

Protein-protein interactions that are mediated by short linear motifs (SLiMs) in intrinsically disordered regions (IDRs) of proteins are notoriously difficult to study. Recently, pull-downs with synthetic peptides in combination with quantitative mass spectrometry emerged as a powerful screening approach. Here, we briefly highlight the relevance of SLiMs for protein-protein interactions, outline existing screening technologies, discuss unique advantages of peptide-based interaction screens, and provide practical suggestions for setting up such peptide-based screens.
Keywords: Peptide array, protein-protein interactions, affinity proteomics, peptide interactions, peptides, post-translational modifications, high throughput screening, mass spectrometry, intrinsically disordered regions, short linear motifs
Graphical Abstract

Highlights
Peptide-based screens provide a scalable approach to study protein-protein interactions.
These screens help to characterize the function of structurally disordered regions.
The impact of posttranslational modifications can be directly investigated.
Abstract
Protein-protein interactions are often mediated by short linear motifs (SLiMs) that are located in intrinsically disordered regions (IDRs) of proteins. Interactions mediated by SLiMs are notoriously difficult to study, and many functionally relevant interactions likely remain to be uncovered. Recently, pull-downs with synthetic peptides in combination with quantitative mass spectrometry emerged as a powerful screening approach to study protein-protein interactions mediated by SLiMs. Specifically, arrays of synthetic peptides immobilized on cellulose membranes provide a scalable means to identify the interaction partners of many peptides in parallel. In this minireview we briefly highlight the relevance of SLiMs for protein-protein interactions, outline existing screening technologies, discuss unique advantages of peptide-based interaction screens and provide practical suggestions for setting up such peptide-based screens.
Most proteins interact with others to exert their biological functions. Therefore, studying protein-protein interactions (PPIs) provides mechanistic insights into the molecular processes underlying health and disease. Traditionally, PPIs were thought to be mediated by folded globular domains. Interactions formed this way tend to be stable, often forming complexes to fulfill tasks together as one molecular machinery. However, cells also need to react to stimuli and adapt to their environment in a flexible and versatile manner. To this end, proteins pass on information in signaling cascades, interact with each other for trafficking or are marked for degradation. In all these cases proteins interact, but the system can only function if these interactions are reversible and transient.
In the last decades it has become clear that also parts of proteins that do not acquire a fixed three-dimensional structure can be involved in PPIs (1). In multicellular organisms, around 40% of amino acid residues are in so-called intrinsically disordered regions (IDRs) (2). In recent years, the interest in IDRs has been rising because they were found to play critical roles in many biological processes. For example, IDRs are particularly abundant in so-called hub proteins (i.e. proteins with high connectivity in PPI networks), suggesting that these regions are important mediators of PPIs (3). Indeed, IDRs often harbor short linear motifs (SLiMs) - short regions of typically less than ten amino acids that can mediate PPIs, posttranslational modifications, or both (4, 5). The small information content of SLiMs makes bioinformatic prediction solely based on peptide sequences error-prone and care must be taken while trying to establish new peptide-domain connections (6). In addition, IDRs play a central role in liquid-liquid phase separation—a process in which proteins come together to form organelles without membrane confinement (7, 8). The assembly of these organelles is driven by transient, multivalent interactions (8, 9) and the liquid property is maintained through the constant formation and separation of these interactions.
Studying PPIs that are mediated by IDRs is of paramount importance for our understanding of cell biology. At the same time, their reversible and transient nature makes it hard to study such interactions experimentally (Fig. 1): Classical affinity purification mass spectrometry (AP-MS) is a very versatile approach, especially when it is combined with quantification (q-AP-MS) (10, 11). However, although AP-MS works very well for stable interactions, it may fail to detect weak binding mediated by SLiMs. Alternatively, proximity labeling approaches can reveal also weak interactions and thus be used to map the proteomes of membraneless organelles (12, 13). However, proximity labeling detects protein colocalization and not necessarily PPIs. Also, any given protein contains many potential SLiMs, and both AP-MS and proximity labeling do not provide sufficient information to assign an observed interaction to a specific motif.
Fig. 1.
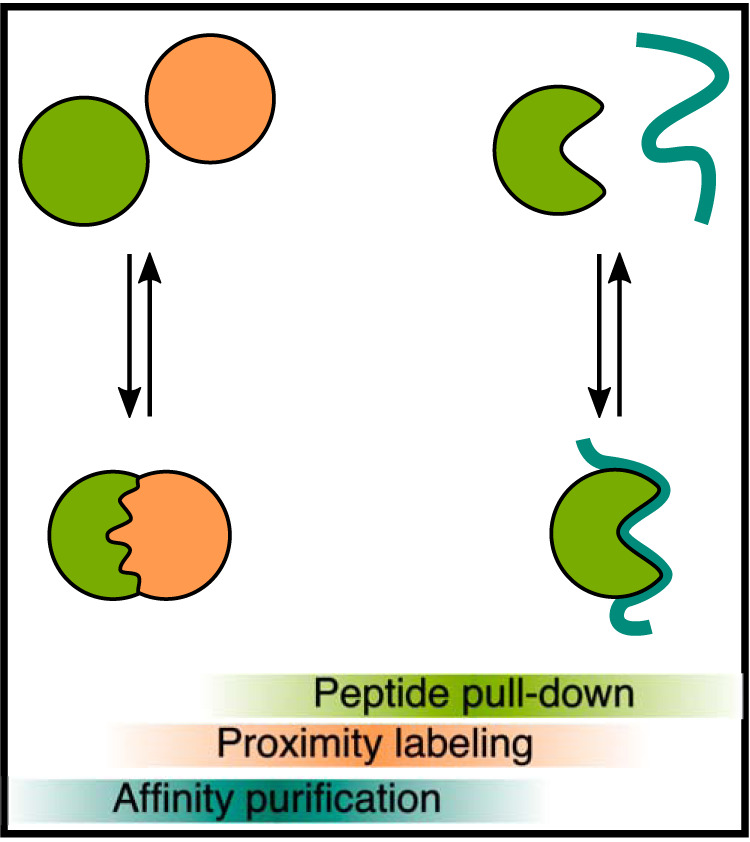
Studying protein-protein interactions experimentally. Protein-protein interactions range from stable interactions involving structured regions (left) to transient interactions involving intrinsically disordered regions (right). Different experimental methods are more suitable for specific types of interactions, also depending on binding affinities.
High throughput peptide-based interaction proteomics recently emerged as a powerful technology to study PPIs mediated by IDRs. Here we review this methodology, discuss practical implications involved in it and outline future perspectives. To set the scene, we will begin with a brief overview of existing methods to study peptide-mediated interactions.
Overview of Methods To Study Protein-Protein Interactions Mediated by SLiMs
Despite the challenges outlined above, IDRs have one inherent property that actually facilitates their experimental analysis: They can exert their function independently of the context of the full-length protein. This means that interactions mediated by SLiMs can be studied using short peptides. This enables the design of screening methods to systematically probe peptide-mediated interactions (Table I). The methods to study peptide-mediated interactions can be broadly categorized into genetic and biochemical screens (14).
Table I. Overview of screening methods to study peptide-protein interactions.
| Peptide library size | Domains/Proteins | Source | |
|---|---|---|---|
| Proteomic peptide-phage display (ProP-PD) | 10,000–1,000,000 | ∼10 | (18, 20) |
| Y2H | >100,000,000 | <50 | (21, 22) |
| Yeast surface display | 100,000,000 | <5 | (14, 23) |
| Protein microarray | <100 | <200 | (24, 25) |
| Peptide array (classical) | <1,000; < 1,000,000 (high-density array) | <10 | (26, 27) |
| Peptide array (+ mass spec) | <1,000 | Whole proteome | (28, 29) |
Phage display, yeast two-hybrid (Y2H) and yeast surface display are popular genetic screening methods to study peptide-protein interactions. However, these methods suffer from difficulties to identify interactions that rely on post-translational modifications (PTMs). This is an important limitation because some PTMs are enriched in IDRs and often play an important regulatory role (4, 15–17). Phage display libraries covering all disordered regions of the human proteome have been constructed (18). However, with few notable exceptions (19), such libraries are typically screened against a rather small set of preselected bait proteins. This is because of the practical challenges associated with producing many individual bait proteins in parallel. Therefore, these approaches typically have a limited throughput (few protein baits against a library of peptide preys).
Peptide-mediated interactions can also be studied biochemically. A prerequisite for these assays was the development of solid phase peptide synthesis, which enables the chemical production of peptides with defined amino acid sequences (30). Interaction assays with synthetic peptides have the advantage that posttranslational modifications can be directly incorporated into peptides during synthesis. Also, instead of incorporating a defined amino acid at a given position, it is possible to use a mix of different amino acids. This yields a degenerate peptide library (that is, a peptide mixture with random amino acids at specific positions). Already almost 30 years ago, the Lewis Cantley lab used such a degenerate phosphopeptide library with an invariant phosphotyrosine in the central position as prey in interaction screens with immobilized Src Homology 2 (SH2) domains as baits (31). Edman degradation of the bound peptides then revealed the average amino acid composition at different positions along the peptides and thus the “consensus” binding motif recognized by the SH2 domain. This approach is limited to the analysis of selected protein domains (one bait against a library of peptide preys). The throughput on the bait side can be increased by immobilizing the proteins/domains on a protein microarray (24, 25). This however reduces the throughput on the peptide side, because now individual peptides (rather than a degenerate peptide library) must be tested. Protein microarrays are also quite difficult to produce and limited to the detection of relatively high affinity interactions.
Instead of immobilizing protein baits to pull-down peptides, it is also possible to immobilize peptides as baits to pull down interacting proteins. A key advantage of this set-up is that peptides can be immobilized at high density. The resulting high local peptide concentration allows enrichment of proteins with low binding affinities. Another advantage is the increased throughput: Multiple peptides with defined amino acid composition can be synthesized in parallel via the so-called SPOT synthesis (32). SPOT synthesis yields arrays of thousands of different peptides immobilized on cellulose membranes, even though quality control is more difficult in this format (see below). Cellulose is an attractive support as it is biocompatible, hydrophilic, inexpensive, environmentally friendly, and stable under a wide range of reaction conditions (33). Traditionally, peptide arrays on cellulose are probed with a single prey protein that is subsequently detected by specific antibodies or by fluorescent/radioactive labels on the prey protein of interest (34). A common application for such peptide arrays is to map the linear epitopes recognized by antibodies (26) or to identify peptides that are recognized by recombinantly expressed protein domains (35). This approach is limited to a single prey protein that is studied at a time (many peptide baits, single candidate prey).
The prey proteins interacting with immobilized peptides can also be identified using mass spectrometry. In this case peptides were usually coupled to beads. Importantly, because peptide-protein interactions typically have low affinities, such pull-down experiments require low stringency conditions, which often results in many nonspecific binders. Therefore, it is crucial to use quantitative mass spectrometry to assess which interactions are truly specific. The general idea is to perform parallel control pull-downs with suitable negative controls (e.g. mutated or unmodified peptides). Quantitative proteomics can then distinguish specific interaction partners from unspecific background binders. This combination of peptide pull-downs with quantitative proteomics was pioneered in 2004 by the Matthias Mann laboratory and has since been successfully employed to study the impact of PTMs and sequence motifs on peptide-protein interactions (36–39). In these experiments, the number of preys is huge and corresponds to the complexity of protein lysate employed. In contrast, peptide baits are typically used in individual pull-down experiments. This limits the number of peptide baits that can be used in parallel (few peptide baits against a complex proteome).
High Throughput Peptide-based Interaction Proteomics
Recently, the advantageous parts of both above-mentioned peptide pull-down approaches have been combined (Fig. 2): Instead of probing a peptide array with a single prey protein, peptide arrays were probed with whole cell lysates and interacting proteins were identified using quantitative mass spectrometry (28, 29, 40). In all three cases, 14–18 mer peptides were synthesized on cellulose membranes via the SPOT technology that were then incubated with cell extracts to identify binding partners via mass spectrometry. This set-up, coined “Protein Interaction Screen on Peptide Matrix” (PRISMA) by Dittmar and co-workers, has the advantage that it enables interaction screens with high-throughput for both baits and preys (many peptide baits against a complex proteome as prey).
Fig. 2.

Peptide-based interaction proteomics. Interaction partners of short disordered peptide regions can be determined using peptide-pulldowns from a complex protein mixture. To this end, peptides are synthesized on a cellulose membrane, which allows for either N- or C-terminal coupling, analyses of mutated sequences or insertion of various posttranslational modifications. After incubation of the membrane with a protein extract, interacting proteins can be identified via mass spectrometry. Quantification can then reveal which interactions are specific for a given peptide sequence or modification.
The three above-mentioned publications employed the overall concept to address biological questions from different angles. Okada et al. incorporated photoactivatable amino acids into their peptides and used photocrosslinking to covalently trap interacting proteins (40). Although this enabled high stringency washing conditions, the list of identified proteins still contained numerous unspecific contaminants. The two other studies therefore employed quantification to distinguish specific from nonspecific binders. Dittmar and co-workers used a sliding window approach (overlapping “tiled” peptides) to map binding partners along the sequence of C/EBPbeta (28). They also assessed the impact of PTMs because SPOT synthesis permits inclusion of phosphorylation, methylation (mono, di, tri), citrullination, acetylation, crotonylation, sumoylation and many more. In our own work, we used PRISMA to study how disease-causing mutations in disordered regions of proteins lead to changes in protein-protein interactions (29). Here, we combined label-free quantification (41) and stable isotope labeling by amino acids in cell culture (SILAC) (42) to determine specific interaction partners and the impact of the mutation on the interaction, respectively.
Practical Considerations
Several practical points should be considered when designing a PRISMA screen. First, an overall experimental design and a strategy for data analysis and quantification are needed: Because the proteins identified in peptide pull-down experiments will be dominated by nonspecific interactors, it is critical to design a sound quantification strategy to single out specific interactors. Quantification methods such as label-free quantification, SILAC and chemical labeling are all possible options (43). Which of these methods is best suited for a given PRISMA screen depends on the aims of this screen. Key questions in this context are: To which control(s) will any given peptide pull-down be compared? Which statistical strategy will be used to identify specific binding? How many replicates are needed? Which positive and negative controls should be included in the screen?
The second question is how the cellulose arrays will be obtained. Although SPOT synthesis can be performed manually (44), it is more convenient to use a dedicated machine like the MultiPep synthesizer (Intavis AG, Köln, Germany) for this purpose (34). Alternatively, custom peptide arrays can be directly ordered from companies such as JPT Peptide Technologies (Berlin, Germany). Those peptide spots have a diameter of ∼2–3 mm and a typical yield between 5–10 nmol (8–17 μg for an average 15-mer peptide). This high local concentration facilitates identification of low affinity interactions.
Third, irrespective of how the SPOT synthesis is carried out, several points should be considered when designing peptide sequences. Fmoc based solid phase peptide synthesis proceeds from the C terminus toward the N terminus, i.e. in the opposite direction of biological peptide synthesis (45). Standard SPOT synthesis therefore results in peptides that are immobilized via their C terminus and a free N terminus. It is usually beneficial to synthesize the peptides with an acetylated N terminus, because this (i) eliminates the positive charge and thus better reflects the situation within a protein sequence in which the N terminus participates in an amide bond and (ii) makes the peptide more stable (46). Because peptides immobilized via their C termini are not suitable to screen for interaction partners of free C termini, methods to invert the peptide on the membrane have also been developed (47). It is also important to keep in mind that the yield of individual coupling steps is not 100%. Therefore, the yield of correctly synthesized full-length peptides decreases with increasing peptide length, which is why peptides should not be longer than about 15 amino acids. Fortunately, this is in good agreement with the fact that SLiMs are typically less than 10 amino acids in length (4, 5). Also, specific combinations of amino acids are notoriously difficult to synthesize (48, 49). The failure to incorporate an amino acid in one coupling cycle leads to a peptide with a deletion at this position. Such deletions can be prevented by incorporating a capping step, which blocks defective peptide chains in subsequent synthesis cycles (50). Although this results in truncated peptides, it should reduce the risk of false positive identifications arising from peptides with erroneous sequences.
The amino acid composition of the peptides is another important factor. If the peptide contains cysteines, the formation of disulfide bonds between the peptide and a protein might lead to false-positive results. In these cases, adding a reducing agent during the incubation step should be considered, which however must be compatible with the chemistry used for peptide immobilization. Also, although peptides are typically not eluted from the cellulose and therefore not identified in the mass spectrometer, sequences which contain a cleavage site for the used protease (e.g. R/K for trypsin) will also be digested and hence become an abundant peptide species in the mass spec measurement. To avoid misidentifications, they should therefore be included in the protein sequence database against which mass spectra are later searched.
For the actual pull-down experiment, it is advantageous to use a cell extract that contains the putative interaction partners. For example, for interactions in the neuronal context it makes sense to use extract from neuronal cell lines (29), and for interaction partners of nuclear proteins nuclear extracts are a good choice (28). Whatever extract is used, it should be rather concentrated (5 mg protein/ml) to increase the sensitivity of the screen (28, 29). Approximately 10 ml of lysate are needed to cover an array with 200 spots (∼30 cm2, peptide grid: 0.37 cm × 0.37 cm, JPT). It is generally recommended to perform the pull-downs at 4 °C and to use protease inhibitors. However, the final washing buffer should not contain protease inhibitors to ensure efficient subsequent digestion. After the pulldown washing, the cellulose membranes can be air dried and stored. For sample preparation individual spots need to be excised from the cellulose membrane and transferred into individual wells of a 96-well plate. We found that a mouse ear puncher is a helpful tool for this step and that dry membranes are easier to handle than wet ones. Because of electrostatic effects, the excised spots tend to be repelled from the wells of 96-well plates, hence it has proven useful to already fill the wells with the digestion buffer. Reduction, alkylation, and digestion can then all be performed in 96-well format.
A key factor for the mass spectrometric measurements is the length of the chromatographic gradient and thus the total measurement time needed for the screen. In our experience, the complexity of individual pull-down samples is rather low (a few hundred proteins), which permits relatively short analysis times on the mass spectrometer. The length of the gradient should be adapted to the required depth of proteomic coverage, for example based on the positive control samples.
For the interpretation of results, it is important to keep in mind that PRISMA is an in vitro screening method: The combination of cell lysates with the bait might bring together interaction partners that never meet under physiological conditions and thus lead to false positive identifications. Hence, before any biological conclusion can be drawn from the data, specific follow-up experiments under more physiological conditions are required for validation.
Conclusions and Outlook
As outlined above, PRISMA screening is a powerful technology to study PPIs that are mediated by short linear motifs (SLiMs) in disordered protein regions (IDRs). This will help characterize new SLiM-domain interaction pairs, by providing valuable insight on sequence recognition specificity. Getting better insight into the rules that guide these transient interactions might help us answer different questions in molecular/cellular biology, as for example how protein-protein interaction leads to formation of condensates via liquid-liquid phase separation (8) or provide hints on the functions of newly emerging miniproteins (51). Some detected peptides might also activate or inhibit the activity of their interaction partner and hence serve diagnostic or therapeutic purposes.
The PRISMA methodology is scalable and can already produce data for dozens of peptides per day. In the future, further automation of sample preparation, faster mass spectrometers and HPLC systems like the Evosep system (52) will enable analysis of over 100 samples per day. Combination with multiplexing via TMTpro (53) enables quantification and combined measurement of up to 16 samples, which effectively leads to about 1600 analyses per day. For example, using 16plex TMTpro would allow for alanine scanning of a 15 amino acid long peptide in every position and comparing it to the interactome of the wild type peptide in a single run. We thus predict that peptide-based interaction proteomics will continue to provide us with invaluable insights in the exciting realm of disordered regions, short linear motifs, and their function.
Author contributions—K.M. and M.S. wrote the paper.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- PPI
- protein-protein interaction
- IDR
- intrinsically disordered region
- SLiMS
- short linear motifs
- AP-MS
- affinity purification mass spectrometry
- PTMs
- post-translational modifications
- PRISMA
- Protein Interaction Screen on Peptide Matrix
- SILAC
- stable isotope labeling by amino acids in cell culture
- Y2H
- yeast two-hybrid.
REFERENCES
- 1. Keskin O., Gursoy A., Ma B., and Nussinov R. (2008) Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem. Rev. 108, 1225–1244 [DOI] [PubMed] [Google Scholar]
- 2. Xue B., Keith Dunker A., and Uversky V. N. (2012) Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Structure Dynamics 30, 137–149 [DOI] [PubMed] [Google Scholar]
- 3. Haynes C., Oldfield C. J., Ji F., Klitgord N., Cusick M. E., Radivojac P., Uversky V. N., Vidal M., and Iakoucheva L. M. (2005) Intrinsic Disorder is a Common Feature of Hub Proteins from Four Eukaryotic Interactomes. PLoS Comput. Biol. 2, e100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., and Gibson T. J. (2012) Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 5. Tompa P., Davey N. E., Gibson T. J., and Madan Babu M. (2014) A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 [DOI] [PubMed] [Google Scholar]
- 6. Gibson T. J., Dinkel H., Van Roey K., and Diella F. (2015) Experimental detection of short regulatory motifs in eukaryotic proteins: tips for good practice as well as for bad. Cell Commun. Signal. 13, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J., Grishin N. V., Frantz D. E., Schneider J. W., Chen S., Li L., Sawaya M. R., Eisenberg D., Tycko R., and McKnight S. L. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Banani S. F., Lee H. O., Hyman A. A., and Rosen M. K. (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li P., Banjade S., Cheng H.-C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J. V., King D. S., Banani S. F., Russo P. S., Jiang Q.-X., Nixon B. T., and Rosen M. K. (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer K., and Selbach M. (2015) Quantitative affinity purification mass spectrometry: a versatile technology to study protein-protein interactions. Front. Genet. 6, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang J., Wagner S. A., and Beli P. (2015) Illuminating Spatial and Temporal Organization of Protein Interaction Networks by Mass Spectrometry-Based Proteomics. Front. Genet. 6, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trinkle-Mulcahy L. (2019) Recent advances in proximity-based labeling methods for interactome mapping. F1000Res. 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Youn J.-Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Chen G. I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Côté J.-F., and Gingras A.-C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532.e11 [DOI] [PubMed] [Google Scholar]
- 14. Blikstad C., and Ivarsson Y. (2015) High-throughput methods for identification of protein-protein interactions involving short linear motifs. Cell Commun. Signal. 13, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Roey K., Gibson T. J., and Davey N. E. (2012) Motif switches: decision-making in cell regulation. Curr. Opinion Structural Biol. 22, 378–385 [DOI] [PubMed] [Google Scholar]
- 16. Bah A., and Forman-Kay J. D. (2016) Modulation of Intrinsically Disordered Protein Function by Post-translational Modifications. J. Biol. Chem. 291, 6696–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darling A. L., and Uversky V. N. (2018) Intrinsic disorder and posttranslational modifications: the darker side of the biological dark matter. Front. Genet. 9, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davey N. E., Seo M., Yadav V. K., Jeon J., Nim S., Krystkowiak I., Blikstad C., Dong D., Markova N., Kim P. M., and Ivarsson Y. (2017) Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 284, 485–498 [DOI] [PubMed] [Google Scholar]
- 19. Teyra J., Huang H., Jain S., Guan X., Dong A., Liu Y., Tempel W., Min J., Tong Y., Kim P. M., Bader G. D., and Sidhu S. S. (2017) Comprehensive analysis of the human SH3 domain family reveals a wide variety of non-canonical specificities. Structure 25, 1598–1610.e3 [DOI] [PubMed] [Google Scholar]
- 20. Ivarsson Y., Arnold R., McLaughlin M., Nim S., Joshi R., Ray D., Liu B., Teyra J., Pawson T., Moffat J., Li S. S.-C., Sidhu S. S., and Kim P. M. (2014) Large-scale interaction profiling of PDZ domains through proteomic peptide-phage display using human and viral phage peptidomes. Proc. Natl. Acad. Sci. U.S.A. 111, 2542–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang M., Wu Z., and Fields S. (1995) Protein-peptide interactions analyzed with the yeast two-hybrid system. Nucleic Acids Res. 23, 1152–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mu Y., Cai P., Hu S., Ma S., and Gao Y. (2014) Characterization of diverse internal binding specificities of PDZ domains by yeast two-hybrid screening of a special peptide library. PLoS ONE 9, e88286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bidlingmaier S., and Liu B. (2006) Construction and application of a yeast surface-displayed human cDNA library to identify post-translational modification-dependent protein-protein interactions. Mol. Cell. Proteomics 5, 533–540 [DOI] [PubMed] [Google Scholar]
- 24. Jones R. B., Gordus A., Krall J. A., and MacBeath G. (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439, 168–174 [DOI] [PubMed] [Google Scholar]
- 25. Stiffler M. A., Chen J. R., Grantcharova V. P., Lei Y., Fuchs D., Allen J. E., Zaslavskaia L. A., and MacBeath G. (2007) PDZ domain binding selectivity is optimized across the mouse proteome. Science 317, 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buus S., Rockberg J., Forsström B., Nilsson P., Uhlen M., and Schafer-Nielsen C. (2012) High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Mol. Cell. Proteomics 11, 1790–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carmona S. J., Nielsen M., Schafer-Nielsen C., Mucci J., Altcheh J., Balouz V., Tekiel V., Frasch A. C., Campetella O., Buscaglia C. A., and Agüero F. (2015) Towards High-throughput Immunomics for Infectious Diseases: Use of Next-generation Peptide Microarrays for Rapid Discovery and Mapping of Antigenic Determinants. Mol. Cell. Proteomics 14, 1871–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dittmar G., Hernandez D. P., Kowenz-Leutz E., Kirchner M., Kahlert G., Wesolowski R., Baum K., Knoblich M., Hofstätter M., Muller A., Wolf J., Reimer U., and Leutz A. (2019) PRISMA: protein interaction screen on peptide matrix reveals interaction footprints and modifications- dependent interactome of intrinsically disordered C/EBPβ. iScience 13, 351–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyer K., Kirchner M., Uyar B., Cheng J.-Y., Russo G., Hernandez-Miranda L. R., Szymborska A., Zauber H., Rudolph I.-M., Willnow T. E., Akalin A., Haucke V., Gerhardt H., Birchmeier C., Kühn R., Krauss M., Diecke S., Pascual J. M., and Selbach M. (2018) Mutations in disordered regions can cause disease by creating dileucine motifs. Cell 175, 239–253.e17 [DOI] [PubMed] [Google Scholar]
- 30. Merrifield R. B. (1963) Solid phase peptide synthesis. I. the synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 [Google Scholar]
- 31. Songyang Z., Shoelson S. E., Chaudhuri M., Gish G., Pawson T., Haser W. G., King F., Roberts T., Ratnofsky S., and Lechleider R. J. (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72, 767–778 [DOI] [PubMed] [Google Scholar]
- 32. Frank R. (1992) Spot-synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48, 9217–9232 [Google Scholar]
- 33. Klemm D., Heublein B., Fink H.-P., and Bohn A. (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed Engl. 44, 3358–3393 [DOI] [PubMed] [Google Scholar]
- 34. Volkmer R., Tapia V., and Landgraf C. (2012) Synthetic peptide arrays for investigating protein interaction domains. FEBS Lett. 586, 2780–2786 [DOI] [PubMed] [Google Scholar]
- 35. Tapia V. E., Nicolaescu E., McDonald C. B., Musi V., Oka T., Inayoshi Y., Satteson A. C., Mazack V., Humbert J., Gaffney C. J., Beullens M., Schwartz C. E., Landgraf C., Volkmer R., Pastore A., Farooq A., Bollen M., and Sudol M. (2010) Y65C missense mutation in the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J. Biol. Chem. 285, 19391–19401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schulze W. X., and Mann M. (2004) A novel proteomic screen for peptide-protein interactions. J. Biol. Chem. 279, 10756–10764 [DOI] [PubMed] [Google Scholar]
- 37. Selbach M., Paul F. E., Brandt S., Guye P., Daumke O., Backert S., Dehio C., and Mann M. (2009) Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 5, 397–403 [DOI] [PubMed] [Google Scholar]
- 38. Vermeulen M., Mulder K. W., Denissov S., Pijnappel W. W. M. P., van Schaik F. M. A., Varier R. A., Baltissen M. P. A., Stunnenberg H. G., Mann M., and Timmers H. T. M. (2007) Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 [DOI] [PubMed] [Google Scholar]
- 39. Zhao S., Yue Y., Li Y., and Li H. (2019) Identification and characterization of “readers” for novel histone modifications. Current Opinion Chem. Biol. 51, 57–65 [DOI] [PubMed] [Google Scholar]
- 40. Okada H., Uezu A., Soderblom E. J., MAMoseley 3rd, Gertler F. B., and Soderling S. H. (2012) Peptide array X-linking (PAX): a new peptide-protein identification approach. PLoS ONE 7, e37035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 43. Ankney J. A., Astor Ankney J., Muneer A., and Chen X. (2018) Relative and absolute quantitation in mass spectrometry–based proteomics. Ann. Rev. Anal. Chem. 11, 49–77 [DOI] [PubMed] [Google Scholar]
- 44. Hilpert K., Winkler D. F. H., and Hancock R. E. W. (2007) Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protocols 2, 1333–1349 [DOI] [PubMed] [Google Scholar]
- 45. Fields G. B., and Noble R. L. (1990) Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 35, 161–214 [DOI] [PubMed] [Google Scholar]
- 46. Katz C., Levy-Beladev L., Rotem-Bamberger S., Rito T., Rüdiger S. G. D., and Friedler A. (2011) Studying protein-protein interactions using peptide arrays. Chem. Soc. Rev. 40, 2131–2145 [DOI] [PubMed] [Google Scholar]
- 47. Boisguerin P., Leben R., Ay B., Radziwill G., Moelling K., Dong L., and Volkmer-Engert R. (2004) An improved method for the synthesis of cellulose membrane-bound peptides with free C termini is useful for PDZ domain binding studies. Chem. Biol. 11, 449–459 [DOI] [PubMed] [Google Scholar]
- 48. Tickler A. K., and Wade J. D. (2007) Overview of solid phase synthesis of “difficult peptide” sequences. Curr. Protoc. Protein Sci. Chapter 18, Unit 18.8 [DOI] [PubMed] [Google Scholar]
- 49. Zander N., and Gausepohl H. (2002) Chemistry of Fmoc peptide synthesis on membranes. Peptide Arrays Membrane Supports, 23–39 [Google Scholar]
- 50. Hilper K., Winkler D. F. H., and Hancock R. E. W. (2007) Cellulose-bound peptide arrays: preparation and applications. Biotechnol. Genet. Eng. Rev. 24, 31–106 [DOI] [PubMed] [Google Scholar]
- 51. Leslie M. (2019) New universe of miniproteins is upending cell biology and genetics. Science [Google Scholar]
- 52. Bache N., Geyer P. E., Bekker-Jensen D. B., Hoerning O., Falkenby L., Treit P. V., Doll S., Paron I., Müller J. B., Meier F., Olsen J. V., Vorm O., and Mann M. (2018) A novel LC system embeds analytes in pre-formed gradients for rapid, ultra-robust proteomics. Mol. Cell. Proteomics 17, 2284–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thompson A., Wölmer N., Koncarevic S., Selzer S., Böhm G., Legner H., Schmid P., Kienle S., Penning P., Höhle C., Berfelde A., Martinez-Pinna R., Farztdinov V., Jung S., Kuhn K., and Pike I. (2019) TMTpro: design, synthesis, and initial evaluation of a proline-based isobaric 16-plex tandem mass tag reagent set. Anal. Chem. 91, 15941–15950 [DOI] [PubMed] [Google Scholar]