

Summary
The proteasome is a therapeutic target in cancer, but resistance to proteasome inhibitors often develops owing to the induction of compensatory pathways. Through a genome-wide siRNA screen combined with RNA sequencing analysis, we identified hexokinase and downstream O-GlcNAcylation as cell survival factors under proteasome impairment. The inhibition of O-GlcNAcylation synergistically induced massive cell death in combination with proteasome inhibition. We further demonstrated that O-GlcNAcylation was indispensable for maintaining proteasome activity by enhancing biogenesis as well as proteasome degradation in a manner independent of Nrf1, a well-known compensatory transcription factor that upregulates proteasome gene expression. Our results identify a pathway that maintains proteasome function under proteasome impairment, providing potential targets for cancer therapy.
Subject Areas: Biological Sciences, Cell Biology, Functional Aspects of Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
O-GlcNAcylation suppresses cell death under proteasome impairment
-
•
Combined inhibition of O-GlcNAcylation and proteasome induces massive tumor cell death
-
•
O-GlcNAcylation maintains proteasome activity independently of Nrf1
-
•
O-GlcNAcylation enhances proteasome turnover under the proteasome impairment
Biological Sciences; Cell Biology; Functional Aspects of Cell Biology; Cancer
Introduction
The proteasome is responsible for the degradation of regulatory and misfolded proteins that are ubiquitinated within eukaryotic cells, and it plays pivotal roles in various cellular events such as the cell cycle, DNA repair, signal transduction, and protein quality control (Murata et al., 2009). Cancer cells often have high proteasome activity to improve their growth and survival (Arlt et al., 2009; Chen and Madura, 2005; Kumatori et al., 1990; Schwartz and Ciechanover, 2009). Inhibition of the proteasome causes proteotoxic stress that leads to cell-cycle arrest and apoptosis in cancer cells, and it is now widely accepted that the proteasome is an attractive therapeutic target in cancer (Manasanch and Orlowski, 2017).
The US Food and Drug Administration-approved proteasome inhibitors bortezomib, carfilzomib, and ixazomib are used for the treatment of multiple myeloma and have led to significant improvements in the clinical outcomes of patients with this condition (Kubiczkova et al., 2014). Unfortunately, most patients ultimately relapse because surviving cancer cells acquire drug resistance (Wallington-Beddoe et al., 2018). In patients with multiple myeloma, several drugs are available for overcoming drug resistance, including immunomodulatory agents, histone deacetylase inhibitors, and monoclonal antibodies (Kubiczkova et al., 2014; Kurtin and Bilotti, 2013). Although these drugs improve clinical outcomes, they do not completely overcome bortezomib resistance, probably because they do not directly target the pathways that confer resistance to proteasome inhibitors (Manasanch and Orlowski, 2017).
Recent studies have shown that multiple myeloma cells acquire resistance to proteasome inhibitors in several ways, such as mutation of the β5 subunit, a direct target of bortezomib (Lü et al., 2008; Oerlemans et al., 2008); induction of multidrug transporters (Besse et al., 2018); suppression of endoplasmic reticulum (ER) stress sensors (Leung-Hagesteijn et al., 2013); activation of the bounce-back response mediated by Nrf1, which induces proteasome subunits (Radhakrishnan et al., 2010; Steffen et al., 2010); upregulation of antioxidant responses (Riz et al., 2016; Weniger et al., 2011); and alteration of metabolic pathways, including glycolysis (Maiso et al., 2015; Zaal et al., 2017). However, the mechanisms by which cancer cells escape the cytotoxic effects of proteasome inhibitors remain controversial because proteasome inhibitor-resistant cancer cells accompany various genomic mutations and alterations.
In this study, we performed a nonbiased genome-wide analysis and identified hexokinase 1 (HK1), and subsequently O-GlcNAc transferase (OGT), as cell death suppressors under proteasome impairment. We demonstrated that combined inhibition of both O-GlcNAcylation and the proteasome resulted in synergistic cell death and suppression of tumor growth with deterioration of proteasome activity, suggesting that enhanced protein O-GlcNAcylation plays a cytoprotective role under proteasome impairment. We further showed that O-GlcNAcylation promotes turnover of the proteasome by enhancing both its degradation and biogenesis and thus recovers proteasome activity under proteasome impairment, demonstrating a new mechanism by which cells maintain proteasome activity.
Results
Identification of Hexokinase 1 as a Cell Death Suppressor under Proteasome Impairment
To understand the compensatory pathways that maintain cell survival under proteasome impairment, we first determined the bortezomib concentration that inhibits proteasome activity but does not induce massive cell death. We employed p53-intact U2OS cells to comprehensively understand the cellular responses to proteasome dysfunction. We treated U2OS cells with various concentrations of bortezomib and found that 10 nM bortezomib caused only modest cell death (Figure 1A). Compared with 50 nM bortezomib, the concentration that kills most cells, 10 nM bortezomib decreased the peptidase activity of the proteasome by 25% (Figure 1B) and resulted in modest accumulation of ubiquitinated proteins (Figure 1C).
Figure 1.

Genome-wide siRNA Screen for Cell Death Suppressors under Proteasome Inhibition
(A) Viability assay of U2OS cells treated with bortezomib (BTZ) (0–100 nM) for 48 h.
(B) Proteasome peptidase activity of lysates from U2OS cells treated with 10 nM BTZ for 48 h.
(C) Immunoblot analysis of lysates from U2OS cells with antibodies against the indicated proteins. U2OS cells were treated with BTZ (0–50 nM) for 16 h before harvest.
(D) Viability assay of U2OS cells transfected with Nrf1 and Ump1 siRNAs or control siRNA and then treated with 10 nM BTZ for 48 h.
(E) B score of all samples in the first genome-wide siRNA screen. The results are ordered from lowest to highest. The dashed red line shows the cutoff value of positive hits in the first screen.
(F) The workflow of siRNA screens. In the first screen, U2OS cells were treated with a pooled siRNA library containing four siRNA constructs per gene. In the second screen, U2OS cells were treated with four individual siRNAs.
(G) Enrichment analysis of 2,322 genes upregulated by BTZ treatment for 60 h was performed using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The gene expression values were log2 transformed, and GO analysis was performed. Representative data are shown.
(H) Venn diagram showing positive hits from the siRNA screens (red) and genes that were upregulated by BTZ treatment (blue). Five genes were common to the results of both screens (white).
(I) The final list of five hits and the scores of each assay in the two screens. The percentage of dead cells was assessed by FACS after staining with Hoechst 33,342 and propidium iodide (PI) in (A) and (D). Data in (A), (B), and (D) are the mean ± SEM (n = 3). Data in (A) and (B) were analyzed by two-tailed Student's t test (∗p < 0.05, ∗∗∗p < 0.001). Data in (D) were analyzed by two-way ANOVA followed by Tukey's test (∗∗∗p < 0.001). See also Figure S1 and Table S1.
We presumed that cells barely survive exposure to 10 nM bortezomib and thus expected that inhibiting compensatory pathways that maintain survival under proteasome impairment would induce cell death. Upregulation of de novo proteasome synthesis is a well-known compensatory mechanism for proteasome impairment. Nrf1 is a transcription factor that is activated to induce the expression of proteasome subunit genes upon proteasome inhibition (Radhakrishnan et al., 2010; Steffen et al., 2010), and Ump1 is a critical molecule for proteasome assembly (Murata et al., 2009). Indeed, knockdown of Nrf1 and Ump1 in the presence of 10 nM bortezomib markedly induced cell death (Figures 1D and S1A).
Thus, we comprehensively screened for genes involved in the compensatory response to proteasome impairment under 10 nM bortezomib. Of the cell lines tested in a preliminary investigation, including 293T, HeLa, and U2OS cells, we obtained most robust and reproducible results by using U2OS cells. Therefore, we performed a genome-wide siRNA screen in U2OS cells in the presence of 10 nM bortezomib by monitoring cell death. A total of 1,146 genes with a B score >3 in the primary screen, in which each well contained a mixture of four siRNAs targeting one gene, were further tested using independent siRNAs (Figures 1E and 1F). We obtained 28 genes with positive results for at least three of the four siRNAs (Figure 1F and Table S1).
To further narrow the candidate gene list, we performed RNA sequencing (RNA-seq) analysis based on the assumption that compensatory pathways might be upregulated by proteasome inhibition. We identified 2,322 genes for which the mRNA levels increased by more than 1.8-fold in the presence of 10 nM bortezomib; five of these genes overlapped with the candidates obtained from the siRNA screen (Figures 1G and 1H). This gene list included reasonable factors such as an antiapoptotic factor (BCL2L1) and a stress-inducible ubiquitin gene (UBC), both of which has been known to be involved in resistance to proteotoxic stress, validating our screening approach (Bianchi et al., 2018; Hagenbuchner et al., 2010) (Figure 1I). In addition, a glucose phosphorylating enzyme (HK1), a ubiquitin ligase (RNF181), and a putative transcription factor (ZNF770) of unknown function were identified (Figure 1I). From this list of genes, we chose to focus on HK1. Mammals have four hexokinase isoforms, of which HK1 is dominantly expressed in U2OS cells (Figure S1B). HK1 catalyzes the initial step in glucose utilization and is a rate-limiting enzyme in glycolysis, but the role of glucose metabolism in proteasome dysfunction remains incompletely understood.
Combined Inhibition of the Proteasome and Hexokinase Promotes Cell Death
To confirm that attenuation of HK1 activity promotes cell death in the presence of bortezomib in other cell types, we treated B16 cells with 2-deoxy-D-glucose (2-DG), a hexokinase inhibitor, in combination with bortezomib. Separate treatment with either 2-DG or bortezomib weakly induced cell death, whereas the simultaneous presence of the two reagents markedly enhanced cell death (Figure 2A). We further confirmed the synergistic cytotoxicity of bortezomib and 2-DG as calculated by the Bliss independence model (Figure 2B). No synergistic effect of 2-DG and bortezomib was observed in HK1-knockdown cells, consistent with the notion that 2-DG shows an effect primarily through HK1 inhibition, at least in our experimental conditions (Figure S2), although we cannot exclude the possibility that 2-DG interferes with biological processes other than HK1.
Figure 2.

Combined Inhibition of the Proteasome and Hexokinase Promotes Cancer Cell Death
(A) Viability assay of B16 cells treated with 10 nM BTZ alone or in combination with 3 mM 2-DG for 48 h. Data are presented as the mean ± SEM (n = 3).
(B) Viability assay of B16 cells treated with various concentrations of BTZ and 2-DG for 48 h. Data are presented as the mean (n = 3).
(C) Representative tumor allografts are shown.
(D) Mouse allograft model to confirm the effect of combined treatment with BTZ and 2-DG. B16 cells were subcutaneously transplanted into C57BL/6N mice. Mice were intraperitoneally injected with control (PBS), BTZ (0.5 mg kg−1), 2-DG (1,500 mg kg−1), or BTZ (0.5 mg kg−1) in combination with 2-DG (1,500 mg kg−1) three times per week. Tumor size was measured using a digital caliper. Data are presented as the mean ± SEM (n = 5 mice per group). The percentage of dead cells was assessed by FACS after staining with Hoechst 33,342 and PI in (A) and (B). Data in (A) and (D) were analyzed by two-way ANOVA followed by Tukey's test (∗∗p < 0.01, ∗∗∗p < 0.001). See also Figure S2.
This synergistic effect was further confirmed in an in vivo model. Treatment of B16 melanoma-bearing mice with an intraperitoneal injection of bortezomib (0.5 mg kg−1) and 2-DG (1,500 mg kg−1) showed synergistic antitumor activity (Figures 2C and 2D). These results suggest that a pathway involving HK1 plays a crucial role in preventing cell death under proteasome impairment.
Increase in Protein O-GlcNAcylation under Proteasome Inhibition
HK1 is a key enzyme in downstream metabolic pathways, including glycolysis, the pentose phosphate pathway, and protein glycosylation. To determine which downstream pathway is responsible for the protective effect under proteasome impairment, we evaluated the result of knocking down or inhibiting downstream enzymes and measured their mRNA expression and also assessed the stress response induced by bortezomib.
Although knockdown of HK1 induced cell death in combination with bortezomib, knockdown of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and enolase 1 (ENO1) did not show obvious synergistic cytotoxicity (Figures S3A and S3B). In addition, synergistic treatment with bortezomib and either the pyruvate dehydrogenase kinase (PDK) inhibitor sodium dichloroacetate (DCA) or the lactate dehydrogenase (LDH) inhibitor oxamate had modest effects on cell death compared with those of treatment with bortezomib and 2-DG (Figures S3C and S3D), indicating that glycolysis is not important for suppressing cell death under proteasome inhibition. Indeed, intracellular ATP levels were not decreased by combined treatment with bortezomib and 2-DG, demonstrating that glycolysis was not strongly inhibited in our experimental conditions (Figure S3E).
The pentose phosphate pathway was also dispensable for cell survival under proteasome inhibition, as knockdown of glucose-6-phosphate dehydrogenase (G6PD) did not show synergistic cytotoxicity with bortezomib (Figure S3B).
When we organized the RNA-seq data according to the potentially involved pathways, we found that many of the genes involved in O-GlcNAcylation, including OGT, were coordinately upregulated in response to proteasome inhibition, whereas genes involved in glycolysis and glycosylation in the ER and Golgi were not (Figure 3A). We confirmed the induction of HK1 or OGT mRNA expression under bortezomib treatment in B16, HEK293T, and RPMI8226 cells (Figure S3F). O-GlcNAcylation is one of the most abundant types of eukaryotic intracellular glycosylation; OGT catalyzes polypeptide glycosylation by adding N-acetylglucosamine (GlcNAc) to serine and threonine residues, whereas O-GlcNAcase (OGA) removes O-GlcNAc moieties from polypeptides (Bond and Hanover, 2015). Previous studies reported that O-GlcNAcylation of proteasome subunits inhibited proteasome activity (Keembiyehetty et al., 2011; Zhang et al., 2003). On the other hand, recent studies showed that enhanced O-GlcNAcylation of Nrf1 promoted the expression of proteasome subunits (Sekine et al., 2018) and that increased O-GlcNAcylation of FOXA1 was involved in bortezomib resistance in breast cancer cells (Liu et al., 2019). Thus, the biological significance of O-GlcNAcylation under proteasome inhibition remains elusive.
Figure 3.

Increased Protein O-GlcNAcylation Is Required for Cell Survival under Proteasome Inhibition Both In Vitro and In Vivo
(A) Expression heatmap for genes associated with glucose transport, HK, glycolysis, the N-linked and O-linked glycosylation pathways, and O-GlcNAcylation. Expression data were derived from RNA-seq data.
(B) Immunoblot analysis of lysates from B16 cells with antibodies against the indicated proteins. B16 cells were treated with 10 nM BTZ and 50 mM GlcNAc for 48 h and with 1 mM 2-DG for 24 h before harvest. Relative band intensity values for O-GlcNAcylated proteins or OGT were normalized to those for β-actin.
(C) Viability assay of B16 cells treated with 10 nM BTZ and 1 mM 2-DG with or without 20 mM GlcNAc for 48 h.
(D and E) Viability assay of U2OS cells transfected with OGT siRNAs (#01 and #02) or control siRNA in the presence of 15 nM (D) or various concentrations of BTZ (E) for 48 h. (F) Viability assay of B16 cells treated with the indicated combination of 20 μM OSMI-1 and 10 nM BTZ for 48 h.
(G) Viability assay of B16 cells treated with various concentrations of BTZ and OSMI-1 for 48 h. Data are presented as the mean (n = 3).
(H) Viability assay of B16 cells treated with 25 nM BTZ with or without 20 μM Thiamet-G for 48 h.
(I) Mouse allograft model to confirm the effect of combined treatment with BTZ and OSMI-1. B16 cells were subcutaneously transplanted into C57BL/6N mice. Mice were intravenously injected with control (PBS), BTZ (0.5 mg kg−1), OSMI-1 (0.56 mg kg−1), or BTZ (0.5 mg kg−1) in combination with OSMI-1 (0.56 mg kg−1) three times per week. Representative tumor allografts are shown.
(J) Tumor size was measured using a digital caliper. Data are presented as the mean ± SEM (n = 5 mice per group).
(K) Tumor weight. Data are presented as the mean ± SEM (n = 5 mice per group). The percentage of dead cells was assessed by FACS after staining with Hoechst 33,342 and PI in (C–H).
Data in (B)–(F) and (H) are presented as the mean ± SEM (n = 3). Data in (C) and (H) were analyzed by two-tailed Student's t test (∗∗∗p < 0.001). Data in (B) and (D) were analyzed by Tukey's test (∗p < 0.05, ∗∗∗p < 0.001). Data in (F), (J), and (K) were analyzed by two-way ANOVA followed by Tukey's test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). See also Figures S3–S5.
To determine whether protein O-GlcNAcylation increases in response to proteasome inhibition, we quantified intracellular protein O-GlcNAcylation levels. O-GlcNAcylated proteins increased in cells treated with bortezomib, and this increase was reversed by the addition of 2-DG and by HK1 knockdown (Figures 3B and S3G). OGT protein levels also increased in response to bortezomib treatment (Figure 3B). The addition of GlcNAc, which is known to promote O-GlcNAcylation (Zhang et al., 2017), restored intracellular O-GlcNAcylation levels (Figures 3B and S3G). These results are consistent with a previous study reporting increased O-GlcNAcylated proteins in bortezomib-treated cells (Liu et al., 2019).
Increased O-GlcNAcylation Is Required for Cell Survival under Proteasome Inhibition
In the primary siRNA screen, the Β-score of OGT (2.04) fulfilled the commonly used criterion (Β-score >1.65, corresponding to p < 0.05). To investigate whether an increase in O-GlcNAcylation catalyzed by OGT is involved in cell survival under proteasome impairment, we examined cell viability under conditions that either enhance or inhibit O-GlcNAcylation. The addition of GlcNAc restored cell viability after combined treatment with bortezomib and 2-DG in B16 and U2OS cells (Figures 3C and S4A). In addition, knockdown of OGT and inhibition of OGT activity by the specific OGT inhibitor OSMI-1 enhanced cell death in the presence of bortezomib (Figures 3D–3F), with synergistic cytotoxicity (Figure 3G). OGT knockdown and OSMI-1 also showed a synergistic effect with another proteasome inhibitor, carfilzomib (Figures S4B and S4C). Conversely, enhancing protein O-GlcNAcylation by treatment with Thiamet-G, an OGA inhibitor, reduced the cytotoxicity of bortezomib (Figure 3H). Synergistic cytotoxicity was also observed in U2OS and RPMI8226 multiple myeloma cells (Figure S4D and S4E), suggesting that the cytoprotection mediated by O-GlcNAcylation under proteasome impairment is a general event.
Since data from genome-wide siRNA screens in the Cancer Dependency Map (depmap.org) indicate that many cell lines exhibit decreased viability when OGT is knocked down, we examined whether the synergistic effect of OSMI-1 was also observed with other anti-cancer drugs. We tested thapsigargin, nocodazole, cisplatin, and 5-fluorouracil, but none of the drugs showed synergistic cytotoxicity with OSMI-1, suggesting that the synergistic effect of OSMI-1 is at least relatively specific to proteasome inhibitors (Figure S5).
Although bortezomib is used as a first-line drug for the treatment of multiple myeloma, recent studies reported that multiple myeloma cells usually acquire resistance to bortezomib within several months or years (Dispenzieri et al., 2010; Moreau et al., 2011; Ruschak et al., 2011). We therefore examined whether inhibition of O-GlcNAcylation sensitized bortezomib-resistant cells to bortezomib. We generated bortezomib-resistant U2OS (U2OS/BTZ) and B16 (B16/BTZ) cells by culturing cells with bortezomib at stepwise increasing concentrations for 3 months (Figure S6A and S6B). Combined treatment with OSMI-1 and bortezomib demonstrated synergistic cell death in both U2OS/BTZ and B16/BTZ cells (Figure S6C and S6D). These results indicate that enhanced protein O-GlcNAcylation is an important mechanism even in bortezomib-resistant cancer cells.
We next examined the effects of bortezomib and OSMI-1 in combination using a mouse allograft model. Concomitant administration of bortezomib and OSMI-1 to B16 melanoma-bearing mice markedly suppressed tumor development compared with either drug alone (Figures 3I–3K), demonstrating that OGT inhibition enhances the antitumor effect of bortezomib in vivo.
O-GlcNAcylation Is Involved in Maintaining Proteasome Function upon Proteasome Impairment
We next addressed the mechanism by which O-GlcNAcylation plays a protective role against proteasome impairment. We found that combined treatment with bortezomib and OSMI-1 markedly enhanced the accumulation of ubiquitinated proteins (Figure 4A).
Figure 4.

Enhanced O-GlcNAcylation Compensates for Proteasome Activity upon Proteasome Inhibition in an Nrf1-Independent Manner
(A and B) Immunoblot analysis with antibodies against the indicated proteins (A) and proteasome peptidase activity (B) of lysates from U2OS cells treated with 10 nM BTZ for 48 h and with 20 μM OSMI-1 for 24 h.
(C and D) Proteasome activity assay with U2OS cells stably expressing ZsGreen-mODC treated with 20 μM OSMI-1 (C) or transfected with OGT siRNAs (siOGT#01 and siOGT#02) or control siRNA (D) and then treated with 10 nM BTZ for 48 h.
(E) Proteasome activity assay with HEK293T cells stably expressing ZsGreen-mODC treated with 25 nM BTZ with or without 20 μM Thiamet-G.
(F) Immunoblot analysis of lysates from U2OS cells with antibodies against the indicated proteins. U2OS cells were treated with 20 μM OSMI-1 or transfected with OGT siRNAs (siOGT#01 and siOGT#02) or control siRNA and then treated with 10 nM BTZ for 48 h.
(G) Relative mRNA expression of proteasome gene PSMB7 in U2OS cells transfected with target siRNAs (siNrf1 or siOGT#02) or control siRNA and then treated with 10 nM BTZ for 48 h. mRNA levels of target genes were normalized to GUSB mRNA levels.
(H) Viability assay of wild-type (WT) and Nrf1-knockout (KO; KO#01 and KO#02) HCT116 cells treated with 10 nM BTZ for 48 h with or without 20 μM OSMI-1 for 24 h.
(I) Proteasome peptidase activity of lysates from WT and Nrf1-KO (KO#01 and KO#02) HCT116 cells treated with 10 nM BTZ for 48 h with or without 20 μM OSMI-1 for 24 h.
Data in (B)–(E), (G), (H), and (I) are presented as the mean ± SEM (n = 3). Data in (B)–(E), (H), and (I) were analyzed by two-way ANOVA followed by Tukey's test (∗∗p < 0.01, ∗∗∗p < 0.001). Data in (G) were analyzed by Tukey's test (∗∗∗p < 0.001, n.s.: not significant). See also Figure S6.
To examine whether this accumulation was caused by increased ubiquitination or defects in proteasomal degradation, we measured the peptidase activity of the proteasome. Compared with single treatments, the combined treatment with bortezomib and OSMI-1 decreased proteasome activity (Figure 4B). We further monitored the fluorescence intensity of U2OS cells expressing ZsGreen-mODC, a model proteasomal substrate that is degraded in a ubiquitin-independent manner. Although monotherapy with bortezomib or OSMI-1 did not affect ZsGreen-mODC degradation, combined treatment with bortezomib and OSMI-1 caused the accumulation of ZsGreen-mODC (Figure 4C). Knockdown of OGT also caused ZsGreen-mODC accumulation when combined with bortezomib treatment (Figure 4D). Conversely, enhancement of O-GlcNAcylation by Thiamet-G reduced the accumulation of ZsGreen-mODC caused by bortezomib treatment (Figure 4E). Taken together, these results suggest that inducible O-GlcNAcylation plays an important role in maintaining proteasome function upon proteasome impairment.
Nrf1 Is Not Involved in O-GlcNAcylation-Mediated Proteasome Maintenance
It is well known that compromised proteasome activity is restored by the Nrf1-mediated upregulation of proteasome subunits (Radhakrishnan et al., 2010; Steffen et al., 2010). It has been reported that Nrf1 is stabilized by O-GlcNAcylation (Sekine et al., 2018), raising the possibility that OGT inhibition attenuates Nrf1 function and thus proteasome function. However, neither OGT knockdown nor OSMI-1 treatment dampened the bounce-back increase in proteasome subunits induced by bortezomib (Figure 4F). Furthermore, OGT knockdown did not affect the bortezomib-induced increase in the mRNA levels of the proteasome subunit gene PSMB7, an increase that was abolished by Nrf1 knockdown (Figure 4G), indicating that OGT inhibition does not disrupt the bounce-back response.
We also showed that the synergistic effect of OSMI-1 and bortezomib on cytotoxicity and proteasome dysfunction persisted in Nrf1-knockout cells (Figures 4H, 4I, and S6E–S6G). These results indicate that the bounce-back transcriptional upregulation of proteasome subunits is not altered by the inhibition of O-GlcNAcylation and that O-GlcNAcylation-mediated proteasome maintenance occurs independently of Nrf1.
O-GlcNAcylation Maintains Proteasome Activity by Enhancing Turnover of the Proteasome upon Proteasome Impairment
The observation that proteasome activity was decreased while the proteasome amount was unaffected indicates a decrease in specific proteasome activities by the inhibition of O-GlcNAcylation in the presence of bortezomib (Figures 4A–4F). To investigate the mechanism by which cells maintain proteasome functionality, we monitored the half-life of the proteasome by using HeLa cells in which HaloTag was fused at the C terminus of the endogenous Rpn11 subunit (Tomita et al., 2018). First, we confirmed the synergistic toxicity of bortezomib with O-GlcNAcylation inhibition in HeLa cells by conducting a series of experiments (Figures S7A–S7F). After treatment with or without bortezomib, proteasomes were pulse-labeled by tetramethylrhodamine (TMR) ligand and chased in the presence of OSMI-1 or bafilomycin A1, an autophagy inhibitor (Figure 5A). In yeast and plants, impaired proteasomes are known to be degraded by a selective form of autophagy called proteaphagy (Marshall and Vierstra, 2019). Treatment of bafilomycin A1 extended the proteasome half-life both in the absence and presence of bortezomib, suggesting that the proteasome is constitutively degraded by autophagy regardless of proteasome impairment (Figure 5B). Intriguingly, OSMI-1 treatment and knockdown of OGT also extended the proteasome half-life in the absence or presence of bortezomib, indicating that O-GlcNAcylation plays a role in degradation of the proteasome (Figures 5B and S7G).
Figure 5.

O-GlcNAcylation Enhances Turnover of the Proteasome
(A and B) Schematic view of measuring half-life of the proteasome (A).
HeLa Rpn11-Halo knockin cells were treated with 200 nM BTZ or 20 μM OSMI-1 cells. After 4 h, BTZ was washed out and cells were stained with TMR ligand and subsequently subjected to the blocking ligand. Cells were incubated at 37°C for the indicated times (B).
(C and D) Schematic view of measuring the amounts of newly synthesized proteasomes (C). HeLa Rpn11-Halo knockin cells were treated with 200 nM BTZ or 20 μM OSMI-1. After 4 h, BTZ were washed out and cells were treated with the blocking ligand. Cells were incubated at 37°C for 6 h and then stained with TMR ligand (D).
(E and F) Schematic view of measuring proteasome peptidase activity of newly synthesized proteasomes (E). HeLa Rpn11-Halo knockin cells were treated with 30 nM CFZ or 20 μM OSMI-1. After 4 h, CFZ was washed out and then cells were incubated at 37°C for 18 h. Cells were lysed and immunoprecipitated with HaloLink Resin (F).
(G) Immunoblot analysis of lysates from HeLa Rpn11-Halo knockin cells treated as in (E) with antibodies against the indicated proteins. Relative band intensity values for α6 and Rpn8 were normalized to those for β-actin.
Data in (B), (D), (F), and (G) are presented as the mean ± SEM (n = 3). Data in (B), (D), and (F) were analyzed by Tukey's test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n.s.: not significant). Data in (G) were analyzed by two-tailed Student's t test (∗p < 0.05, ∗∗p < 0.01). See also Figures S7–S10.
Despite decelerated degradation of the proteasome by OSMI-1, the amounts of proteasome subunits were not increased under combined treatment with bortezomib and OSMI-1 or under OGT knockdown (Figure 4F). We then examined whether O-GlcNAcylation was involved in proteasome biogenesis. After blocking pre-existing proteasomes by succinimidyl ester ligands, we quantified newly synthesized proteasomes by labeling them with TMR ligands in the presence or absence of bortezomib and OSMI-1 (Figure 5C). OSMI-1 decreased newly synthesized proteasomes, and the effect was more profound in the presence of bortezomib (Figure 5D), suggesting that O-GlcNAcylation promotes proteasome biogenesis, especially under proteasome impairment.
Next, we measured the activity of newly synthesized proteasomes. To this end, we first treated cells with the irreversible proteasome inhibitor carfilzomib to minimize the activity of pre-existing proteasomes. We then blocked pre-existing proteasomes by succinimidyl ester ligands and immunoprecipitated newly synthesized proteasomes with HaloLink Resin (Figure 5E). We also measured the activity of “total proteasomes” by using cell lysates before immunoprecipitation. The activity of total proteasomes decreased under combined treatment with carfilzomib and OSMI-1 (Figure 5F, left), similar to the results obtained by bortezomib treatment (Figure 4B). We found that OSMI-1 decreased the activity of newly synthesized proteasomes, and the effect was more profound in the presence of carfilzomib (Figure 5F, right), further confirming the role of O-GlcNAcylation in proteasome biogenesis under proteasome impairment (Figure 5D). The immunoprecipitated newly synthesized proteasomes were further subjected to immunoblot analyses of proteasome subunits. This confirmed the decrease in the amount of newly synthesized proteasomes in OSMI-1-treated cells (Figure 5G), consistent with the results shown in Figures 5D and 5F. An extended half-life of the proteasome and a decrease in newly synthesized proteasomes were also observed in Rpn11-Halo knock-in U2OS and B16 cells (Figure S8), validating the involvement of O-GlcNAcylation in proteasome turnover.
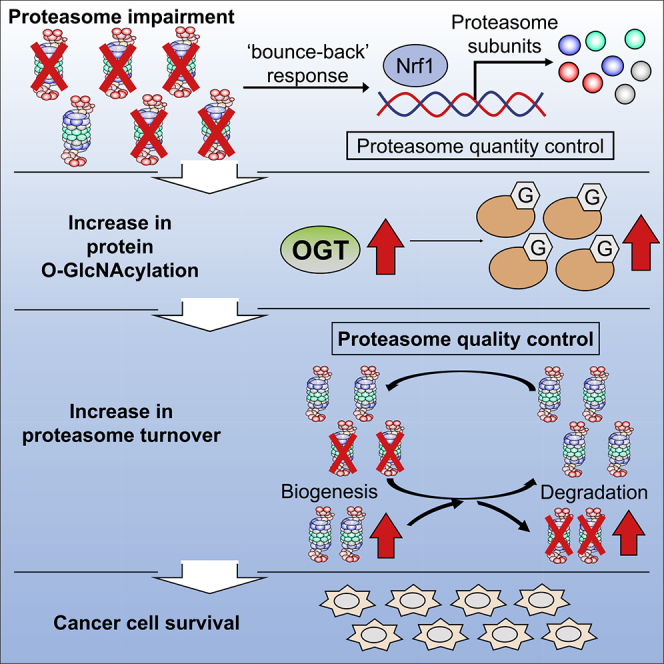
Taken together, we conclude that O-GlcNAcylation plays a role in proteasome turnover by promoting both the degradation and biogenesis of proteasomes to maintain proteasome activity and thus cellular homeostasis under proteasome impairment (Figure 6).
Figure 6.

Proposed Model by Which Enhanced Protein O-GlcNAcylation Mediates the Cytoprotective Response to Proteasome Impairment
Proposed schematic models by which enhanced protein O-GlcNAcylation mediates the cytoprotective response to proteasome impairment. Cells upregulate OGT expression to enhance O-GlcNAcylation of cellular proteins upon proteasome impairment. O-GlcNAcylation enhances biogenesis and degradation of the proteasome and then maintains proteasome activity and suppresses cell death. This mechanism may play a redundant role in the Nrf1-mediated bounce-back response that maintains proteasome function upon proteasome impairment.
Discussion
In the present study, we demonstrated that enhanced protein O-GlcNAcylation was involved in maintaining proteasome function under proteasome inhibition. Prior large-scale genome-wide screens identified genes that are involved in the sensitization or resistance to proteasome inhibitors, but none identified HK1 or OGT (Acosta-Alvear et al., 2015; Tsvetkov et al., 2015). This is probably due to differences between our study and these previous ones in terms of experimental conditions such as cell lines and inhibitor concentrations. Prior studies reported that various mechanisms are involved in the acquired resistance to proteasome inhibitors, including the Nrf1-mediated expression of proteasome subunits (Radhakrishnan et al., 2010; Steffen et al., 2010) and the activity of drug transporters (Besse et al., 2018). Although these reports raise the possibility that enhanced O-GlcNAcylation contributes to the regulation of these compensatory mechanisms, our data demonstrated that O-GlcNAcylation mediated the maintenance of proteasome activity independently of Nrf1 (Figures 4G–4I). Unlike a previous report (Sekine et al., 2018), our study showed that O-GlcNAcylation was not involved in the Nrf1-mediated compensatory increase in proteasomes. This discrepancy is probably due to differences in experimental conditions, including cell types and the degree of proteasome inhibition. However, our finding demonstrated a new compensatory role of O-GlcNAcylation in proteasome impairment that is independent of Nrf1. We also showed that the activity of ABCB1, which is known to efflux bortezomib and carfilzomib (Ao et al., 2012; O'Connor et al., 2013; Verbrugge et al., 2012), was not attenuated under O-GlcNAcylation inhibition (Figure S9).
We demonstrated that enhanced O-GlcNAcylation promoted turnover of the proteasome under proteasome impairment, although further analyses are required to elucidate the detailed mechanisms underlying enhanced proteasome degradation and the biogenesis of the proteasome by O-GlcNAcylation. Considering that neither mRNA levels of proteasome subunits nor the assembly pathway of the proteasome was affected by OGT knockdown or OSMI-1 treatment (Figures 4G and S10), the translation efficacy of proteasome subunit mRNAs and the stability of proteasome subunits following their translation might be improved by O-GlcNAcylation. Identification of the O-GlcNAcylation target proteins responsible for enhanced proteasome turnover is critical to clarify the mechanisms involved. To date, Nrf1-mediated upregulation of proteasome gene expression is widely known to quantitatively compensate for proteasome impairment, whereas the mechanism by which cells maintain proteasome quality control is poorly understood. Our data suggest that O-GlcNAcylation plays a role in turnover of the proteasome, even in the steady state (Figure 5). Therefore, it is logical to speculate that O-GlcNAcylation constitutively contributes to proteasome quality control, and its significance becomes apparent under conditions involving proteasome defects.
Recent studies showed that O-GlcNAcylation is important in both cancer and neurodegeneration (Ma et al., 2017). Enhanced O-GlcNAcylation is thought to prevent protein phosphorylation by competing with target Ser/Th residues, and thus the administration of Thiamet-G, an OGA inhibitor, is expected to protect against Alzheimer's disease progression by suppressing the phosphorylation of tau proteins (Yuzwa et al., 2008). It has also been shown that increased O-GlcNAcylation by Thiamet-G decelerates neurodegeneration by stabilizing tau against aggregation, although the detailed mechanism remains unclear (Yuzwa et al., 2012). Since the proteasome plays a pivotal role in the degradation of tau proteins, the effect of Thiamet-G might be mediated in part by maintaining proteasome activity though O-GlcNAcylation (Götzl et al., 2016; Opoku-Nsiah and Gestwicki, 2018).
Proteasome inhibitors have succeeded in improving the clinical outcomes of patients with multiple myeloma, but acquired resistance to proteasome inhibitors eventually emerges (Wallington-Beddoe et al., 2018). Furthermore, the clinical response to proteasome inhibitors in solid tumors remains poor (Manasanch and Orlowski, 2017; Ruschak et al., 2011). Upregulated OGT and enhanced O-GlcNAcylation have been reported in some solid tumors, such as breast and pancreatic cancer (Champattanachai et al., 2013; Krześlak et al., 2012; Ma et al., 2013). These findings raise the possibility that enhanced O-GlcNAcylation confers resistance to proteasome inhibitors in solid cancer cells. Our results in cells and mice reveal that O-GlcNAcylation, in combination with proteasome inhibitors, is a promising target for cancer treatment.
In conclusion, we identified enhanced O-GlcNAcylation as a regulator of proteasome function under proteasome inhibition. Further analysis is required to elucidate the detailed mechanism that maintains proteasome activity, as this will provide insight into the design of drugs against cancer and possibly neurodegeneration.
Limitations of the Study
In the present study, we demonstrated that enhanced O-GlcNAcylation suppressed cell death upon proteasome impairment by maintaining proteasome activity. However, the distinct targets of O-GlcNAcylation and the precise mechanisms of O-GlcNAcylation-mediated biogenesis and degradation of the proteasome remain to be addressed in future studies. Mass spectrometry analysis to identify O-GlcNAcylated proteins by developing a new method for their purification will provide clues regarding the mechanistic role of O-GlcNAcylation. We also demonstrated that the proteasome was constitutively degraded by autophagy in mammalian cells. However, the mechanistic interaction between proteaphagy and O-GlcNAcylation remains unclear. Further investigations are required to characterize the detailed mechanism whereby O-GlcNAcylation achieves proteasome quality control.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Shigeo Murata (smurata@mol.f.u-tokyo.ac.jp).
Materials Availability
All unique materials generated from this study are available from the Lead Contact with a complete Materials Transfer Agreement.
Data and Code Availability
The accession number for the RNA-seq data and processed data reported in this paper is DDBJ Sequence Read Archive (DRA): DRA008318 and Genomic Expression Archive (GEA): E-GEAD-342.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by JSPS KAKENHI (Grant Numbers JP18H05500, JP18H04022, and JP25221102), AMED (Grant Number JP18gm1110003 AMED-CREST), and the Tokyo Biochemical Research Foundation.
Author Contributions
E.H. designed and performed the experiments, analyzed and interpreted the data, prepared the figures, and wrote the manuscript. S.O. performed the experiments and analyzed and interpreted the data. S.H., Y.A., and T.G. analyzed and interpreted the data. J.H. designed the experiments, analyzed and interpreted the data, and wrote the manuscript. H.K. performed the experiments and analyzed and interpreted the data. S.M. supervised the project, designed the experiments, analyzed and interpreted the data, and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101299.
Supplemental Information
References
- Acosta-Alvear D., Cho M.Y., Wild T., Buchholz T.J., Lerner A.G., Simakova O., Hahn J., Korde N., Landgren O., Maric I. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife. 2015;4:e08153. doi: 10.7554/eLife.08153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao L., Wu Y., Kim D., Jang E.R., Kim K., Lee D.M., Kim K.B., Lee W. Development of peptide-based reversing agents for P-glycoprotein-mediated resistance to carfilzomib. Mol. Pharm. 2012;9:2197–2205. doi: 10.1021/mp300044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt A., Bauer I., Schafmayer C., Tepel J., Muerkoster S.S., Brosch M., Roder C., Kalthoff H., Hampe J., Moyer M.P. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2) Oncogene. 2009;28:3983–3996. doi: 10.1038/onc.2009.264. [DOI] [PubMed] [Google Scholar]
- Besse A., Stolze S.C., Rasche L., Weinhold N., Morgan G.J., Kraus M., Bader J., Overkleeft H.S., Besse L., Driessen C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia. 2018;32:391–401. doi: 10.1038/leu.2017.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M., Crinelli R., Arbore V., Magnani M. Induction of ubiquitin C (UBC) gene transcription is mediated by HSF1: role of proteotoxic and oxidative stress. FEBS Open Biol. 2018;8:1471–1485. doi: 10.1002/2211-5463.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M.R., Hanover J.A. A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champattanachai V., Netsirisawan P., Chaiyawat P., Phueaouan T., Charoenwattanasatien R., Chokchaichamnankit D., Punyarit P., Srisomsap C., Svasti J. Proteomic analysis and abrogated expression of O-GlcNAcylated proteins associated with primary breast cancer. Proteomics. 2013;13:2088–2099. doi: 10.1002/pmic.201200126. [DOI] [PubMed] [Google Scholar]
- Chen L., Madura K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005;65:5599–5606. doi: 10.1158/0008-5472.CAN-05-0201. [DOI] [PubMed] [Google Scholar]
- Dispenzieri A., Jacobus S., Vesole D.H., Callandar N., Fonseca R., Greipp P.R. Primary therapy with single agent bortezomib as induction, maintenance and re-induction in patients with high-risk myeloma: results of the ECOG E2A02 trial. Leukemia. 2010;24:1406–1411. doi: 10.1038/leu.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götzl J.K., Lang C.M., Haass C., Capell A. Impaired protein degradation in FTLD and related disorders. Ageing Res. Rev. 2016;32:122–139. doi: 10.1016/j.arr.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Hagenbuchner J., Ausserlechner M.J., Porto V., David R., Meister B., Bodner M., Villunger A., Geiger K., Obexer P. The anti-apoptotic protein BCL2L1/Bcl-xL is neutralized by pro-apoptotic PMAIP1/noxa in neuroblastoma, thereby determining bortezomib sensitivity independent of prosurvival MCL1 expression. J. Biol. Chem. 2010;285:6904–6912. doi: 10.1074/jbc.M109.038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keembiyehetty C.N., Krzeslak A., Love D.C., Hanover J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011;124:2851–2860. doi: 10.1242/jcs.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krześlak A., Forma E., Bernaciak M., Romanowicz H., Bryś M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin. Exp. Med. 2012;12:61–65. doi: 10.1007/s10238-011-0138-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiczkova L., Pour L., Sedlarikova L., Hajek R., Sevcikova S. Proteasome inhibitors - molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014;18:947–961. doi: 10.1111/jcmm.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumatori A., Tanaka K., Inamura N., Sone S., Ogura T., Matsumoto T., Tachikawa T., Shin S., Ichihara A. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. U S A. 1990;87:7071–7075. doi: 10.1073/pnas.87.18.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtin S.E., Bilotti E. Novel agents for the treatment of multiple myeloma: proteasome inhibitors and immunomodulatory agents. J. Adv. Pract. Oncol. 2013;4:307–321. [PMC free article] [PubMed] [Google Scholar]
- Leung-Hagesteijn C., Erdmann N., Cheung G., Keats J.J., Stewart A.K., Reece D.E., Chung K.C., Tiedemann R.E. Xbp1s-Negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24:289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wang X., Zhu T., Zhang N., Wang L., Huang T., Cao Y., Li W., Zhang J. Resistance to bortezomib in breast cancer cells that downregulate Bim through FOXA1 O-GlcNAcylation. J. Cell. Physiol. 2019;234:17527–17537. doi: 10.1002/jcp.28376. [DOI] [PubMed] [Google Scholar]
- Lü S., Yang J., Song X., Gong S., Zhou H., Guo L., Song N., Bao X., Chen P., Wang J. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J. Pharmacol. Exp. Ther. 2008;326:423–431. doi: 10.1124/jpet.108.138131. [DOI] [PubMed] [Google Scholar]
- Ma X., Li H., He Y., Hao J. The emerging link between O-GlcNAcylation and neurological disorders. Cell. Mol. Life Sci. 2017;74:3667–3686. doi: 10.1007/s00018-017-2542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z., Vocadlo D.J., Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J. Biol. Chem. 2013;288:15121–15130. doi: 10.1074/jbc.M113.470047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiso P., Huynh D., Moschetta M., Sacco A., Aljawai Y., Mishima Y., Asara J.M., Roccaro A.M., Kimmelman A.C., Ghobrial I.M. Metabolic Signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015;75:2071–2082. doi: 10.1158/0008-5472.CAN-14-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasanch E.E., Orlowski R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017;14:417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.S., Vierstra R.D. Dynamic regulation of the 26S proteasome: from synthesis to degradation. Front. Mol. Biosci. 2019;6:40. doi: 10.3389/fmolb.2019.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P., Pylypenko H., Grosicki S., Karamanesht I., Leleu X., Grishunina M., Rekhtman G., Masliak Z., Robak T., Shubina A. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12:431–440. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- Murata S., Yashiroda H., Tanaka K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009;10:104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- O’Connor R., Ooi M.G., Meiller J., Jakubikova J., Klippel S., Delmore J., Richardson P., Anderson K., Clynes M., Mitsiades C.S. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013;71:1357–1368. doi: 10.1007/s00280-013-2136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R., Franke N.E., Assaraf Y.G., Cloos J., Van Zantwijk I., Berkers C.R., Scheffer G.L., Debipersad K., Vojtekova K., Lemos C. Molecular basis of bortezomib resistance: proteasome subunit25 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- Opoku-Nsiah K.A., Gestwicki J.E. Aim for the core: suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Transl. Res. 2018;198:48–57. doi: 10.1016/j.trsl.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan S.K., Lee C.S., Young P., Beskow A., Chan J.Y., Deshaies R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riz I., Hawley T.S., Marsal J.W., Hawley R.G. Noncanonical SQSTM1/p62-Nrf2 pathway activation mediates proteasome inhibitor resistance in multiple myeloma cells via redox, metabolic and translational reprogramming. Oncotarget. 2016;7:66360–66385. doi: 10.18632/oncotarget.11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak A.M., Slassi M., Kay L.E., Schimmer A.D. Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 2011;103:1007–1017. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- Schwartz A.L., Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- Sekine H., Okazaki K., Kato K., Alam M.M., Shima H., Katsuoka F., Tsujita T., Suzuki N., Kobayashi A., Igarashi K. O -GlcNAcylation signal mediates proteasome inhibitor resistance in cancer cells by stabilizing NRF1. Mol. Cell. Biol. 2018;38 doi: 10.1128/MCB.00252-18. e00252–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen J., Seeger M., Koch A., Krüger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell. 2010;40:147–158. doi: 10.1016/j.molcel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Tomita T., Hirayama S., Sakurai Y., Ohte Y., Yoshihara H., Saeki Y., Hamazaki J., Murata S. Specific modification of aged proteasomes revealed by tag-exchangeable knock-in mice. Mol. Cell. Biol. 2018;39 doi: 10.1128/MCB.00426-18. e00426–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P., Mendillo M.L., Zhao J., Carette J.E., Merrill P.H., Cikes D., Varadarajan M., van Diemen F.R., Penninger J.M., Goldberg A.L. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. Elife. 2015;4:e08467. doi: 10.7554/eLife.08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbrugge S.E., Assaraf Y.G., Dijkmans B.A.C., Scheffer G.L., Al M., Den Uyl D., Oerlemans R., Chan E.T., Kirk C.J., Peters G.J. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with. J. Pharmacol. Exp. Ther. 2012;341:174–182. doi: 10.1124/jpet.111.187542. [DOI] [PubMed] [Google Scholar]
- Wallington-Beddoe C.T., Sobieraj-Teague M., Kuss B.J., Pitson S.M. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br. J. Haematol. 2018;182:11–28. doi: 10.1111/bjh.15210. [DOI] [PubMed] [Google Scholar]
- Weniger M.A., Rizzatti E.G., Peŕez-Galań P., Liu D., Wang Q., Munson P.J., Raghavachari N., White T., Tweito M.M., Dunleavy K. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 2011;17:5101–5112. doi: 10.1158/1078-0432.CCR-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa S.A., Macauley M.S., Heinonen J.E., Shan X., Dennis R.J., He Y., Whitworth G.E., Stubbs K.A., McEachern E.J., Davies G.J. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa S.A., Shan X., MacAuley M.S., Clark T., Skorobogatko Y., Vosseller K., Vocadlo D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zaal E.A., Wu W., Jansen G., Zweegman S., Cloos J., Berkers C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017;5:7. doi: 10.1186/s40170-017-0169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Su K., Yang X., Bowe D.B., Paterson A.J., Kudlow J.E. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–725. doi: 10.1016/s0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- Zhang X., Qiao Y., Wu Q., Chen Y., Zou S., Liu X., Zhu G., Zhao Y., Chen Y., Yu Y. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017;8:15280. doi: 10.1038/ncomms15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data and processed data reported in this paper is DDBJ Sequence Read Archive (DRA): DRA008318 and Genomic Expression Archive (GEA): E-GEAD-342.