column. Although it is possible to share chromatogram libraries between multiple columns, we typically do not recommend it because slight variations between columns can change peptide retention time ordering. This reordering may be correctable with peptide-by-peptide alignment using tools like DIAlignR (61), but cannot be corrected using standard global alignment.
CONCLUSIONS
DIA is a powerful technique for analyzing quantitative experiments, and GPF-DIA can be a useful, low-effort method for deep fractionation for the purposes of detecting peptides. Here we present “best practices” for DIA methods for Orbitrap mass spectrometers and the intuition needed to modify them to suit specific types of experiments. In addition, we discuss how to collect DIA-only libraries using GPF-DIA from a pool with an example developed as a tutorial in supplementary Note 1, and when it makes sense to deploy this approach. We also provide supplementary Note 2, which contains answers to frequently asked questions about this approach. This DIA-only method to library generation can make DIA an attractive alternative to DDA, even when the sample is limited or spectrum library building is impractical. Finally, library-free data analysis and workflows can be performed using open-source, GUI-based tools as discussed here, or with vendor-supported commercial software.
DATA AVAILABILITY
The raw data are available at MassIVE (MSV000084531) available at https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=b0c33f6caba04dd7a17a18e01e35071d.
Supplementary Material
This article contains supplementary data.
Funding and additional information—This work is supported in part by National Institutes of Health Grants F31 AG055257 (to L.K.P.), R01 GM133981 (to B.C.S.), P41 GM103533 (to M.J.M.), RF1 AG053959 (to M.J.M.), R01 GM103551 (to M.J.M.), and IARPA Proteos. B.C.S. is supported by the Translational Research Fellows Program (TRFP) from the Institute for Systems Biology.
Author contributions—L.K.P., M.J.M., and B.C.S. designed research; L.K.P. and B.C.S. performed research; L.K.P. and B.C.S. analyzed data; L.K.P., S.C.J., M.J.M., and B.C.S. wrote the paper; S.C.J. and B.C.S. contributed new reagents/analytic tools.
Conflict of interest—The authors have declared a conflict of interest. B.C.S. is a founder and shareholder in Proteome Software, which operates in the field of proteomics. The MacCoss Lab at the University of Washington has a sponsored research agreement with Thermo Fisher Scientific, the manufacturer of the instrumentation used in this research. Additionally, M.J.M. is a paid consultant for Thermo Fisher Scientific.
Abbreviations—The abbreviations used are:
- DDA
- data dependent acquisition
- DIA
- data independent acquisition
- GPF
- gas-phase fractionation
- PRM
- parallel reaction monitoring
- iRT
- indexed retention time
- AGC
- automatic gain control
- ToF
- time-of-flight
- TIC
- total ion current
- IIT
- ion inject time
- FDR
- false discovery rate
- CV
- coefficient of variation
- PTM
- post-translational modification.
REFERENCES
- 1. Wu C. C., and MacCoss M. J. (2002) Shotgun proteomics: tools for the analysis of complex biological systems. Curr. Opin. Mol. Ther. 4, 242–250 [PubMed] [Google Scholar]
- 2. Aebersold R., and Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature. 537, 347–355 [DOI] [PubMed] [Google Scholar]
- 3. Stahl D. C., Swiderek K. M., Davis M. T., and Lee T. D. (1996) Data-controlled automation of liquid chromatography/tandem mass spectrometry analysis of peptide mixtures. J. Am. Soc. Mass Spectrom. 7, 532–540 [DOI] [PubMed] [Google Scholar]
- 4. Venable J. D., Dong M.-Q., Wohlschlegel J., Dillin A., and Yates J. R. (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods. 1, 39–45 [DOI] [PubMed] [Google Scholar]
- 5. Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., and Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doerr A. (2015) DIA mass spectrometry. Nat. Methods. 12, 35–35 [Google Scholar]
- 7. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schilling B., Gibson B. W., and Hunter C. L. (2017) acquisition data for label-free quantitative proteomics studies using tripleTOF (R) Mass Spectrometers. Methods Mol. Biol. 1550, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B. C., and Aebersold R. (2018) Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, e8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Houel S., Abernathy R., Renganathan K., Meyer-Arendt K., Ahn N. G., and Old W. M. (2010) Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 9, 4152–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saito M. A., Bertrand E. M., Duffy M. E., Gaylord D. A., Held N. A., Hervey W. J. 4th, Hettich R. L., Jagtap P. D., Janech M. G., Kinkade D. B., Leary D. H., McIlvin M. R., Moore E. K., Morris R. M., Neely B. A., Nunn B. L., Saunders J. K., Shepherd A. I., Symmonds N. I., and Walsh D. A. (2019) Progress and challenges in ocean metaproteomics and proposed best practices for data sharing. J. Proteome Res. 18, 1461–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deutsch E. W., Perez-Riverol Y., Chalkley R. J., Wilhelm M., Tate S., Sachsenberg T., Walzer M., Käll L., Delanghe B., Böcker S., Schymanski E. L., Wilmes P., Dorfer V., Kuster B., Volders P.-J., Jehmlich N., Vissers J. P. C., Wolan D. W., Wang A. Y., Mendoza L., Shofstahl J., Dowsey A. W., Griss J., Salek R. M., Neumann S., Binz P.-A., Lam H., Vizcaíno J. A., Bandeira N., and Röst H. (2018) Expanding the use of spectral libraries in proteomics. J. Proteome Res. 17, 4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Egertson J. D., MacLean B., Johnson R., Xuan Y., and MacCoss M. J. (2015) Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 10, 887–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reubsaet L., Sweredoski M. J., and Moradian A. (2019) Data-independent acquisition for the Orbitrap Q Exactive HF: A tutorial. J. Proteome Res. 18, 803–813 [DOI] [PubMed] [Google Scholar]
- 15. Bruderer R., Bernhardt O. M., Gandhi T., Xuan Y., Sondermann J., Schmidt M., Gomez-Varela D., and Reiter L. (2017) Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteomics 16, 2296–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li G.-Z., Vissers J. P. C., Silva J. C., Golick D., Gorenstein M. V., and Geromanos S. J. (2009) Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics. 9, 1696–1719 [DOI] [PubMed] [Google Scholar]
- 17. Tsou C.-C., Avtonomov D., Larsen B., Tucholska M., Choi H., Gingras A.-C., and Nesvizhskii A. I. (2015) DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods. 12, 258–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ting Y. S., Egertson J. D., Bollinger J. G., Searle B. C., Payne S. H., Noble W. S., and MacCoss M. J. (2017) PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods. 14, 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Searle B. C., Pino L. K., Egertson J. D., Ting Y. S., Lawrence R. T., MacLean B. X., Villén J., and MacCoss M. J. (2018) Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 9, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M.-Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amodei D., Egertson J., MacLean B. X., Johnson R., Merrihew G. E., Keller A., Marsh D., Vitek O., Mallick P., and MacCoss M. J. (2019) Improving precursor selectivity in data-independent acquisition using overlapping windows. J. Am. Soc. Mass Spectrom. 30, 669–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenberger G., Koh C. C., Guo T., Röst H. L., Kouvonen P., Collins B. C., Heusel M., Liu Y., Caron E., Vichalkovski A., Faini M., Schubert O. T., Faridi P., Ebhardt H. A., Matondo M., Lam H., Bader S. L., Campbell D. S., Deutsch E. W., Moritz R. L., Tate S., and Aebersold R. (2014) A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data. 1, 140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lawrence R. T., Searle B. C., Llovet A., and Villén J. (2016) Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry. Nat. Methods. 13, 431–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matthews D. E., and Hayes J. M. (1976) Systematic errors in gas chromatography-mass spectrometry isotope ratio measurements. Anal. Chem. 48, 1375–1382 [Google Scholar]
- 26. Zhang Y., Bilbao A., Bruderer T., Luban J., Strambio-De-Castillia C., Lisacek F., Hopfgartner G., and Varesio E. (2015) The use of variable Q1 isolation windows improves selectivity in LC-SWATH-MS acquisition. J. Proteome Res. 14, 4359–4371 [DOI] [PubMed] [Google Scholar]
- 27. Donoho D. L. (2006) Compressed sensing. IEEE Trans. Inf. Theory. 52, 1289–1306 [Google Scholar]
- 28. Egertson J. D., Kuehn A., Merrihew G. E., Bateman N. W., MacLean B. X., Ting Y. S., Canterbury J. D., Marsh D. M., Kellmann M., Zabrouskov V., Wu C. C., and MacCoss M. J. (2013) Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods. 10, 744–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moseley M. A., Hughes C. J., Juvvadi P. R., Soderblom E. J., Lennon S., Perkins S. R., Thompson J. W., Steinbach W. J., Geromanos S. J., Wildgoose J., Langridge J. I., Richardson K., and Vissers J. P. C. (2018) Scanning quadrupole data-independent acquisition, Part A: qualitative and quantitative characterization. J. Proteome Res. 17, 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Messner C., Demichev V., Bloomfield N., Ivosev G., Wasim F., Zelezniak A., Lilley K., Tate S., and Ralser M. (2019) ScanningSWATH enables ultra-fast proteomics using high-flow chromatography and minute-scale gradients. bioRxiv. 10.1101/656793 [Google Scholar]
- 31. Senko M. W., Beu S. C., and McLaffertycor F. W. (1995) Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 6, 229–233 [DOI] [PubMed] [Google Scholar]
- 32. Frahm J. L., Howard B. E., Heber S., and Muddiman D. C. (2006) Accessible proteomics space and its implications for peak capacity for zero-, one- and two-dimensional separations coupled with FT-ICR and TOF mass spectrometry. J. Mass Spectrom. 41, 281–288 [DOI] [PubMed] [Google Scholar]
- 33. Scheltema R. A., Hauschild J.-P., Lange O., Hornburg D., Denisov E., Damoc E., Kuehn A., Makarov A., and Mann M. (2014) The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics 13, 3698–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nefedov A. V., Mitra I., Brasier A. R., and Sadygov R. G. (2011) Examining troughs in the mass distribution of all theoretically possible tryptic peptides. J. Proteome Res. 10, 4150–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egertson J. D., Eng J. K., Bereman M. S., Hsieh E. J., Merrihew G. E., and MacCoss M. J. (2012) De novo correction of mass measurement error in low resolution tandem MS spectra for shotgun proteomics. J. Am. Soc. Mass Spectrom. 23, 2075–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 37. Weisbrod C. R., Eng J. K., Hoopmann M. R., Baker T., and Bruce J. E. (2012) Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. J. Proteome Res. 11, 1621–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Röst H. L., Rosenberger G., Navarro P., Gillet L., Miladinović S. M., Schubert O. T., Wolski W., Collins B. C., Malmström J., Malmström L., and Aebersold R. (2014) OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 [DOI] [PubMed] [Google Scholar]
- 39. Bruderer R., Bernhardt O. M., Gandhi T., Miladinović S. M., Cheng L.-Y., Messner S., Ehrenberger T., Zanotelli V., Butscheid Y., Escher C., Vitek O., Rinner O., and Reiter L. (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang J., Tucholska M., Knight J. D. R., Lambert J.-P., Tate S., Larsen B., Gingras A.-C., and Bandeira N. (2015) MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nat. Methods. 12, 1106–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Searle B. C., Swearingen K. E., Barnes C. A., Schmidt T., Gessulat S., Küster B., and Wilhelm M. (2020) Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 11, 1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., Stein S. E., and Aebersold R. (2008) Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods. 5, 873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang M., Wang J., Carver J., Pullman B. S., Cha S. W., and Bandeira N. (2018) Assembling the community-scale discoverable human proteome. Cell Syst. 7, 412–421.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Desiere F., Deutsch E. W., King N. L., Nesvizhskii A. I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S. N., and Aebersold R. (2006) The PeptideAtlas project. Nucleic Acids Res. 34, D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., and Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gessulat S., Schmidt T., Zolg D. P., Samaras P., Schnatbaum K., Zerweck J., Knaute T., Rechenberger J., Delanghe B., Huhmer A., Reimer U., Ehrlich H.-C., Aiche S., Kuster B., and Wilhelm M. (2019) Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 16, 509–518 [DOI] [PubMed] [Google Scholar]
- 47. Tiwary S., Levy R., Gutenbrunner P., Salinas Soto F., Palaniappan K. K., Deming L., Berndl M., Brant A., Cimermancic P., and Cox J. (2019) High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods. 16, 519–525 [DOI] [PubMed] [Google Scholar]
- 48. Panchaud A., Scherl A., Shaffer S. A., von Haller P. D., Kulasekara H. D., Miller S. I., and Goodlett D. R. (2009) Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ting Y. S., Egertson J. D., Payne S. H., Kim S., MacLean B., Käll L., Aebersold R., Smith R. D., Noble W. S., and MacCoss M. J. (2015) Peptide-centric proteome analysis: an alternative strategy for the analysis of tandem mass spectrometry data. Mol. Cell. Proteomics 14, 2301–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Käll L., Canterbury J. D., Weston J., Noble W. S., and MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 51. Spahr C. S., Davis M. T., McGinley M. D., Robinson J. H., Bures E. J., Beierle J., Mort J., Courchesne P. L., Chen K., Wahl R. C., Yu W., Luethy R., and Patterson S. D. (2001) Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I. Profiling an unfractionated tryptic digest. Proteomics. 1, 93–107 [DOI] [PubMed] [Google Scholar]
- 52. Meier F., Geyer P. E., Virreira Winter S., Cox J., and Mann M. (2018) BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods. 15, 440–448 [DOI] [PubMed] [Google Scholar]
- 53. Trachsel C., Panse C., Kockmann T., Wolski W. E., Grossmann J., and Schlapbach R. (2018) rawDiag: An R package supporting rational LC-MS method optimization for bottom-up proteomics. J. Proteome Res. 17, 2908–2914 [DOI] [PubMed] [Google Scholar]
- 54. Rosenberger G., Bludau I., Schmitt U., Heusel M., Hunter C. L., Liu Y., MacCoss M. J., MacLean B. X., Nesvizhskii A. I., Pedrioli P. G. A., Reiter L., Röst H. L., Tate S., Ting Y. S., Collins B. C., and Aebersold R. (2017) Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat. Methods. 14, 921–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., and Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics. 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 56. Pino L. K., Searle B. C., Yang H.-Y., Hoofnagle A. N., Noble W. S., and MacCoss M. J. (2020) Matrix-matched calibration curves for assessing analytical figures of merit in quantitative proteomics. J. Proteome Res. 19, 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rosenberger G., Liu Y., Röst H. L., Ludwig C., Buil A., Bensimon A., Soste M., Spector T. D., Dermitzakis E. T., Collins B. C., Malmström L., and Aebersold R. (2017) Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat. Biotechnol. 35, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meyer J. G., Mukkamalla S., Steen H., Nesvizhskii A. I., Gibson B. W., and Schilling B. (2017) PIQED: automated identification and quantification of protein modifications from DIA-MS data. Nat. Methods. 14, 646–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Searle B. C., Lawrence R. T., MacCoss M. J., and Ville′n J. (2019) Thesaurus: quantifying phosphopeptide positional isomers. Nat. Methods. 16, 703–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pino L. K., Searle B. C., Huang E. L., Noble W. S., Hoofnagle A. N., and MacCoss M. J. (2018) Calibration using a single-point external reference material harmonizes quantitative mass spectrometry proteomics data between platforms and laboratories. Anal. Chem. 90, 13112–13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gupta S., Ahadi S., Zhou W., and Röst H. (2019) DIAlignR provides precise retention time alignment across distant runs in DIA and targeted proteomics. Mol. Cell. Proteomics 18, 806–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data are available at MassIVE (MSV000084531) available at https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=b0c33f6caba04dd7a17a18e01e35071d.