

Abstract
The vesicular glutamate transporters (VGLUTs) bind and move glutamate (Glu) from the cytosol into the lumen of synaptic vesicles using a H+-electrochemical gradient (ΔpH and Δψ) generated by the vesicular H+-ATPase. VGLUTs show very low Glu binding and to date, no three-dimensional structure has been elucidated. Prior studies have attempted to identify the key residues involved in binding VGLUT substrates and inhibitors using homology models and docking experiments. Recently, the inward and outward oriented crystal structures of D-galactonate transporter (DgoT) emerged as possible structure templates for VGLUT. In this review, a new homology model for VGLUT2 based on DgoT has been developed and used to conduct docking experiments to identify and differentiate residues and binding orientations involved in ligand interactions. This review describes small molecule-ligand interactions including docking using a VGLUT2 homology model derived from DgoT.
Keywords: Vesicular glutamate transporter, Glutamate, Substrate, Inhibitor, Homology models, Docking
1. Introduction
1.1. Vesicular glutamate transporters (VGLUTs): cellular location, isoforms, and function
The excitatory neurotransmitter L-glutamate (Glu) is concentrated in the cytosol of neurons as a result of reuptake of released Glu via membrane transporters or produced precursors [1–7]. Vesicles are then filled with Glu via the H+-dependent vesicular glutamate transporters of which there are three isoforms VGLUTs 1–3 [6,8] belonging to the class SLC17 type I. During glutamatergic synaptic transmission, Glu-filled vesicles fuse with the plasma membrane and release Glu, whereupon ionotropic (iGluR) and metabotropic receptors (mGluR) are activated (Fig. 1). To terminate the transmission, several high-affinity, Na+-dependent protein transporters (e.g., EAAT3) remove Glu from the synapse [5,8,9] whereupon it is repackaged into vesicles. Glu is also synthesized by vesicle-bound aspartate amino transferase from α-ketoglutarate [10] where it is brought into vesicles by VGLUT for additional cycles of release [6].
Fig. 1.

Left: Glu release, recycling and receptor action. Right: Glu transport into vesicles and inhibitor blocking Glu uptake.
The VGLUT1 and VGLUT2 isoforms are largely expressed in brain whereas VGLUT3 is expressed in brain and elsewhere [11–13]. VGLUT1 and 2 are responsible for the majority of glutamatergic neurotransmission but are expressed in different populations of cells [14]. VGLUTs show a large sequence similarity especially in the transmembrane domains where glutamate is recognized and transported, but differ in the cytoplasmic domains. Functionally, VGLUTs transport Glu using a H+ electrochemical gradient (ΔpH and Δψ) generated by the vesicular H+-ATPase [7,15,16] but with low (1 mM) Glu binding [17]. A comparison of vesicular neurotransmitter mechanisms was recently covered [18]. VGLUTs have a biphasic dependence on Cl− [19] identified as an allosteric activator [20]. The bioenergetics and role of Cl− in the regulation of VGLUT function was reviewed [2,21]. Using the structure of E. coli D-galactonate transporter (DgoT), Edwards and co-workers effectively delineated a mechanism in which VGLUT concentrates anions opposite to the translocation of H+ [22] [Edwards, this issue].
1.2. VGLUT pharmacology: current limitations
Despite significant advances in our understanding of VGLUT mechanism and function, these proteins are one of the few remaining in glutamatergic signaling that has yet to be pharmacologically characterized and routinely manipulated by small molecule ligands. Not surprisingly, there has been far greater interest in glutamate receptors and EAATs as druggable targets [23,24] because VGLUTs are more challenging as intracellular, membrane bound, low expression [25] proteins with relatively low affinity for the natural substrate. Pharmacologic assessment of VGLUT ligands [26] also has been a procedural limitation, in part, because Glu is transported using a H+ electrochemical gradient (ΔpH and Δψ) generated by the vesicular H+-ATPase [7,15,16,27]. As a result, assays are mostly limited to radiochemical uptake (3H-Glu) in intact vesicles although liposomal preparations have been reported that may furnish screening options [17,20,28–30].
Because potent and highly specific regulators of glutamate vesicle loading and synaptic release are lacking it is unclear if ligand development would be of therapeutic benefit in CNS disorders where normal excitatory signaling has been disrupted (e.g., epilepsy, Alzheimer’s, schizophrenia, stroke, etc.). Still, high affinity, selective ligands targeted to a specific protein makes it possible to isolate and/or regulate a single process in the cycle of synaptic transmission and of great value. Ligands targeted to VGLUT, for example, could control the amount of released Glu during neurotransmission by altering the amount of Glu that can be packaged in vesicles prior to a stimulus. VGLUT-selective ligands that change vesicle filling of Glu could regulate pre-synaptic quantal size and cause a corresponding change in extracellular, Glu-mediated events. But, one of the more pressing impediments to the development of VGLUT ligands has been a lack of structural information that in turn, diminishes activity in ligand development. Recently, however, there have been a number of indispensable advances in VGLUT mechanism and function [22,30,31] that will stimulate new investigations, and would benefit from better ligands.
2. VGLUT homology models and structure correlations
2.1. Previous VGLUT homology models
Computational analyses of VGLUT have been conducted including predictions of the transmembrane domains, homology models, binding domains, pharmacophores and mechanism [17,22,32–37]. Lacking a crystal or solution structure, homology models have helped understand the relationship between VGLUT structure and function. Juge et al. [17] combined mutagenesis experiments with computational approaches to verify key binding residues in a homology model of VGLUT2 based on the glycerol-3-phosphate transporter (GltP; pdb version 1PW4): a common structure template for the major facilitator superfamily (MFS). The resultant VGLUT2 model was a 12-transmembrane structure with R184 noted as critical to function showing a complete loss of transport activity as R184K. That R184 was conserved across all SLC17 transporters implicates it as a candidate for glutamate anion binding. Four additional residues, R88, H128, E191 and R322, were also identified as important to Glu binding and transport. The E191A, E191Q and E191D mutations all showed markedly reduced activity, which indicated not only the importance of the negatively charged E191 residue but also the importance of longer reach into the binding domain. Indeed, the homology model shows that the three charged residues required for glutamate transport (H128 in TM2 and R184 and E191 in TM4) were found at the closed end of the cavity between the N and C domains. These residues were clustered and proposed to be where Glu recognition was likely to occur. Surprisingly, the mutations at R184, H128 and E191 did not affect inorganic phosphate transport suggesting different residues may be involved in this function.
Almqvist et al. [32] developed a 3D model for human VGLUT1 based of GlpT but refined within a phospholipid bilayer. Docking studies showed that substrates bound the model at two different sites within a pore or cavity (27 Å long) that were also a shared location for inhibitors. The hVGLUT1 model revealed eight residues including R80, H120, E183, R176, R314, H348, E388 and H479 with side chains extended into the putative binding domain (note: VGLUT1 residues are shifted by eight less relative to VGLUT2). Residues R80 and R314 defined one substrate binding cluster and R176 and H120 defined the second. Docking of the azo-dye inhibitors of VGLUT, Evans Blue (EB) and Chicago Sky Blue (CSB) showed in level binding to R80 and R314 whereas Trypan Blue (TB) bound these residues and R176 possibly explaining the more potent inhibition. Interestingly, the pore or channel is large enough to accommodate the entirety of the azo-dye structures. The Juge [17] and Almqvist [32] VGLUT models were highly informative providing useful insights and a deeper understanding of substrate and inhibitor binding despite low sequence identity to GlpT (10–15%).
A parallel model to investigate VGLUT structure-function is the homologue sialin: an SLC17 family co-transporter that removes sialic acid from lysosomes. Pietrancosta et al. [38] combined experimental and computational approaches to show that sialin contains a single central binding site by docking high-affinity sialic acid analogs to the GlpT-based model [38]. In the study, a single binding site was found at a specific depth and validated by site specific mutation and high-throughput screening of structurally unrelated, high-affinity competitive inhibitors.
In addition to the exceptional mechanistic understanding of VGLUTs provided by the aforementioned models [17,32,38], the inward/outward conformer crystal structures of D-galactonate transporter (DgoT) [22] and sequence similarity support the notion that DgoT could serve as a useful template for a computer-aided homology model and structure-function analyses. Although VGLUTs rely on membrane potential to accumulate Glu in vesicles (inside positive) whereas other SLC17 members use H+ symport to drive anions outward, there were numerous consistencies between the two proteins. Most notably, VGLUTs contain residues key to Glu transport such as R88, R184 and E191 (TM4) with E191 potentially serving as a possible protonation site for allosteric activation rather than coupling to H+ as found with DgoT. In turn, protonation of E191 from the outside may permit R88 to bind and translocate the substrate Glu.
In the sections to follow, the interaction of VGLUT2 with substrates, non-substrates, and inhibitors is presented using computational models and docking experiments to identify strategic binding locations and residues. Foremost, an updated VGLUT2 homology model based on DgoT has been advanced to compare with prior GlpT models and in this review used to assess ligand interactions. It is hoped that the models and depictions can provide insightful measures that can help investigators develop new substrates, alternate substrates and potent inhibitors that can be eventually translated into useful therapeutics.
2.2. A VGLUT2 structure homology model based on the D-galactose transporter (DgoT)
The finding that mechanistic and structural similarities exist between DgoT and VGLUT [22] that encourages a new homology models based on DgoT to assess substrate and inhibitor interactions especially since both inward open (pdb: 6E9N) and outward substrate bound (pdb: 6E9O) conformations are available. The sequence from mouse VGLUT2 was chosen to produce a homology model because it is widely expressed in brain, shares 98% sequence conservation with rat (rat brain is the most widely used to assay VGLUT activity), and mouse is among the easiest organisms to mutate genetically for validation. Given the sequence similarities, selecting a homology model based on mouse VGLUT2 was expected to be representative of the three isoforms across mammalian subtypes. The VGLUT1–3 isoforms differ substantially in the N- and C-terminal regions and a VGLUT1 homology model based on DgoT was also examined to establish that no changes in transmembrane domain regions occurred. Thus, the mVGLUT2 FASTA sequence was used to build a homology model using DgoT as a structural template (Scheme 1). T-coffee alignment [39] indicates extensive similarity in the transmembrane regions between VGLUT2 and DGOT (Scheme 1) further supporting selection of this model. Fifty-seven residues from the N-terminal domain and seventy-five from the C-terminal domain of VGLUT2 were skipped and are not part of the model.
Scheme 1.

LEFT: mVGLUT2 (top line) and DGOT (lower line) sequence similarity alignment using T-Coffee (score 891/1000) [40]. Legend: inside or cytoplasmic domains = yellow; outside or lumenal domains = blue; helical membrane regions = pink. Single, fully conserved residue = asterisk(*); conservation between groups of strongly similar properties = colon(:); conservation between groups of weakly similar properties = period(.).
The resultant homology models derived from multiple PSI-BLAST (3 iterations) and BLAST searches conducted separately or within YASARA build command [41] further identified DgoT with an alignment score/coverage/quality score of 264/70%/0.684 (6E9N) and 235/68%/0.584 (6E9O) based on a hybrid of twenty-one ranked models. The final model had an overall Z-score of −1.096. The VGLUT2 homology model is shown in side- and end-views with the R88, H128 and R322 residues marked to orient the displays (see Fig. 2).
Fig. 2.
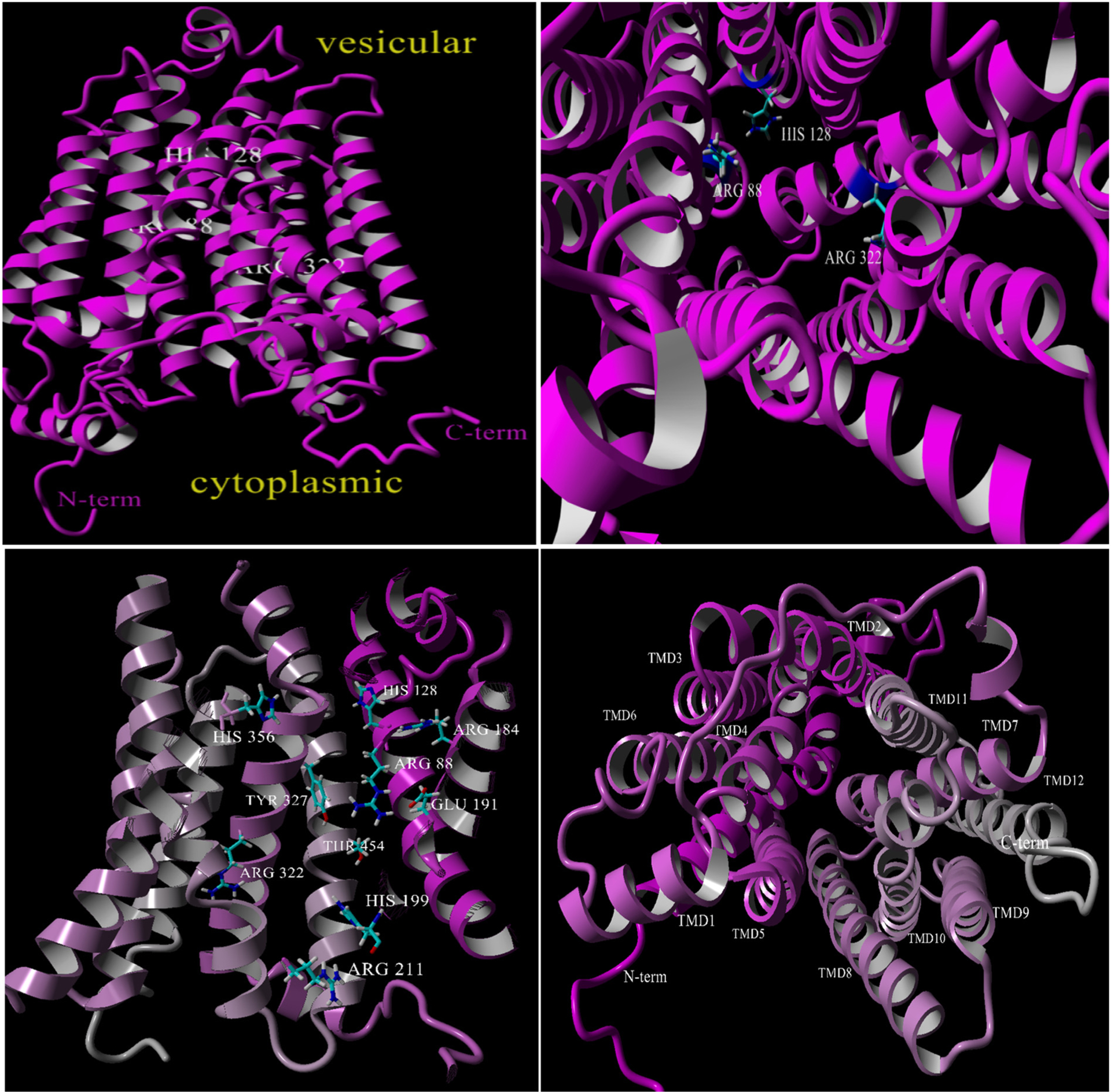
Top left: VGLUT2 homology model based on DgoT (magenta) as a side view. Top right: VGLUT2 homology model view from cytoplasmic side showing the position of R88, H128 and R322 for clarity. Bottom left: VGLUT2 (cutaway) showing previously identified residues and as included in docking studies. Bottom right: assignment of the VGLUT2-DgoT homology model transmembrane domains (TMDs).
The VGLUT2-DgoT homology model is in structural agreement with those constructed prior using rat VGLUT2 [17] and human VGLUT1 [32] with GlpT as template including the N- and C-terminus positioned cytosolic, twelve transmembrane domains, and most positions of the ten residues previously implicated in binding and transport were largely conserved in topology (Fig. 2; bottom left). The alignment of VGLUT1 and VGLUT2 homology models using DgoT as template yielded an RMSD = −13.1 (total backbone alignment) using the MUSTANG algorithm in YASARA [42], indicating high similarity among the isoform structures (not shown). In sum, a VGLUT2 homology model based on a DgoT has been produced that adequately represents the structure of mammalian VGLUTs with positioning of residues that enable preliminary assessment of substrate and inhibitor binding.
To bring a greater understanding of substrate, non-substrate and inhibitor binding phenomena, the VGLUT2 homology model was then used to conduct docking studies in YASARA (Autodock). The substrate and inhibitor binding domains in the VGLUT2 homology model were identified by comparison with reported models [17,22,32] and key residues defined in these and other studies used to define the binding sites. The simulation cell coordinates spanned two-thirds the length of VGLUT2 (Y-axis ~50 Å) with X-axis = 30 Å and Z-axis = 40 Å inclusive of each of the following ten residues: R88, H128, R184, E191, H199, R211, R322, Y327, H356 and T454. The backbone of VGLUT2 was fixed while the ten residues were free to move along with ligand in the simulation. Functional groups in the ligands were minimized in their ionized form prior to docking runs. Each docking model is the result of twenty-five VINA runs yielding results sorted by binding energy clusters that afforded 8–12 distinct complex conformations that differ by at least 5.0 Å heavy atom RMSD after superposing on the receptor. The results from the docking runs were typically acquired and presented as the top energy binding pose (except as noted), the pose exported into 2D protein ligand map programs (PLIP, LigPlus, etc.) to identify the interactions, and visualized in PyMol ver 2.1 (Schrodinger) or YASARA. Each of the individual ligands for which docking was conducted appears in a subsection along with the top binding energy value in parentheses as an indication of the relative stability. Some of the binding orientations, docked molecules and structure overlays for VGLUT2-ligand displays had domains removed to assist with clarity and to more easily identify participating residues that would have been occluded.
3. Docking of substrates and non-substrates
VGLUTs are highly selective transporters and recognize very few compounds as substrates (Fig. 3). L-Glutamate, of course, is recognized and transported as substrate but even the highly similar structures D-glutamate, D- and L-aspartate and GABA are not. Likewise, neither serotonin nor acetylcholine are substrates for VGLUT. Erythro-(2S,4R)-4-methyl-DL-glutamate, 4-methylene-L-glutamate, and the conformationally-restricted amino acid analog 1S,3R-trans-aminocyclopentane dicarboxylic acid (trans-ACPD) are competitive substrates [43]. trans-ACPD also acts as an inhibitor, and as with the D-glutamate, the stereoisomer cis-ACPD is inactive. The uptake of glutamate is regulated allosterically by protons and at low concentration of chloride but at higher concentrations of Cl− uptake is inhibited [20,30,44,45]. The Km values for Glu at VGLUT is 100- to 1000-fold lower (~1–2 mM) than the excitatory amino acid transporters (EAATs) with Km ~4–40 μM. Currently, there are few VGLUT substrate analogs and none are specific for any VGLUT isoform. It would be useful to identify alternate substrates and also substrates that are selective for the VGLUT isoforms to separate and potentially regulate their physiologic function, and docking models that reveal differences in substrate and non-substrate binding may be helpful in this effort. In the following sections, results of the docking between some substrates and non-substrates with the VGLUT2 homology model derived from DgoT are presented.
Fig. 3.

Structures of substrates and non-substrates.
3.1. L-Glutamate (5.5 kcal/mol) and D-glutamate (5.0 kcal/mol)
Docking of the stereoisomers of glutamate into the homology model will be helpful as a starting point toward understanding why VGLUT recognizes and transports very few substrates. When L-glutamate was docked into VGLUT2 (Fig. 4), several poses ranging in binding energy from 2.5 to 5.5 kcal/mol were obtained. The top two poses were nearly equal in binding energy and positioned L-glutamate with either the α-amino acid group inward toward R88 or with the γ-carboxylate facing R88. Distances calculated for the top pose place the α-carboxylate oxyanion of L-glutamate in a salt bridge distance of ~5 Å with R88, the amino group binding Y327 at ~2.3 Å and T454 in a hydrogen bond with the γ-carboxylate oxyanion. When the α-amino acid head group was oriented away from R88 the γ-carboxylate group is < 2.5 Å from R88 and protonated amino group binding to Y327 at ~3.4 Å. The docking also showed that T454 had a hydrogen bond to the protonated amino moiety of L-glutamate. Interestingly, R322 was not found to bind substrate in either of these two poses but was evident in the lower energy poses consistent with a possible second substrate binding site proposed for VGLUT1 [32] and VGLUT2 [17].
Fig. 4.

L-Glutamate (left) and D-glutamate (right) docked into VGLUT2 homology model showing nearby interactions.
The D-glutamate docking poses differed from L-glutamate (Fig. 4). The two top poses were about 5.0 kcal/mol in binding energy and again positioned in VGLUT2 with the α-amino acid group or the γ-carboxylate pointing inward into the luminal side. The α-carboxylate inward orientation placed it within ~3.5–4.0 Å to R88 but also binding Y327 (2.6 Å) but neither the α-amino nor the γ-carboxylate showed binding to a residue with a favorable distance. The outward (pointing toward the cytosolic side) orientation likewise placed the γ-carboxylate within 2.5 Å of R88 and Y327 but again showed that the α-amino and γ-carboxylate were without nearby interactions. T454 was not hydrogen bound to D-glutamate in either instance. Views of the two luminal facing poses for L- and D-glutamate in the VGLUT2 homology model are shown. Overall, the difference in docking of the two stereoisomers shows that L-glutamate and not D-glutamate forms an interaction with Y327. Thus, it appears that Y327 may be important for the recognition of L-Glu or possibly selecting against D-glutamate. Docking analyses of other substrate and non-substrates may help interpret this difference further (Fig. 4).
3.2. L-Aspartate (5.3 kcal/mol)
L-Aspartate, the lower homologue of glutamate, is not a VGLUT substrate. Docking provided a number of binding energy poses from 2.2–5.3 kcal/mol but the protein-ligand maps revealed only two key interactions: (a) salt bridge (4.4–4.9 Å) between the aspartate β-carboxylate with R88 and (b) hydrogen bond between T454 and the amino moiety. Weak hydrophobic interactions to Y195 and H-bonding to Y327 were also observed. The α-carboxylate group faced downward and away from R88 forming weak interactions with S323 and Y453. Overall, L-aspartate showed few of the favorable interactions with residues found for the substrate L-glutamate, in particular poor interactions with Y327.
3.3. Trans-aminocyclopentane dicarboxylic acid (trans-ACPD; 6.5 kcal/mol) and cis-aminocyclopentane dicarboxylic acid (cis-ACPD; 6.5 kcal/mol)
1S,3R-trans-Aminocyclopentane dicarboxylic acid (trans-ACPD) is a competitive substrate at low concentration and an inhibitor at high concentration [43] but cis-ACPD is inactive as either substrate or inhibitor despite identical binding energies (6.5 kcal/mol). Docking of both isomers (Fig. 5) was attempted to show differences in the binding orientations of these isomers. The high energy poses placed trans-ACPD with the α-alpha amino acid facing toward R88 and showed the same three interactions as L-glutamate including the characteristic 4.0 Å salt bridge interaction with R88, the hydrophobic interaction with Y195, and hydrogen bond to Y327 (~3.0 Å). Additional weaker interactions included hydrogen bonds to S323 and Q319 to the γ-carboxylate (Fig. 5).
Fig. 5.
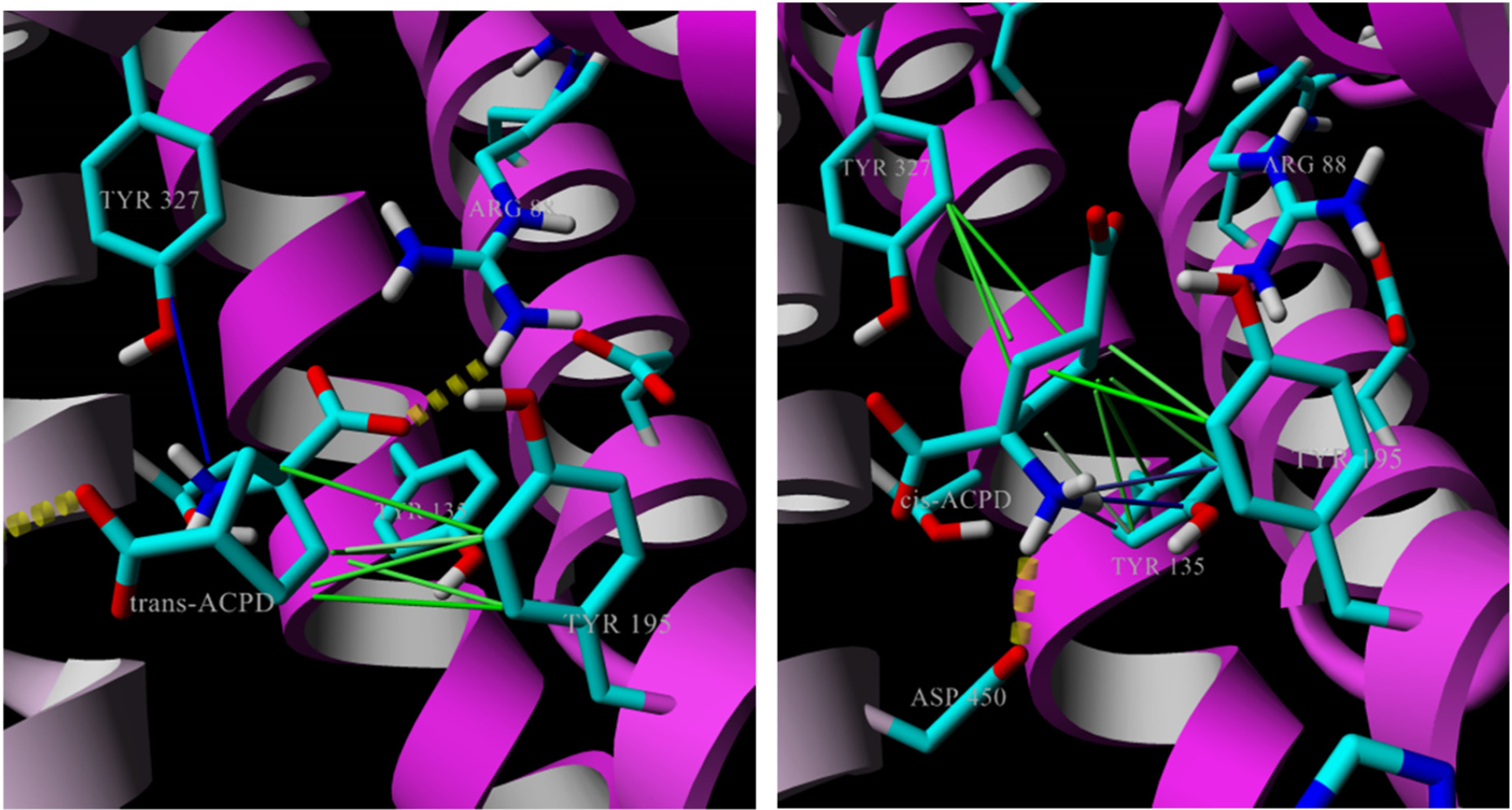
Trans-ACPD (left) and cis-ACPD (right) docked into VGLUT2 homology model showing nearby interactions. Trans-ACPD orients similarly to L-Glu with charged amino toward Y327 and carboxylate interaction with R88 whereas the amino of cis-ACPD faces toward Y135/D450. S323 and Q319 not shown.
Conversely, the most favorably binding energy poses positioned cis-ACPD opposite in orientation with the γ-carboxylate forming a salt bridge to R88 (4.4 Å), and placed the α-alpha amino acid in interactions with S323 and D450, and hydrophobic interactions to Y195 were not found. As indicated, trans-ACPD and cis-ACPD show nearly identical binding energies (~6.5 kcal/mol), however, trans-ACPD shares a number of interactions with that of L-glutamate whereas cis-ACPD forms interactions with residues that presumably do not favor transport. From these docking studies, it is clear that the binding energy alone is not sufficient to define the differences between substrates and non-substrates, and that key interactions with specific residues within VGLUT2 are needed to initiate the mechanism. It is also possible that the inhibition of VGLUT observed at higher trans-ACPD concentrations is due to binding specific residues and appear to be those in common with those that bind Glu.
3.4. Erythro-(2S,4R)-4-methyl-L-glutamic acid (5.9 kcal/mol)
As with prior examples, docking of erythro-(2S,4R)-4-methyl-L-glutamate (E4MeGlu) into VGLUT2 led to two top poses with close binding energies but again opposite in binding direction. The E4MeGlu pose more closely matching that of glutamate depicts the α-COOH group interacting with R88 (4.2 Å), the amino with Y327 (3.1 Å), and a weak interaction between the γ-carboxylate and T454 (Fig. 6). The 4-methyl group that distinguishes E4MeGlu from Glu is positioned such that there are minimal contacts pointed downward and away from the cluster of R88/Y195/Y327. A contrasting second pose has the γ-carboxylate interacting with R88 and the α-carboxylate forming a hydrogen bond to Y327. Since E4MeGlu acts as substrate, the pose with the α-amino acid group facing R88 (Fig. 6) seems the more likely orientation for transport.
Fig. 6.

Left: Docking image of E-4-methylglutamate binding to VGLUT2. Right: Key interactions between E-4-methyl glutamate and nearby residues along with distances. Binding to R88/Y327 is similar to that found for the natural substrate L-Glu.
3.5. Comparison of substrate and non-substrate binding
VGLUTs are highly selective with respect to substrate recognition and transport. When the substrate and non-substrate poses were combined in the VGLUT2-DgoT homology model using MUSTANG (YASARA) [42] some differences were identified (Fig. 7). The substrates L-Glu, E4MeGlu and trans-ACPD (left panel) form salt bridges between the α-carboxylate and R88 along with H-bonding or cation-pi interaction between the amine and Y327. For the non-substrates, L-aspartate and cis-ACPD use the γ-carboxylate while D-Glu uses the α-carboxylate to form a salt bridge to R88. All the non-substrates, however, tend to form a cation-pi interaction with Y135 rather than with Y327 as do substrates. These differences do not explain how molecules are recognized for transport or excluded from transport by VGLUTs, but provide some differences to explore in new structures.
Fig. 7.

Left: Overlay of the substrates glutamate, E4MeGlu and trans-ACPD in VGLUT2. Right: Overlay of L-aspartate, D-glutamate, and cis-ACPD non-substrate binding modes in VGLUT2. Cation-pi interactions in blue; hydrophobic interactions in green; gold dotted cylinders are salt bridges and hydrogen bonds.
Site specific mutation in VGLUT2 revealed that H128, R184, and E191 were found as critical residues in the substrate binding site [17]. The added residues identified for substrate binding in our model, R322 and R88, may or may not be critical for transport but possibly important for recognition. Although the differences in homology models from GlpT [17] to DgoT were small the position of certain residues in VGLUT2 may have been altered and binding interactions changed, whereas the residues actually required for function allow for transient binding. The prior homology model developed for VGLUT1 [32] did however, indicate involvement of arginines R80/R176 (R88/R184 VGLUT2) in transport. Inorganic phosphate, an alternative substrate, binds at a second, undefined binding domain [17,20] whose co-occupation in the DgoT-based model was not considered but could play a role in the development of a more precise structure-function homology model. Likewise, Cl− was not docked in coordination with the VGLUT2-DgoT model and its absence could affect VGLUT2 conformation and the residues involved.
The docking studies with substrates and non-substrates also provided a glimpse into the alternate binding locations including the finding that ‘central’ and ‘upper’ sites occur as first hypothesized by Almqvist et al. [32] in the cytoplasmic open conformation. Although the differences in binding energy typically varied by 2–3 kcal/mol, the central (main) and upper binding locations were localized to the arginine residues R80 and R176. Still, identification of a second substrate domain would be a unique discovery for VGLUT2 although vestibule sites have been reported for other transporters [46–49].
4. Docking of competitive inhibitors
The demonstration that ligands can be designed that selectively regulate Glu uptake into vesicles without interfering with other proteins in the Glu neurotransmitter cycle would be highly useful. Selective ligands could control vesicle filling, synaptic release and reuptake, and in turn, pharmacologically manipulate post-synaptic response(s) including co-transmission and spill-over. Inhibitors, for example, could be used to advance our understanding of VGLUT structure-function and mechanism, radiolabeled to serve in assays, modified into affinity labels or imaging agents (e.g., fluorescence), and optimized and developed for potential clinical use. The structure-activity relationships of VGLUT inhibitors have been reviewed [6,34] but a number of new pharmacologic studies that along with advances in computational access warrant a re-examination. In contrast to the scant number of substrates and non-substrates, there have been large number of structurally diverse VGLUT competitive inhibitors including amino acid and amino acid analogs [50,51], quinolone-2,4-dicarboxylates (QDCs; [52,53]), tetrapeptides [54], hydantoins [55], azo dyes and their analogs [31,56–61], fatty acids [62], kynurenate and xanthurenate [57,63], the alkaloid bromocriptine [64], endogenous blockers bilirubin and biliverdin [65], and the non-competitive inhibitor Rose Bengal and its analogs [66] (see Fig. 8). Also known to block VGLUT function is the protein IPF [67] and a clever approach using nanobodies [29]. The inhibitors covered in this section do not include proteins [67] or those that act indirectly to block VGLUT transport by lowering the vesicular H+-ATPase activity or by altering the membrane potential such as DIDS, FCCP, nigericin and valinomycin.
Fig. 8.

Structures of representative VGLUT inhibitors.
Despite the advances, potent and selective VGLUT inhibitors for routine pharmacologic and/or physiologic use remain lacking. In part, this is because the substrate and/or inhibitor binding domain(s) had not been well-defined, the transport mechanism was not fully delineated, and the structure-function role of the VGLUT isoforms was still evolving. But the tempo of VGLUT research activity has increased dramatically over the past ten years and beginning to remove some of these obstacles. One of the biggest remaining challenge to VGLUT-selective ligands is that they must be able to cross the cell membrane, access the vesicle pool before being metabolized, and block VGLUT without affecting other proteins.
Some of the most potent inhibitors are the azo dyes, for example Trypan Blue (TB) (Ki ~50 nM) (Fig. 8) [56–60]. TB and similar azo dyes were presumed effective inhibitors because they contain two, substituted aminonaphthalene sulfonates that resemble or mimic glutamate (Fig. 9; one of several possible overlays shown) that could bind the substrate domain and the overall size would then block VGLUT. However, substituted naphthalene sulfonic acids lacking azo linker groups and/or second glutamate mimic were largely inactive including the glutamate isostere of Trypan Blue, 8-aminonaphthol-3,6-disulfonic acid (~1 mM) indicating the importance of the azo-group or at least the attachment of a lipophilic moiety to an azo-linkage [50,59,63,68]. A number of interesting inhibitor structures have been reported based on modifications to the azo-dyes or using the azo-dyes to produce related structures. Still, the wide variety in inhibitor structure types provides an opportunity to interrogate the different binding orientations and possibly identify residues involved in VGLUT2 inactivation that are conserved. Using the docking analyses, it may also reveal differences in non-competitive binding modes and the aforementioned second binding domain [32]. In the following sections, results of docking competitive inhibitors and non-competitive inhibitor with the VGLUT2 homology model derived from DgoT are presented.
Fig. 9.
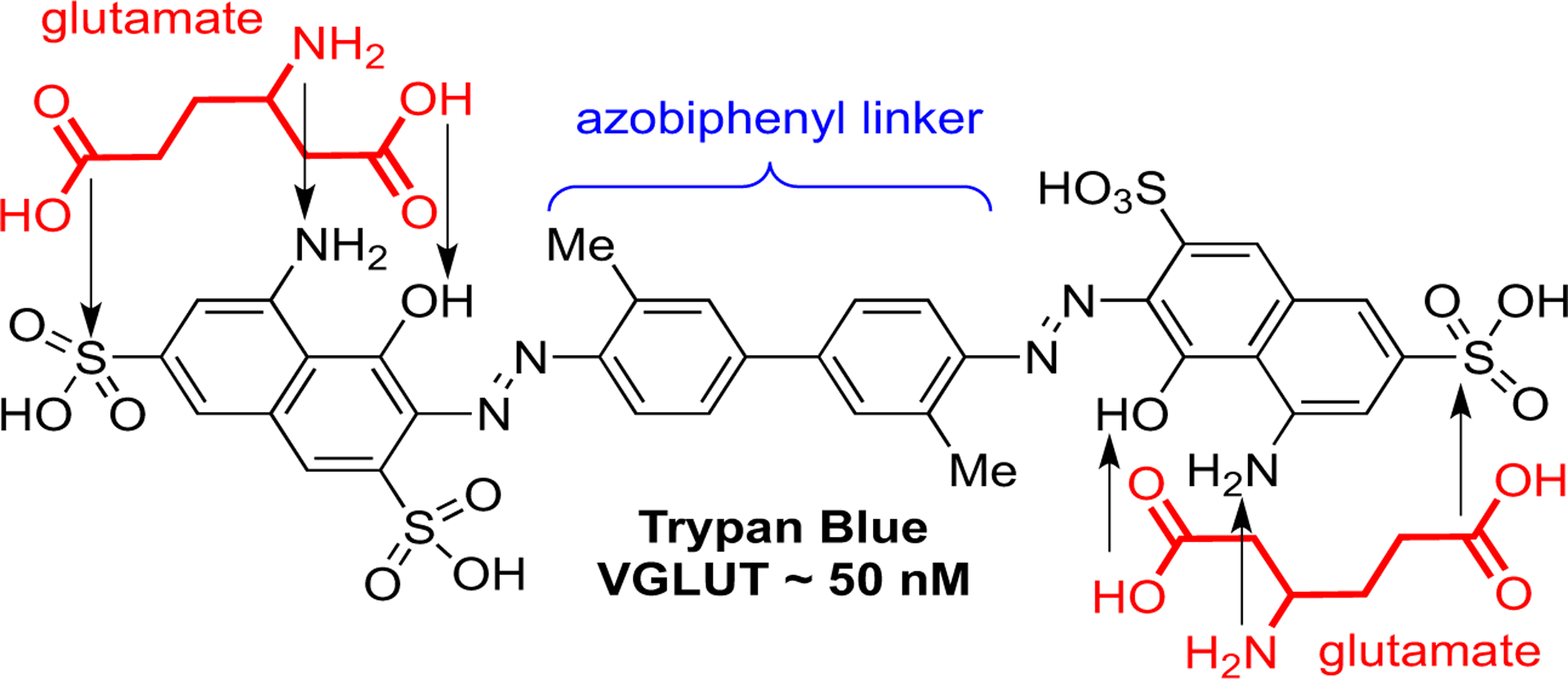
Overlay of glutamates onto matching functional groups of Trypan Blue.
4.1. Bromocriptine (11.1 kcal/mol)
Of all the VGLUT inhibitors identified, bromocriptine (BC) is somewhat of a structural outlier containing no discernable carboxylates that mimic glutamate but shown to be a strong competitive inhibitor at 25 nM [64]. Even several decades after its discovery, a clear understanding of which VGLUT residues bind BC are unclear. Therefore, docking of this unique and potent inhibitor some would furnish a preliminary idea of where and how it might bind. It is also important to consider that BC lacks ionizable groups making it a possible template for development of new VGLUT inhibitors capable of crossing membranes although the molecular complexity of BC renders it a daunting scaffold.
The results of the BC docking led to a small number of unique docking clusters with the top two highest binding energy poses (11 kcal/mol) positioned in the channel with the bromo indole facing Y327 and R88 (Fig. 10). The remainder of the poses showed BC docked lower in the channel or in a few instances where BC was inverted such that the bromoindole group faced the cytosolic side. In the lower energy poses, BC interacted with residues near the cytosolic entry, outside the channel or alongside transmembrane helices via a network of hydrophobic interactions. These latter poses would not directly interfere with Glu binding but could enable conformer changes to VGLUT2 and would be more representative of non-competitive inhibitors. The higher energy poses show interactions with Y195 and Y327 (Fig. 10) that are more consistent with its perceived mechanism as a competitive inhibitor. Two-dimensional protein ligand analysis (PLIP; [69]) showed that BC interacted with VGLUT2 via multiple hydrophobic interactions, hydrogen bonds to Y327 and N431 and a salt bridge between the hemiketal alcohol moiety and H199. The interesting interaction between the cyclic hemiketal and H199 suggests either a hydrogen bond or ionic attraction with the latter suggesting a unique carboxylate mimic. Overall, BC potently inhibits VGLUT but likely via interactions with a unique set of residues.
Fig. 10.
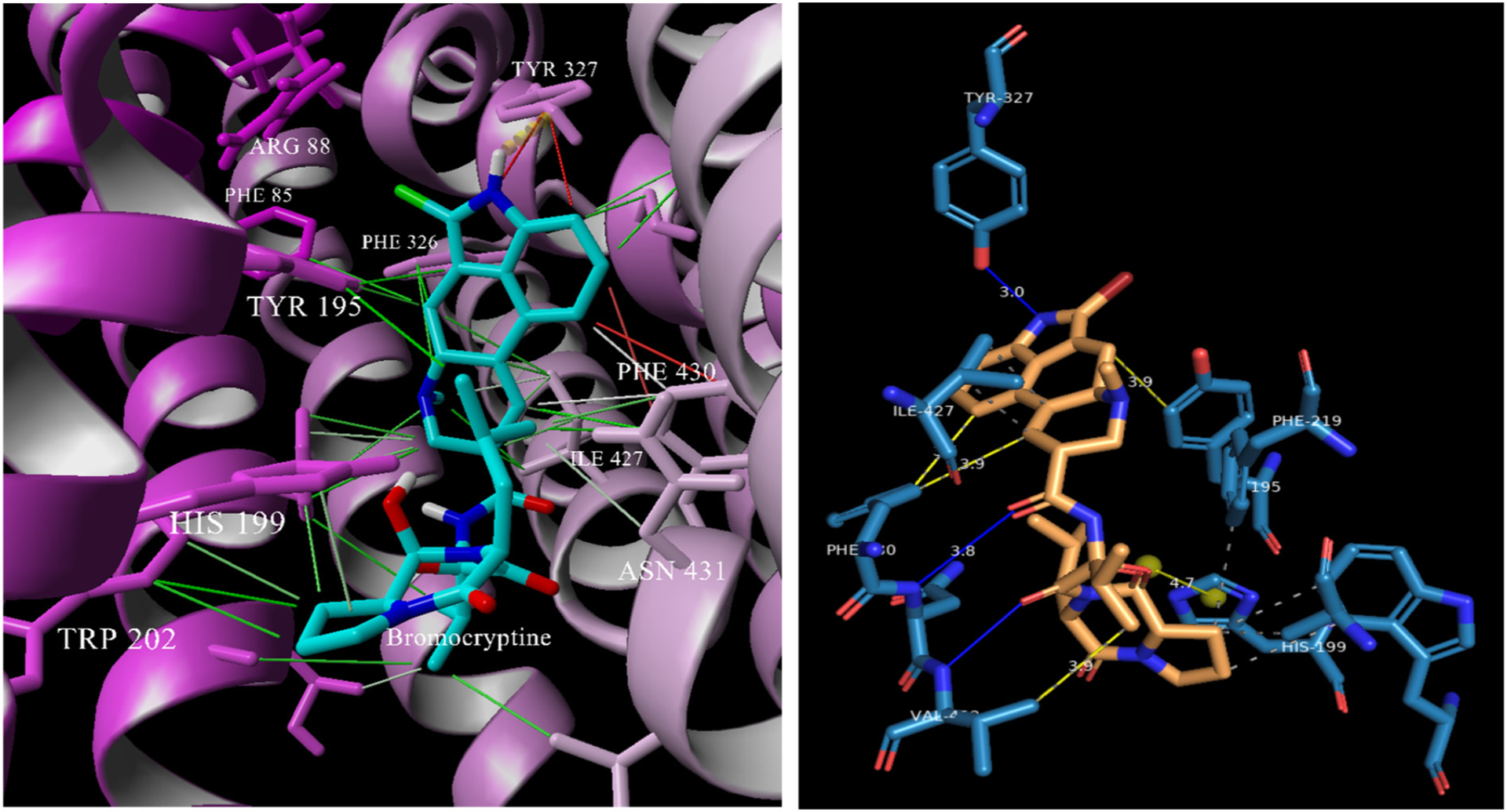
Left: Bromocriptine docked in VGLUT2 showing an array of interactions with residues identified as binding the substrate Glu. Right: A 2D ligand map showing the interactions and distances in the absence of protein.
4.2. Trypan Blue (15.9 kcal/mol)
Trypan Blue (TB) is one of the strongest inhibitors of VGLUT known with a Ki ~50 nM and resembles the dyes Evans Blue and Chicago Sky Blue. As shown in Fig. 9, TB is a long, symmetric molecule containing two glutamate isosteres linked via azo linkages to a central biphenyl. The role of the biphenyl group was not immediately understood but theorized to serve as a spacer between two possible glutamate binding sites. Docking of TB led to three predominant binding clusters: (a) high binding energy poses (15.1–15.8 kcal/mol) that are positioned within a channel with one end exiting the protein, (b) poses intermediate in energy (12.7–13.5 kcal/mol) that are positioned lengthwise within the channel, and (c) lower energy poses that block channel entry through binding of the His199 and R211 residues. The intermediate cluster of poses also formed a number of hydrogen bonds with the same residues engaged in substrate recognition, i.e., R88, Y195 and Y327. Residue Y195 was positioned such that it formed a pi-stacking interaction with the naphthylene and form a hydrogen bond with a sulfonate oxygen. Another feature that favors the intermediate cluster over the other two was a number of interactions that stabilize the second naphthalene moiety. The depiction of the intermediate pose of TB is shown as both stick and VDW surface (Fig. 11) along with a two-dimensional ligand-protein schematic derived from the intermediate pose (LigPlot [70,71] and PLIP [69]) that identifies surrounding residues.
Fig. 11.

Left: Docking of TB in VGLUT2 showing the span length of the inhibitor in a pose entering from the cytosolic side. Center: TB in a transparent VDW surface docked in VGLUT2. Right: two-dimensional schematic of TB (in orange) without protein showing the residue interactions (salt-bridges or H-bonding shown as cyan bars).
The docked images (Fig. 11; left and middle) support the notion that TB enters and spans the channel until it comes into its highly favorable pose about halfway into the chamber with residues R88, Y195, and H199 orienting the docking. The 2D ligand map (Fig. 11; right) shows sulfonic acid groups on the head naphthalene stabilized by hydrogen bonding interactions (cyan) to R88 (3.65 Å) and Y223 and the aminonaphthalene (aniline-type) through D450 (2.85 Å). The biphenyl linker is flanked by residues that offer hydrophobic interactions including F219, Y223, F430 and V432. The tail or trailing naphthalene group interacts at the hydroxyl and amino with T216 and at the sulfonic acid with R385. Other residues also appear to be involved in the stabilization but do not appear in all poses. Overall, the high binding energy and docking poses support TB as an excellent inhibitor because it is able to enter the VGLUT2 channel and form a number of favorable interactions with residues over the span of the entire molecule including interactions with those suspected of binding substrate.
4.3. Congo Red (13.9 kcal/mol)
Congo Red (CR) is a symmetrical, bis-azo dye linked via a biphenyl similar to TB but approximately ten-fold less effective as an inhibitor (Ki ~550 nM). CR possesses only one amino and sulfonic acid group on each naphthyl ring and bears no substituents on the biphenyl; a feature unlike the other known VGLUT inhibitor dyes such as TB, Chicago Sky Blue and Evans Blue. Therefore, CR is minimally substituted and the analysis could be very useful in an effort to identify binding determinants for the azo dyes. Due to the fewer number of functional groups, a large number of similar binding energy docking poses were found for CR and most involved the formation of a strong ionic interaction between the naphthalene sulfonate and R88 and hydrophobic interactions of the naphthylene with Y135. As with TB, CR enters and docks with VGLUT2 residues to occupy the chamber with the naphthyl head group near the substrate binding residues (Fig. 12) with the other naphthyl group near the cytosolic side entry point. With the sulfonate facing R88, the para-NH2 moiety does not appear to engage in any specific interactions with VGLUT2. The CR diazo –N=N- group forms hydrogen-bonding interactions – the more inward facing diazo group with Y195 and the more outward diazo moiety with S212. The biphenyl forms a pi-stacking interaction with F219 but the residues that formed interactions with the biphenyl varied slightly depending on the pose. The docking of CR is made stronger by an ionic interaction between R211 and the second naphthylsulfonate group at or near the cytosolic access point (see Fig. 12; right). The dual arginine interactions that span ‘head and tail’ naphthylene sulfonates are noteworthy and could be one explanation for the inhibitory activity of CR and other related azo dyes.
Fig. 12.

Left: Docking of CR in VGLUT2 homology model. Right: two-dimensional ligand-protein interaction map (PLIP; [69]) showing the key residues that interact.
4.4. Brilliant Yellow (12.2 kcal/mol) and the Brilliant Yellow analog BYA2 (10.9 kcal/mol)
Brilliant Yellow (BY) is another important addition to the azo-dye family of inhibitors [31,59,72]. Unlike TB and CR, the azo-linkages in BY connect an E-stilbene to two phenol groups and the sulfonate groups occupy positions on the stilbene linker rather than on the end groups. BY is a unique presentation to VGLUT2 as it is unlikely to contort to bind the residues proposed for substrate interaction without isomerization of an azo group from E to Z. Likewise, if a phenol end group of BY docks near the substrate domain, the stilbene-sulfonates would need to partner with residues different from TB and CR compounds that lack any ionizable moiety on the biphenyl. It was therefore of great interest to investigate the manner in which BY binds to VGLUT2 and if any of the interactions overlap with the other azo dyes. A second critically important element to this docking study was the recent finding that Brilliant Yellow analogs (BYA) in which the sulfonates were replaced with carboxylates and the azo groups with amides or benzylethers were good inhibitors [31]. Docking of BY and the benzyl ether analog of Brilliant Yellow known as BYA2 (Fig. 8) were conducted and the binding residues that are shared or differ from the other azo dyes identified (Fig. 13).
Fig. 13.
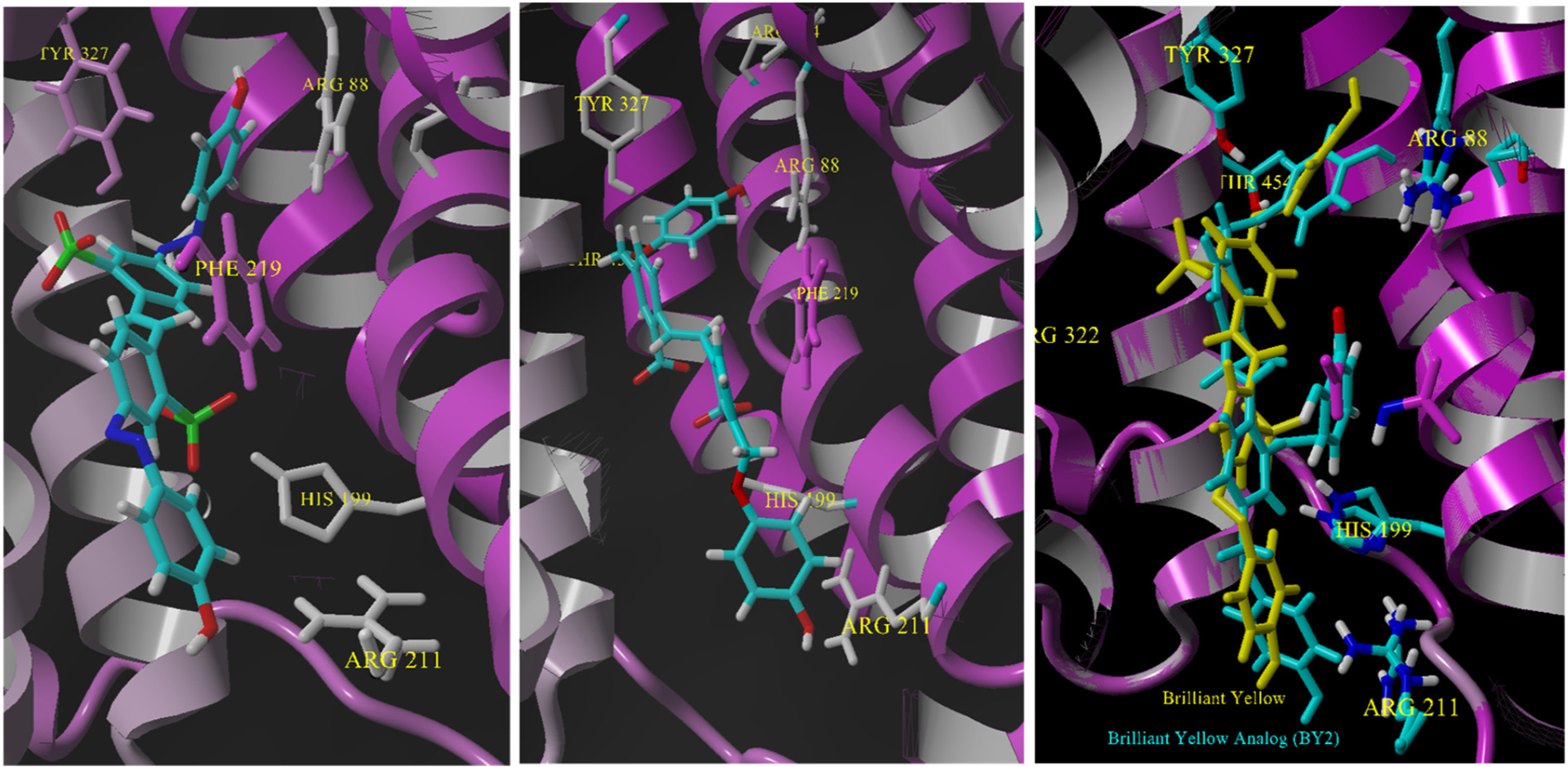
Left: docking of Brilliant Yellow. Center: Docking of Brilliant Yellow analog 2 (BYA2). Right: an overlay of the two structures in VGLUT2 using the Mustang program [42] in YASARA.
BY binds within VGLUT2 in a similar overall pose to the other azo dyes but unlike the other azo dyes there is no acid head group to form an interaction with R88 (Fig. 13). In fact, 2D ligand-protein analyses showed hydrogen bonds between the phenol-OH of BY and Y135 and Y 327 rather than interact with R88. Also noted were unique interactions with the sulfonate groups. The inward sulfonate closer to the substrate binding domain forms a hydrogen bond to N319 and the lower sulfonate binds H199 in a strong ionic interaction at ~4.0 Å. A greater number of pi-stacking and hydrophobic interactions occurred with the outward half of the molecule near the cytosolic entry.
The structure changes that distinguish the Brilliant Yellow analog 2 (BY2) from BY are the substitution of the sulfonates for carboxylates and azo (-N=N-) linkages exchanged for a methylene ether (-CH2O-). When BY2 was minimized and docked into the VGLUT2 homology model, the top binding energy poses (~11 kcal/mol) were similar to those found for BY including interactions with many of the same residues (Fig. 13). Conversely, the added conformational flexibility of the benzyl ether as compared to the azo linkage led to a greater number of lower energy poses (9–9.5 kcal/mol) distributed throughout the cavity of the protein. The high energy pose for BY2 showed key interactions between Y327 and the phenol and also between the lower positioned carboxylate and H199 interactions also observed with BY. An additional and perhaps strategic interaction was found between R211 and the end phenol-OH near the cytosolic entry (Fig. 13).
4.5. Xanthurenate (7.6 kcal/mol)
The quinolines kynurenic acid (KA), xanthurenic acid (XA) and 7-chlorokynurenic acid (7ClKA) are competitive VGLUT inhibitors initially screened because of their known inhibition at glutamate receptors [63]. The flat-fused aromatic structure lacking a second carboxylic acid group indicated that VGLUT may tolerate a range of inhibitor structures. These quinoline structures are considered conformationally-restricted analogs because the 4-hydroxy-quinoline-2-carboxylate resembles a flat, rigid Glu. Xanthurenic showed slightly higher inhibition of VGLUT (Ki ~0.19 mM) than KA (Ki ~1.3 mM) and 7ClKA (Ki ~0.59 mM) and was selected for docking.
Not surprising, the highest energy poses (7.6 kcal/mol) were far lower in value than the azo-dyes and place find the carboxylic acid of XA in close proximity to R88 (see Fig. 14). Unlike many of the other docking experiments with multiple poses with similar energy, however, a large drop in binding energy for the remaining poses was observed (4.1–5.4 kcal/mol) seemingly because they lacked a carboxylate to R88 interaction. Most of the low energy clusters showed XA forming hydrogen-bond interactions throughout the channel without a clear preference for any particular orientation. In addition to the ionic interaction with R88, the high binding energy pose showed some additional interactions at Y327 and T454 that are in common with Glu. Based on the interactions and binding energy, it is not surprising that XA is a weak inhibitor because at minimum it can bind R88 but is stabilized by different functional groups than L-Glu and the azo-dyes. Moreover, XA may be a weaker inhibitor than azo dyes as it lacks lipophilic groups that form hydrophobic interactions.
Fig. 14.

Left: xanthurenic acid docked in VGLUT2 near the substrate residues. Right: The key residues that bind xanthurenic acid without protein.
4.6. 6-(4-Biphenyl)quinoline-2,4-dicarboxylic acid (6BP-QDC) (8.35 kcal/mol)
The quinoline-dicarboxylic acid (QDC) class of VGLUT inhibitors was prepared as conformationally-restricted mimics of glutamate. The rationale for the QDC emerged from the finding that kynurenic/xanthurenic acids showed some activity as VGLUT inhibitors with a co-planar arrangement of amine and oxyanion groups on pyridine. It was reasoned that QDC’s would be superior inhibitors to XA by replacing the p-hydroxy with a second carboxylate while increasing the lipophilicity via an attached aryl groups to facilitate membrane permeability. Since QDCs are readily prepared from anilines and dimethyl-4-oxoglutarate, a focused structure-activity investigation of VGLUT inhibitors was possible [52,53,73]. Over 75 QDC analogs were prepared and the most potent inhibitors were those that contained aryl or biaryl substituents or had fused aromatic rings to the quinolone [73]. One of the more active inhibitors to emerge from the study was 6-[4-biphenyl]-QDC (Ki ~50 μM) that structurally overlaps in part with the biphenyl moiety present in azo dyes. Lacking sulfonate and azo groups, the properties of 6-biphenyl-QDC make it membrane permeable similar to the strategy indicated for BYA2 [31,72], however, the docking of 6-biphenyl-QDC with VGLUT2, did not demonstrate the same clear binding interactions as BYA2. While a majority of the highest energy binding poses placed the quinoline-dicarboxylic acid group facing toward the substrate binding residues R88, Y135 and Y327 many of the docked structures appeared in the opposite orientation with the lipophilic tail facing toward R88 and the dicarboxylic acids forming interactions with H199. Although the oppositely-oriented poses were typically 2–3 kcal/mol lower in energy than the inward-oriented QDC, the near equal population of these poses could not be ignored. Still, 6-biphenyl-QDC was demonstrated to show competitive kinetics and therefore, the predominant pose with the carboxylate facing R88 appears likely with the caveat that highly lipophilic QDCs may bind VGLUT2 in alternate orientations. This 6-biphenyl-QDC pose placed the 2-carboxylic acid in an interaction with R88 (~5 Å) but closer to T454 (~3 Å) (see Fig. 15). Again, Y195 (pi-stacking to quinoline) and Y327 (hydrogen bond to nitrogen) were involved in the heterocyclic ring stabilization whereas the biphenyl substituent was stabilized via hydrophobic interactions to F219 and F326 and others.
Fig. 15.

Left: Pose of 6-(4-biphenyl)QDC docked in VGLUT in which the 2-carboxylic acid faces R88 and Y327. Right: Ligand-interaction map showing the residues and distances for binding.
4.7. Trypan Blue-QDC hybrid structures (12.7 kcal/mol)
An important approach in the design of any potent ligand is to combine the optimal components of two or more molecules into a single structure. Acher and coworkers smartly used the naphthalene disulfonic core of Trypan Blue and combined it with substituted aryl and biaryl groups (via an azo linkage) similarly to the quinoline dicarboxylic acids (QDCs) [61]. Among the best inhibitors were those containing the 1-amino-8-hydroxy naphthalene-3,6-disulfonic acid as found in TB although a number of new chemical entities were found to inhibit VGLUT with IC50 ~1.5–7 μM. Docking revealed that the 3,6-disulfonic acids are positioned on the naphthalene to form ionic interactions with R88 and H199 while placing the azo-biphenyl downward toward the cytosolic entry. It should be noted that the 8-hydroxy azo-naphthalene structure can exist in an ortho-iminoquinone tautomer structure [61]. This isomer was also docked in VGLUT2 showing a slightly lower binding energy (12.0 kcal/mol) with a greater proportion of poses (not shown) inverted but overall the principle interactions between the sulfonates and VGLUT2 remained the same.
4.8. Arachidonic acid (6.6 kcal/mol) and pregnanolone sulfate (10.9 kcal/mol)
These inhibitors represent endogenous structures containing a single acid group that have been shown to inhibit VGLUT to about 20% of control [62]. Docking of arachidonic acid afforded somewhat expected results with the carboxylate forming an interaction with R88 or T454 with the remainder of the tetra-cis-unsaturated molecule coiled in the channel. Pregnanolone sulfate (PS) was initially screened because a number of benzo- and naphtho-fused QDCs showed enhanced activity as VGLUT inhibitors [73]. The fused QDCs resembled a sulfated steroid (metabolite) but unlike arachidonic acid, the docking of PS led to two dominant binding energy (~11 kcal/mol) poses with the sulfate group facing toward R88 within 2.0–2.5 Å of R88 or facing toward the cytosol with the sulfate group interacting with H199. The sulfosteroids like PS and estrone sulfate blocked Glu uptake > 75% with IC50 values in the range ~100–150 μM.
4.9. Tetrapeptides (x-x-EF and x-x-EW) (9.5–10.5 kcal/mol)
Since the QDCs and the inhibitor bromocriptine contained amino acids embedded in their structures (e.g., glutamate, tryptophan, isoleucine) libraries of tetrapeptides that vary in two positions were prepared and tested at VGLUT [54]. Each of the tetrapeptide libraries contained a glutamic acid at position three and amino acids with aromatic sidechains were positioned at the N- and C-terminus positions due to the improvement in inhibitory activity in QDC when lipophilic residues were added [34,52,73]. Deconvolution of the libraries identified QIEW and WNEF tetrapeptides as weak VGLUT inhibitors with Ki values ~ 700–900 μM. As expected, docking of WNEF and QIEW showed an array of possible poses with close binding energies likely due to the conformational flexibility of the peptides. Most of the top energy binding poses for WNEF positioned the tetrapeptide within the channel but placed the phenylalanyl C-term inward toward R88 and a binding interaction between the glutamate side chain carboxylic acid and T454. The lower binding poses for WNEF placed the tryptophan inward toward R88 but with an interaction between the side chain Glu-COOH and H199 was identified. A similar distribution of high and low energy binding clusters was found for QIEW except a greater number of poses placing the tryptophan inward occurred. However, few if any poses placed the Glu-COOH sidechain in the vicinity of H199. Overall, docking of the tetrapeptide structures did not reveal highly favored poses, which may be consistent with the weak inhibitory activity.
4.10. Benzothiophene-3-glycine (7.7 kcal/mol) and bis-2,5-hydantoin thiophene (8.1 kcal/mol)
Potential VGLUT inhibitors were studied in which various groups and isosteres were substituted for the acid or amino acid moiety of glutamate [50,74]. A number of sulfated phenylalanine, sulfated phenylglycine, and sulfated thiophenylglycine analogs were prepared. R/S-benzothiophene-3-glycine showed ~ 70% reduction in Glu uptake but none of the sulfated phenylalanine nor sulfated phenylglycine analogs showed significant activity [50]. In a study aimed to determine if a hydantoin would serve as an acid isostere in VGLUT inhibitors, a panel of aryl and thiophenyl structures bearing one or two hydantoins were prepared and tested at VGLUT [74]. Three structures emerged as inhibitors: (a) phenylhydantoin-2,4-disulfonic acid (PHDS; blocked 89% uptake), (b) thiophene-2,5-bis-hydantoin (TBH; blocked 94% uptake), and (c) 5-hydantoin thiophene-2-carboxaldehyde (HTC; blocked 95% uptake). The IC50 values were determined to be 350–500 μM. Inhibition by PHDS is understandable owing to the presence of the sulfonic acid groups but the activity of TBH and HTC suggest that a hydantoin moiety can mimic an acid group at least when interacting with VGLUT2. Docking of R/S-benzothiophene-3-glycine and TBH showed a broad mix of binding orientations presumably due to a lack of secondary interactions, small size and weak ionization (TBH).
5. Docking of non-competitive inhibitors
5.1. Rose Bengal (RB; 8.4 kcal/mol) and Rose Bengal analogs (RBAs)
RBs and RBAs represent the only well-characterized class of non-competitive inhibitors at VGLUT [66,75]. Because these structures act at different location in VGLUT than the competitive inhibitors it was instructive to conduct docking studies to attempt to differentiate between binding sites to aid the future development of similar compounds. RB is non-competitive but exhibits little to no inhibition of the vacuolar-ATPase and does not dissipate the membrane potential [75]. The unique inhibitory action of RB was shown to decrease the amount exocytotically-released 3H-Glu from vesicles in a depolarization and Ca+2-dependent manner; a mechanism not shared by the non-inhibitory, but structurally similar fluorescein [75]. RB is an interesting compound that equilibrates between lactone (closed) and carboxylic acid (open) forms and it is unclear if both forms would bind at the same location. RB was minimized in both open and closed forms prior to docking each form. The open form was minimized at pH 7.4 to set the ionization of the carboxylic acid.
Docking of the closed and open forms led to a clear overlap in binding locations and orientation despite the open form bearing an oxyanion and the closed form being neutral. Both forms docked relatively close to the cytosolic entry domain far removed from the presumed substrate binding site (Fig. 16; H199 added in image to orient docking location to prior illustrations). The residue interactions with RB in open and closed form were M296, L293, and P196 and docking of the lactone form showed nearly no interactions suggesting that the open carboxylate form is the likely structure blocking VGLUT uptake. The unique docking location points to a possible allosteric binding site for RB and could be cause for some excitement since allosteric ligands can offer a selectivity advantage. It is also possible that RB indirectly blocks Glu uptake by interacting with one or more proteins upstream in the Glu uptake pathway [66]. However, the binding energy for this proposed allosteric site was 8.4 kcal/mol, which is weaker than the azo dye inhibitors (> 11 kcal/mol) but stronger than substrates binding (~5 to 6 kcal/mol).
Fig. 16.

Left: Rose Bengal lactone (cyan; closed) and Rose Bengal carboxylic acid (yellow; open) docked near the cytosolic entry regions of the VGLUT2 model. Right: Expansion of the docking pose showing interactions of RB with H199, P196, M296 and L293.
Using the initial discovery that RB blocked VGLUT by a non-competitive mechanism, Acher and co-workers [66] produced a number of analogs based on the RB scaffold including halogenated analogs, chemically-trimmed analogs lacking the pyran oxygen, and some in which the benzolactone was replaced with a propionic acid. The RB analog study also took into account the various protonation states of RB residing in the open and closed forms. None of the new analogs reported showed greater inhibition of VGLUT than RB but furnished excellent SAR boundaries for future development [66].
6. Miscellaneous VGLUT inhibitors
6.1. Biliverdin (11.5 kcal/mol)
The endogenous compounds bilirubin and biliverdin inhibit VGLUT but also the uptake of dopamine and GABA at physiologically relevant concentrations. Biliverdin is a more potent inhibitor of VGLUT glutamate uptake than bilirubin [76], and been shown to act at the transporter and not on the vesicular H + -ATPase [62]. The unique structure of these polypyrroles that assume an overall U-shape and possibly too large to enter VGLUT suggested they would be an important comparator in the docking experiments with VGLUT2 (Fig. 17). Surprisingly, docking showed that biliverdin was able to bind VGLUT2 and form a number of favorable poses that were distributed equally between the carboxylates facing inward toward the substrate binding region and also outward with carboxylates facing the cytosolic entry. The inward facing pose showed ionic interactions between either carboxylate with R88 whereas outward poses facing the cytosol interacted with R211. Although occurring at lower energy, some poses of biliverdin occupied the similar non-competitive domain and interactions found for Rose Bengal.
Fig. 17.

Structure of biliverdin.
6.2. Proteins
Not included in the computational analysis but of importance to understanding the Glu uptake mechanism are the protein inhibitors of VGLUT including inhibitory protein factor [67,77] and VGLUT nanobodies [29].
7. Comparison of inhibitor binding
The docking studies revealed some interesting binding phenomena and trends that may assist with the design and evaluation of new inhibitors. First, the binding energies provided for each inhibitor within the VGLUT2 homology model are only a relative indicator but show that, in general, azo dyes bind more strongly than other inhibitor structure subtypes except bromocriptine. The binding energies of the competitive inhibitors also correlate strongly (r = −0.822) with the IC50 values reported (Fig. 18), which indirectly supports the docking approach and homology model. When the non-competitive inhibitor Rose Bengal is included, the trend line value decreases to r = −0.70 and becomes moderately correlated. Overall, the binding energies strongly align to the pharmacology reported suggesting that the homology model and docking studies reflect, in part, the experimental observations.
Fig. 18.
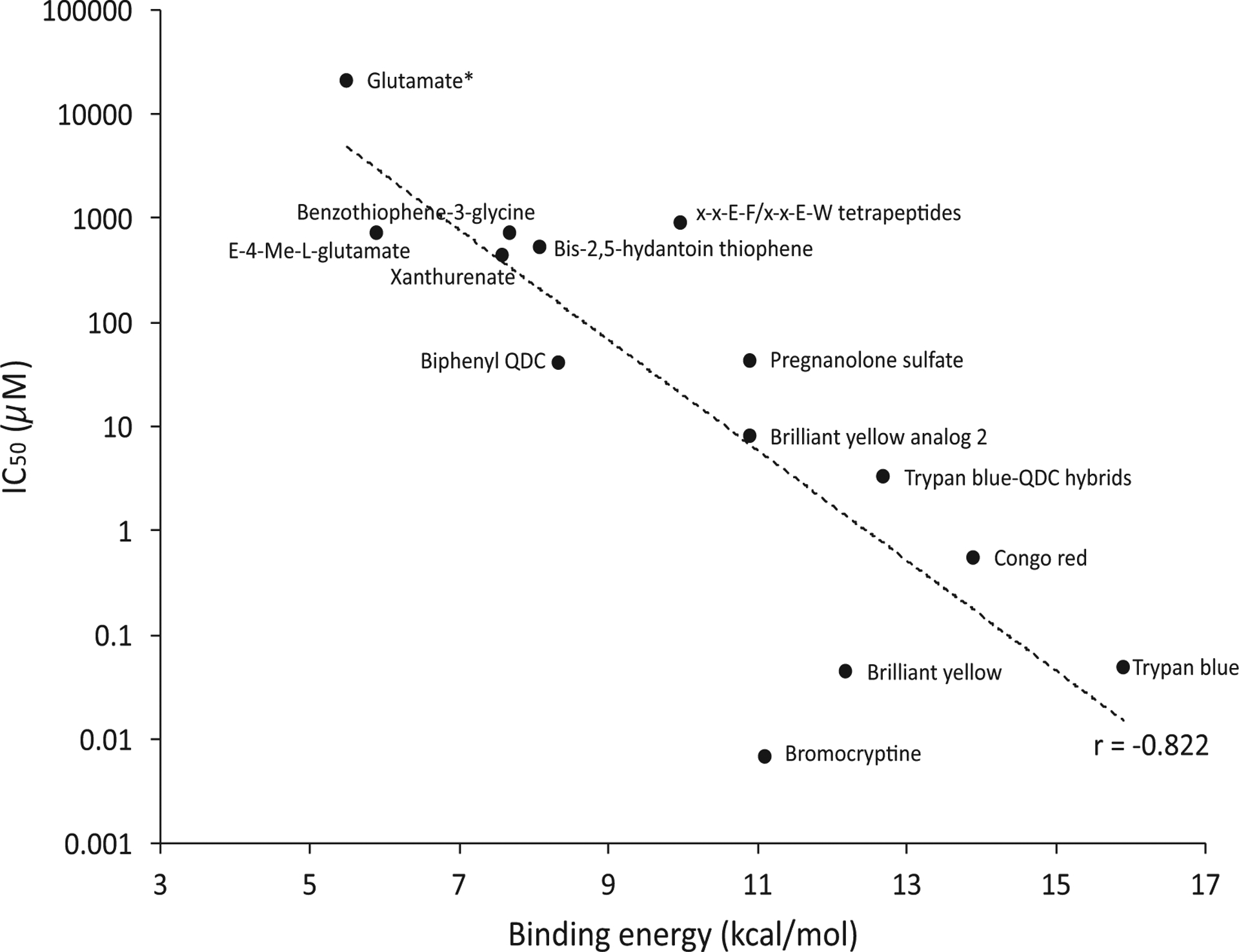
Plot of IC50 values of competitive inhibitors versus their calculated binding energies. Position of the substrate glutamate shown but not included in the correlation. Rose Bengal was not included as a non-competitive inhibitor. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The docking showed that the best competitive inhibitors bound residues that were also shown to interact with substrate, in particular R88, H128 and H199. A difference found in these docking studies as compared to prior homology models [17,32] was the finding that tyrosine residues Y195, Y223 and Y327 played a role in binding. From a VGLUT2 topology standpoint, the residues most involved in binding appeared approximately halfway into the VGLUT2 channel and not deeper toward R322 and the luminal side. In part, this may be due to the initial DgoT structure used to produce the VGLUT2 model. Although the conformer accepting approaching ligands from the cytosol differs from that facing the luminal compartment a follow up study using multiple conformers of VGLUT2 would be beneficial to identify differences in substrate and inhibitor binding. Doubtless, the presence of sulfonates and lipophilic groups add inhibitory potency although the sulfonates deter membrane penetration. Still, the utility of sulfonates has been helpful to identify strategic interactions in VGLUT2. As shown by Ueda, Acher and the authors group, new inhibitors structures can be generated via alterations to existing structures [31,52,66]. Docking of Rose Bengal, shown to be a non-competitive inhibitor, appears to bind a new undetermined site although this needs to be experimentally validated. Taken together, docking studies of VGLUT2 inhibitors showed that a number of the same residues are involved despite a broad diversity in the structure. The relative orientation of these residues may be helpful in the design of new inhibitors.
To control for overall docking errors in the VGLUT2 model, the binding energies of L-Glu and TB were conducted with just DgoT in a docking simulation in which the key binding residues Y44, R47, Y79, F137, Q164, Q264, S370, W373 and N393 were initially made flexible followed by the H+-transport residues D46 and Q146E (exchanged in simulation). L-Glutamate binding energies varied from 3.1–5.8 kcal/mol but only four of twenty-five poses were located at the galactonate binding domain. The top poses for L-Glu showed the γ-COOH in ionic bonding with R47 and Y44, α-amino H-bonding with Q164 and α-COOH also with Q164, and hydrophobic binding between the L-Glu chain and F137. Binding to R47 was expected as this residue position is conserved between VGLUT2 and DgoT and no change in binding orientation was observed after the addition of D46 and Q146E (site mutation) to the docking. TB docking into DgoT led to binding energies that were 7 to 9 kcal/mol less than found with VGLUT2, and there was no evidence of a single binding pose within the protein or with any interactions with the flexible residues. Also, no evidence of a pose mi-micking the position of galactonate was observed. In sum, there appears to be some favorable binding orientations within DgoT for L-Glu but few if any poses showed TB docking near the galactonate site. Lacking any pharmacology to validate these observations, these results need to be viewed with some uncertainty but the finding that TB binds the VGLUT2 model and not DgoT is important.
8. Conclusions
The aim of this review was to summarize VGLUT substrates and inhibitors using a computational-based approach that to our knowledge has not yet been conducted. Importantly, this review would not have been possible without the critical finding that aligned key mechano-structural overlaps between VGLUT and DgoT [22]. Relative to other glutamatergic targets, the preparation and screening of new VGLUT substrates and inhibitors has neither grown as quickly nor to the same level of development. One possible reason for the lower production of VGLUT substrate and inhibitors has been a lack of three-dimensional computer homology models that when combined with docking studies can interrogate and inform on ligand-protein interactions, and assist with the rational design of selective and potent compounds. To partially overcome this void, this review created a homology model for VGLUT2 derived from the structures of D-galactonate/H+ symporter D-galactonate transporter (DgoT; 6E9O/6E9N). The resultant model was used in docking simulations to interpret the possible location and type of residues involved in substrates, non-substrate and inhibitors binding. The VGLUT2-DgoT homology model shares an extensive amount of structural overlap with the GlpT models including most residues involved in substrate/inhibitor binding. Those earlier models and the recent advance of DgoT structure provided tremendous insights into the VGLUT2 machinery allowing our docking studies to proceed with greater focus. Still, computational models and simulations that aim to extend the knowledge of ligand-protein interactions lack the precision of x-ray or solution structures are limited and require experimental validation. Moreover, interpretation of the docking results is necessarily biased by selection of the putative residues used and the binding region or simulation cell size. Such approaches can be further flawed by the assumption that the homology model itself is correct.
Several findings in this review of small molecule ligands acting at VGLUT2 were noteworthy: (a) the residues R88, Y195 and Y327 interact with substrates; (b) substrates and inhibitors share interactions with residues in a binding domain located midway in VGLUT2 consistent with prior models; (c) docking of symmetrical azo-dyes showed interactions with residues over the entire length of inhibitor suggesting their potency may represent a combination of precisely positioned interactions; (d) addition of lipophilic groups improve inhibition; (e) a separate binding region was identified for the non-competitive inhibitor Rose Bengal. Some of the findings presented were consistent with an inhibitor pharmacophore model for VGLUT [34] that suggested the need for a γ-COOH but not α-COOH, and that a lipophilic domain or pocket be 13–15 Å from the γ-COOH binding moiety. These pharmacophore model criteria are present in the docking studies with Congo Red, and Trypan Blue that lack a specific alpha amino acid group but position their biphenyl or equivalent lipophilic groups 12–19 Å from R88; the residue that binds a sulfonic acid or equivalent isostere. Some elements of a pharmacophore model presented for Brilliant Yellow, TB and related dyes [31,72] are also consistent with the docking results. The 9.3 Å interatomic distance noted between sulfonate oxyanion and azenyl nitrogen groups of TB and BY was noted as key to inhibition [72] and matches the distance between R88 and H199 or Y135 and H199 (8.9 to 10.5 Å) that potentially interact with the sulfonate and nitrogens, respectively. However, the direct involvement of H199 with the azenyl group of these dyes was not found in a majority of poses. It is hoped that insights derived from this review will assist with the development of new substrates, non-substrates and inhibitors.
Acknowledgements
Research in the authors’ laboratories is funded by a grant to The Center for Biomolecular and Structural Dynamics from the National Institute of General Medical Sciences of the National Institutes of Health, United States under grant number P20GM103546.
Abbreviations:
- VGLUT
vesicular glutamate transporter
- YASARA
yet another scientific artificial reality application
- PLIP
protein ligand interaction profiler
- Glu
glutamate
- TB
trypan blue
- BY
brilliant yellow
- CR
congo red
- QDC
quinolone-2,4-dicarboxylic acid
- BC
bromocriptine
- ACPD
aminocyclopentane-1,3,-dicarboxylic acid
- RB
Rose Bengal
- E4MeGlu
erythro-(2S,4R)-4-methyl-L-glutamate
- VINA
Vina is not autodock
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Zhang FX, Ge SN, Dong YL, Shi J, Feng YP, Li Y, Li YQ, Li JL, Vesicular glutamate transporter isoforms: the essential players in the somatosensory systems, Prog. Neurobiol 171 (2018) 72–89. [DOI] [PubMed] [Google Scholar]
- [2].Omote H, Miyaji T, Juge N, Moriyama Y, Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport, Biochemistry 50 (2011) 5558–5565. [DOI] [PubMed] [Google Scholar]
- [3].El Mestikawy S, Wallen-Mackenzie A, Fortin GM, Descarries L, Trudeau LE, From glutamate co-release to vesicular synergy: vesicular glutamate transporters, Nat. Rev. Neurosci 12 (2011) 204–216. [DOI] [PubMed] [Google Scholar]
- [4].Liguz-Lecznar M, Skangiel-Kramska J, Vesicular glutamate transporters (VGLUTs): the three musketeers of glutamatergic system, Acta Neurobiol. Exp. (Wars) 67 (2007) 207–218. [DOI] [PubMed] [Google Scholar]
- [5].Takamori S, VGLUTs: ‘exciting’ times for glutamatergic research? Neurosci. Res 55 (2006) 343–351. [DOI] [PubMed] [Google Scholar]
- [6].Shigeri Y, Seal RP, Shimamoto K, Molecular pharmacology of glutamate transporters, EAATs and VGLUTs, Brain Res. Brain Res. Rev 45 (2004) 250–265. [DOI] [PubMed] [Google Scholar]
- [7].Shigeri Y, Shimamoto K, Pharmacology of excitatory amino acid transporters (EAATs and VGLUTs), Nippon Yakubutsugaku Zasshi 122 (2003) 253–264. [DOI] [PubMed] [Google Scholar]
- [8].Moriyama Y, Yamamoto A, Glutamatergic chemical transmission: look! Here, there, and anywhere, J. Biochem 135 (2004) 155–163. [DOI] [PubMed] [Google Scholar]
- [9].Reimer RJ, Fremeau RT Jr., E.E. Bellocchio, R.H. Edwards, The essence of excitation, Curr. Opin. Cell Biol 13 (2001) 417–421. [DOI] [PubMed] [Google Scholar]
- [10].Takeda K, Ishida A, Takahashi K, Ueda T, Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from alpha-ketoglutarate for vesicular loading, J. Neurochem 121 (2012) 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fremeau RT Jr., Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH, The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 14488–14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S, Third Vesicular A, Glutamate transporter expressed by cholinergic and serotoninergic neurons, J. Neurosci 22 (2002) 5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vigneault E, Poirel O, Riad M, Prud’homme J, Dumas S, Turecki G, Fasano C, Mechawar N, El Mestikawy S, Distribution of vesicular glutamate transporters in the human brain, Front. Neuroanat 9 (2015) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fremeau RT Jr., Voglmaier S, Seal RP, Edwards RH, VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate, Trends Neurosci. 27 (2004) 98–103. [DOI] [PubMed] [Google Scholar]
- [15].Tabb JS, Kish PE, Van Dyke R, Ueda T, Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH, J. Biol. Chem 267 (1992) 15412–15418. [PubMed] [Google Scholar]
- [16].Moriyama Y, Yamamoto A, Vesicular L-glutamate transporter in microvesicles from bovine pineal glands. Driving force, mechanism of chloride anion activation, and substrate specificity, J. Biol. Chem 270 (1995) 22314–22320. [DOI] [PubMed] [Google Scholar]
- [17].Juge N, Yoshida Y, Yatsushiro S, Omote H, Moriyama Y, Vesicular glutamate transporter contains two independent transport machineries, J. Biol. Chem 281 (2006) 39499–39506. [DOI] [PubMed] [Google Scholar]
- [18].Anne C, Gasnier B, Vesicular neurotransmitter transporters: mechanistic aspects, Curr. Top. Membr 73 (2014) 149–174. [DOI] [PubMed] [Google Scholar]
- [19].Wolosker H, de Souza DO, de Meis L, Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient, J. Biol. Chem 271 (1996) 11726–11731. [DOI] [PubMed] [Google Scholar]
- [20].Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, Uneyama H, Edwards RH, Nicoll RA, Moriyama Y, Metabolic control of vesicular glutamate transport and release, Neuron 68 (2010) 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Omote H, Miyaji T, Hiasa M, Juge N, Moriyama Y, Structure, function, and drug interactions of neurotransmitter transporters in the postgenomic era, Annu. Rev. Pharmacol. Toxicol 56 (2016) 385–402. [DOI] [PubMed] [Google Scholar]
- [22].Leano JB, Batarni S, Eriksen J, Juge N, Pak JE, Kimura-Someya T, Robles-Colmenares Y, Moriyama Y, Stroud RM, Edwards RH, Structures suggest a mechanism for energy coupling by a family of organic anion transporters, PLoS Biol. 17 (2019) e3000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hinoi E, Takarada T, Tsuchihashi Y, Yoneda Y, Glutamate transporters as drug targets, Curr. Drug Targets CNS Neurol. Disord 4 (2005) 211–220. [DOI] [PubMed] [Google Scholar]
- [24].Jahn R, VGLUTs—potential targets for the treatment of seizures? Neuron 68 (2010) 6–8. [DOI] [PubMed] [Google Scholar]
- [25].Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A, A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle, Neuron 49 (2006) 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fonnum F, Fykse EM, Roseth S, Uptake of glutamate into synaptic vesicles, Prog. Brain Res 116 (1998) 87–101. [DOI] [PubMed] [Google Scholar]
- [27].Naito S, Ueda T, Characterization of glutamate uptake into synaptic vesicles, J. Neurochem 44 (1985) 99–109. [DOI] [PubMed] [Google Scholar]
- [28].Maycox PR, Deckwerth T, Hell JW, Jahn R, Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes, J. Biol. Chem 263 (1988) 15423–15428. [PubMed] [Google Scholar]
- [29].Schenck S, Kunz L, Sahlender D, Pardon E, Geertsma ER, Savtchouk I, Suzuki T, Neldner Y, Stefanic S, Steyaert J, Volterra A, Dutzler R, Generation and characterization of anti-VGLUT nanobodies acting as inhibitors of transport, Biochemistry 56 (2017) 3962–3971. [DOI] [PubMed] [Google Scholar]
- [30].Preobraschenski J, Cheret C, Ganzella M, Zander JF, Richter K, Schenck S, Jahn R, Ahnert-Hilger G, Dual and direction-selective mechanisms of phosphate transport by the vesicular glutamate transporter, Cell Rep. 23 (2018) 535–545. [DOI] [PubMed] [Google Scholar]
- [31].Kehrl J, Althaus JC, Showalter HD, Rudzinski DM, Sutton MA, Ueda T, Vesicular glutamate transporter inhibitors: structurally modified brilliant yellow analogs, Neurochem. Res 42 (2017) 1823–1832. [DOI] [PubMed] [Google Scholar]
- [32].Almqvist J, Huang Y, Laaksonen A, Wang DN, Hovmoller S, Docking and homology modeling explain inhibition of the human vesicular glutamate transporters, Protein Sci. 16 (2007) 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hirai T, Heymann JA, Maloney PC, Subramaniam S, Structural model for 12-helix transporters belonging to the major facilitator superfamily, J. Bacteriol 185 (2003) 1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM, Inhibitor of the glutamate vesicular transporter (VGLUT), Curr. Med. Chem 12 (2005) 2041–2056. [DOI] [PubMed] [Google Scholar]
- [35].Tsigelny IF, Greenberg J, Kouznetsova V, Nigam SK, Modeling of glycerol-3-phosphate transporter suggests a potential ‘tilt’ mechanism involved in its function, J. Bioinforma. Comput. Biol 6 (2008) 885–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fei H, Karnezis T, Reimer RJ, Krantz DE, Membrane topology of the Drosophila vesicular glutamate transporter, J. Neurochem 101 (2007) 1662–1671. [DOI] [PubMed] [Google Scholar]
- [37].Acher FC, Selvam C, Pin JP, Goudet C, Bertrand HO, A critical pocket close to the glutamate binding site of mGlu receptors opens new possibilities for agonist design, Neuropharmacology 60 (2011) 102–107. [DOI] [PubMed] [Google Scholar]
- [38].Pietrancosta N, Anne C, Prescher H, Ruivo R, Sagne C, Debacker C, Bertrand HO, Brossmer R, Acher F, Gasnier B, Successful prediction of substrate-binding pocket in SLC17 transporter sialin, J. Biol. Chem 287 (2012) 11489–11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chang JM, Di Tommaso P, Taly JF, Notredame C, Accurate multiple sequence alignment of transmembrane proteins with PSI-coffee, BMC Bioinf. 4 (13 Suppl) (2012) S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sievers F, Higgins DG, Clustal omega for making accurate alignments of many protein sequences, Protein Sci. 27 (2018) 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Krieger E, Vriend G, YASARA view - molecular graphics for all devices - from smartphones to workstations, Bioinformatics 30 (2014) 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM, MUSTANG: a multiple structural alignment algorithm, Proteins 64 (2006) 559–574. [DOI] [PubMed] [Google Scholar]
- [43].Winter HC, Ueda T, Glutamate uptake system in the presynaptic vesicle: glutamic acid analogs as inhibitors and alternate substrates, Neurochem. Res 18 (1993) 79–85. [DOI] [PubMed] [Google Scholar]
- [44].Hartinger J, Jahn R, An anion binding site that regulates the glutamate transporter of synaptic vesicles, J. Biol. Chem 268 (1993) 23122–23127. [PubMed] [Google Scholar]
- [45].Eriksen J, Chang R, McGregor M, Silm K, Suzuki T, Edwards RH, Protons regulate vesicular glutamate transporters through an allosteric mechanism, Neuron 90 (2016) 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA, Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue, Nature 474 (2011) 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shan J, Javitch JA, Shi L, Weinstein H, The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter, PLoS One 6 (2011) e16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sarker S, Weissensteiner R, Steiner I, Sitte HH, Ecker GF, Freissmuth M, Sucic S, The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin transporter, Mol. Pharmacol 78 (2010) 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Celik L, Schiott B, Tajkhorshid E, Substrate binding and formation of an occluded state in the leucine transporter, Biophys. J 94 (2008) 1600–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Etoga JL, Ahmed SK, Patel S, Bridges RJ, Thompson CM, Conformationally-restricted amino acid analogues bearing a distal sulfonic acid show selective inhibition of system x(c)(−) over the vesicular glutamate transporter, Bioorg. Med. Chem. Lett 20 (2010) 2680–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dunlop J, Fear A, Griffiths R, Glutamate uptake into synaptic vesicles—inhibition by sulphur amino acids, Neuroreport 2 (1991) 377–379. [DOI] [PubMed] [Google Scholar]
- [52].Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM, Synthesis and in vitro pharmacology of substituted quinoline-2,4-dicarboxylic acids as inhibitors of vesicular glutamate transport, J. Med. Chem 45 (2002) 2260–2276. [DOI] [PubMed] [Google Scholar]
- [53].Carrigan CN, Esslinger CS, Bartlett RD, Bridges RJ, Thompson CM, Quinoline-2,4-dicarboxylic acids: synthesis and evaluation as inhibitors of the glutamate vesicular transport system, Bioorg. Med. Chem. Lett 9 (1999) 2607–2612. [DOI] [PubMed] [Google Scholar]
- [54].Patel SA, Nagy JO, Bolstad ED, Gerdes JM, Thompson CM, Tetrapeptide inhibitors of the glutamate vesicular transporter (VGLUT), Bioorg. Med. Chem. Lett 17 (2007) 5125–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ahmed SAE, G JL, Patel SA, Bridges RJ, Thompson CM, Use of the hydantoin isostere to produce inhibitors showing selectivity toward the vesicular glutamate transporter versus the obligate exchange transporter system xc, Bioorg. Med. Chem. Lett 21 (14) (2011) 4358–4362, 10.1016/j.bmcl.2011.05.018 In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fykse EM, Fonnum F, Amino acid neurotransmission: dynamics of vesicular uptake, Neurochem. Res 21 (1996) 1053–1060. [DOI] [PubMed] [Google Scholar]
- [57].Fykse EM, Iversen EG, Fonnum F, Inhibition of L-glutamate uptake into synaptic vesicles, Neurosci. Lett 135 (1992) 125–128. [DOI] [PubMed] [Google Scholar]
- [58].Roseth S, Fykse EM, Fonnum F, Uptake of L-glutamate into rat brain synaptic vesicles: effect of inhibitors that bind specifically to the glutamate transporter, J. Neurochem 65 (1995) 96–103. [DOI] [PubMed] [Google Scholar]
- [59].Roseth S, Fykse EM, Fonnum F, Uptake of L-glutamate into synaptic vesicles: competitive inhibition by dyes with biphenyl and amino- and sulphonic acid-substituted naphthyl groups, Biochem. Pharmacol 56 (1998) 1243–1249. [DOI] [PubMed] [Google Scholar]
- [60].Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM, Inhibitors of the glutamate vesicular transporter (VGLUT), Curr. Med. Chem 12 (2005) 2041–2056. [DOI] [PubMed] [Google Scholar]
- [61].Favre-Besse FC, Poirel O, Bersot T, Kim-Grellier E, Daumas S, El Mestikawy S, Acher FC, Pietrancosta N, Design, synthesis and biological evaluation of smallazo-dyes as potent vesicular glutamate transporters inhibitors, Eur. J. Med. Chem 78 (2014) 236–247. [DOI] [PubMed] [Google Scholar]
- [62].Roseth S, Fykse EM, Fonnum F, The effect of arachidonic acid and free fatty acids on vesicular uptake of glutamate and gamma-aminobutyric acid, Eur. J. Pharmacol 341 (1998) 281–288. [DOI] [PubMed] [Google Scholar]
- [63].Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ, Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles, Neuropharmacology 37 (1998) 839–846. [DOI] [PubMed] [Google Scholar]
- [64].Carlson MD, Kish PE, Ueda T, Glutamate uptake into synaptic vesicles: competitive inhibition by bromocriptine, J. Neurochem 53 (1989) 1889–1894. [DOI] [PubMed] [Google Scholar]
- [65].Roseth S, Hansen TW, Fonnum F, Walaas SI, Bilirubin inhibits transport of neurotransmitters in synaptic vesicles, Pediatr. Res 44 (1998) 312–316. [DOI] [PubMed] [Google Scholar]
- [66].Pietrancosta N, Kessler A, Favre-Besse FC, Triballeau N, Quentin T, Giros B, El Mestikawy S, Acher FC, Rose Bengal analogs and vesicular glutamate transporters (VGLUTs), Bioorg. Med. Chem 18 (2010) 6922–6933. [DOI] [PubMed] [Google Scholar]
- [67].Ozkan ED, Ueda T, Glutamate transport and storage in synaptic vesicles, Jpn. J. Pharmacol 77 (1998) 1–10. [DOI] [PubMed] [Google Scholar]
- [68].Bartlett RD, Identification and Characterization of Inhibitors of L-Glutamate Transport into Rat Brain Synaptic Vesicles (Ph.D. Thesis), The University of Montana, Missoula MT., 1999. [Google Scholar]
- [69].Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M, PLIP: fully automated protein-ligand interaction profiler, Nucleic Acids Res. 43 (2015) W443–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Laskowski RA, Swindells MB, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery, J. Chem. Inf. Model 51 (2011) 2778–2786. [DOI] [PubMed] [Google Scholar]
- [71].Wallace AC, Laskowski RA, Thornton JM, LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein Eng. 8 (1995) 127–134. [DOI] [PubMed] [Google Scholar]
- [72].Tamura Y, Ogita K, Ueda T, A new VGLUT-specific potent inhibitor: pharmacophore of brilliant yellow, Neurochem. Res 39 (2014) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Carrigan CN, Patel SA, Cox HD, Bolstad ES, Gerdes JM, Smith WE, Bridges RJ, Thompson CM, The development of benzo- and naphtho-fused quinoline-2,4-dicarboxylic acids as vesicular glutamate transporter (VGLUT) inhibitors reveals a possible role for neuroactive steroids, Bioorg. Med. Chem. Lett 24 (2014) 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ahmed SK, Etoga JL, Patel SA, Bridges RJ, Thompson CM, Use of the hydantoin isostere to produce inhibitors showing selectivity toward the vesicular glutamate transporter versus the obligate exchange transporter system x(c)(−), Bioorg. Med. Chem. Lett 21 (2011) 4358–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ogita K, Hirata K, Bole DG, Yoshida S, Tamura Y, Leckenby AM, Ueda T, Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal, J. Neurochem 77 (2001) 34–42. [DOI] [PubMed] [Google Scholar]
- [76].Chaudhry FA, Edwards RH, Fonnum F, Vesicular neurotransmitter transporters as targets for endogenous and exogenous toxic substances, Annu. Rev. Pharmacol. Toxicol 48 (2008) 277–301. [DOI] [PubMed] [Google Scholar]
- [77].Tamura Y, Ozkan ED, Bole DG, Ueda T, IPF, a vesicular uptake inhibitory protein factor, can reduce the Ca(2+)-dependent, evoked release of glutamate, GABA and serotonin, J. Neurochem 76 (2001) 1153–1164. [DOI] [PubMed] [Google Scholar]