

Abstract
Under stress, the heart undergoes extensive remodeling resulting in cardiac fibrosis and hypertrophy, ultimately contributing to chronic heart failure (CHF). Alterations in microRNA levels are associated with dysfunctional gene expression profiles involved in the pathogenesis of heart failure. We previously showed that myocardial infarction-induced microRNA-enriched extracellular vesicles (EVs) contribute to the reduction in antioxidant enzymes by targeting Nrf2 signaling in CHF. MicroRNA-27a (miRNA-27a) is the predominant microRNA contained in cardiac fibroblast-derived EVs contributing to oxidative stress along with hypertrophic gene expression in cardiomyocytes. In the present study, we observed that miRNA-27a passenger strand (miRNA-27a*) was markedly upregulated in the non-infarcted area of the left ventricle of rats with CHF and encapsulated into EVs and secreted into the circulation. Bioinformatic analysis revealed that PDZ and LIM domain 5 (PDLIM5) is one of the major targets of miRNA-27a*, playing a major role in cardiac structure and function, and potentially contributing to the progression of cardiac hypertrophy. Our in vivo data demonstrate that PDLIM5 is down-regulated in the progression of heart failure, accompanied with the upregulation of hypertrophic genes and consistent with alterations in miRNA-27a*. Moreover, exogenous administration of miRNA27a* mimics inhibit PDLIM5 translation in cardiomyocytes whereas a miRNA27a* inhibitor enhanced PDLIM5 expression. Importantly, we confirmed that infarcted hearts have higher abundance of miRNA-27a* in EVs compared to normal hearts and further demonstrated that cultured cardiac fibroblasts secrete miRNA27a*-enriched EVs into the extracellular space in response to Angiotensin II stimulation, which inhibited PDLIM5 translation, leading to cardiomyocyte hypertrophic gene expression. In vivo studies suggest that the administration of a miRNA-27a* inhibitor in CHF rats partially blocks endogenous miR-27a* expression, prevents hypertrophic gene expression and improves myocardial contractility. These findings suggest that cardiac fibroblast-secretion of miRNA27a*-enriched EVs may act as a paracrine signaling mediator of cardiac hypertrophy that has potential as a novel therapeutic target.
Keywords: Chronic heart failure, Extracellular vesicles, miRNA-27a*, PDLIM5, Cardiac hypertrophy
1. Introduction
Chronic Heart failure (CHF) is a major public health issue, and causes substantial morbidity, mortality and healthcare expenditures worldwide [1–3], often caused by cardiac remodeling including cardiomyocyte hypertrophy and cardiac fibrosis after a primary insult or a genetic abnormality. The heart is composed of multiple cell types, such as cardiomyocytes, cardiac fibroblasts and endothelial cells [4, 5], each of which plays an important role in both physiological and pathophysiological conditions. Increasing evidence suggests that various cells within the heart are not isolated from each other but communicate with each other through either direct cell-cell interactions or paracrine signaling, contributing to highly organized and complex structure and physiological functions of the heart [6, 7].
Recently, extracellular vesicles (EVs), such as exosomes and other microvesicles, have emerged as important paracrine mediators that modulate intercellular communication related to the pathogenesis of cardiovascular diseases [8–12]. EV-mediated intercellular communications rely on the transfer of their bioactive components to recipient cells. In particular, EV-enriched microRNAs play important roles in the pathophysiological phenotypic transfer between cell types in the progression of cardiovascular disease [13–16]. Cardiomyocytes contribute 30–40% of the cellular elements in the heart, and cardiac fibroblasts are the predominant non-muscle cell type in the remaining cells. Thus, cross-talk between cardiomyocytes and cardiac fibroblasts are likely to play an important role in the pathogenesis of heart failure. Myocyte-secreted bioactive exosomal cargo are released following myocardial injury leading to cardiomyocyte hypertrophy and death. This cargo includes heat shock protein 90 (HSP90) and interleukin 6 (IL-6); both involved in the activation of STAT-3 signaling in cardiac fibroblasts, leading to increased collagen production and deposition [17]. Cardiac fibroblasts also secrete miRNA-enriched EVs whose cargo contribute to cardiomyocyte hypertrophy by either regulating cardiac structure or myocyte function [16] or by regulating oxidative stress in the chronic heart failure (CHF) state [15]. In general, during miRNA biogenesis, one strand of the RNA duplex is preferentially selected for entry into a silencing complex, whereas the complementary strand, known as the passenger strand or miRNA star strand, normally undergoes intracellular degradation [18]. However, bioinformatic analyses demonstrated that miRNA star species also exerts a demonstrable impact on vertebrate regulatory networks, and may contribute to disease states [19], such as cancer [20, 21]. Recently, cardiac fibroblast-derived exosomal miRNA-21* has been identified as a potent paracrine-acting RNA molecule that induces cardiomyocyte hypertrophy [16].
In a recent study we showed that myocardial infarction in the rodent model upregulated miRNA-27a levels in both infarcted heart tissue and in the circulation and that miRNA-27a was also significantly upregulated in cultured cardiac fibroblasts following pro-inflammatory stimulation. This miRNA was preferentially incorporated into EVs and secreted into the extracellular space and were taken up by cardiomyocytes contributing to oxidative stress and hypertrophic gene expression in cardiomyocytes by targeting the Nuclear factor (erythroid-derived 2)-like 2/Kelch-like ECH-associated protein 1 (Nrf2/keap1) signaling pathway [15]. In addition, clinical studies have demonstrated that miRNA-27a levels are upregulated in failing hearts and in the circulation [22–24], suggesting that miRNA-27a may potentially serve as a biomarker for diagnosis and prognosis of HF. Strikingly, in the present study, we observed that miRNA-27a* does not undergo degradation, and exhibits a similar expression profile as miRNA-27a in both the failing heart and the circulation in rats with CHF. However, the roles of miRNA-27a* in CHF are not fully understood. It has been reported that multiple Z-line-associated proteins, such as Cypher, Calsarcin-1 and Enigma Homolog Protein (ENH or PDLIM5) play an important role in the pathogenesis of human cardiomyopathy [25, 26]. In particularly, ENH, one of the PDZ-LIM domain protein family, is highly expressed in the heart, and silencing or cardiac-specific deletion of PDLIM5 causes dilated cardiomyopathy [16, 26]. Here, we hypothesize that miRNA-27a* is highly expressed in cardiac cells in response to cardiac stress, abundantly packaged into EVs and secreted into the extracellular space where they are taken up by cardiomyocytes and target PDLIM5 and then contribute to hypertrophic gene expression.
2. Methods and Materials
2.1. Rat Model of Heart Failure
Because male animals more easily develop HF due, in part, to an influence of estrogens on inflammatory processes and remodeling following MI [27], male Sprague-Dawley rats (180–200g) were used in this study. They were subjected to permanent left coronary artery ligation as previously described [28, 29]. Animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Experiments were carried out as recommended in the NIH Guide for the Care and Use of Laboratory Animals. Animal care was provided by the Department of Comparative Medicine at University of Nebraska Medical Center. Sham-operated rats were subjected to all procedures, excluding permanent coronary artery ligation. Cardiac function in experimental rats was evaluated by high-frequency echocardiography (Vevo 3100; MX201 probe, 15 MHz center frequency; Visual Sonics, Inc. Toronto, Canada) as previously described [30]. Rats were considered to be in CHF when their ejection fractions (EF, %) were less than 40%. Infarct size was determined post mortem by tracing the scar area and the whole left ventricle using Image J software (Bethesda, MD, USA). The percentage of scar area to the whole left ventricle was used to quantify infarct size.
2.2. Primary cardiomyocyte and fibroblast isolation and culture
Neonatal cardiomyocytes and fibroblasts were isolated from postnatal day 3 (P3) rat pups using a Thermo Scientific™ Pierce™ Primary Cardiomyocyte Isolation Kit (Cat. 88281) per the manufacturer’s protocol with modifications. Briefly, left ventricles of hearts from P3 rat pups were minced and washed with ice cold HBSS, and then digested with Cardiomyocyte Isolation Enzyme 1 (with papain) and Cardiomyocyte Isolation Enzyme 2 (with thermolysin) in a 37°C incubator for 30 minutes. The enzyme solution was removed and cardiac tissue was washed twice with ice cold HBSS, and then disrupted by pipetting up and down in Complete DMEM medium. Cells were plated on 75 cm2 flasks with complete DMEM medium and cultured in a 37°C incubator for 3–4 hours. Floating cells were transferred into other flasks and maintained with fresh complete DMEM containing Cardiomyocyte Growth Supplement for 5–7 days. Attached cells were switched to fresh Fibroblast Medium-2 (FM-2, Cat. #2331, Carlsbad, CA) supplemented with 2% FBS and Fibroblast Growth Supplement-2.
2.3. Tissue collection and Western blot analysis
At 6 weeks post-MI, rats were anesthetized with an intraperitoneal injection of α-chloralose (100 mg/kg) and urethane (500 mg/kg) dissolved in 2% borax solution. The non-infarcted region and border zone of the left ventricles were rapidly sampled and frozen in liquid nitrogen. All samples were homogenized with RIPA buffer (Thermo Scientific, Rockford, lL) containing a complete protease inhibitor (Roche, Indianapolis, IN). Lysates were cleared by centrifugation at 14,000 g for 20 min, and sample protein concentrations were normalized with Pierce™ BCA Protein Assay Kit (Thermo Scientific), separated on a 10% SDS-PAGE gel and transferred onto a PVDF membrane (Millipore, Billerica, MA). Membranes were blocked with 5% milk for 1 h and then incubated with primary antibodies overnight. Blots were rinsed with PBS-Tween 20 (PBST) three times and then incubated with horseradish peroxidase-conjugated secondary antibodies (Thermo Scientific) for 1h at RT. After being washed with PBST three times, blots were applied to the chemiluminescent substrate per the manufacturer’s recommendations (Thermo Scientific) and then subjected to UVP Bioimaging Systems (Upland, CA) for quantification.
2.4. EV isolation and characterization
2.4.1. EV isolation and characterization of plasma and conditioned medium
Extracellular vesicles were collected and purified from cardiac fibroblast supernatants or rat plasma by filtration with a 0.22 μm filter and then serial ultracentrifugation at 10,000 × g for 30 min and 100,000 × g for 2 h at 4°C, respectively. The EV pellets were either directly lysed in RIPA buffer (Thermo Scientific) containing complete protease inhibitors (Roche, Indianapolis, IN) and then subjected to western blotting analysis for CD63 (sc-15363), TSG101 [EPR7130 (B)] (ab125011), CD9 (sc-13118), CD81 (sc-23962) and HSC70 (sc-7298), respectively, or subjected to morphology and size distribution analysis using transmission electron microscopy (TEM, FEI Tecnai G2 Spirit TWIN, Hillsboro, OR) and the NanoSight NS300 system (Malvern Instruments), respectively, as previously described [15].
2.4.2. Cardiac derived EV isolation
Heart tissue EVs were isolated from frozen left ventricles of Sham and CHF rats as previously described with modifications [31, 32]. Briefly, rats were anesthetized with a urethane/chloralose mixture by i.p injection, and then perfused with ice-cold PBS. The left ventricles were minced quickly on ice using fine sterile scissors in PBS. Samples were centrifuged twice at 400g for 20 minutes, and the resulting supernatants were then centrifuged at 10,000g for 30 minutes at 4°C. The supernatants were further centrifuged at 20,500 g for 1 hour to pellet large EVs, which were suspended in 100 μl PBS and stored at −80°C. The remaining supernatants were finally subjected to 100,000g at 4°C for 2 hours to collect the small EV (sEVs) pellets which were suspended in 100 μl PBS and stored at −80°C.
2.5. Quantitative real-time PCR (qRT-PCR)
Total RNAs were isolated with TRIzol Reagent (Invitrogen, Carlsbad, CA) and a miRNeasy Mini Kit (QIAGEN, Germantown, MD) per manufacturer’s recommendations. Reverse transcription was performed using 5X All-In-One RT MasterMix (Applied Biological Materials Inc., CANADA). The quantitative RT-PCR analyses were performed using SYBR Select Master Mix (Life Technologies, Los Angeles, CA) with primer pairs (Integrated DNA Technologies) (PDLIM5: 5’- GGCTCCACCTGGCATGAC-3’, 5’-AAAATGTCTGCCCTTCCAAACTT-3’; ANP: 5’-ATCACCAAGGGCTTCTTCCT-3’, 5’-TGTTGGACACCGCACTGTAT-3’; BNP: 5’-ACAATCCACGATGCAGAAGCT-3’, 5’-GGGCCTTGGTCCTTTGAGA-3’; β-MHC: 5’-TTGGCACGGACTGCGTCATC-3, 5’-GAGCCTCCAGAGTTTGCTGAAGGA-3’; GAPDH: 5’-AGGTCGGTGTGAACGGATTTG-3’, 5’-TGTAGACCATGTAGTTGAGGTCA-3’). GAPDH was used as an internal control. To allow for normalization of sample to sample variations in RNA isolation from EVs, synthetic cel-miRNA-39 by Integrated DNA Technologies (Coralville, IA) was added to each denatured sample as an endogenous control. 10 ng of total RNAs were reversely transcribed using a TaqMan microRNA reverse transcription kit (Applied Biosystems, Carlsbad, CA) per the manufacturer’s protocol. MiRNA-27a*, microRNA-21–3p, cel-microRNA-39, and U6 snRNA were detected using Taqman microRNA assays (Applied Biosystems) on a StepOne Real-Time PCR systems (Applied Biosystems). The 2−ΔΔCt method was used to quantify relative mRNA and microRNA expression.
2.6. Cell transfections and co-culture
Plasmid transfections were performed using Lipofectamine 3000 (Cat. L3000015; ThermoFisher Scientific) according to the manufacturer’s instructions. In brief, primary cardiomyocytes (CMs) and cardiac fibroblasts (RCFs) were transfected with 500 ng pEF6.mCherry-TSG101 (Addgene plasmid 38318) and CD63-pEGFP (Addgene plasmid 62964) plasmids, respectively, mixed with 2 μL of Lipofectamine 3000 diluted in 100 μL of Opti-MEM (catalog no. 31985062; Life Technologies). The resulting plasmid-lipid complexes were added to the cells, incubated for 6 hr, and the medium changed into fresh DMEM. Next, the medium was changed to 10% FBS-containing medium for 20-hr incubation. The transfected RCFs were transferred into a trans-well cell culture insert, the bottom of which was comprised of a membrane with a 0.4-μm pore size (Millipore, USA). The transwell inserts were then placed into the 6-well culture plate containing CMs. DMEM medium supplemented with Ang II (1 μM) was applied to Transwell 6-well plate. After 24 h of incubation, the transwell inserts were removed, CMs cultured on 6-well plate were washed with PBS and fixed with 4% paraformaldehyde, and DAPI was used for counterstaining of nuclei (blue), and fluorescent images were obtained using a Zeiss 710 Confocal Laser Scanning Microscope (Carl Zeiss, Oberkochen, Germany).
2.7. Transfection of microRNA mimic and inhibitor
Primary cardiomyocytes were cultured on 6-well plates at a density of 2 × 105 cells/ml with complete DMEM containing Cardiomyocyte Growth Supplement for 5 days. Cardiomyocytes were transfected with microRNA mimic and inhibitor, respectively, using Xfect™ RNA Transfection Reagent (Clontech Labortories, Inc., Mountain View, CA) following the manufacturer’s protocol. Seventy-two-hour post-transfection, western blotting was performed with ENH1 (PDLIM5) primary antibody (Cat. 10530–1-AP, Proteintech™, Rosemont, IL). β-actin was used as a loading control.
2.8. Delivery of miRNA-27a* inhibitor in vivo
Rno-miRNA-27a* inhibitor (mU/ZEN/mGmCmUmCmAmCmAmAmGmCmAmGmCmUmAmA mGmCmCmC/3ZEN/) and negative control (mG/ZEN/mCmGmAmCmUmAmUmAmCmG mCmGmC mAmAmU mAmUmG mG/3ZEN/) (designed and synthesized by IDT) were dissolved in saline and delivered by intraperitoneal (i.p) injections as previously described [33, 34] with some modifications. In brief, Sham and CHF rats were injected weekly with 200 μg/kg of the miR-27a* inhibitor or a comparable volume of vehicle starting at 3 weeks post-MI continuously for 4 weeks. At the conclusion of the treatment, animals were echocardiographed and tissue was collected after euthanization with an intraperitoneal injection of α-chloralose (100 mg/kg) and urethane (500 mg/kg) dissolved in 2% borax solution.
2.9. Statistical analyses
All statistical analyses were performed using GraphPad Prism 8.0 (GraphPad software, La Jolla, CA). Data are expressed as means ± S.E.M. Statistical significance was determined by Student’s t test or One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. A value of p<0.05 was considered statistically significant.
3. Results
3.1. miRNA-27a* is up-regulated in infarcted heart and in extracellular vesicles derived from plasma of CHF rats
In a previous study, we showed high abundance of miRNA-27a in non-infarcted areas and in the border zone of the post MI heart [15]. In the current study, we observed that miRNA-27a* was also upregulated in these regions (Figure 1A). To determine miRNA-27a* levels in plasma, we generated CHF and sham rats that were evaluated by echocardiographic analyses at 6 weeks post-myocardial infarction as shown in Table 1. These data clearly demonstrate that left ventricular end-diastolic and end-systolic diameters (LVEDD and LVESD) and volumes (LVEDV and LVESV) were significantly increased, whereas ejection fraction (EF %) and fractional shortening (FS %) were significantly reduced. Average infarct size was approximately 33.4% of left ventricle in CHF rats. Extracellular vesicles (EVs) from plasma of sham and CHF rats were isolated by differential ultracentrifugation and subjected to extensive characterization. Transmission electron microscopy (Figure 1B) and NanoSight analysis (Figure 1C) revealed a typical size of 50 to 300 nm and a characteristic cup-shaped morphology of EVs (exosomes and microvesicles). Western blotting data showed the presence of EV markers, including CD63 and TSG101 (Figure 1D), and qRT-PCR analysis further demonstrated that miRNA-27a* was also enriched in circulating EVs of CHF rats (Figure 1E), consistent with that of miRNA-27a.
Figure 1. miRNA-27a* is up-regulated in the infarcted heart and circulating extracellular vesicles.
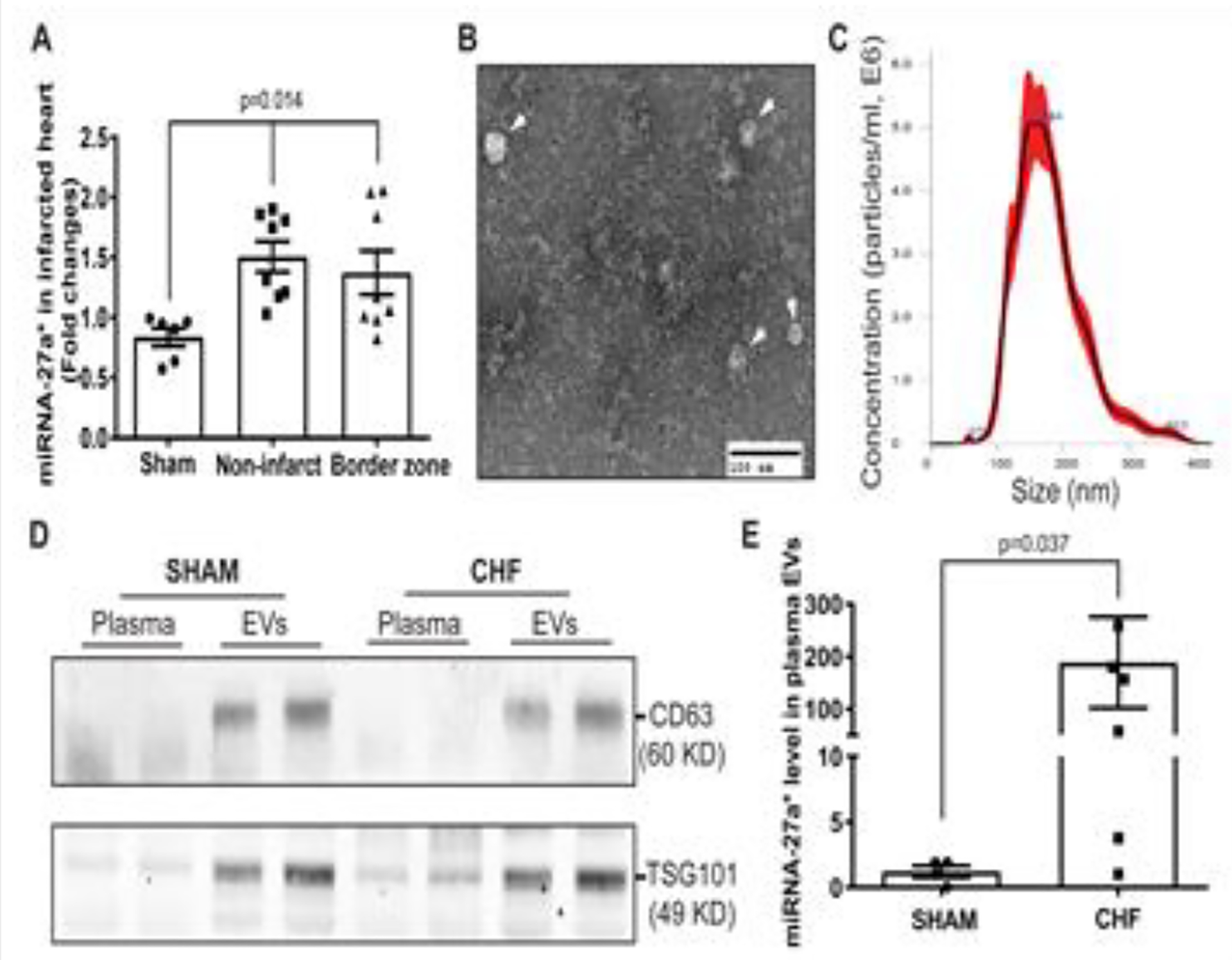
qRT-PCR shows that miRNA-27a* is up-regulated in the both non-infarcted and border zone tissues of the left ventricle compared to that in Sham rats (A), U6 snRNA was used as an internal control (±SEM, n=6, Sham; n=8, CHF); EVs isolated from rat plasma by differential centrifugation were subjected to negative staining and electron microscopy (B). Scale bar is 100 nm; EVs were also subjected to Malvern NanoSight analysis show the size distribution of circulating EVs. Five captures were collected and averaged (±SEM, n=5) (C); Western blotting data shows the typical EV markers (CD63 and TSG101) in EV pellets derived from plasma of Sham and CHF rats, respectively (D); qRT-PCR results showing increased miRNA-27a* in plasma EVs from CHF rats compared to that of Sham rats (E), cel-mir-39 was used as a spike-in control (±SEM, n=6).
Table 1.
Six-Week Echocardiographic Data for Sham and CHF rats. Values are mean ± SE (n=10–11).
| Parameters | Sham (10) | CHF (11) |
|---|---|---|
| LVEDD (mm) | 7.2 ± 0.6 | 10.5 ± 0.8* |
| LVESD (mm) | 4.0 ± 0.7 | 8.8 ± 0.8* |
| LVEDV (μl) | 276.5 ± 48.6 | 631.1 ± 113.3* |
| LVESV (μl) | 71.8 ± 28.4 | 427.9 ± 90.5* |
| Ejection Fraction (EF, %) | 74.8 ± 6.3 | 32.5 ± 5.5* |
| Fraction Shortening (FS, %) | 45.3 ± 6.0 | 16.4 ± 3.0* |
| Infarct size (% of LV) | – | 33.4 ± 6.1 |
VEDD, left ventricle end-diastolic diameter; LVESD, left ventricle end-systolic diameter; LVEDV, left ventricle end-diastolic volume; LVESV, left ventricle end-systolic volume; EF, ejection fraction; FS, fractional shortening.
p < 0.0001 vs SHAM
3.2. Bioinformatic analysis shows that miRNA-27a* is related to cardiac development and function
To investigate the potential roles of miRNA-27a* in the pathogenesis of CHF, we searched the predicted targets of miRNA-27a* using TargetScan 7.1 [35], and performed a ClueGO analysis showing that most target genes were related to multicellular organismal development (Fig. 2A) and were involved in the heart development, the response to hypoxia and cardiac muscle hypertrophy in response to stress, etc. (Fig. 2B). In particular, some of these genes regulate myocardial contraction and cardiac hypertrophy in response to stress (Supplemental Table 1). One of these, PDZ and LIM domain 5 (PDLIM5) has been shown to play a major role in cardiac structure and function and contribute to cardiac hypertrophy [16, 26].
Figure 2. Target prediction and Bioinformatic Analysis of MiRNA-27a* using TargetScan 7.1 and ClueGO platforms.
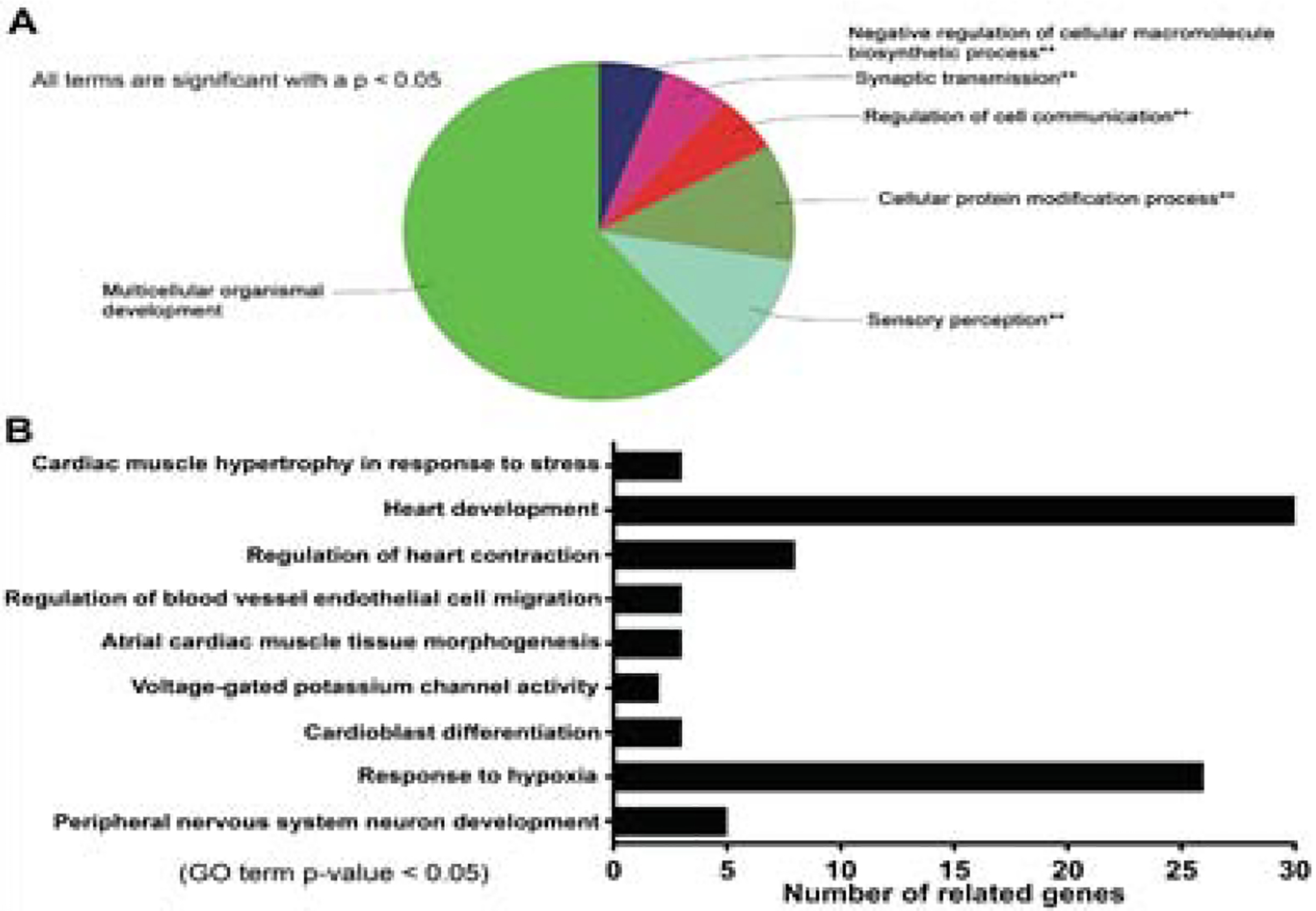
ClueGo analysis of miRNA-27a* gene ontology enrichment for biological processes, all terms are significant with p<0.05 (A); Go terms of miRNA-27a* targets are related to cardiac development and contraction (B).
3.3. PDLIM5 is down-regulated in the infarcted heart and associated with miRNA-27a* upregulation
To determine the transcription and translation levels of PDLIM5 in the progression of CHF, we collected heart tissues from non-infarcted areas at 3, 6 and 12 weeks post-MI, respectively, and performed qRT-PCR and western blotting analyses. To validate the molecular weight of PDLIM5 protein detected by ENH1 (PDLIM5) antibody in this stud y, we performed PDLIM5 siRNA transfections in rat myoblast cells (H9C2), qRT-PCR and western blot analyses are shown in Supplemental figure 3, and confirmed that PDLIM5 siRNA can successfully block the PDLIM5 transcription at 48 hours post-transfection (Supplemental figure 3A and 3B). Moreover, we observed that PDLIM5 mRNA was significantly upregulated in response to myocardial infarction at 3 wks post-MI, and then was slightly decreased in the progression of CHF (Fig. 3A), whereas the expression of hypertrophic genes, including ANP, BNP and β-MHC was significantly increased compared with that of the sham group (Fig. 3B, 3C and 3D). BNP and β-MHC levels peaked at 6 wks post-MI. Consistently, PDLIM5 protein at 3 wks post-MI was slightly increased, and then significantly decreased at 6 wks and 12 wks post-MI (Fig. 3E and 3F), suggesting the translation of PDLIM5 mRNA may be inhibited by abundant miR-27a* in the infarcted heart. To further confirm the translational inhibition of PDLIM5 by miRNA-27a* in cardiomyocytes, we isolated primary cardiomyocytes from 3-day old rats, and transfected cells with a miRNA-27a* mimic and its inhibitor, respectively. Western blot data suggest that miRNA-27a* inhibits PDLIM5 translation, whereas miRNA-27a* inhibitor enhances the PDLIM5 translation (Fig. 3G, 3H and 3I).
Figure 3. MiRNA-27a* down-regulates PDLIM5 in CHF.
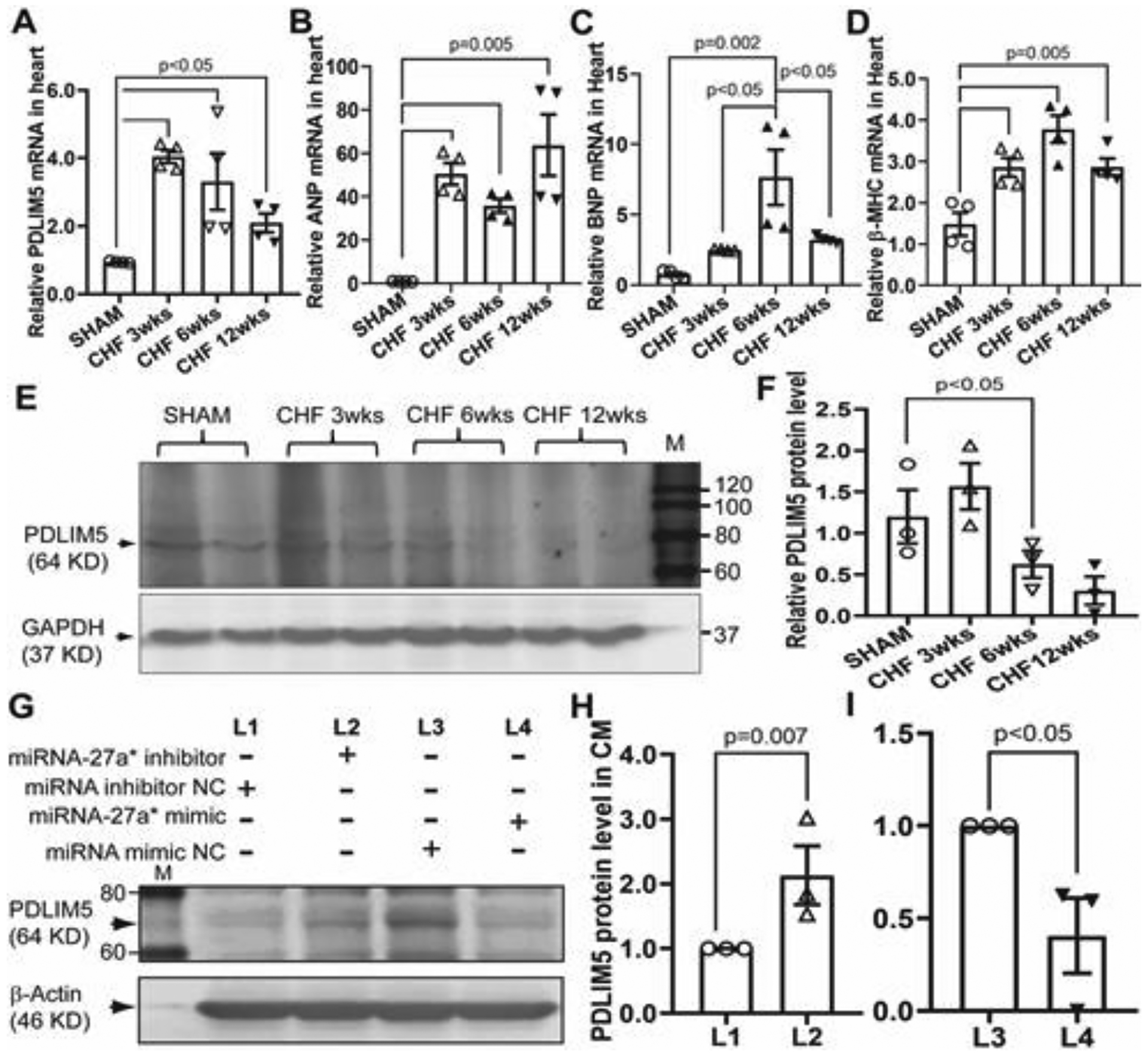
At three, six and twelve weeks post myocardial infarction (MI), respectively, qRT-PCR was performed with specific primers for PDLIM5 (A), ANP (B), BNP (C) and β-MHC (D), GAPDH was used as an internal control; Western blotting analysis was carried out with primary PDLIM5 antibody (E) and pooled data (±SEM) (F), GAPDH was used as a loading control; Primary cardiomyocytes were transfected with miRNA-27a* mimic and inhibitor, respectively and then subjected to western blotting analysis with PDLIM5 primary antibody, β-actin was used as a loading control (G). Data is representative of three independent experiments (±SEM) (H and I).
3.4. Cardiac-derived extracellular vesicles are enriched with miRNA-27a*
To determine if miRNA-27a* can be incorporated into extracellular vesicles and secreted into the extracellular space, we isolated and classified EVs from the non-infarct area of hearts as large EVs (lEVs) and small EVs (sEVs) by differential ultracentrifugation. Although lEVs and sEVs showed different size ranges (Fig. 4C and 4D), transmission electron microscopic analysis demonstrated that both lEVs and sEVs exhibited similar characteristic cup-shaped morphology and size overlap (Fig. 4A–4D). Moreover, sEVs shared the characteristic markers of EVs, including CD9, CD81, TSG101 and HSC70 with lEVs except for CD63 (Fig. 4G). However, qRT-PCR analysis showed that miRNA-27a* level was abundant in sEVs of infarcted hearts rather that in lEVs compared to that of normal hearts, suggesting miRNA-27a* may be upregulated and selectively packaged into sEVs and then secreted into extracellular space.
Figure 4. Cardiac-Derived EVs enrich miRNA-27a* post myocardial infarction.
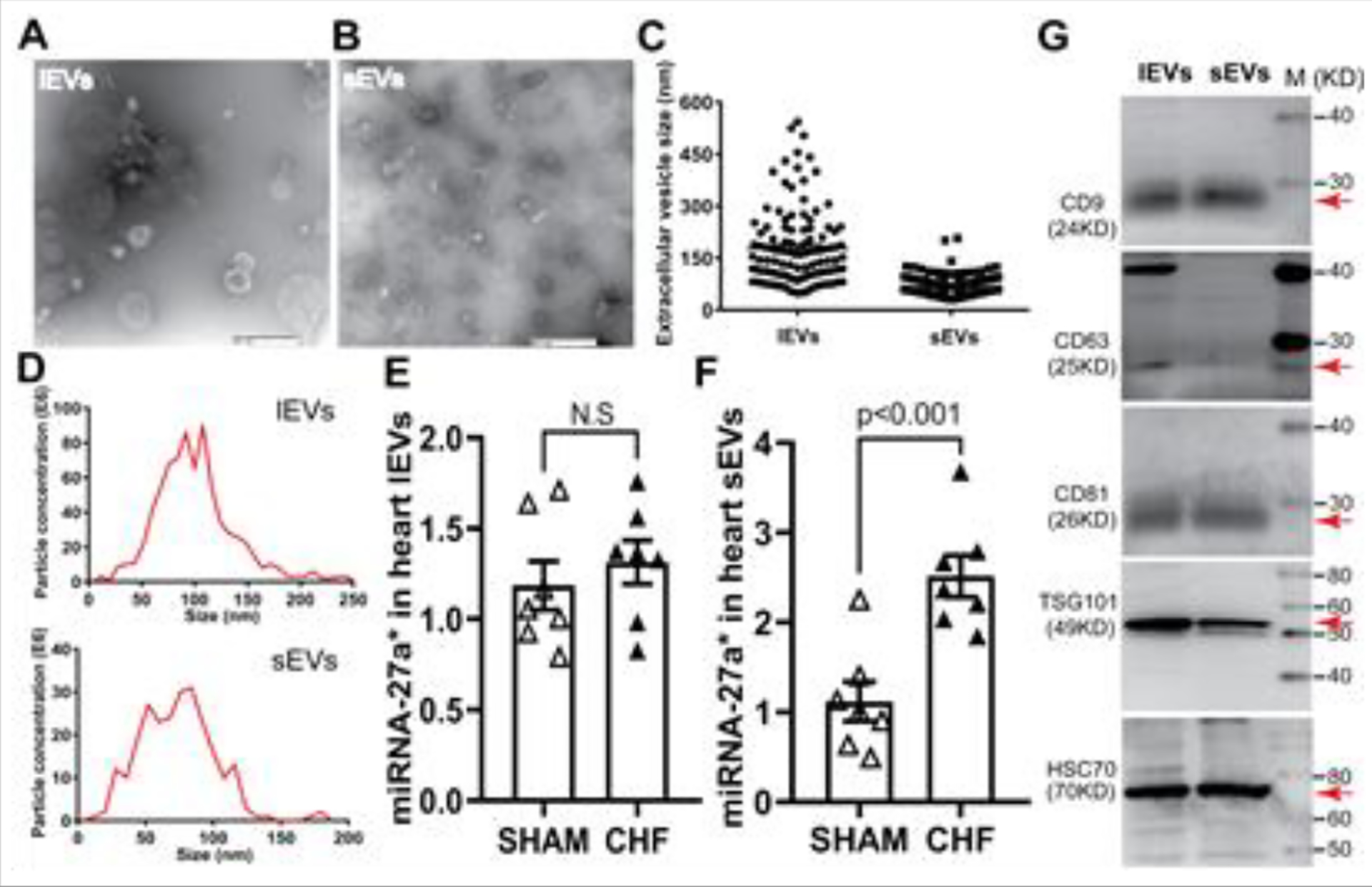
Transmission electron microscopy show the typical cup-shaped morphology of lEVs (A) and sEVs (B) scale bar is 500 nm; mean data are shown in C; EV characterizations by NanoSight analysis (D) and western blotting analysis; (G) qRT-PCR data show miRNA-27a* levels in both lEVs (E) and sEVs (F), cel-mir-39 was used as a spike-in control for EVs (±SEM, n=7).
3.5. miRNA-27a* is highly expressed in cardiac fibroblasts and preferentially secreted into extracellular space via EVs in response to angiotensin II stimulation
Cardiac fibroblasts are one of the largest cell populations in the heart, contributing to structural, biochemical, mechanical and electrical properties of the myocardium [4]. To determine if cardiac fibroblasts secrete miRNA-27a*-enriched EVs in pathological conditions, we cultured primary rat cardiac fibroblasts (RCF) and treated cells with 1 μM angiotensin II (Ang II) overnight. Conditioned media were collected and subjected to EV isolation by differential ultracentrifugation. We observed that cardiac fibroblast-secreted EVs not only demonstrated a typical cup-shaped morphology (Fig. 5A), but also expressed the characteristic markers of EVs, including CD63, TSG101 and HSC70 (Fig. 5B, 5C and 5D). NanoSight analysis shows the size distribution profiles of EVs secreted from cultured cardiac fibroblasts with peak diameters of 80 nm (Fig. 5E). In addition, qRT-PCR results showed that Ang II stimulated the upregulation of miRNA-27a* in RCF (Fig. 5G), which was predominantly secreted into the extracellular space via EVs (Fig. 5F).
Figure 5. Isolation and characterization of extracellular vesicles (EVs) derived from cardiac fibroblasts.
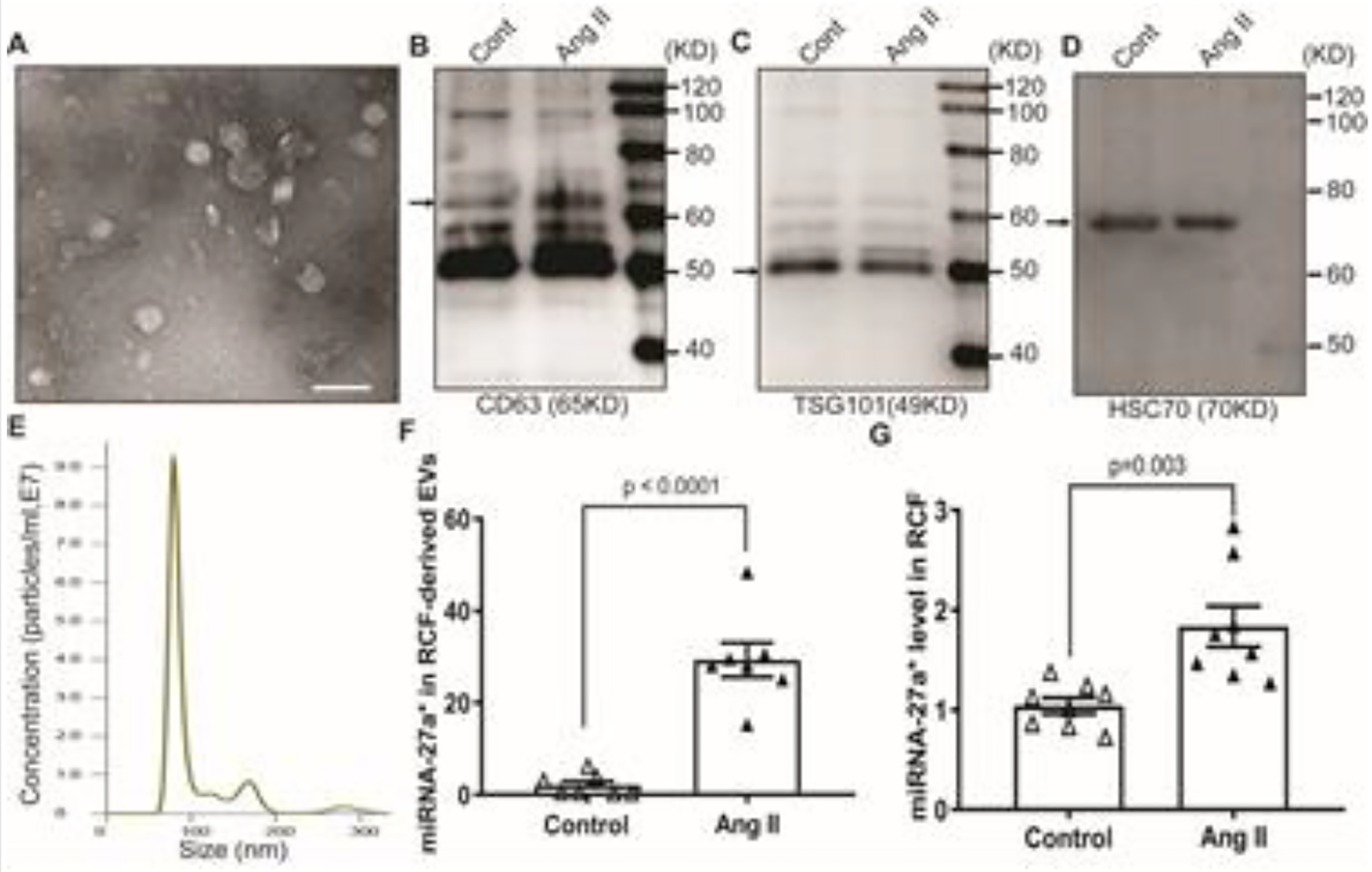
TEM shows the typical cup-shaped morphology of EVs (A) , scale bar is 100 nm; Western blotting analysis show the characteristic makers of EVs: CD63 (B), TSG101(C) and HSC70 (D); NanoSight analysis show the size distribution of EVs (E); qRT-PCR results show the miRNA-27a* level in cardiac fibroblast-derived EVs (F) and cardiac fibroblasts (G), cel-mir-39 was used as a spike-in control for EVs, and U6 snRNA was used as an internal control for cells (± S.E.M. n=8).
3.6. Ang II-induced RCF EVs can be taken up by cardiomyocytes and promote hypertrophic gene expression
To determine a direct communication between cardiomyocytes (CM) and RCF, we transfected CM (Fig. 6A) and RCF (Fig. 6B) with TSG1010-mCherry and CD63-EGFP plasmids, respectively. We then performed a co-culture of transfected cells as illustrated (Fig. 6C–a). After treatment with Ang II overnight, RCF were subjected to nuclear staining with DAPI and confocal microscopy. The results show the perinuclear distribution of GFP+ EVs in mCherry+ CM (Fig 6C, b-d). A mixed culture of mCherry+ CM and GFP+ RCF also confirmed Ang II-induced GFP+ EV uptake by mCherry+ CM (Supplemental figure 1). Moreover, we cultured primary CM from left ventricle which not only show mature morphology, and express cardiac troponin T (cTnT) (Fig. 7A, a, b), but also exhibit pulsatility (Supplemental video). We treated cells with EVs from cultured RCF without (EVs-Cont) and with Ang II stimulation (EVs-Ang II), respectively. Western blot analysis showed that EVs-Ang II treatment decreased PDLIM5 expression compared to EVs-Cont (Fig.7B and 7C). Furthermore, we incubated primary mature CM with EVs-Ang II from RCF as illustrated in Figure 8A, and analyzed the transcription of PDLIM5 and hypertrophic genes in CM by qRT-PCR. The results clearly demonstrated that EVs secreted from Ang II-treated RCF significantly decreased PDLIM5 mRNA (Fig. 8C), along with the increase of miRNA-27a* (Fig. 8B), whereas the transcription of hypertrophic marker genes in CM, including ANP (Fig. 8D), BNP (Fig.8E) and β-MHC (Fig.8F) were markedly increased after EVs-Ang II treatment.
Figure 6. Ang II promotes EV secretion from RCF and EV uptake by CM.
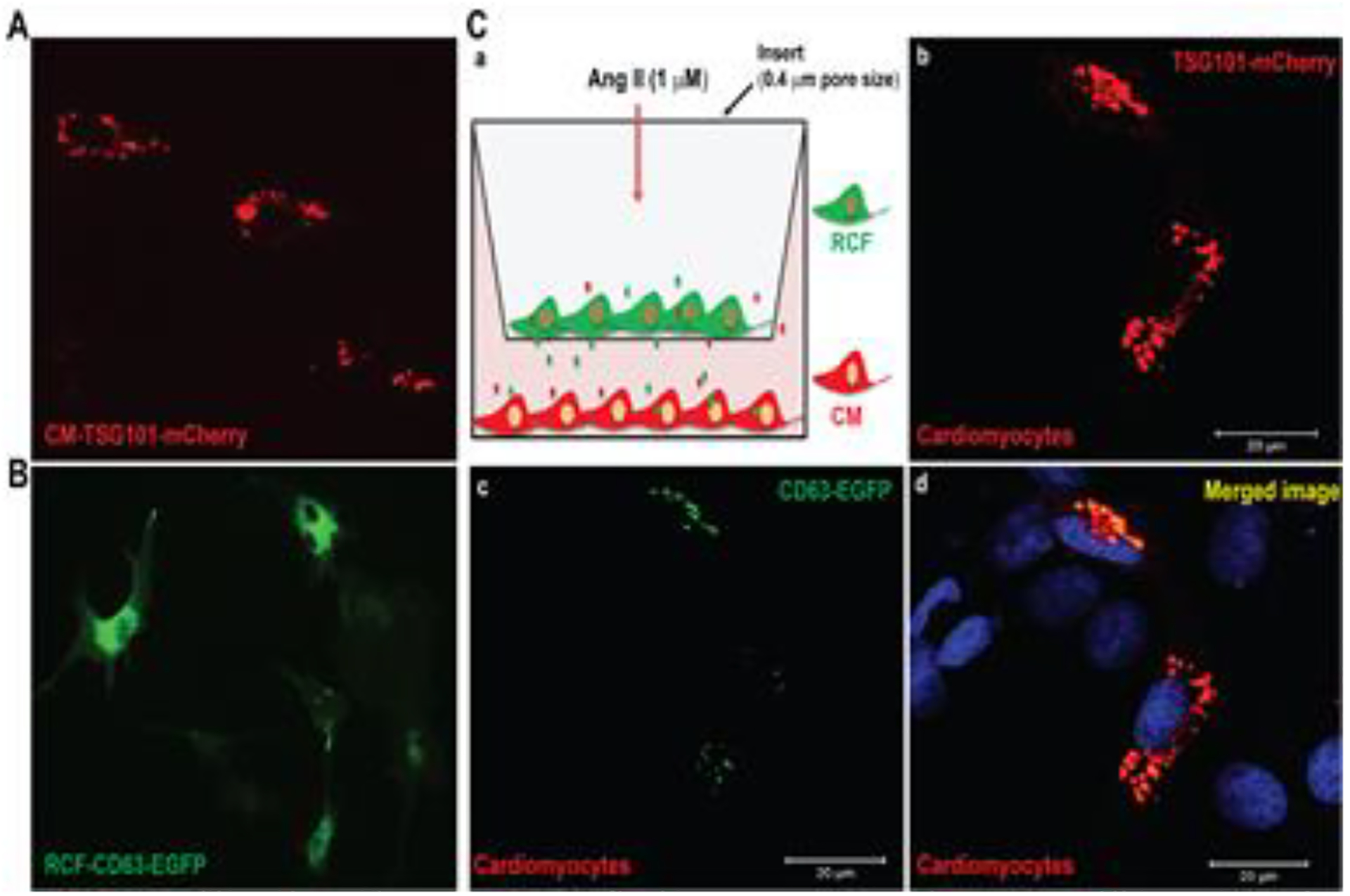
Primary CM and RCF were transfected with mCherry-TSG101 (A) and CD63-EGFP (B), respectively, and co-cultured as illustrated (C, a). Cells were treated with 1μM Ang II for 12 hours, and then subjected to confocal microscopy. Nuclei were stained with DAPI. Scale bar is 20 μm.
Figure 7. EV-enriched miRNA-27a* released from Ang II-treated RCF contribute s to PDLIM5 translational inhibition.
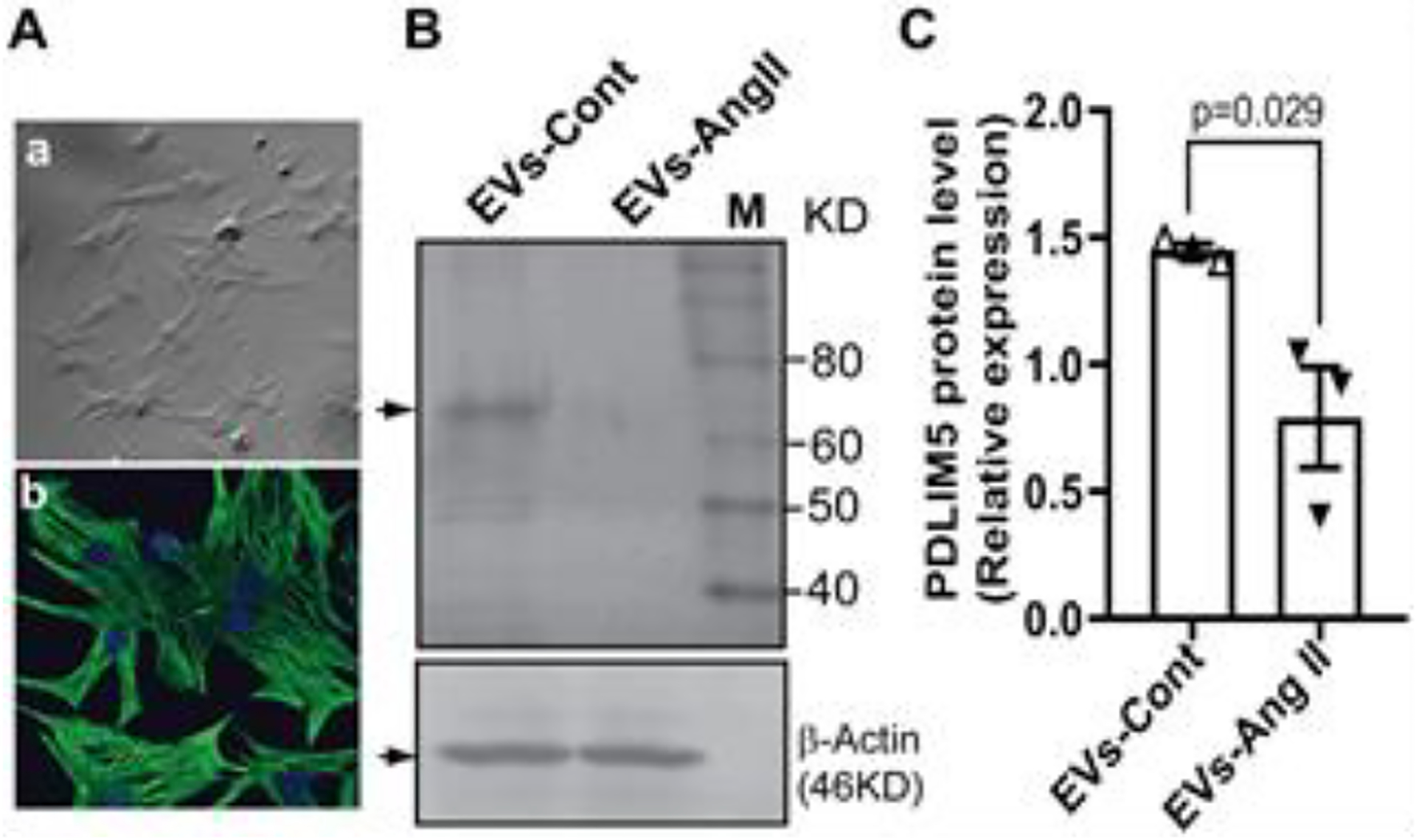
Primary cardiomyocytes show typical morphology (A-a) and cardiac troponin expression (cTnT, A-b). Nuclei are stained with DAPI. Scale bar is 20 μm; Western blot analysis show the down-regulation of PDLIM5 by EVs derived from Ang II-treated cardiac fibroblasts (EVs-Ang II) (B) and pool data were shown in (C), and β-actin was used as a loading control. Data represent the mean (± S.E.M) of three independent experiments,
Figure 8. EV-enriched miRNA-27a* from Ang II-treated RCFs promote s the expression of hypertrophic markers in CMs.
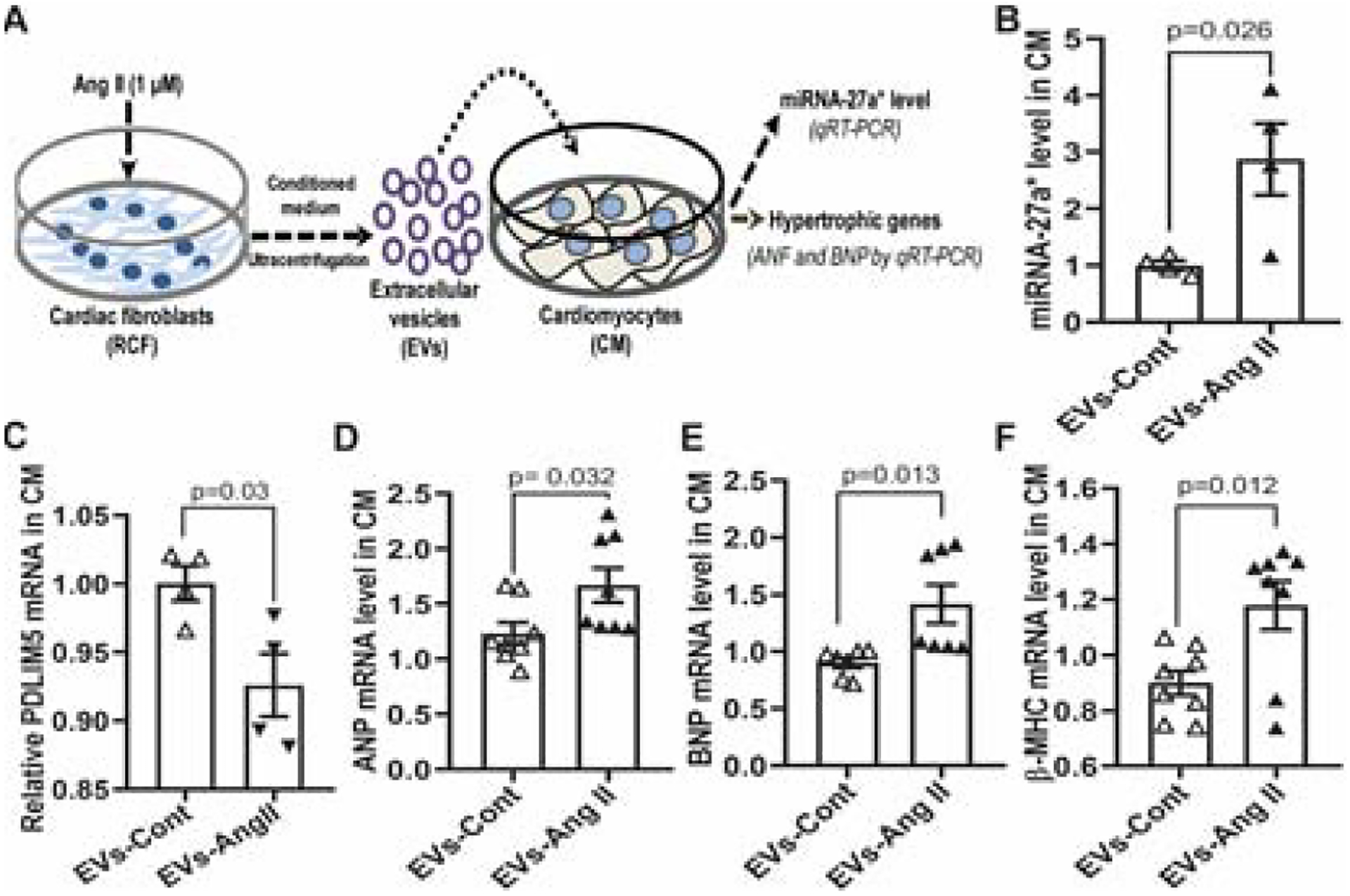
Schematic diagram of EV transfer experiments (A); Rat mature CM were treated with EVs from control RCF (EVs-Cont) and Ang II-treated RCF (EVs-Ang II) for 24 hours, respectively, and then subjected to qRT-PCR analysis for miRNA-27a* (B), U6 snRNA was used as an internal control (mean ±S.E.M); PDLIM5 (C); ANP (D); BNP (E); β-MHC (F), GAPDH was used as an internal control (mean ±S.E.M).
3.7. Systemic administration of miRNA-27a* inhibitor in CHF rats partially improves cardiac function
To evaluate the therapeutic effects of miRNA-27a* in vivo, we synthesized the miRNA-27a* inhibitor and administrated it weekly by i.p injections starting at 3 wks post-MI for 4 weeks as illustrated in Fig.9A. At 7 wks post-MI, CHF rats were subjected to echocardiography, qRT-PCR and western blot analyses. qRT-PCR data demonstrated that cardiac miRNA-27a* was significantly higher in CHF+NC (negative control) than that in Sham groups whereas the endogenous miRNA-27a* was significantly decreased after the administration of the miRNA-27a* inhibitor (Fig.9B). Although PDLIM5 mRNA was not different between CHF+NC and CHF+inhibitor groups (Fig.9C), of the hypertrophic genes in the CHF+NC group post-MI, BNP was significantly blocked (Fig.9D), and ANP and BNP levels were partially decreased (Fig.9E and 9F). Consistently, PDLIM5 protein in the CHF+inhibitor group also showed a trend towards improvement compared with that of CHF+NC group (Supplemental figure 4A and 4B). Importantly, CHF rats exhibited the expected decrease in stroke volume (Fig.9G) and cardiac output (Fig.9I) pre-treatment with miRNA-27a* inhibitor. However, following administration of the miRNA-27a* inhibitor stroke volume was significantly improved (Fig.9H), and the cardiac output trended to increase in the miRNA-27a* inhibitor treated-CHF rats compared to the pre-treatment group with miRNA-27a* inhibitor (Fig.9J). These data suggest that the systemic administration of a miRNA-27a* inhibitor potentially targets cardiac miRNA-27a* to recover PDLIM5 expression and improve myocardial function.
Figure.9. Systemic administration of miRNA-27a* inhibitor partially improve heart contractility.
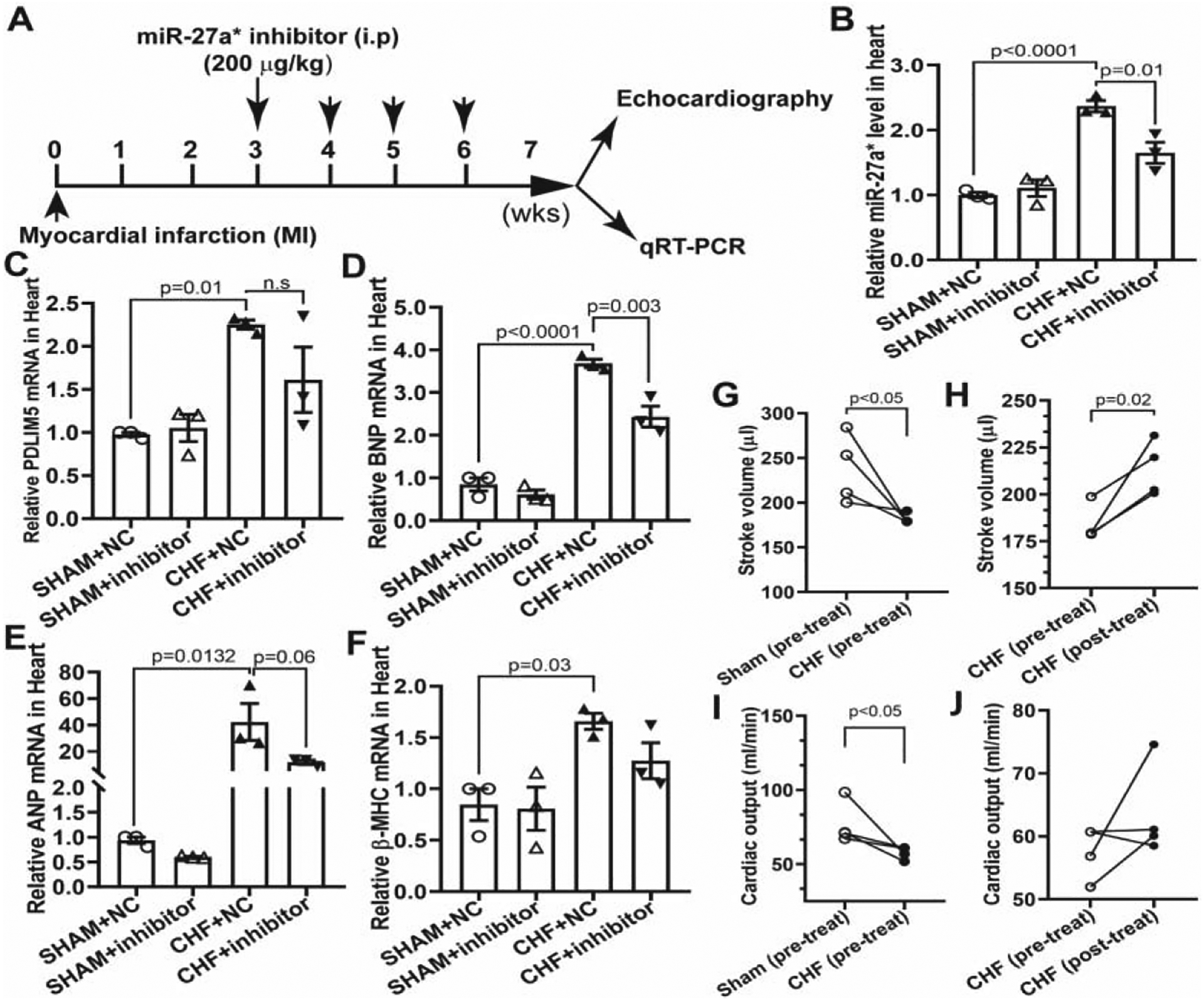
The experimental designs for the systemic delivery of miRNA-27a* inhibitor in CHF rats (A); qRT-PCR analysis of mature miRNA-27a* using specific TaqMan miRNA assay, U6 snRNA was used as an internal control (± SEM) (B); qRT-PCR analyses were performed with specific primers: PDLIM5 (C), BNP (D), ANP (E) and β-MHC (F), GAPDH was used as an internal control; Stroke volume (G and H) and cardiac output (I and J ) were analyzed by echocardiographic analyses (Mean ± S.E.M).
4. Discussion
In this study, we identified one miRNA passenger strand, miRNA-27a*, which shares a similar expression profile as miRNA-27a in the pathogenesis of CHF. Under cardiac stress, such as Ang II stimulation, miRNA-27a* was upregulated in cardiac fibroblasts, and preferentially packaged into extracellular vesicles and then secreted into the extracellular space, where miRNA-27a*-enriched EVs were taken up by CM leading to a hypertrophic phenotype. Moreover, we administrated a miRNA-27a* inhibitor to CHF rats and observed that systemic administration of miRNA-27a* inhibitor decreased cardiac miRNA-27a* and inhibited the expression of hypertrophic genes, partially improving cardiac function. Recently, miRNAs have emerged as paracrine signaling mediators in the pathogenesis of cardiovascular diseases [36–38]. In a previous study, we demonstrated that Nrf2/Keap1 signaling in CM can be dysregulated post myocardial infarction by RCF-secreted EV miRNAs, including miRNA-27a, miRNA-28a and miRNA-34a, all contributing to oxidative stress-mediated hypertrophic gene expression [15]. MiRNA-27a has been shown to have potential as a biomarker for diagnosis and prognosis of CHF as well as a therapeutic target for CHF. Clinical studies have demonstrated that miRNA-27a is upregulated in failing hearts and in the circulation of patients with CHF [22–24]. We have also demonstrated high abundance of miRNA-27a in EVs either from the circulation or from in vitro cultured cardiac fibroblasts [15]. In general, it has been assumed that the passenger strand of the miRNA duplex is degraded during miRNA biogenesis and only the guide strand of the duplex selectively becomes the mature functional miRNA. However, increasing evidence suggest that passenger strand miRNAs can also target mRNAs and have biological functions in the pathogenesis of diseases, such cancers [20, 21] and cardiovascular disease [16]. Interestingly, we observed that the passenger strand of miRNA-27a (also called miRNA-27a*) was not degraded during miRNA biogenesis, but significantly enriched in the infarcted heart, in the circulation and in cardiac-derived EVs (Fig. 1 and 4). It has been reported that miRNA passenger strands (star miRNAs) are produced by cardiac fibroblasts and secreted into the extracellular space via extracellular vesicles, such as miRNA21* which contributes to cardiomyocyte hypertrophy by targeting SORBS2, PDLIM5 and histone deacetylase-8 [16, 39]. We also observed that in vitro cardiac fibroblasts also produce and secrete EV-enriched miRNA-27a* in response to cardiac stress such as Ang II stimulation (Figure 5). Although both miRNA-27a and miRNA-27a* were predominantly incorporated into EVs of cardiac fibroblasts and extracellularly secreted, they can regulate the expression of hypertrophic genes by differently targeting Nrf2/ARE signaling [15] and PDLIM5 (Figure 3 and 7), respectively. miRNA-21 was also increased in the failing hearts, but it was only abundant in cardiac fibroblasts rather than in cardiac fibroblast-derived EVs and had no effect on cardiomyocyte function [40]. Moreover, we also observed that miRNA-27a* was only significantly encapsuled into small extracellular vesicles (sEVs) rather than large extracellular vesicles (lEVs) which matches the size range of cardiac fibroblast-derived EVs (Figure 4, Figure 5). However, miRNA-21* was enriched in both sEVs and lEVs (Supplemental figure 2), suggesting that miRNA-27a* may be predominantly produced and secreted by cardiac fibroblasts while miRNA-21* may be derived from various cell types other than cardiac fibroblasts. Using bioinformatic analysis software, we predicted the targets of miRNA-27a* and performed GO analysis to identify the molecular functions of potential target genes (Fig. 2). This analysis showed that some targets are involved in multicellular organismal development and contribute to cardiac development, contraction and pathological hypertrophy (Supplemetnal table 1). In particular, one potential target of miRNA-27a* is PDLIM5, also called Enigma homolog protein (ENH), which is one of the important Z-line-associated components at the boundary between sarcomeres to maintain muscle structure and function, and plays an increasingly important role in the pathogenesis of dilated cardiomyopathy [25]. PDLIM5 has been reported to contribute to impaired cardiac contraction and dilated cardiomyopathy after cardiac-specific knockout [26]. Interestingly, consistent with the abundance of miRNA-27a* in the infarcted heart, we observed that PDLIM5 protein was down-regulated in the progression of CHF (Fig. 3E), which was accompanied by the dysregulation of hypertrophic genes (Fig. 3B, 3C and 3D). Moreover, while PDLIM5 mRNA was significantly increased in the CHF group compared to that in the sham group at 3 weeks post-MI, there was no difference in PDLIM5 protein between the two groups, suggesting that a compensatory mechanism may be at play in the early stages (3wks post-MI) to enhance the transcription of pdlim5 in response to MI, but the existence of miRNAs, such as miRNA-21* and miRNA-27a* may lead to dysregulation of PDLIM5 protein by either mRNA degradation or translational repression, which are general mechanisms of miRNA action and dependent on the degree of complementarity between miRNA and target sequences [41]. This possibility was supported by the evidence that the PDLIM5 protein was significantly decreased at 6 weeks post-MI whereas the PDLIM5 mRNA level was still higher in CHF group compared to the sham group (Fig. 3A and 3F). Furthermore, PDLIM5 as a potential target of miR-27a* was confirmed by transfecting primary cardiomyocytes (CM) with miRNA-27a* mimic and inhibitor, respectively (Fig. 3G and 3H), suggesting that miRNA-27a* may also contribute to PDLIM5 translational inhibition.
Cardiac remodeling is a hallmark of chronic heart failure, including cardiomyocyte hypertrophy and cardiac fibrosis. The major cardiac cell involved in the remodeling process is the cardiomyocytes [42]. Left ventricular hypertrophy is an initial compensatory mechanism reducing cardiac stress ultimately resulting in heart failure. Recently, increasing evidence suggest that miRNAs are involved in several cardiovascular disorders [37, 43–45]. A single miRNA may regulate many genes as its targets, while one gene may be targeted by many miRNAs. In this study, we suggest that miRNA-27a* and miRNA-21* share the same target, PDLIM5. Both were abundant in cardiac fibroblast-derived EVs suggesting the existence of a general transport mechanism of star miRNAs. Moreover, cardiac fibroblast-derived EVs in response to cardiac stress, such as Ang II induce cardiac hypertrophy through the down-regulation of PDLIM5 probably attributed to both miRNA-27a* and miRNA-21* (Fig. 7, 8), suggesting that cardiac fibroblast secreted miRNA*-enriched EVs play a role in cardiac hypertrophy in the CHF state. miRNAs, in particular circulating EV-enriched miRNAs have recently been developed as therapeutic targets and biomarkers of cardiovascular disorders [12, 38, 46, 47]. Pharmacological inhibition of miR-21* has been shown to attenuate cardiac hypertrophy [16]. In this study, we administrated a miRNA-27a* inhibitor to CHF rats at 3 wks post-MI. We observed that this exogenous inhibitor significantly decreased the endogenous miRNA-27a* level induced by MI, partially prevented the expression of hypertrophic genes by increasing PDLIM5 expression which may consequently improve the myocardial function (Fig. 1). PDLIM5 as one of multiple Z-line-associated proteins is highly expressed in the heart and plays an important role in the pathogenesis of human cardiomyopathy [16, 25, 26]. Our findings suggest that the combination of pharmacological inhibition of miRNAs*, such as miRNA-21* and miRNA-27a* may provide a more efficient therapeutic strategy for CHF. Furthermore, these miRNAs* may be good potential biomarkers for early diagnosis of CHF.
Limitations.
Despite the potential involvement of cardiac fibroblast-derived EV-miRNA-27a* in myocardial infarction-induced cardiac hypertrophy, there are several limitations to this study that should be discussed. First, cardiac fibroblasts may produce and secrete various miRNA*s, such as miRNA-21* and miRNA-27a*, that target the same gene contributing to cardiac hypertrophy. It will be difficult to determine which miRNA* dominates in this pathological process thus the transfection of cardiac fibroblast-derived EVs with individual miRNA inhibitors following cardiac stress will be helpful to elucidate differences. Second, the therapeutic effects of the miRNA-27a* inhibitor in an animal model of CHF is tentative. It is not clear at the present time what dose and route of administration are most effective. It may be more effective to develop a cardiac homing peptide-based EV delivery system for miRNA-27a* inhibitor administration as therapy for CHF. Moreover, we administrated the miRNA-27a* inhibitor at 3 weeks post-MI. It remains to be investigated whether it is more effective to prevent myocardial dysfunction when administrated at early time points post-MI. Furthermore, the group size may need to be increased in this experiment in order to make the data significant. Third, other potential targets of miRNA-27a* as listed in Supplemental Table 1, may play more important roles in the pathogenesis of CHF in addition to PDLIM5. Their roles need to be clarified.
5. Conclusions
Taken together, under stress miRNA-27a* is upregulated in cardiac fibroblasts, and preferentially packaged into extracellular vesicles and secreted into the extracellular space. These star miRNAs are taken up by cardiomyocytes in which they target PDLIM5, leading to cardiac hypertrophy (Fig. 10). Our findings suggest that cardiac fibroblast-derived miRNA-27a* may be one of several star RNAs selectively incorporated into extracellular vesicles to act as a crucial paracrine signaling mediator during cardiac remodeling and may act as a potential biomarker and therapeutic target for CHF.
Figure 10. Schematic diagram illustrates the potential for EV-enriched miRNA27a* to contribute to cardiac hypotrophy through intercellular communication with cardiac fibroblasts.
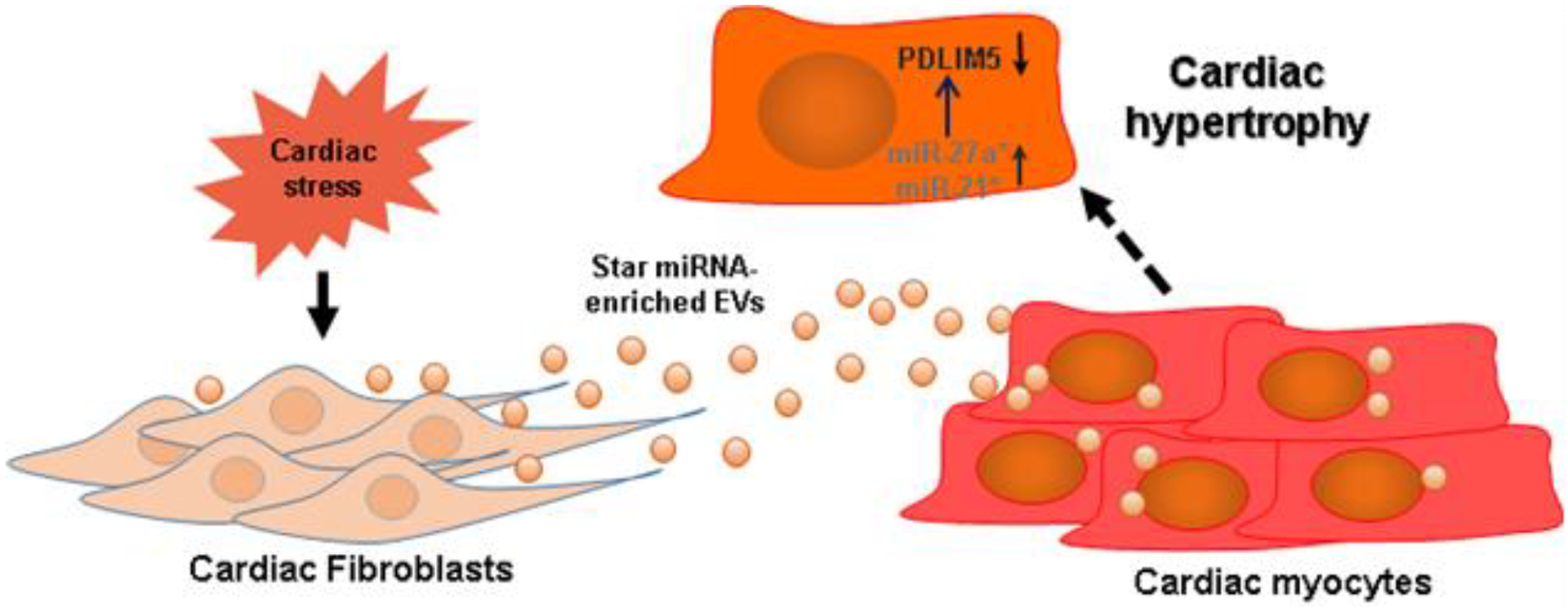
Under cardiac stress, cardiac fibroblasts secrete EV-enriched miRNA-27a* and/or miRNA-21*, which are transported to cardiomyocytes and regulates the expression of PDLIM5, leading to cardiac hypertrophy.
Supplementary Material
Highlights.
MicroRNA-27a passenger strand (miRNA-27a*) does not undergo intracellular degradation, but is highly upregulated and enriched in extracellular vesicles (EVs) in heart failure.
LIM domain 5 (PDLIM5) is one of the targets of miRNA-27a*, which is down-regulated in post MI heart failure.
Under cardiac stress, miRNA-27a* is highly expressed in cardiac fibroblasts and preferentially packaged into EVs, which are secreted into the extracellular space and taken up by cardiomyocytes.
EV-enriched miRNA-27a* may contribute to cardiomyocyte hypertrophy by targeting PDLIM5, and systemic administration of miRNA-27a* inhibitor may improve cardiac function.
Acknowledgements
The authors thank Ms Kaye Talbitzer for her expert technical and surgical assistance in these experiments. This work was supported by the National Institution of Health Grant P01 HL62222 to IHZ; CT was supported by American Heart Association (AHA) Career Development Award (19CDA34520004). IHZ was supported, in part, by the Theodore F. Hubbard Professorship for Cardiovascular Research and the University of Nebraska Foundation.
Abbreviations
- Ang II
angiotensin II
- CM
cardiomyocytes
- CHF
chronic heart failure
- cTnT
cardiac troponin
- EVs
extracellular vesicles
- HSC70
heat shock chaperone
- lEVs
large extracellular vesicles
- MicroRNA
miRNA
- NC
negative control
- PDLIM5
PDZ and LIM Domain 5
- RCF
rat cardiac fibroblasts
- sEVs
small extracellular vesicles
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TEM
transmission electron microscopy
- RIPA
radioimmunoprecipitation assay
- miRNA-21*
miRNA-21–3p
- miRNA-21
miRNA-21–5p
- miRNA-27a*
miRNA-27a-5p
- miRNA-27a
miRNA-27a-3p
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
None.
References
- [1].Bui AL, Horwich TB, Fonarow GC, Epidemiology and risk profile of heart failure, Nature reviews. Cardiology 8(1) (2011) 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roger VL, Epidemiology of heart failure, Circulation research 113(6) (2013) 646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Savarese G, Lund LH, Global Public Health Burden of Heart Failure, Cardiac failure review 3(1) (2017) 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Camelliti P, Borg TK, Kohl P, Structural and functional characterisation of cardiac fibroblasts, Cardiovascular research 65(1) (2005) 40–51. [DOI] [PubMed] [Google Scholar]
- [5].Zhou P, Pu WT, Recounting Cardiac Cellular Composition, Circulation research 118(3) (2016) 368–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tirziu D, Giordano FJ, Simons M, Cell communications in the heart, Circulation 122(9) (2010) 928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Talman V, Kivela R, Cardiomyocyte-Endothelial Cell Interactions in Cardiac Remodeling and Regeneration, Frontiers in cardiovascular medicine 5 (2018) 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lyu L, Wang H, Li B, Qin Q, Qi L, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL, Cui T, A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes, Journal of molecular and cellular cardiology 89(Pt B) (2015) 268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jiang X, Sucharov J, Stauffer BL, Miyamoto SD, Sucharov CC, Exosomes from pediatric dilated cardiomyopathy patients modulate a pathological response in cardiomyocytes, American journal of physiology. Heart and circulatory physiology 312(4) (2017) H818–h826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Boulanger CM, Loyer X, Rautou PE, Amabile N, Extracellular vesicles in coronary artery disease, Nature reviews. Cardiology 14(5) (2017) 259–272. [DOI] [PubMed] [Google Scholar]
- [11].Sluijter JPG, Davidson SM, Boulanger CM, Buzas EI, de Kleijn DPV, Engel FB, Giricz Z, Hausenloy DJ, Kishore R, Lecour S, Leor J, Madonna R, Perrino C, Prunier F, Sahoo S, Schiffelers RM, Schulz R, Van Laake LW, Ytrehus K, Ferdinandy P, Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology, Cardiovascular research 114(1) (2018) 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bei Y, Das S, Rodosthenous RS, Holvoet P, Vanhaverbeke M, Monteiro MC, Monteiro VVS, Radosinska J, Bartekova M, Jansen F, Li Q, Rajasingh J, Xiao J, Extracellular Vesicles in Cardiovascular Theranostics, Theranostics 7(17) (2017) 4168–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Das S, Halushka MK, Extracellular vesicle microRNA transfer in cardiovascular disease, Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology 24(4) (2015) 199–206. [DOI] [PubMed] [Google Scholar]
- [14].Zou X, Wang J, Chen C, Tan X, Huang Y, Jose PA, Yang J, Zeng C, Secreted Monocyte miR-27a, via Mesenteric Arterial Mas Receptor-eNOS Pathway, Causes Hypertension, American journal of hypertension (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tian C, Gao L, Zimmerman MC, Zucker IH, Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure, American journal of physiology. Heart and circulatory physiology 314(5) (2018) H928–h939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T, Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy, The Journal of clinical investigation 124(5) (2014) 2136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Datta R, Bansal T, Rana S, Datta K, Datta Chaudhuri R, Chawla-Sarkar M, Sarkar S, Myocyte-Derived Hsp90 Modulates Collagen Upregulation via Biphasic Activation of STAT-3 in Fibroblasts during Cardiac Hypertrophy, Molecular and cellular biology 37(6) (2017) e00611–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ha M, Kim VN, Regulation of microRNA biogenesis, Nature reviews. Molecular cell biology 15(8) (2014) 509–24. [DOI] [PubMed] [Google Scholar]
- [19].Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC, Widespread regulatory activity of vertebrate microRNA* species, RNA (New York, N.Y.) 17(2) (2011) 312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang X, Du WW, Li H, Liu F, Khorshidi A, Rutnam ZJ, Yang BB, Both mature miR-17–5p and passenger strand miR-17–3p target TIMP3 and induce prostate tumor growth and invasion, Nucleic Acids Res 41(21) (2013) 9688–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shan SW, Fang L, Shatseva T, Rutnam ZJ, Yang X, Du W, Lu WY, Xuan JW, Deng Z, Yang BB, Mature miR-17–5p and passenger miR-17–3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways, J Cell Sci 126(Pt 6) (2013) 1517–30. [DOI] [PubMed] [Google Scholar]
- [22].Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW, 2nd, Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support, Circulation 119(9) (2009) 1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuosmanen SM, Hartikainen J, Hippelainen M, Kokki H, Levonen AL, Tavi P, MicroRNA profiling of pericardial fluid samples from patients with heart failure, PloS one 10(3) (2015) e0119646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ovchinnikova ES, Schmitter D, Vegter EL, Ter Maaten JM, Valente MA, Liu LC, van der Harst P, Pinto YM, de Boer RA, Meyer S, Teerlink JR, O’Connor CM, Metra M, Davison BA, Bloomfield DM, Cotter G, Cleland JG, Mebazaa A, Laribi S, Givertz MM, Ponikowski P, van der Meer P, van Veldhuisen DJ, Voors AA, Berezikov E, Signature of circulating microRNAs in patients with acute heart failure, European journal of heart failure 18(4) (2016) 414–23. [DOI] [PubMed] [Google Scholar]
- [25].Mu Y, Jing R, Peter AK, Lange S, Lin L, Zhang J, Ouyang K, Fang X, Veevers J, Zhou X, Evans SM, Cheng H, Chen J, Cypher and Enigma Homolog Protein Are Essential for Cardiac Development and Embryonic Survival, Journal of the American Heart Association 4(5) (2015) e001950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cheng H, Kimura K, Peter AK, Cui L, Ouyang K, Shen T, Liu Y, Gu Y, Dalton ND, Evans SM, Knowlton KU, Peterson KL, Chen J, Loss of enigma homolog protein results in dilated cardiomyopathy, Circulation research 107(3) (2010) 348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Klocke R, Tian W, Kuhlmann MT, Nikol S, Surgical animal models of heart failure related to coronary heart disease, Cardiovascular research 74(1) (2007) 29–38. [DOI] [PubMed] [Google Scholar]
- [28].Wang HJ, Pan YX, Wang WZ, Gao L, Zimmerman MC, Zucker IH, Wang W, Exercise training prevents the exaggerated exercise pressor reflex in rats with chronic heart failure, Journal of applied physiology (Bethesda, Md. : 1985) 108(5) (2010) 1365–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gao L, Schultz HD, Patel KP, Zucker IH, Wang W, Augmented input from cardiac sympathetic afferents inhibits baroreflex in rats with heart failure, Hypertension (Dallas, Tex. : 1979) 45(6) (2005) 1173–81. [DOI] [PubMed] [Google Scholar]
- [30].Tian C, Gao L, Zhang A, Hackfort BT, Zucker IH, Therapeutic Effects of Nrf2 Activation by Bardoxolone Methyl in Chronic Heart Failure, J Pharmacol Exp Ther 371(3) (2019) 642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Loyer X, Zlatanova I, Devue C, Yin M, Howangyin KY, Klaihmon P, Guerin CL, Kheloufi M, Vilar J, Zannis K, Fleischmann BK, Hwang DW, Park J, Lee H, Menasche P, Silvestre JS, Boulanger CM, Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction, Circulation research 123(1) (2018) 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Leroyer AS, Ebrahimian TG, Cochain C, Recalde A, Blanc-Brude O, Mees B, Vilar J, Tedgui A, Levy BI, Chimini G, Boulanger CM, Silvestre JS, Microparticles from ischemic muscle promotes postnatal vasculogenesis, Circulation 119(21) (2009) 2808–17. [DOI] [PubMed] [Google Scholar]
- [33].Eding JE, Demkes CJ, Lynch JM, Seto AG, Montgomery RL, Semus HM, Jackson AL, Isabelle M, Chimenti S, van Rooij E, The Efficacy of Cardiac Anti-miR-208a Therapy Is Stress Dependent, Molecular therapy : the journal of the American Society of Gene Therapy 25(3) (2017) 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E, Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure, Circulation 124(14) (2011) 1537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Agarwal V, Bell GW, Nam J-W, Bartel DP, Predicting effective microRNA target sites in mammalian mRNAs, eLife 4 (2015) e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dutka M, Bobinski R, Korbecki J, The relevance of microRNA in post-infarction left ventricular remodelling and heart failure, Heart failure reviews 24(4) (2019) 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Das A, Samidurai A, Salloum FN, Deciphering Non-coding RNAs in Cardiovascular Health and Disease Frontiers in cardiovascular medicine, 5(73) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barwari T, Joshi A, Mayr M, MicroRNAs in Cardiovascular Disease, Journal of the American College of Cardiology 68(23) (2016) 2577–2584. [DOI] [PubMed] [Google Scholar]
- [39].Yan M, Chen C, Gong W, Yin Z, Zhou L, Chaugai S, Wang DW, miR-21–3p regulates cardiac hypertrophic response by targeting histone deacetylase-8, Cardiovascular research 105(3) (2015) 340–52. [DOI] [PubMed] [Google Scholar]
- [40].Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S, MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts, Nature 456(7224) (2008) 980–4. [DOI] [PubMed] [Google Scholar]
- [41].Ryan B, Joilin G, Williams JM, Plasticity-related microRNA and their potential contribution to the maintenance of long-term potentiation, Frontiers in molecular neuroscience 8 (2015) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cohn JN, Ferrari R, Sharpe N, Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling, Journal of the American College of Cardiology 35(3) (2000) 569–82. [DOI] [PubMed] [Google Scholar]
- [43].Indolfi C, Curcio A, Stargazing microRNA maps a new miR-21 star for cardiac hypertrophy, The Journal of clinical investigation 124(5) (2014) 1896–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sucharov CC, Kao DP, Port JD, Karimpour-Fard A, Quaife RA, Minobe W, Nunley K, Lowes BD, Gilbert EM, Bristow MR, Myocardial microRNAs associated with reverse remodeling in human heart failure, JCI insight 2(2) (2017) e89169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang Q, Yu X, Dou L, Huang X, Zhu K, Guo J, Yan M, Wang S, Man Y, Tang W, Shen T, Li J, miR-154–5p Functions as an Important Regulator of Angiotensin II-Mediated Heart Remodeling, Oxidative medicine and cellular longevity 2019 (2019) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jaquenod De Giusti C, Santalla M, Das S, Exosomal non-coding RNAs (Exo-ncRNAs) in cardiovascular health, Journal of molecular and cellular cardiology 137 (2019) 143–151. [DOI] [PubMed] [Google Scholar]
- [47].Jansen F, Nickenig G, Werner N, Extracellular Vesicles in Cardiovascular Disease: Potential Applications in Diagnosis, Prognosis, and Epidemiology, Circulation research 120(10) (2017) 1649–1657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.