

ABSTRACT
The valves of the heart are crucial for ensuring that blood flows in one direction from the heart, through the lungs and back to the rest of the body. Heart valve development is regulated by complex interactions between different cardiac cell types and is subject to blood flow-driven forces. Recent work has begun to elucidate the important roles of developmental pathways, valve cell heterogeneity and hemodynamics in determining the structure and function of developing valves. Furthermore, this work has revealed that many key genetic pathways involved in cardiac valve development are also implicated in diseased valves. Here, we review recent discoveries that have furthered our understanding of the molecular, cellular and mechanosensitive mechanisms of valve development, and highlight new insights into congenital and acquired valve disease.
KEY WORDS: Heart valve development, Congenital valve disease, Hemodynamics
Summary: This Review discusses recent discoveries of molecular, cellular and mechanosensitive mechanisms of valve development in mammals and zebrafish, along with relevance to human congenital and acquired valve disease.
Introduction
As the first organ to form, the heart has the essential role of pumping blood through the developing embryo. The resulting pattern of blood flow and fluid forces contribute to the development of heart chambers, including the formation of heart valves (Hove et al., 2003). The atria and ventricles of the vertebrate heart are separated by the atrioventricular (AV) valves (the mitral valve and the tricuspid valve; see Glossary, Box 1) while the semilunar (SL) valves at the base of the aorta and pulmonary artery (the aortic valve and the pulmonary valve; see Glossary, Box 1) separate the ventricles from the outflow tract (OFT; see Glossary, Box 1) (Fig. 1A). Together, these valves ensure that blood flows in the correct direction through the heart. Abnormal valve development contributes to the most common congenital heart malformations, including defects in valve leaflet anatomy and function (Loffredo, 2000; Schoen, 2008). In addition, congenitally malformed valves are predisposed to heart valve disease later in life (LaHaye et al., 2014). Heart valve disease is an increasing health-care burden, with an overall incidence of ∼2.5% in the USA and an incidence of over 10% in individuals older than 65 years of age in the USA and Europe (Nkomo et al., 2006; Virani et al., 2020; Alfieri and Vahanian, 2017). Thus, understanding valve development has important implications for identifying the mechanisms that underlie heart valve disease and for developing new therapeutic approaches to treat it.
Box 1. Glossary.
Atrioventricular canal (AVC). The channels between the atria and the ventricles where the atrioventricular valves develop.
Atrioventricular (AV) valve. A classification referring to the mitral valve and the tricuspid valve: two cardiac valves that ensure blood moves from the atrium to the ventricle.
Bicuspid aortic valve (BAV). An aortic valve that has two leaflets instead of the typical three, as two of the leaflets have fused to become one or one leaflet has failed to develop.
Calcific aortic valve disease (CAVD). A disease in which the aortic valve begins to thicken, aortic sclerosis, before becoming calcified, aortic stenosis. As the valve begins to calcify, it becomes stiff, limiting its movement and eventually leading to heart failure.
Diastole. One of two cardiac phases in which the heart relaxes after it contracts, allowing blood to flow in.
Endothelial-to-mesenchymal transition (EndoMT). A process by which an endothelial cell loses its connection with neighboring endothelial cells and transitions into a mesenchymal cell, accompanied with a decrease in endothelial cell markers and an increase in mesenchymal cell markers.
Laminar shear stress. The frictional force exerted on the endothelium by high unidirectional flow.
Myxomatous mitral valve disease (MMVD). A disease characterized by the accumulation of proteoglycans, and the disruption of the collagen and elastin fibers that ultimately results in mitral valve thickening and regurgitation.
Oscillatory shear stress (OSS). The frictional force exerted on the endothelium by low bidirectional flow.
Outflow tract (OFT). The region of the ventricle in which blood passes through to the great arteries (either the pulmonary artery or the aorta).
Parietal leaflet. Leaflets of the mitral and tricuspid valves located on the free wall of the heart in contrast to leaflets adjacent to the interventricular septum.
Semilunar (SL) valve. A classification referring to the pulmonary valve and aortic valve: two cardiac valves that ensure blood moves from the ventricle to the large vessel leaving the heart.
Systole. One of two cardiac phases in which the heart contracts to pump blood out.
Valve endothelial cell (VEC). A specialized endocardial endothelial cell subpopulation lining the outside of cardiac valves.
Valve interstitial cell (VIC). A specialized subpopulation of mesenchymal cells that are dispersed throughout the three layers of concentrated extracellular matrix (ECM) of cardiac valves.
Fig. 1.

Cardiac valves in the adult mammalian and the zebrafish heart. (A) The mammalian heart consists of four chambers – the left and right atria, and the left and right ventricles – separated by four valves. The AV valves, which include the mitral valve (green) and the tricuspid valve (dark blue), lie between the atria and the ventricles; the SL valves, which include the pulmonary valve (dark red) and the aortic valve (yellow), lie between the ventricles and the outflow tracts. (B) The zebrafish heart consists of two chambers – one atrium and one ventricle – and two valves. The atrioventricular valve (cyan), which is similar to the mammalian tricuspid and mitral valves, lies between the atrium and the ventricle; the bulboventricular valve (orange), which is similar to the mammalian aortic valve, lies between the ventricle and the bulbus arteriosus (purple). AV, atrioventricular; SL, semilunar.
In this Review, we summarize recent findings related to the molecular, cellular and mechanosensitive mechanisms of heart valve development. In addition, we compare valve development in the four-chambered hearts of birds and mammals with that in the two-chambered hearts of zebrafish (Fig. 1B), in which the role of blood flow in valve development has been recently investigated. Finally, we highlight how some of the molecular pathways involved in heart valve development have been implicated in heart valve disease and, potentially, regeneration. Together, these recent studies provide new knowledge of molecular and mechanosensitive mechanisms in valvulogenesis, thus establishing important connections between developmental pathways and blood flow during valve homeostasis and the progression of heart valve disease.
An introduction to the structure and function of cardiac valves
Both types of valves – the AV valves and the SL valves – control unidirectional blood flow through the heart. With the contraction of the ventricles during systole (see Glossary, Box 1), blood is expelled across the pulmonary and aortic SL valves, while closing of the tricuspid and mitral AV valves prevents backflow of blood into the atria. Likewise, during diastole (see Glossary, Box 1), blood flows across the tricuspid and the mitral valves as the ventricles fill, while the closed SL valves prevent blood from returning to the heart (Schoen, 2008). Unlike the SL valves, the AV valves are tethered to the ventricles by chordae tendineae, thin tendinous cords that attach the leaflets to the ventricular papillary muscles (Fig. 2) (Schoen, 2008). Thus, mature valves experience a wide range of biomechanical forces, including back pressure when closed, fluid shear stress, bending stress and axial stretch, during every cardiac cycle (Balachandran et al., 2011). Furthermore, the valves must maintain their structure as they open and close more than 3 billion times in the typical lifespan of a human (Schoen, 2008).
Fig. 2.
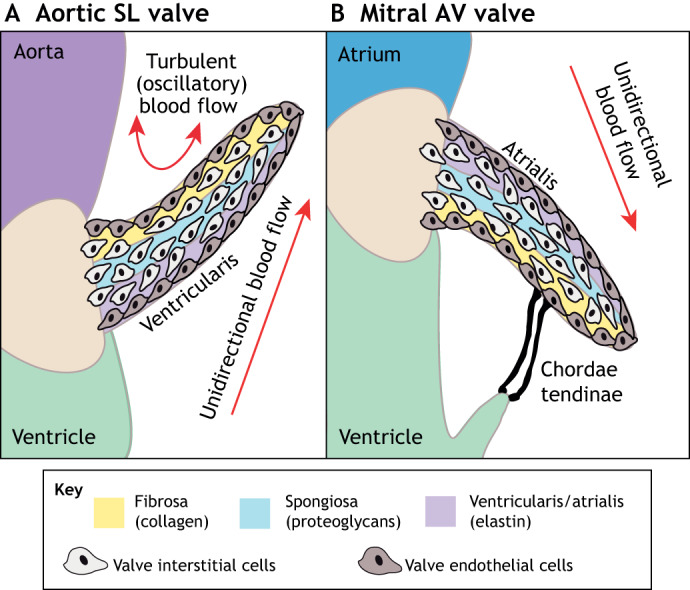
The microarchitecture of adult cardiac valves. (A,B) The stratified structure of aortic (A) and mitral (B) cardiac valves ensures unidirectional blood flow. Valve leaflets are composed of three layers of stratified extracellular matrix, including an elastin-rich ventricularis layer in the SL valves or the atrialis layer in the AV valves (purple), a proteoglycan-rich spongiosa layer (light blue) and a collagen-rich fibrosa layer (yellow), interspersed with valve interstitial cells (VICs) and sheathed in a monolayer of valve endothelial cells (VECs). The direction of pulsatile blood flow in relation to valve leaflets is indicated (red arrows). The microarchitecture of an aortic valve leaflet ensures blood moves from the ventricle to the aorta. The microarchitecture of a mitral valve leaflet is supported by the chordae tendineae, which ensure blood flow from the atrium to the ventricle. AV, atrioventricular; SL, semilunar.
The AV and SL valves in the four-chambered hearts of humans, mice and chickens share a similar microarchitecture composed of three layers of extracellular matrix (ECM) interspersed with valve interstitial cells (VICs; see Glossary, Box 1) and sheathed in a monolayer of endocardial valve endothelial cells (VECs; see Glossary, Box 1) (Fig. 2) (Gross and Kugel, 1931; Hinton et al., 2006). These layers function together, providing strength and durability to the valves. The central spongiosa layer of proteoglycans provides leaflet compressibility, while a layer of predominantly fibrillar collagen in the fibrosa layer provides strength. Elastin in the atrialis layer of SL valves or the ventricularis layer of AV valves then provides elasticity (Fig. 2) (Yalcin et al., 2011). VECs are in direct contact with blood flow and communicate with VICs to modulate the structure of the valve and maintain unidirectional blood flow (Schoen, 1997). In a healthy human valve, the majority of VICs are quiescent, exhibiting little cell proliferation and low levels of ECM gene expression (Aikawa et al., 2006). By contrast, in congenitally malformed valves, which are predisposed to valve disease, VICs can become activated and start to overproduce ECM proteins, leading to valve stenosis or myxomatous degeneration (Aikawa et al., 2007; LaHaye et al., 2014).
The initiation of valve development: endocardial cushion formation and endothelial-to-mesenchymal transition
In the developing vertebrate embryo, the primitive heart initially consists of an outer myocardial cell layer surrounding an inner layer of endocardial endothelial cells, with a layer of hyaluronan-rich cardiac jelly in between (Person et al., 2005b). As the heart tube loops to the right, the primitive atria and ventricles of the heart begin to contract. Just after looping, cardiac valve development initiates [on embryonic day (E)3 in chick, E9.5 in mouse and E31 to E35 in human]. The valves begin as swellings of cardiac jelly at the junction between the atrium and ventricle (the atrioventricular canal, AVC; see Glossary, Box 1) and also in the OFT (Person et al., 2005b; Martinsen, 2005; Fishman and Chien, 1997; Moorman et al., 2003). These swellings of cardiac jelly then become populated with mesenchymal cells that are derived from a subpopulation of endocardial endothelial cells that undergo endothelial-to-mesenchymal transition (EndoMT; see Glossary, Box 1) and migrate internally (Markwald et al., 1977).
Formation of endocardial cushions is dependent on bone morphogenetic protein (BMP) signaling in the AVC and OFT myocardium (Fig. 3A). BMPs are members of the transforming growth factor (TGF) β superfamily. Bmp2, which is produced in the myocardium, promotes the expression of hyaluronan synthase 2 (Has2) to produce the cardiac jelly required to initiate EndoMT (Camenisch et al., 2000). AVC myocardial Bmp2 and endocardial endothelial BMP type 1A receptor (Bmpr1a) are required for the formation of endocardial cushion cells that express mesenchymal markers, including Twist1, Msx1/2 and Snail transcription factors (Fig. 3B). Bmp4 has a similar role in initiating EndoMT in the AVC and OFT. Mice with a hypomorphic allele of Bmp4 have smaller AV cushions and the targeted knockout of myocardial Bmp4 results in reduced SL endocardial cushion expansion due to insufficient cells (Jiao et al., 2003; McCulley et al., 2008). Finally, BMP-induced transcription factors Tbx2 and Tbx3 are also required to suppress the expression of atrial and ventricular chamber-specific genes in the myocardium underlying the site of future AV cushion formation (Fig. 3B) (Ma et al., 2005; Rivera-Feliciano and Tabin, 2006; Singh et al., 2012). Together, these data demonstrate the important role of the myocardium in the initial swelling of cardiac jelly, as well as for the initiation of EndoMT in the overlaying population of endocardial cells.
Fig. 3.

The molecular pathways regulating endocardial cushion formation, endothelial-to-mesenchymal transition (EndoMT) and valve primordia growth. (A) E9-10: endocardial cushion formation is initiated when myocardial Bmp2 promotes the expression of hyaluronan, Tbx2 and Tbx3 to stimulate cardiac jelly formation in endocardial swellings in the atrioventricular canal (AVC). (B) E10-12: myocardial BMP signaling, through endocardial Bmpr1a and endocardial Notch1, and Wnt/β-catenin, TGFβ and Hippo/Yap1 signaling promote EndoMT and the expression of the mesenchymal markers Twist, Msx1/2 and Snail. (C) E14-18: the distal proliferation of valve endothelial cells (VECs) and valve interstitial cells (VICs) stimulates the outgrowth of leaflet primordia. VEC proliferation is induced by Wnt/β-catenin and VEGF signaling. VIC proliferation is induced by BMP and FGF4 signaling, and is inhibited by EGF and Notch signaling.
Other TGFβ family members, along with Wnt/β-catenin, Hippo/Yap and Notch signaling proteins, have been implicated in endocardial cushion EndoMT. Endocardial cushion formation also requires the diversification of VECs from atrial, ventricular and OFT endothelial cells. This process is first evident in the restricted expression of TGFβ type III receptor in VECs, which is also required for the initiation of EndoMT (Brown et al., 1999). TGFβ2 promotes the expression of the transcription factor Slug to induce EndoMT in the AVC (Romano and Runyan, 2000). Yes-associated protein 1 (YAP1), phosphorylated through the Hippo signaling pathway, contributes to TGFβ mediated EndoMT by interacting with the Smad2/3/4 complex to activate expression of Snail, Twist1 and Slug (Zhang et al., 2014). TGFβ2 signaling also promotes EndoMT through the Wnt/β-catenin signaling pathway in vitro, and Wnt/β-catenin signaling is required for VIC proliferation in vivo (Liebner et al., 2004). Notch signaling also has been implicated in endocardial cushion EndoMT, acting upstream of TGFβ signaling and intersecting with BMP signaling. Enhanced activation of Notch1 signaling in immortalized endothelial cells in vitro promotes EndoMT by inducing Snail expression and repressing VE-cadherin expression (Timmerman et al., 2004). The Notch ligand DELTA-like ligand 4 (DLL-4) is also required for EndoMT (Fig. 3B) (MacGrogan et al., 2016). Interestingly, only a subpopulation of VECs undergoes EndoMT but the mechanisms that govern this selectivity are unknown. Together, these studies demonstrate that the TGFβ, BMP, Wnt/β-catenin, Hippo/Yap and Notch signaling pathways coordinate endocardial cushion EndoMT.
Endocardial cushion mesenchyme proliferation and valve primordia formation
After the endocardial cushions become populated by mesenchymal cells through EndoMT, the valve primordia elongate and stratify into mature valve leaflets (Fig. 3C) through a combination of cell proliferation and ECM synthesis (Hinton et al., 2006; Martinsen, 2005; Armstrong and Bischoff, 2004). Cell proliferation is initially active in the distal regions of the endocardial cushions and acts to promote outgrowth of valve primordia, decreasing as the valve primordia remodel into leaflets (Lincoln et al., 2004). BMP and epidermal growth factor (EGF) signaling play key roles in VIC proliferation and maturation. Specifically, EGF signaling acts to limit VIC proliferation by inhibiting BMP signaling, usually through Smad1 (Person et al., 2005b). Accordingly, mice with a hypomorphic mutation in the EGF receptor have enlarged SL valves due to increased VIC numbers (Chen et al., 2000). Similarly, while DLL4-mediated Notch signaling is initially required for EndoMT, jagged 1-mediated Notch activation negatively regulates VIC proliferation by inhibiting BMP signaling (MacGrogan et al., 2016). Fibroblast growth factors (FGFs) are also implicated in valve primordia growth, as overexpressing Fgf4 in developing mouse embryos results in increased VIC proliferation (Sugi et al., 2003). These studies demonstrate that BMP signaling, its negative regulators and FGFs play important roles in controlling the growth of valve primordia.
The VECs overlaying the VICs in the endocardial cushions and valve primordia are also highly proliferative (Hinton et al., 2006; Markwald et al., 1977; Martinsen, 2005). One of the driving forces of VEC proliferation during valve primordia growth is vascular endothelial growth factor (VEGF), which acts with the transcription factor nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) (Miquerol et al., 2000; Dor et al., 2001; Lee et al., 2006; Combs and Yutzey, 2009b). Wnt9a also promotes VEC proliferation, while the secreted Wnt antagonist Frzb, which is produced by both VECs and VICs, inhibits VIC proliferation (Fig. 3C) (Person et al., 2005a). Thus, Wnt/β-catenin signaling and VEGF signaling pathways are essential for promoting VEC proliferation during valve development.
The diversification of VICs within valve primordia initiates ECM compartmentalization. In vitro studies demonstrate that Bmp2-treated valve cell progenitors express Sox9, which subsequently promotes the expression of aggrecan, an important component of the central spongiosa ECM layer; accordingly, the embryonic loss of Sox9 in mice results in abnormal ECM organization (Lincoln et al., 2006, 2007). Less is known about the diversification of VIC subpopulations in the collagen-rich and elastin-rich ECM layers. Wnt signaling has been implicated in the collagen-rich fibrosa ECM layer, and Notch1 has been implicated in the elastin-rich atrialis/ventricularis ECM layer (Combs and Yutzey, 2009a). These data support a role for diversified VICs in ECM compartmentalization and valve primordia morphogenesis. However, further studies are required to determine the plasticity and specific contributions of VIC subpopulations to ECM compartmentalization in vivo.
The VEC populations likewise begin to diversify during fetal development, exhibiting distinct profiles of gene expression in localized subpopulations. There is evidence that VEC diversification is dictated by physical forces, including blood flow. For example, VECs on the atrialis/ventricularis side of the valves begin to express the shear stress-responsive transcription factor kruppel-like factor 2 (KLF2) by E10.5 (Goddard et al., 2017). Similarly, Notch1 activation is restricted to the VECs on the atrialis/ventricularis side of developing valve leaflets during elongation (Goddard et al., 2017; MacGrogan et al., 2016). Likewise, loss of the endothelial-specific receptor tyrosine kinase Tie1, which is upregulated by oscillatory shear stress (OSS; see Glossary, Box 1) and is expressed by VECs on the fibrosa side, leads to enlarged aortic valves with increased glycosaminoglycan (GAG) and decreased collagen content (Qu et al., 2019). Thus, flow-responsive signaling pathways are involved in VEC diversification.
It is speculated that blood flow-mediated VEC diversification also influences VIC diversification. Atrialis/ventricularis KLF2-positive VECs secrete Wnt9b to activate Wnt/β-catenin signaling in neighboring VICs. Furthermore, loss of endocardial KLF2 or Wnt9b results in valves that fail to elongate and thin in the direction of blood flow (Goddard et al., 2017). Tools for VEC- or VIC-specific gene targeting have recently been reported and will be useful for dissecting these interactions in vivo (Wu et al., 2011; Kanisicak et al., 2016). However, the ability to manipulate fluid flow on developing heart valves in utero remains a significant challenge. Understanding how subpopulations of VICs and VECs are specified, and how these subpopulations contribute to ECM compartmentalization during development could provide valuable insight into alterations in valve structure and function in disease.
Postnatal valve remodeling and valve cell heterogeneity
During postnatal development, valve primordia remodel into valve leaflets with three distinct layers of stratified ECM (Fig. 2) (Hinton et al., 2006). Although genes essential for elastin fiber formation and immature elastin are found early in valve development, mature elastin is not present until after birth (Votteler et al., 2013). During this period, elastin expression is induced on the atrialis/ventricularis side of the AV and SL valves, respectively, and periostin-dependent collagen fiber remodeling is active in the fibrosa ECM compartment (Box 2) (Snider et al., 2008; Lincoln et al., 2004). ECM remodeling enzymes, including matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family of zinc metalloproteinases also have crucial roles in postnatal heart valve remodeling (Lockhart et al., 2011). For example, embryonic mice haploinsufficient for ADAMTS9 do not display SL valve abnormalities, but adult haploinsufficient ADAMTS9 mice have increased accumulation of versican in SL and AV valves (Kern et al., 2010). Recently, ADAMTS19 has been found to also be involved in valve homeostasis. Adamts19 knockout mice demonstrate early onset aortic valve regurgitation and/or stenosis accompanied with a disorganized ECM (Wünnemann et al., 2020). These data highlight how the regulation of ECM compartmentalization is an important feature of postnatal valve remodeling and valve homeostasis.
Box 2. Cardiac valve development in humans.
Valve endocardial cushions in humans are derived from four major cushions: the posterior and anterior atrioventricular cushions, and the parietal and outflow tract (OFT) septal cushions. These cushions initially form as cardiomyocytes secrete extracellular matrix (ECM) between the endocardial and myocardial layers of the atrioventricular canal (AVC) and the outflow tract (OFT). The cushions then protrude into the lumen and fuse, separating the different compartments of the heart (reviewed by Buijtendijk et al., 2020). Valve endocardial cushions appear in the OFT by 5 weeks gestation (Maron and Hutchins, 1974). Similar to mouse valve development, there is evidence of endothelial-to-mesenchymal transition (EndoMT), including a role for NFATc1 and BMP signaling (Monaghan et al., 2016; Neri et al., 2019).
The distinct trilaminar ECM structure found in rodents is also conserved in human valves (Hinton et al., 2006). Later stages of human gestation include matrix organization and remodeling. At 14 weeks gestation, the SL valves are composed of glycosaminoglycans before becoming bilayered at 20 weeks of gestation after a collagen-rich layer forms (Aikawa et al., 2006). By 36 weeks gestation, the SL valves are roughly stratified into the three ECM layers, which become more distinct in infants and children as the ECM strata matures (Aikawa et al., 2006). This indicates that, as in mice, human valves continue to develop postnatally (Aikawa et al., 2006). Throughout fetal development, VECs and VICs demonstrate activation, indicating extensive valve remodeling, when compared with the more quiescent phenotype of adult valve cells (Aikawa et al., 2006).
Although not all developmental pathways governing mouse, chicken and zebrafish valve development have been observed in humans, many genes related to these pathways have been linked to inherited congenital heart valve defects and acquired valve disease in human patients (Balistreri et al., 2018; Zaidi and Brueckner, 2017; Garg et al., 2005; LaHaye et al., 2014).
Advances in single cell RNA sequencing (scRNAseq) have begun to elucidate how distinct VEC and VIC subpopulations, as well as other cardiac valve cell types, contribute to valve remodeling. For example, a recent study (Hulin et al., 2019) used scRNAseq on remodeling murine mitral and aortic valves, and found distinct populations of VECs, VICs, immune cells and melanocytes. Furthermore, at postnatal day (P) 7 when the cardiac valves are undergoing remodeling, subpopulations of these cells localize in distinct regions of the valves and display unique transcriptional profiles (Fig. 4). One VEC subpopulation is found solely on the fibrosa side of the valve leaflet exposed to turbulent (oscillatory) blood flow and expresses the transcription factor Prox1, a lymphatic marker influenced by turbulent (oscillatory) flow (Fig. 4, yellow cells). Another VEC subpopulation can be found on the tip of the valve where the leaflets coapt as the valve closes and expresses hyaluronan and proteoglycan link protein 1 (Hapln1) (Fig. 4, orange cells). Similarly, at P7 a collagen-expressing VIC subpopulation (Fig. 4, pink cells) exists underneath the Prox1-positive VEC subpopulation, and a GAG-expressing VIC subpopulation (Fig. 4, green cells) is localized largely in the tip and hinge region under the Hapln1-positive VEC subpopulation (Hulin et al., 2019). Whereas the VEC subpopulations are similarly localized at P7 and P30, the VIC subpopulations undergo significant maturation in the month after birth, which is demonstrated by differential ECM gene expression during active remodeling at P7 and relative quiescence in mature valves at P30. Additional studies are required to better understand the plasticity and specific functions of these subpopulations in valve development and homeostasis.
Fig. 4.
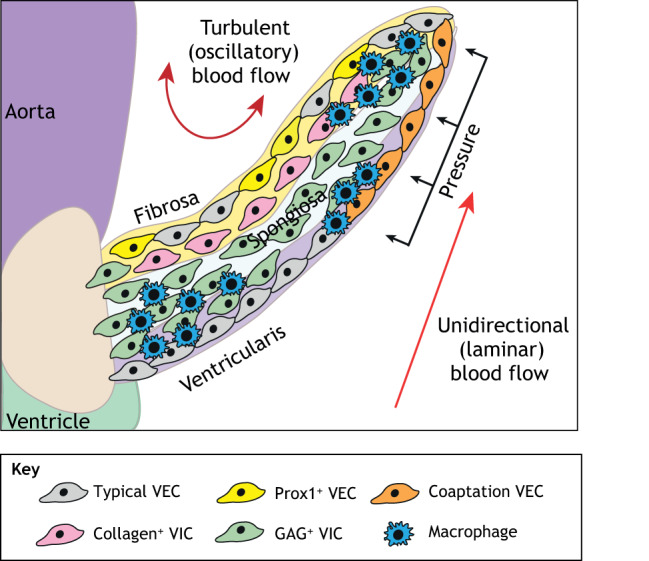
Postnatal aortic valve endothelial and interstitial subpopulations. Postnatal aortic valves contain localized subpopulations of valve endothelial cells (VECs), valve interstitial cells (VICs) and immune cells that exhibit distinct transcriptional profiles. These populations are present in regions of the valve exposed to unique combinations of mechanical forces. The typical VEC subpopulation (gray) can be found on both sides of the valve leaflet, while a VEC subpopulation expressing Prox1 (yellow), a lymphatic marker influenced by oscillatory shear stress, is found solely on the side away from laminar flow. A coaptation VEC population (orange) is found where the leaflets meet. A collagen-expressing subpopulation of VICs (pink) is found in the fibrosa layer and a GAG-expressing subpopulation of VICs (green) is in the hinge and tip of the leaflet. Similarly, macrophages are present in the hinge and tip regions of the valve leaflet, which are subject to high mechanical stress. GAG, glycosaminoglycan.
In addition to VECs and VICs, cells expressing immune system markers are found in postnatal valves and may play a role in valve maturation and homeostasis. Immune cells make up 8-10% of cells in postnatal SL and AV valves and consist of T cells, dendritic cells, mast cells and two populations of macrophages (Hulin et al., 2019). In mice, valve macrophages arise from hemogenic endocardial cells beginning at E10.5 and are found just under the endocardium (Shigeta et al., 2019). In addition, macrophage subpopulations are present in regions of high biomechanical stress, beginning as soon as 1 week after birth (Fig. 4, blue cells) (Hulin et al., 2018). While the role of immune cells in valve development remains unclear, macrophages have been linked to valve remodeling. Specifically, the loss of endocardial-derived macrophages in developing mouse valves results in defects in valve remodeling, including hypercellularity and increased ECM content. These phenotypes are thought to be linked to the phagocytic and antigen presenting roles of macrophages observed in wild-type valves (Shigeta et al., 2019). However, additional studies are required to determine the specific functions of macrophage subtypes and other immune cells in ECM maturation and valve leaflet morphogenesis during late gestation and postnatally.
scRNAseq has also been performed on whole hearts at earlier stages of development, although subpopulations of VECs and VICs were not identified in this study (DeLaughter et al., 2016). This is possibly due to the small number of valve cells relative to other cell types in the rest of the heart, or to the lack of distinctive markers for valve subpopulations at this stage. Additional scRNAseq studies focused on individual cardiac valves followed by in vivo studies are therefore needed to better identify specific markers of individual valve cell subpopulations and to determine the roles these subpopulations play during valve development, homeostasis and disease.
Embryonic sources of cardiac valve cells
As mentioned above, the majority of mature cardiac VICs are derived from endothelial endocardial cushion cells through the process of EndoMT (de Lange et al., 2004; Lincoln et al., 2004). However, another important embryonic source of valve progenitor cells, particularly for SL valves, is the neural crest. Neural crest contributions to the developing heart, in particular the OFT and aortic arch arteries, were first reported in studies using avian embryos (Kirby et al., 1983). Subsequent cell lineage studies in mice showed neural crest-derived cells in the OFT cushions, septum and valve primordia (Jiang et al., 2000). Lineage tracing using an endothelial cell-specific Tie2Cre, compared with a neural crest Wnt1Cre, found that, although endothelial-derived cells are distributed evenly throughout all three leaflets of the aortic valve, neural crest-derived cells are present predominantly in the hinge region of adult murine aortic valves (Gomez-Stallons et al., 2016). Neural crest ablation studies support abnormal neural crest cell migration as a mechanism connecting SL valve malformations with aortic arch and craniofacial abnormalities. Inhibiting neural crest migration by disrupting Rho kinase signaling in neural crest cells results in incorrect positioning of the outflow tract cushions as well as future abnormalities in leaflet patterning (Phillips et al., 2013). Likewise, loss of BMP type IA receptor (BMPRIA) in neural crest-derived cells results in the disorganized distribution of neural crest cells in the OFT septum and endocardial cushions. This phenotype is accompanied by an OFT septum defect, reversed arterial blood flow and abnormal SL valve development, suggesting that neural crest cells play a key role in SL endocardial cushion growth (Nomura-Kitabayashi et al., 2009). Although neural crest cells differentiate into multiple cell types in the craniofacial region, it is not clear whether their ultimate fates within the valve leaflets are distinct from VICs that arise from other sources. However, it is clear that neural crest cells in the OFT are crucial for septation of the aortic and pulmonic root, as well as for the correct number and morphogenesis of SL valve leaflets.
A small number of cells in AV and SL valves are not derived from endothelial or neural crest cells. In avians, but not mammals, one leaflet of the tricuspid valve is highly muscularized, suggesting some contribution from myocardial cells (Lincoln et al., 2004; Sedmera et al., 1997). In line with this, a lineage tracing study using a cardiomyocyte-specific Tnnt2Cre in mice revealed that a small number of myocardial Isl1+ cells also contribute to the SL valves. Interestingly, the disruption of this population leads to an increase in the incidence of bicuspid SL valves, particularly in the aortic valve (Eley et al., 2018). Epicardial-derived cells were identified as a source of AV valve progenitor cells in quail-chicken transplantation experiments (Gittenberger-de Groot et al., 1998). Subsequent lineage analyses in mice found that epicardial-derived cells predominantly populate the parietal leaflets (see Glossary, Box 1) of mitral and tricuspid valves, but it is not known whether epicardial-derived VICs have specific functions (Wessels et al., 2012; Liu et al., 2018). These studies highlight how multiple sources of embryonic cells contribute to the complexity of individual valve leaflet morphologies and might explain why some valve leaflets are more commonly affected by disease.
The role of blood flow and biomechanical forces in valve development
As many morphological changes in cardiac valves correspond to changes in the hemodynamic environment, blood flow has long been thought to influence valve development (Hu and Clark, 1989; Yalcin et al., 2011). An early study using nonliving material demonstrated that fluid flow through a putty-lined channel causes the formation of cushions and valve-like structures, recapitulating the earliest aspects of embryonic valve development (Rodbard, 1956). Adult heart valves primarily experience three types of mechanical forces: flexure as the valves open and close, shear stress as blood flows through the valve, and tension as the closed valve prevents retrograde blood flow. Furthermore, the AV and SL valves experience different types of mechanical load due to the presence or absence of chordae tendineae, respectively (Fig. 2) (Sacks et al., 2009). Fluid shear stress, in particular, appears to be an important force in valve development since the atrialis/ventricularis sides of cardiac valves experience high unidirectional shear stress (or laminar shear stress; see Glossary, Box 1), while the fibrosa side experiences low bidirectional shear stress (turbulent or oscillatory shear stress; see Glossary, Box 1) (Fig. 2) (Simmons et al., 2005). However, owing to the difficulty of manipulating blood flow in vivo without causing lethality, it has been challenging to elucidate the mechanisms by which fluid forces dictate valve development in mammalian species.
Owing to their accessibility, avian embryos have been used to study the role of biomechanical forces at early stages of endocardial cushion development, prior to formation of the body wall. Abnormal flow produced by mechanical manipulations in developing avian hearts results in different valve abnormalities, including valve thickening (Colvee and Hurle, 1983). Similarly, ligation of the right lateral vitelline vein in a developing chicken embryo alters the intracardiac flow pattern, leading to SL valve abnormalities, including a bicuspid aortic valve and a quadricuspid pulmonary valve (Hogers et al., 1997). Likewise, increasing the hemodynamic load on avian OFT cushions, through banding of the proximal aorta, leads to increased cell density and an enhanced EndoMT phenotype (Midgett et al., 2017). Computational modeling has also been used to demonstrate how pressure and shear stress can influence valve development. For example, the highest wall shear stress is found in the valve forming region of the AVC (Yalcin et al., 2011; Buskohl et al., 2012). Interestingly, another study found that ECM composition also changes with increasing fluid forces acting on the valve, suggesting a role for biomechanics in ECM stratification (Butcher et al., 2007). Together, these studies demonstrate how shear stress and blood flow can affect avian valve development.
It has proven more challenging to manipulate blood flow during mammalian valve development (Wakimoto et al., 2000). Consequently, disruption of downstream mechanosensitive genes has been used to examine biomechanically regulated molecular pathways of valve development. Indeed, this approach has been used to identify many mechanosensitive proteins that are involved in VEC diversification, including KLF2, Notch1 and Tie2 (Goddard et al., 2017; MacGrogan et al., 2016; Qu et al., 2019); however, additional studies are required to identify novel mechanosensitive proteins present in the valve. One avenue for identifying potential biomechanically regulated mechanisms is to study the development of other types of vascular valves, including lymphatic valves, venous valves and lymphovenous valves (Petrova et al., 2004; Bazigou et al., 2011; Geng et al., 2016). These valves all form by localized outgrowth of endothelial cells subject to fluid flow, and a number of shear-stress sensitive genes expressed during luminal valve formation are also expressed in developing SL heart valves (Geng et al., 2017). These include genes encoding the gap junction protein connexin 43 (Cx43 or Gja1), which is expressed on the ventricularis/atrialis side of the valves exposed to directional (laminar) blood flow, as well as connexin 37 (Cx37 or Gja4), Gata2, Foxc2 and Prox1, which are expressed on the fibrosa side of the valve exposed to turbulent (oscillatory) flow (Geng et al., 2017). Thus, studies of luminal valves in cardiovascular system may prove instructive for identifying mechanosensitive regulatory mechanisms involved in cardiac valve development, although further characterization of these mechanisms in the context of cardiac valves will also be needed.
The development of cardiac valves: insights from zebrafish
Given their accessibility and ease of manipulation, zebrafish have been used to identify novel mechanisms of valve development, including those subject to biomechanical regulation. The zebrafish heart begins to contract around 22 h post fertilization (hpf) and loops between 30 and 54 hpf (Lombardo et al., 2019). The two valves of the zebrafish heart include the AV valve, which is located between the atrium and the ventricle, and the bulboventricular (BV) valve, which lies between the ventricle and the bulbus arteriosus. Most studies have focused on the AV valve, the development of which occurs in three steps (Fig. 5): (1) specification of the AVC between the atrium and the ventricle; (2) formation of endocardial cushion-like valve primordia; and (3) elongation and maturation of the valve leaflet primordia (Walsh and Stainier, 2001; Lombardo et al., 2019; Gunawan et al., 2019; Beis et al., 2005; Steed et al., 2016b; Pestel et al., 2016). These studies have highlighted conserved and specialized mechanisms of valve formation. In addition, a number of recent studies have leveraged the strength of zebrafish as a model to examine how biomechanical forces affect valve development.
Fig. 5.

Molecular pathways regulating zebrafish valve development. (A) At ∼36-48 hpf, the restriction of bmp4, versican and tbx2 in the myocardium and notch1b in the endocardium initiates atrioventricular canal (AVC) specification between the atrium (A) and the ventricle (V). (B) At ∼50-60 hpf, endocardial cushion-like valve primordia then form through partial endothelial-to-mesenchymal transition (EndoMT), apparent by endocardial invagination and collective migration. klf2a- and notch1b-expressing valve endothelial cell (VEC) progenitors on the ventricular side of the AVC undergo EndoMT or invaginate under notch1b-expressing valve progenitors on the atrial side of the AVC. The NFATc1, Wnt, Notch, EGF and focal adhesion (FA) signaling pathways have also been implicated in the formation of valve primordia. (C) At ∼100 hpf-adult, valve primordia then elongate and thin in response to TGFβ signaling, and remain thin with little extracellular matrix until 28 dpf. The direction of pulsatile blood flow in relation to developing valve structures is indicated (red arrow). hpf, hours post fertilization.
Atrioventricular canal specialization
The zebrafish AVC forms between the atrium and the ventricle at about 36 hpf (Beis et al., 2005). AVC specification coincides with the restriction of bmp4, versican and tbx2b expression to the AV myocardium, and of notch1b expression to the AV endocardium, similar to the initial phases of mouse endocardial cushion formation (Fig. 5A) (Walsh and Stainier, 2001; Westin and Lardelli, 1997; Chi et al., 2008). A recent study found asymmetric phospho-Smad1/5/8 activity at the future superior AVC region as early as 30 hpf, suggesting that BMP signaling could be playing a role in the initial specification of the AVC (Lombardo et al., 2019). Live imaging of zebrafish AV valve formation has shown that, at ∼48 hpf, cuboidal endocardial cells line the inside of the AVC between the atrium and the ventricle (Fig. 5A) (Gunawan et al., 2019). Once the AVC is established, cushion-like endocardial endothelial valve primordia begin to form.
Formation of endocardial endothelial valve primordia
Zebrafish AV valve development begins when the endocardial cells of the AVC coalesce and protrude into the heart lumen starting around 50 hpf (Fig. 5B) (Gunawan et al., 2019). This has been termed collective migration as it is not clear whether the valve-forming endothelial cells lose complete contact with each other to form mesenchymal cells and achieve full EndoMT (Pestel et al., 2016; Steed et al., 2016b). A recent study found that the collective migration needed for AVC endothelial cells to coalesce and protrude into the AVC lumen is driven by integrin α5β1- and talin 1-mediated integrin-based focal adhesion signaling (FA signaling) (Gunawan et al., 2019). In contrast, a separate study that carefully examined primitive valve outgrowths did not show full disconnection of mesenchymal cells from endothelial cells, supporting a partial EndoMT mechanism during early valve development (Pestel et al., 2016). However, it has recently been determined that there are endothelial-derived VICs present within the AV valve by 20 days post fertilization (dpf), demonstrating that full EndoMT does occur later in valve development (Gunawan et al., 2020).
Several pathways involved in mouse endocardial cushion EndoMT are also active in zebrafish valve development. The inhibition of NFATc1 activation blocks valve primordia formation and leads to decreased expression of EndoMT markers, including twist1b (Beis et al., 2005; Gunawan et al., 2020). Furthermore, injecting a constitutively active form of Nfatc1 into one-cell stage zebrafish embryos results in the upregulation of twist1b, demonstrating how Nfatc1 directly regulates twist1b gene expression (Gunawan et al., 2020). Wnt signaling has also been implicated in early cushion development: overexpression of the Wnt/β-catenin signaling activator adenomatous polyposis coli (Apc) results in hyperplastic endocardial cushions, while overexpression of the Wnt inhibitor dickkopf 1 (Dkk1) results in the failure of endocardial cushions to develop (Hurlstone et al., 2003). Furthermore, expressing activated Notch1 in zebrafish embryos results in increased cardiac valve cellularity, while the loss of Notch signaling elements results in decreased Snai1 gene expression (Timmerman et al., 2004). Moreover, zebrafish larva grown in media containing EGFR kinase inhibitors develop atrioventricular regurgitation, suggesting an AV valve defect (Goishi et al., 2003). Together, these results support conserved functions for Nfatc1, Wnt, Notch and EGF signaling in early stages of valve development in zebrafish and mice (Person et al., 2005a; Hulin et al., 2017; Koenig et al., 2016; Chen et al., 2000; Gunawan et al., 2020).
Elongation and maturation of primordial valves
Zebrafish valve leaflets remain long and thin with little ECM until 28 dpf, when ECM deposition increases (Beis et al., 2005; Martin and Bartman, 2009). Inhibiting TGFβ signaling in zebrafish results in normal endocardial cushion formation but later gives rise to abnormal leaflet morphologies leading to regurgitation (Fig. 5C) (Scherz et al., 2008). Thus, similar to mammalian valve development, TGFβ signaling is involved in zebrafish valve leaflet elongation and maturation.
The ECM compartmentalization of mature zebrafish valves when compared with mammalian heart valves has only recently been studied (Schulz et al., 2019). Histological studies demonstrate that, similar to mammalian valves, both the AV and BV valves are composed of VECs, VICs, proteoglycan, collagen and elastin (Hu et al., 2001; Schulz et al., 2019; Gunawan et al., 2020). However, the stroma of the AV valve is composed of a proteoglycan-rich matrix with a thin layer of collagen- and elastin-rich ECM on the in-flow side of the valve (Schulz et al., 2019). The AV valve is analogous to the mitral or tricuspid valve in the mammalian heart, with its trabecular bands on the ventricular side comparable with the chordae tendineae that prevent retrograde blood flow in mammals (Hu et al., 2000). Similarly, the BV valve is analogous to the aortic valve, but consists of two leaflets, instead of three, and sits at the entrance of the more chamber-like bulbus arteriosus (Fig. 1) (Hu et al., 2000). Therefore, although zebrafish valves share similar developmental mechanisms with mammalian valves, morphological differences also exist (Poon and Brand, 2013). The smaller size of the zebrafish heart, and thus the smaller valves, the absence of lungs and the lack of oxygen dependence early in development might account for these differences. There is also evidence that, unlike avian or mammalian valves, zebrafish valves are capable of regeneration; this process involves TGFβ signaling-mediated proliferation of endothelial- and kidney marrow-derived valve cells (Bensimon-Brito et al., 2020).
The influence of mechanical forces on zebrafish heart valve development
Recently, zebrafish have been used to study the role of biomechanical forces in heart valve development. Their optical clarity, lack of dependence on embryonic blood flow for viability and rapid external development, together with genetic tools and techniques for manipulation of blood flow, have been leveraged to identify conserved mechanosensitive cellular and genetic processes of valve development (Steed et al., 2016a; Stainier, 2001). These approaches have highlighted the importance of blood flow in cardiac valve development. For example, occluding the inflow or outflow tract of the developing zebrafish heart results in impaired cardiac valve development (Hove et al., 2003). Similarly, larvae that lack a heartbeat or blood flow do not develop AV endocardial cushions (Bartman et al., 2004). Recently, a study used quantitative live imaging and mathematical modeling to demonstrate a correlation between reversing blood flow in the AVC and the collective migration of future valve-forming endothelial cells (Boselli et al., 2017). Furthermore, the rate at which valves mature is dependent on the size of the larvae and it was therefore speculated that larval size affects the forces the cardiovascular system exerts on the cardiac valves, thereby affecting valve leaflet elongation (Martin and Bartman, 2009).
Similar to mammals, one of the most-studied mechanosensitive proteins involved in zebrafish valve development is KLF2. One way to alter blood flow in zebrafish is by lowering blood viscosity through knockdown of gata1 and gata2, which results in fewer circulating blood cells and retrograde flow. This reversal of blood flow corresponds with decreased klf2a expression, decreased expression of its target notch1b in the AVC and impaired valve development (Vermot et al., 2009). In wild-type larvae, klf2a and notch1b are expressed in the atrium-derived luminal valve cells on the flow side, while the ventricle-derived abluminal cells away from blood flow express factors involved in Wnt/β-catenin signaling (Fig. 5B) (Pestel et al., 2016). In addition, klf2a expression is regulated by mechanosensitive ion channels expressed in the endocardium, including Trpv4 and its partner Trpp2 (Pkd2) (Gees et al., 2010). The loss of either Trpv4 or Trpp2 results in decreased klf2a expression, as well as abnormal valve development. However, phenotypical differences between klf2a-, trpv4-, and trpp2-deficient valves suggest that other mechanosensitive pathways may also be involved in valve development (Heckel et al., 2015). Furthermore, klf2a and notch1b are negatively regulated by the cerebral cavernous malformations protein complex Krit1 (Ccm1) and its mechanosensitive binding partner Heg1. Loss of Krit1 results in increased klf2a and notch1b expression in abluminal AVC valve cells and prevents valve formation (Donat et al., 2018). Together, these studies demonstrate not only that KLF2 regulates valve development, but also that its localization is dictated by mechanosensitive mechanisms.
Developmental mechanisms implicated in valve disease
Valve disease usually takes the form of stenosis as blood flow is obstructed or regurgitation with retrograde flow. Histologically, valve disease is characterized by abnormal valve structure, particularly with regard to ECM composition. Although valve disease occurs in all four cardiac valves, it is most commonly found in the mitral and aortic valves (Hinton and Yutzey, 2011). There is increasing evidence linking congenitally malformed valves and/or the reactivation of developmental pathways with acquired valve disease (LaHaye et al., 2014). There are many different valve diseases but the most common include bicuspid aortic valve (BAV; see Glossary, Box 1), calcific aortic valve disease (CAVD; see Glossary, Box 1) and myxomatous mitral valve disease (MMVD; see Glossary, Box 1).
BAV is defined by an aortic valve consisting of two leaflets instead of the normal three and has an incidence of at least 1% of all live births (Cripe et al., 2004). Several gene mutations have been associated with BAV in humans and mice, including mutations in NOTCH1, GATA5 and GATA6 (Garg et al., 2005; Laforest et al., 2011; Shi et al., 2014; Xu et al., 2018; Gharibeh et al., 2018). Studies in mice have demonstrated an in vivo genetic link between Notch1, endothelial nitric oxide synthase (eNOS) and BAV (Bosse et al., 2013). Abnormalities in primary cilia, particularly in VICs, have also been associated with BAV and other congenital heart anomalies (Li et al., 2015). Conditional ablation of the ciliogenic gene Ift88 results in loss of primary cilia, increased deposition of ECM proteins including collagen I and versican, and a high penetrance of BAV (Toomer et al., 2017). Individuals with a BAV are also predisposed to develop CAVD; however, it is unclear whether development of CAVD is due to the underlying genetic mutations or to the distorted biomechanical forces caused by the altered geometry of the BAV (Garg et al., 2005).
CAVD is a progressive disease that involves the mineralization of valve leaflets leading to aortic valve stenosis, which often necessitates surgical valve repair and/or replacement, with age being the main risk factor. As the disease progresses, calcium-rich nodules form on the fibrosa side of the valve within the VIC population, eventually impairing ventricular function (Dutta and Lincoln, 2018). Additionally, the stratified ECM of the valve becomes disorganized with increased deposition of collagens and proteoglycans in all three ECM layers (Hinton et al., 2006). CAVD is associated with valve developmental genes, including NOTCH1 (Freeman and Otto, 2005; Garg et al., 2005). Mice with heterozygous Notch1 mutations are at a higher risk of developing aortic valve calcification (Garg et al., 2005). However, it is unclear whether the association between Notch1 and valve disease is due to a disruption in the genetic pathway or to the inability of the valve to respond appropriately to changes in blood flow. A study using human aortic VECs demonstrated that that shear stress may regulate Notch1 signaling (White et al., 2015). Additional in vitro studies have suggested that shear-sensitive microRNAs (miRNAs) may also be involved in CAVD (Holliday et al., 2011; Fernandez Esmerats et al., 2019; Salim et al., 2019). Another study identified two families with early onset progressive heart valve disease caused by a homozygous mutation in ADAMTS19 (Wünnemann et al., 2020). Furthermore, Adamts19 knockout mice demonstrated increased KLF2 expression before developing aortic valve disease characterized by regurgitation and/or valve stenosis (Wünnemann et al., 2020). These studies highlight the connections between valve development, valve disease and mechanical forces.
MMVD can be the result of genetic mutations, aberrant mechanical forces due to structural defects, the reactivation of developmental pathways, or a combination thereof (Levine et al., 2015). Heritable MMVD is characterized by the accumulation of proteoglycans, disruption of collagen and elastin fibers, valve thickening and, ultimately, mitral regurgitation (Aupperle and Disatian, 2012). Defects in the development of primary cilia have also been linked to families with a higher incidence of mitral valve prolapse (Toomer et al., 2017). Mice with a mutation in the cilia gene DZIP1 demonstrate a phenotype similar to that exhibited by individuals presenting with adult MMVD and mitral valve prolapse (Toomer et al., 2019b). MMVD can also result from other genetic causes, including mutations in ECM-encoding genes (Levine et al., 2015). Mutations in filamin A (FLNA) are also associated with MMVD and act via activation of the RAS-MEK-ERK pathway and repression of pSMAD2/3 and ECM production (Toomer et al., 2019a). Moreover, mice with a mutation in the fibrillin 1 gene (Fbn1C1039G/+), which mimics Marfan syndrome, exhibit mitral valve leaflet thickening at birth and progressive myxomatous valve degeneration similar to that seen in human patients (Ng et al., 2004). In addition, these mice demonstrate increased infiltrating and resident macrophages during early stage disease, and loss of these macrophages is protective against disease progression (Kim et al., 2020). Furthermore, although monocytes are present in valve leaflets at birth, MMVD is associated with increasing numbers of monocytes and expansion of macrophage populations within valves (Kim et al., 2020). These data provide initial support for macrophages as a new therapeutic target for treating valve disease.
Increases in TGFβ activation have also been implicated in congenital valve disease of Marfan syndrome, as well as nonsyndromic mitral valve disease (reviewed by Levine et al., 2015). In addition, TGFβ signaling and matrix remodeling enzymes are elevated in canine MMVD (Lu et al., 2015). Recently, a study suggested that the zebrafish AV valve can regenerate after in vivo VIC ablation through TGFβ-mediated cell proliferation, cell differentiation and ECM production (Bensimon-Brito et al., 2020). Although the regenerated valves are functional, they are morphologically distinct from uninjured valves and have characteristics of a more myxomatous valve phenotype. Furthermore, similar to Marfan syndrome myxomatous valves, it has been shown that kidney marrow-derived cells with similarities to infiltrating macrophages infiltrate the injured zebrafish valves during this process (Bensimon-Brito et al., 2020; Kim et al., 2020). Therefore, additional studies are required to determine whether actual regeneration occurs. Other studies have used a combination of zebrafish, human and mouse models to identify genes associated with nonsyndromic mitral valve prolapse. For example, it has been shown that the morpholino knockdown of dchs1 (dchs1b), a homolog of the Drosophila cell polarity gene dachsous involved in the formation of cilia, in zebrafish results in an AVC defect that can be rescued with human wild-type DCHS1 mRNA but not with DCHS1 mRNA with a missense mutation identified in a family with nonsyndromic mitral valve prolapse (Durst et al., 2015). Furthermore, mice heterozygous for a mutation in DCHS1 demonstrate thickened mitral valves, while human pre-valvular endocardial cells derived from pluripotent stem cells from individuals with a DCHS1 mutation exhibit a decreased number of shortened cilia compared with those derived from individuals with wild-type DCHS1 (Durst et al., 2015; Neri et al., 2019). A similar study found that the morpholino knockdown of tensin 1, a focal adhesion protein, in zebrafish results in AV valve regurgitation, and the homozygous loss of tensin 1 in mice results in enlarged posterior mitral valve leaflets (Dina et al., 2015). Therefore, it is possible that valve injury responses, as well as valve disease mechanisms, including TGFβ activation, macrophage infiltration, ECM remodeling and cilia formation, are conserved in zebrafish and mammals.
Conclusions
As we have highlighted here, the coordination of multiple molecular, cellular and biomechanical mechanisms is needed for normal valve development and homeostasis. These complex regulatory networks with multiple inputs and readouts have begun to be elucidated using various animal model systems, including mouse, chicken and zebrafish. Yet crucial mechanisms of valve development, particularly during late fetal and postnatal valve development, remain unclear. Although subpopulations of valve cells have been identified in maturing valves, how and when these subpopulations appear and what roles they play in valve development and valve homeostasis are still largely unknown. Likewise, macrophages have recently been identified as having important, yet unclear, roles in valve development and disease. There is also emerging evidence for the role of biomechanical forces in both valve development and valve disease. However, this area has proven to be difficult to study. Recent advances in zebrafish technology and the discovery of conserved mechanisms of valve development provide new opportunities to discover novel mechanosensitive mechanisms of valve development as well as insights into valve regeneration. Furthermore, single-cell studies can provide valuable insights into how specific subpopulations of valve cells contribute to valve development or respond to mechanical stimuli. Although recent studies have significantly advanced our knowledge of heart valve developmental mechanisms in terms of molecules and cells, new research strategies are needed to fully understand how biomechanical forces contribute to heart valve morphogenesis, homeostasis and disease progression.
The translation of recent findings on heart valve development to the treatment of myxomatous and calcific valve disease has been limited. However, the identification of crucial genes and pathways important for valve development has been informative in identifying causative genes in congenital heart valve disease. In addition, mouse models of valve disease have been used in preclinical testing for therapeutic strategies. For example, we used the Fbn1C1039G mouse model of Marfan syndrome to identify circulating macrophages as being crucial to the progression of myxomatous valve disease (Kim et al., 2020). Future studies will be directed towards targeting these cells in myxomatous valves using available drug therapies. Moreover, understanding the interactions between mechanical stimuli and progression of valve disease at molecular and cellular levels, combined with newly improved in vivo imaging modalities, could be applied to risk stratification and surgical planning for management of congenitally malformed valves. Overall, a better understanding of the molecular and cellular mechanisms, valve cell subpopulations and hemodynamic forces involved in valve development and disease will no doubt aid the development of novel therapies for congenital and acquired valve disease.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
This work was supported by the National Institutes of Health/National Heart, Lung and Blood Institute (R01 HL143881 to K.E.Y. and F31 HL150935 to A.O.). Deposited in PMC for release after 12 months.
References
- Aikawa E., Whittaker P., Farber M., Mendelson K., Padera R. F., Aikawa M. and Schoen F. J. (2006). Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation 113, 1344-1352. 10.1161/CIRCULATIONAHA.105.591768 [DOI] [PubMed] [Google Scholar]
- Aikawa E., Nahrendorf M., Sosnovik D., Lok V. M., Jaffer F. A., Aikawa M. and Weissleder R. (2007). Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 115, 377-386. 10.1161/CIRCULATIONAHA.106.654913 [DOI] [PubMed] [Google Scholar]
- Alfieri O. and Vahanian A. (2017). The year in cardiology 2016: valvular heart disease. Eur. Heart J. 38, 628-633. 10.1093/eurheartj/ehw636 [DOI] [PubMed] [Google Scholar]
- Armstrong E. J. and Bischoff J. (2004). Heart valve development: endothelial cell signaling and differentiation. Circ. Res. 95, 459-470. 10.1161/01.RES.0000141146.95728.da [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aupperle H. and Disatian S. (2012). Pathology, protein expression and signaling in myxomatous mitral valve degeneration: comparison of dogs and humans. J. Vet. Cardiol. 14, 59-71. 10.1016/j.jvc.2012.01.005 [DOI] [PubMed] [Google Scholar]
- Balachandran K., Sucosky P. and Yoganathan A. P. (2011). Hemodynamics and mechanobiology of aortic valve inflammation and calcification. Int. J. Inflam. 2011, 263870 10.4061/2011/263870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balistreri C. R., Crapanzano F., Schirone L., Allegra A., Pisano C., Ruvolo G., Forte M., Greco E., Cavarretta E., Marullo A. G. M. et al. (2018). Deregulation of Notch1 pathway and circulating endothelial progenitor cell (EPC) number in patients with bicuspid aortic valve with and without ascending aorta aneurysm. Sci. Rep. 8, 13834 10.1038/s41598-018-32170-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman T., Walsh E. C., Wen K.-K., Mckane M., Ren J., Alexander J., Rubenstein P. A. and Stainier D. Y. R. (2004). Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol. 2, e129 10.1371/journal.pbio.0020129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazigou E., Lyons O. T. A., Smith A., Venn G. E., Cope C., Brown N. A. and Makinen T. (2011). Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Invest. 121, 2984-2992. 10.1172/JCI58050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beis D., Bartman T., Jin S.-W., Scott I. C., D'amico L. A., Ober E. A., Verkade H., Frantsve J., Field H. A., Wehman A. et al. (2005). Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Development 132, 4193-4204. 10.1242/dev.01970 [DOI] [PubMed] [Google Scholar]
- Bensimon-Brito A., Ramkumar S., Boezio G. L. M., Guenther S., Kuenne C., Helker C. S. M., Sánchez-Iranzo H., Iloska D., Piesker J., Pullamsetti S. et al. (2020). TGF-beta signaling promotes tissue formation during cardiac valve regeneration in adult zebrafish. Dev. Cell 52, 9-20.e7. 10.1016/j.devcel.2019.10.027 [DOI] [PubMed] [Google Scholar]
- Boselli F., Steed E., Freund J. B. and Vermot J. (2017). Anisotropic shear stress patterns predict the orientation of convergent tissue movements in the embryonic heart. Development 144, 4322-4327. 10.1242/dev.152124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse K., Hans C. P., Zhao N., Koenig S. N., Huang N., Guggilam A., Lahaye S., Tao G., Lucchesi P. A., Lincoln J. et al. (2013). Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 60, 27-35. 10.1016/j.yjmcc.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. B., Boyer A. S., Runyan R. B. and Barnett J. V. (1999). Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science 283, 2080-2082. 10.1126/science.283.5410.2080 [DOI] [PubMed] [Google Scholar]
- Buijtendijk M. F. J., Barnett P. and Van Den Hoff M. J. B. (2020). Development of the human heart. Am. J. Med. Genet. C Semin. Med. Genet. 184, 7-22. 10.1002/ajmg.c.31778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskohl P. R., Jenkins J. T. and Butcher J. T. (2012). Computational simulation of hemodynamic-driven growth and remodeling of embryonic atrioventricular valves. Biomech. Model. Mechanobiol. 11, 1205-1217. 10.1007/s10237-012-0424-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher J. T., Mcquinn T. C., Sedmera D., Turner D. and Markwald R. R. (2007). Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ. Res. 100, 1503-1511. 10.1161/CIRCRESAHA.107.148684 [DOI] [PubMed] [Google Scholar]
- Camenisch T. D., Spicer A. P., Brehm-Gibson T., Biesterfeldt J., Augustine M. L., Calabro A. Jr, Kubalak S., Klewer S. E. and Mcdonald J. A. (2000). Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Invest. 106, 349-360. 10.1172/JCI10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Bronson R. T., Klaman L. D., Hampton T. G., Wang J.-F., Green P. J., Magnuson T., Douglas P. S., Morgan J. P. and Neel B. G. (2000). Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 24, 296-299. 10.1038/73528 [DOI] [PubMed] [Google Scholar]
- Chi N. C., Shaw R. M., De Val S., Kang G., Jan L. Y., Black B. L. and Stainier D. Y. R. (2008). Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 22, 734-739. 10.1101/gad.1629408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvee E. and Hurle J. M. (1983). Malformations of the semilunar valves produced in chick embryos by mechanical interference with cardiogenesis. An experimental approach to the role of hemodynamics in valvular development. Anat. Embryol. 168, 59-71. 10.1007/BF00305399 [DOI] [PubMed] [Google Scholar]
- Combs M. D. and Yutzey K. E. (2009a). Heart valve development: regulatory networks in development and disease. Circ. Res. 105, 408-421. 10.1161/CIRCRESAHA.109.201566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs M. D. and Yutzey K. E. (2009b). VEGF and RANKL regulation of NFATc1 in heart valve development. Circ. Res. 105, 565-574. 10.1161/CIRCRESAHA.109.196469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripe L., Andelfinger G., Martin L. J., Shooner K. and Benson D. W. (2004). Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 44, 138-143. 10.1016/j.jacc.2004.03.050 [DOI] [PubMed] [Google Scholar]
- De Lange F. J., Moorman A. F. M., Anderson R. H., Männer J., Soufan A. T., De Gier-De Vries C., Schneider M. D., Webb S., Van Den Hoff M. J. B. and Christoffels V. M. (2004). Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 95, 645-654. 10.1161/01.RES.0000141429.13560.cb [DOI] [PubMed] [Google Scholar]
- Delaughter D. M., Bick A. G., Wakimoto H., Mckean D., Gorham J. M., Kathiriya I. S., Hinson J. T., Homsy J., Gray J., Pu W. et al. (2016). Single-cell resolution of temporal gene expression during heart development. Dev. Cell 39, 480-490. 10.1016/j.devcel.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina C., Bouatia-Naji N., Tucker N., Delling F. N., Toomer K., Durst R., Perrocheau M., Fernandez-Friera L., Solis J., Investigators P. et al. (2015). Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat. Genet. 47, 1206-1211. 10.1038/ng.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donat S., Lourenço M., Paolini A., Otten C., Renz M. and Abdelilah-Seyfried S. (2018). Heg1 and Ccm1/2 proteins control endocardial mechanosensitivity during zebrafish valvulogenesis. eLife 7, e28939 10.7554/eLife.28939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y., Camenisch T. D., Itin A., Fishman G. I., Mcdonald J. A., Carmeliet P. and Keshet E. (2001). A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 128, 1531-1538. [DOI] [PubMed] [Google Scholar]
- Durst R., Sauls K., Peal D. S., Devlaming A., Toomer K., Leyne M., Salani M., Talkowski M. E., Brand H., Perrocheau M. et al. (2015). Mutations in DCHS1 cause mitral valve prolapse. Nature 525, 109-113. 10.1038/nature14670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P. and Lincoln J. (2018). Calcific aortic valve disease: a developmental biology perspective. Curr. Cardiol. Rep. 20, 21 10.1007/s11886-018-0968-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley L., Alqahtani A. M. S., MacGrogan D., Richardson R. V., Murphy L., Salguero-Jimenez A., Sintes Rodriguez San Pedro M., Tiurma S., McCutcheon L., Gilmore A. et al. (2018). A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. eLife 7, e34110 10.7554/eLife.34110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez Esmerats J., Villa-Roel N., Kumar S., Gu L., Salim M. T., Ohh M., Taylor W. R., Nerem R. M., Yoganathan A. P. and Jo H. (2019). Disturbed flow increases UBE2C (Ubiquitin E2 Ligase C) via loss of miR-483-3p, inducing aortic valve calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1α (Hypoxia-Inducible Factor-1α) pathway in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 39, 467-481. 10.1161/ATVBAHA.118.312233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman M. C. and Chien K. R. (1997). Fashioning the vertebrate heart: earliest embryonic decisions. Development 124, 2099-2117. [DOI] [PubMed] [Google Scholar]
- Freeman R. V. and Otto C. M. (2005). Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation 111, 3316-3326. 10.1161/CIRCULATIONAHA.104.486738 [DOI] [PubMed] [Google Scholar]
- Garg V., Muth A. N., Ransom J. F., Schluterman M. K., Barnes R., King I. N., Grossfeld P. D. and Srivastava D. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270-274. 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- Gees M., Colsoul B. and Nilius B. (2010). The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2, a003962 10.1101/cshperspect.a003962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Cha B., Mahamud M. R., Lim K.-C., Silasi-Mansat R., Uddin M. K. M., Miura N., Xia L., Simon A. M., Engel J. D. et al. (2016). Multiple mouse models of primary lymphedema exhibit distinct defects in lymphovenous valve development. Dev. Biol. 409, 218-233. 10.1016/j.ydbio.2015.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Cha B., Mahamud M. R. and Srinivasan R. S. (2017). Intraluminal valves: development, function and disease. Dis. Model. Mech. 10, 1273-1287. 10.1242/dmm.030825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharibeh L., Komati H., Bossé Y., Boodhwani M., Heydarpour M., Fortier M., Hassanzadeh R., Ngu J., Mathieu P., Body S. et al. (2018). GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation 138, 1025-1038. 10.1161/CIRCULATIONAHA.117.029506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-De Groot A. C., Vrancken Peeters M.-P. F. M., Mentink M. M. T., Gourdie R. G. and Poelmann R. E. (1998). Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 82, 1043-1052. 10.1161/01.RES.82.10.1043 [DOI] [PubMed] [Google Scholar]
- Goddard L. M., Duchemin A.-L., Ramalingan H., Wu B., Chen M., Bamezai S., Yang J., Li L., Morley M. P., Wang T. et al. (2017). Hemodynamic forces sculpt developing heart valves through a KLF2-WNT9B paracrine signaling axis. Dev. Cell 43, 274-289.e5. 10.1016/j.devcel.2017.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goishi K., Lee P., Davidson A. J., Nishi E., Zon L. I. and Klagsbrun M. (2003). Inhibition of zebrafish epidermal growth factor receptor activity results in cardiovascular defects. Mech. Dev. 120, 811-822. 10.1016/S0925-4773(03)00068-6 [DOI] [PubMed] [Google Scholar]
- Gomez-Stallons M. V., Wirrig-Schwendeman E. E., Hassel K. R., Conway S. J. and Yutzey K. E. (2016). Bone morphogenetic protein signaling is required for aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 36, 1398-1405. 10.1161/ATVBAHA.116.307526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross L. and Kugel M. A. (1931). Topographic anatomy and histology of the valves in the human heart. Am. J. Pathol. 7, 445-474. [PMC free article] [PubMed] [Google Scholar]
- Gunawan F., Gentile A., Fukuda R., Tsedeke A. T., Jiménez-Amilburu V., Ramadass R., Iida A., Sehara-Fujisawa A. and Stainier D. Y. R. (2019). Focal adhesions are essential to drive zebrafish heart valve morphogenesis. J. Cell Biol. 218, 1039-1054. 10.1083/jcb.201807175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan F., Gentile A., Gauvrit S., Stainier D. Y. R. and Bensimon-Brito A. (2020). Nfatc1 promotes interstitial cell formation during cardiac valve development in zebrafish. Circ. Res. 126, 968-984. 10.1161/CIRCRESAHA.119.315992 [DOI] [PubMed] [Google Scholar]
- Heckel E., Boselli F., Roth S., Krudewig A., Belting H.-G., Charvin G. and Vermot J. (2015). Oscillatory flow modulates mechanosensitive klf2a expression through trpv4 and trpp2 during heart valve development. Curr. Biol. 25, 1354-1361. 10.1016/j.cub.2015.03.038 [DOI] [PubMed] [Google Scholar]
- Hinton R. B. and Yutzey K. E. (2011). Heart valve structure and function in development and disease. Annu. Rev. Physiol. 73, 29-46. 10.1146/annurev-physiol-012110-142145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton R. B. Jr, Lincoln J., Deutsch G. H., Osinska H., Manning P. B., Benson D. W. and Yutzey K. E. (2006). Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 98, 1431-1438. 10.1161/01.RES.0000224114.65109.4e [DOI] [PubMed] [Google Scholar]
- Hogers B., Deruiter M. C., Gittenberger-De Groot A. C. and Poelmann R. E. (1997). Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ. Res. 80, 473-481. 10.1161/01.RES.80.4.473 [DOI] [PubMed] [Google Scholar]
- Holliday C. J., Ankeny R. F., Jo H. and Nerem R. M. (2011). Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 301, H856-H867. 10.1152/ajpheart.00117.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove J. R., Köster R. W., Forouhar A. S., Acevedo-Bolton G., Fraser S. E. and Gharib M. (2003). Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421, 172-177. 10.1038/nature01282 [DOI] [PubMed] [Google Scholar]
- Hu N. and Clark E. B. (1989). Hemodynamics of the stage 12 to stage 29 chick embryo. Circ. Res. 65, 1665-1670. 10.1161/01.RES.65.6.1665 [DOI] [PubMed] [Google Scholar]
- Hu N., Sedmera D., Yost H. J. and Clark E. B. (2000). Structure and function of the developing zebrafish heart. Anat. Rec. 260, 148-157. 10.1002/1097-0185(20001001)260:2<148::AID-AR50>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- Hu N., Yost H. J. and Clark E. B. (2001). Cardiac morphology and blood pressure in the adult zebrafish. Anat. Rec. 264, 1-12. 10.1002/ar.1111 [DOI] [PubMed] [Google Scholar]
- Hulin A., Moore V., James J. M. and Yutzey K. E. (2017). Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc. Res. 113, 40-51. 10.1093/cvr/cvw229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulin A., Anstine L. J., Kim A. J., Potter S. J., Defalco T., Lincoln J. and Yutzey K. E. (2018). Macrophage transitions in heart valve development and myxomatous valve disease. Arterioscler. Thromb. Vasc. Biol. 38, 636-644. 10.1161/ATVBAHA.117.310667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulin A., Hortells L., Gomez-Stallons M. V., O'Donnell A., Chetal K., Adam M., Lancellotti P., Oury C., Potter S. S., Salomonis N. et al. (2019). Maturation of heart valve cell populations during postnatal remodeling. Development 146, dev173047 10.1242/dev.173047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlstone A. F. L., Haramis A.-P. G., Wienholds E., Begthel H., Korving J., Van Eeden F., Cuppen E., Zivkovic D., Plasterk R. H. A. and Clevers H. (2003). The Wnt/β-catenin pathway regulates cardiac valve formation. Nature 425, 633-637. 10.1038/nature02028 [DOI] [PubMed] [Google Scholar]
- Jiang X., Rowitch D. H., Soriano P., Mcmahon A. P. and Sucov H. M. (2000). Fate of the mammalian cardiac neural crest. Development 127, 1607-1616. [DOI] [PubMed] [Google Scholar]
- Jiao K., Kulessa H., Tompkins K., Zhou Y., Batts L., Baldwin H. S. and Hogan B. L. (2003). An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 17, 2362-2367. 10.1101/gad.1124803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanisicak O., Khalil H., Ivey M. J., Karch J., Maliken B. D., Correll R. N., Brody M. J., Lin S.-C. J., Aronow B. J., Tallquist M. D. et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern C. B., Wessels A., Mcgarity J., Dixon L. J., Alston E., Argraves W. S., Geeting D., Nelson C. M., Menick D. R. and Apte S. S. (2010). Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 29, 304-316. 10.1016/j.matbio.2010.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A. J., Xu N., Umeyama K., Hulin A., Ponny S. R., Vagnozzi R. J., Green E. A., Hanson P., Mcmanus B. M., Nagashima H. et al. (2020). Deficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in Marfan syndrome. Circulation 141, 132-146. 10.1161/CIRCULATIONAHA.119.042391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby M. L., Gale T. F. and Stewart D. E. (1983). Neural crest cells contribute to normal aorticopulmonary septation. Science 220, 1059-1061. 10.1126/science.6844926 [DOI] [PubMed] [Google Scholar]
- Koenig S. N., Bosse K., Majumdar U., Bonachea E. M., Radtke F. and Garg V. (2016). Endothelial Notch1 Is required for proper development of the semilunar valves and cardiac outflow tract. J. Am. Heart. Assoc. 5, e003075 10.1161/JAHA.115.003075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B., Andelfinger G. and Nemer M. (2011). Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Invest. 121, 2876-2887. 10.1172/JCI44555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaHaye S., Lincoln J. and Garg V. (2014). Genetics of valvular heart disease. Curr. Cardiol. Rep. 16, 487 10.1007/s11886-014-0487-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. M., Cope J. J., Ackermann G. E., Goishi K., Armstrong E. J., Paw B. H. and Bischoff J. (2006). Vascular endothelial growth factor receptor signaling is required for cardiac valve formation in zebrafish. Dev. Dyn. 235, 29-37. 10.1002/dvdy.20559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine R. A., Hagége A. A., Judge D. P., Padala M., Dal-Bianco J. P., Aikawa E., Beaudoin J., Bischoff J., Bouatia-Naji N., Bruneval P. et al. (2015). Mitral valve disease—morphology and mechanisms. Nat. Rev. Cardiol. 12, 689-710. 10.1038/nrcardio.2015.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Klena N. T., Gabriel G. C., Liu X., Kim A. J., Lemke K., Chen Y., Chatterjee B., Devine W., Damerla R. R. et al. (2015). Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520-524. 10.1038/nature14269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S., Cattelino A., Gallini R., Rudini N., Iurlaro M., Piccolo S. and Dejana E. (2004). β-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 166, 359-367. 10.1083/jcb.200403050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J., Alfieri C. M. and Yutzey K. E. (2004). Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 230, 239-250. 10.1002/dvdy.20051 [DOI] [PubMed] [Google Scholar]
- Lincoln J., Alfieri C. M. and Yutzey K. E. (2006). BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev. Biol. 292, 290-302. 10.1016/j.ydbio.2005.12.042 [DOI] [PubMed] [Google Scholar]
- Lincoln J., Kist R., Scherer G. and Yutzey K. E. (2007). Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 305, 120-132. 10.1016/j.ydbio.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Yu W., Tang M., Tang J., Liu X., Liu Q., Li Y., He L., Zhang L., Evans S. M. et al. (2018). A dual genetic tracing system identifies diverse and dynamic origins of cardiac valve mesenchyme. Development 145, dev167775 10.1242/dev.167775 [DOI] [PubMed] [Google Scholar]
- Lockhart M., Wirrig E., Phelps A. and Wessels A. (2011). Extracellular matrix and heart development. Birth Defects Res. A Clin. Mol. Teratol 91, 535-550. 10.1002/bdra.20810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo C. A. (2000). Epidemiology of cardiovascular malformations: prevalence and risk factors. Am. J. Med. Genet. 97, 319-325. 10.1002/1096-8628(200024)97:4<319::AID-AJMG1283>3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- Lombardo V. A., Heise M., Moghtadaei M., Bornhorst D., Männer J. and Abdelilah-Seyfried S. (2019). Morphogenetic control of zebrafish cardiac looping by Bmp signaling. Development 146, dev180091 10.1242/dev.180091 [DOI] [PubMed] [Google Scholar]
- Lu C.-C., Liu M.-M., Culshaw G., Clinton M., Argyle D. J. and Corcoran B. M. (2015). Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: a microarray study. Vet. J. 204, 23-31. 10.1016/j.tvjl.2015.02.021 [DOI] [PubMed] [Google Scholar]
- Ma L., Lu M.-F., Schwartz R. J. and Martin J. F. (2005). Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 132, 5601-5611. 10.1242/dev.02156 [DOI] [PubMed] [Google Scholar]
- Macgrogan D., D'Amato G., Travisano S., Martinez-Poveda B., Luxán G., del Monte-Nieto G., Papoutsi T., Sbroggio M., Bou V., Gomez-Del Arco P. et al. (2016). Sequential ligand-dependent Notch signaling activation regulates valve primordium formation and morphogenesis. Circ. Res. 118, 1480-1497. 10.1161/CIRCRESAHA.115.308077 [DOI] [PubMed] [Google Scholar]
- Markwald R. R., Fitzharris T. P. and Manasek F. J. (1977). Structural development of endocardial cushions. Am. J. Anat. 148, 85-119. 10.1002/aja.1001480108 [DOI] [PubMed] [Google Scholar]
- Maron B. J. and Hutchins G. M. (1974). The development of the semilunar valves in the human heart. Am. J. Pathol. 74, 331-344. [PMC free article] [PubMed] [Google Scholar]
- Martin R. T. and Bartman T. (2009). Analysis of heart valve development in larval zebrafish. Dev. Dyn. 238, 1796-1802. 10.1002/dvdy.21976 [DOI] [PubMed] [Google Scholar]
- Martinsen B. J. (2005). Reference guide to the stages of chick heart embryology. Dev. Dyn. 233, 1217-1237. 10.1002/dvdy.20468 [DOI] [PubMed] [Google Scholar]
- McCulley D. J., Kang J.-O., Martin J. F. and Black B. L. (2008). BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 237, 3200-3209. 10.1002/dvdy.21743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett M., Lopez C. S., David L., Maloyan A. and Rugonyi S. (2017). Increased hemodynamic load in early embryonic stages alters endocardial to mesenchymal transition. Front. Physiol. 8, 56 10.3389/fphys.2017.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquerol L., Langille B. L. and Nagy A. (2000). Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 127, 3941-3946. [DOI] [PubMed] [Google Scholar]
- Monaghan M. G., Linneweh M., Liebscher S., Van Handel B., Layland S. L. and Schenke-Layland K. (2016). Endocardial-to-mesenchymal transformation and mesenchymal cell colonization at the onset of human cardiac valve development. Development 143, 473-482. 10.1242/dev.133843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman A., Webb S., Brown N. A., Lamers W. and Anderson R. H. (2003). Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart 89, 806-814. 10.1136/heart.89.7.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri T., Hiriart E., Van Vliet P. P., Faure E., Norris R. A., Farhat B., Jagla B., Lefrancois J., Sugi Y., Moore-Morris T. et al. (2019). Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 10, 1929 10.1038/s41467-019-09459-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C. M., Cheng A., Myers L. A., Martinez-Murillo F., Jie C., Bedja D., Gabrielson K. L., Hausladen J. M. W., Mecham R. P., Judge D. P. et al. (2004). TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Invest. 114, 1586-1592. 10.1172/JCI200422715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkomo V. T., Gardin J. M., Skelton T. N., Gottdiener J. S., Scott C. G. and Enriquez-Sarano M. (2006). Burden of valvular heart diseases: a population-based study. Lancet 368, 1005-1011. 10.1016/S0140-6736(06)69208-8 [DOI] [PubMed] [Google Scholar]
- Nomura-Kitabayashi A., Phoon C. K. L., Kishigami S., Rosenthal J., Yamauchi Y., Abe K., Yamamura K., Samtani R., Lo C. W. and Mishina Y. (2009). Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am. J. Physiol. Heart Circ. Physiol. 297, H1617-H1628. 10.1152/ajpheart.00304.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person A. D., Garriock R. J., Krieg P. A., Runyan R. B. and Klewer S. E. (2005a). Frzb modulates Wnt-9a-mediated β-catenin signaling during avian atrioventricular cardiac cushion development. Dev. Biol. 278, 35-48. 10.1016/j.ydbio.2004.10.013 [DOI] [PubMed] [Google Scholar]
- Person A. D., Klewer S. E. and Runyan R. B. (2005b). Cell biology of cardiac cushion development. Int. Rev. Cytol. 243, 287-335. 10.1016/S0074-7696(05)43005-3 [DOI] [PubMed] [Google Scholar]
- Pestel J., Ramadass R., Gauvrit S., Helker C., Herzog W. and Stainier D. Y. R. (2016). Real-time 3D visualization of cellular rearrangements during cardiac valve formation. Development 143, 2217-2227. 10.1242/dev.133272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova T. V., Karpanen T., Norrmén C., Mellor R., Tamakoshi T., Finegold D., Ferrell R., Kerjaschki D., Mortimer P., Ylä-Herttuala S. et al. (2004). Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10, 974-981. 10.1038/nm1094 [DOI] [PubMed] [Google Scholar]
- Phillips H. M., Mahendran P., Singh E., Anderson R. H., Chaudhry B. and Henderson D. J. (2013). Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 99, 452-460. 10.1093/cvr/cvt132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon K. L. and Brand T. (2013). The zebrafish model system in cardiovascular research: a tiny fish with mighty prospects. Glob. Cardiol. Sci. Pract. 2013, 9-28. 10.5339/gcsp.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X., Violette K., Sewell-Loftin M. K., Soslow J., Saint-Jean L. S., Hinton R. B., Merryman W. D. and Baldwin H. S. (2019). Loss of flow responsive Tie1 results in Impaired Aortic valve remodeling. Dev. Biol. 455, 73-84. 10.1016/j.ydbio.2019.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Feliciano J. and Tabin C. J. (2006). Bmp2 instructs cardiac progenitors to form the heart-valve-inducing field. Dev. Biol. 295, 580-588. 10.1016/j.ydbio.2006.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodbard S. (1956). Vascular modifications induced by flow. Am. Heart J. 51, 926-942. 10.1016/0002-8703(56)90236-8 [DOI] [PubMed] [Google Scholar]
- Romano L. A. and Runyan R. B. (2000). Slug is an essential target of TGFβ2 signaling in the developing chicken heart. Dev. Biol. 223, 91-102. 10.1006/dbio.2000.9750 [DOI] [PubMed] [Google Scholar]
- Sacks M. S., David Merryman W. and Schmidt D. E. (2009). On the biomechanics of heart valve function. J. Biomech. 42, 1804-1824. 10.1016/j.jbiomech.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim M. T., Esmerats J. F., Arjunon S., Villa-Roel N., Nerem R. M., Jo H. and Yoganathan A. P. (2019). miR-214 is stretch-sensitive in aortic valve and inhibits aortic valve calcification. Ann. Biomed. Eng. 47, 1106-1115. 10.1007/s10439-019-02206-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz P. J., Huisken J., Sahai-Hernandez P. and Stainier D. Y. R. (2008). High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development 135, 1179-1187. 10.1242/dev.010694 [DOI] [PubMed] [Google Scholar]
- Schoen F. J. (1997). Aortic valve structure-function correlations: role of elastic fibers no longer a stretch of the imagination. J. Heart Valve Dis. 6, 1-6. [PubMed] [Google Scholar]
- Schoen F. J. (2008). Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 118, 1864-1880. 10.1161/CIRCULATIONAHA.108.805911 [DOI] [PubMed] [Google Scholar]
- Schulz A., Brendler J., Blaschuk O., Landgraf K., Krueger M. and Ricken A. M. (2019). Non-pathological chondrogenic features of valve interstitial cells in normal adult zebrafish. J. Histochem. Cytochem. 67, 361-373. 10.1369/0022155418824083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedmera D., Pexieder T., Hu N. and Clark E. B. (1997). Developmental changes in the myocardial architecture of the chick. Anat. Rec. 248, 421-432. 10.1002/(SICI)1097-0185(199707)248:3<421::AID-AR15>3.0.CO;2-R [DOI] [PubMed] [Google Scholar]
- Shi L.-M., Tao J.-W., Qiu X.-B., Wang J., Yuan F., Xu L., Liu H., Li R.-G., Xu Y.-J., Wang Q. et al. (2014). GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 33, 1219-1226. 10.3892/ijmm.2014.1700 [DOI] [PubMed] [Google Scholar]
- Shigeta A., Huang V., Zuo J., Besada R., Nakashima Y., Lu Y., Ding Y., Pellegrini M., Kulkarni R. P., Hsiai T. et al. (2019). Endocardially derived macrophages are essential for valvular remodeling. Dev. Cell 48, 617-630.e3. 10.1016/j.devcel.2019.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C. A., Grant G. R., Manduchi E. and Davies P. F. (2005). Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 96, 792-799. 10.1161/01.RES.0000161998.92009.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Hoogaars W. M., Barnett P., Grieskamp T., Rana M. S., Buermans H., Farin H. F., Petry M., Heallen T., Martin J. F. et al. (2012). Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell. Mol. Life Sci. 69, 1377-1389. 10.1007/s00018-011-0884-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P., Hinton R. B., Moreno-Rodriguez R. A., Wang J., Rogers R., Lindsley A., Li F., Ingram D. A., Menick D., Field L. et al. (2008). Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 102, 752-760. 10.1161/CIRCRESAHA.107.159517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier D. Y. R. (2001). Zebrafish genetics and vertebrate heart formation. Nat. Rev. Genet. 2, 39-48. 10.1038/35047564 [DOI] [PubMed] [Google Scholar]
- Steed E., Boselli F. and Vermot J. (2016a). Hemodynamics driven cardiac valve morphogenesis. Biochim. Biophys. Acta 1863, 1760-1766. 10.1016/j.bbamcr.2015.11.014 [DOI] [PubMed] [Google Scholar]
- Steed E., Faggianelli N., Roth S., Ramspacher C., Concordet J.-P. and Vermot J. (2016b). klf2a couples mechanotransduction and zebrafish valve morphogenesis through fibronectin synthesis. Nat. Commun. 7, 11646 10.1038/ncomms11646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugi Y., Ito N., Szebenyi G., Myers K., Fallon J. F., Mikawa T. and Markwald R. R. (2003). Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev. Biol. 258, 252-263. 10.1016/S0012-1606(03)00099-X [DOI] [PubMed] [Google Scholar]
- Timmerman L. A., Grego-Bessa J., Raya A., Bertran E., Perez-Pomares J. M., Diez J., Aranda S., Palomo S., Mccormick F., Izpisua-Belmonte J. C. et al. (2004). Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 18, 99-115. 10.1101/gad.276304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomer K. A., Fulmer D., Guo L., Drohan A., Peterson N., Swanson P., Brooks B., Mukherjee R., Body S., Lipschutz J. H. et al. (2017). A role for primary cilia in aortic valve development and disease. Dev. Dyn. 246, 625-634. 10.1002/dvdy.24524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomer K., Sauls K., Fulmer D., Guo L., Moore K., Glover J., Stairley R., Bischoff J., Levine R. A. and Norris R. A. (2019a). Filamin-A as a balance between Erk/Smad activities during cardiac valve development. Anat. Rec. 302, 117-124. 10.1002/ar.23911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomer K. A., Yu M., Fulmer D., Guo L., Moore K. S., Moore R., Drayton K. D., Glover J., Peterson N., Ramos-Ortiz S. et al. (2019b). Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 11, eaax0290 10.1126/scitranslmed.aax0290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermot J., Forouhar A. S., Liebling M., Wu D., Plummer D., Gharib M. and Fraser S. E. (2009). Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol. 7, e1000246 10.1371/journal.pbio.1000246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virani S. S., Alonso A., Benjamin E. J., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Delling F. N. et al. (2020). Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation 141, e139-e596. 10.1161/CIR.0000000000000757 [DOI] [PubMed] [Google Scholar]
- Votteler M., Berrio D. A. C., Horke A., Sabatier L., Reinhardt D. P., Nsair A., Aikawa E. and Schenke-Layland K. (2013). Elastogenesis at the onset of human cardiac valve development. Development 140, 2345-2353. 10.1242/dev.093500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto K., Kobayashi K., Kuro-o O. M., Yao A., Iwamoto T., Yanaka N., Kita S., Nishida A., Azuma S., Toyoda Y. et al. (2000). Targeted disruption of Na+/Ca2+ exchanger gene leads to cardiomyocyte apoptosis and defects in heartbeat. J. Biol. Chem. 275, 36991-36998. 10.1074/jbc.M004035200 [DOI] [PubMed] [Google Scholar]
- Walsh E. C. and Stainier D. Y. R. (2001). UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science 293, 1670-1673. 10.1126/science.293.5535.1670 [DOI] [PubMed] [Google Scholar]
- Wessels A., Van Den Hoff M. J. B., Adamo R. F., Phelps A. L., Lockhart M. M., Sauls K., Briggs L. E., Norris R. A., Van Wijk B., Perez-Pomares J. M. et al. (2012). Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366, 111-124. 10.1016/j.ydbio.2012.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin J. and Lardelli M. (1997). Three novel Notch genes in zebrafish: implications for vertebrate Notch gene evolution and function. Dev. Genes Evol. 207, 51-63. 10.1007/s004270050091 [DOI] [PubMed] [Google Scholar]
- White M. P., Theodoris C. V., Liu L., Collins W. J., Blue K. W., Lee J. H., Meng X., Robbins R. C., Ivey K. N. and Srivastava D. (2015). NOTCH1 regulates matrix gla protein and calcification gene networks in human valve endothelium. J. Mol. Cell. Cardiol. 84, 13-23. 10.1016/j.yjmcc.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Wang Y., Lui W., Langworthy M., Tompkins K. L., Hatzopoulos A. K., Baldwin H. S. and Zhou B. (2011). Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ. Res. 109, 183-192. 10.1161/CIRCRESAHA.111.245035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wünnemann F., Ta-Shma A., Preuss C., Leclerc S., Van Vliet P. P., Oneglia A., Thibeault M., Nordquist E., Lincoln J., Scharfenberg F. et al. (2020). Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat. Genet. 52, 40-47. 10.1038/s41588-019-0536-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.-J., Di R.-M., Qiao Q., Li X.-M., Huang R.-T., Xue S., Liu X.-Y., Wang J. and Yang Y.-Q. (2018). GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 663, 115-120. 10.1016/j.gene.2018.04.018 [DOI] [PubMed] [Google Scholar]
- Yalcin H. C., Shekhar A., Mcquinn T. C. and Butcher J. T. (2011). Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 240, 23-35. 10.1002/dvdy.22512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S. and Brueckner M. (2017). Genetics and genomics of congenital heart disease. Circ. Res. 120, 923-940. 10.1161/CIRCRESAHA.116.309140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Von Gise A., Liu Q., Hu T., Tian X., He L., Pu W., Huang X., He L., Cai C.-L. et al. (2014). Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 289, 18681-18692. 10.1074/jbc.M114.554584 [DOI] [PMC free article] [PubMed] [Google Scholar]