

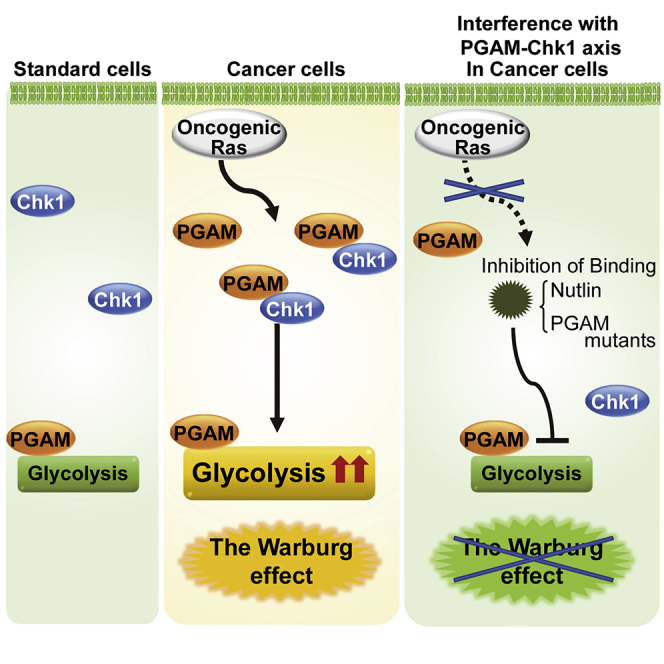
Summary
Dysregulated glycolysis, including the cancerous Warburg effect, is closely involved in pathological mechanisms of diseased states. Among glycolytic enzymes, phosphoglycerate mutase (PGAM) has been known to exert certain physiological impact in vitro, whereas its regulatory role on glycolysis remains unclear. Here, we identified that PGAM plays a key role in regulating glycolysis in cancer cells but not in standard cells. Cancer-prone phenotype by PGAM overexpression in vivo was associated with upregulated glycolytic features. PGAM interacts and cooperates with Chk1 to regulate the enhanced glycolysis in cancer cells, especially under oncogenic Ras expressing conditions. Genetic or chemical interference of the PGAM-Chk1 interaction, with intact PGAM activity, abrogated the maintenance of cancerous enhanced glycolysis. Thus, the nonenzymatic function of PGAM is essential for the Warburg effect that accompanies cancerous proliferation.
Subject Areas: Glycobiology, Molecular Biology, Cancer
Graphical Abstract
Highlights
-
•
PGAM cooperates with Chk1 kinase to boost glycolysis in a non-enzymatic manner
-
•
PGAM interacts with Chk1 in cancer with oncogenic Ras but not in standard cells
-
•
Chemical and genetical interference with PGAM-Chk1 axis abrogates the Warburg effect
Glycobiology; Molecular Biology; Cancer
Introduction
Glycolysis constitutes an essential metabolism among various organisms, serving not only as an energy source but also for the synthesis of macromolecules (Vander Heiden et al., 2009). The regulation of glycolysis depends on several factors including growth conditions, differentiation status, and environmental stress. In mammalian cells, metabolic adaptation in glycolysis is mediated partly by the activation of specific signaling modules, followed by the transcriptional upregulation of glycolytic enzymes (Dang and Semenza, 1999). In addition to transcriptional factors, several other molecules including signals from nutritional stress, oncogenic stimuli, and posttranscriptional regulators are also deeply involved in glycolytic regulation (Mikawa et al., 2015).
Fine-tuning of glycolysis is required to maintain physiological homeostasis in normal cells and tissues, and dysregulated glycolysis is closely related to pathological features. Impaired glycolysis in vivo is associated with dysfunction in various tissues and degenerative disorders (Goyal et al., 2017; Taylor et al., 2001). Conversely, the pathological enhancement of glycolysis is also observed in several diseased states, such as inflammation (Chimenti et al., 2015), ischemia, and the cancerous Warburg effect in association with increase of activity and protein levels for multiple glycolytic enzymes (Warburg, 1956). Although normal cells may adapt to hypoxic conditions by enhancing anaerobic glycolysis and limiting energy demands, cancer cells in vivo continue to grow even in hypoxia, which requires excess glycolysis as a maladaptive metabolism in the core of solid tumors. However, the Warburg effect cannot be explained simply as a consequence of cellular adaptation to hypoxia, as cancer cells maintain enhanced glycolysis even in standard tissue culture conditions (20% oxygen) or in circulating blood (Koppenol et al., 2011). Therefore, a rising question is how the Warburg effect is linked to the other cancerous properties besides adaptation to hypoxia.
Phosphoglycerate mutase (PGAM) is a glycolytic enzyme that converts 3-phosphoglycerate into 2-phosphoglycerate as an isomerase (Rodwell et al., 1957). PGAM consists of two isoforms, PGAM1 and PGAM2, termed also as brain- and muscle-forms, respectively, both of which display a significant similarity in their sequences (79% identity) and enzymatic activities (Kondoh et al., 2005; Mikawa et al., 2014; Zhang et al., 2001). Recent reports suggest that PGAM represents a key factor connecting glycolysis to physiological homeostasis. PGAM supports anti-oxidative defense not only by the reduction of mitochondrial reactive oxygen species (Kondoh et al., 2005, 2007) but also via activation of the pentose phosphate pathway (Hitosugi et al., 2012). Moreover, the p53/Mdm2 axis promotes proteolysis of PGAM during senescence-inducing stress (Mikawa et al., 2014), which is consistent with reports that p53 inactivation enhances glycolysis in cancer (Bensaad and Vousden, 2007). In addition to the ubiquitination, PGAM activity is post transcriptionally modulated by the phosphorylation or acetylation (Wang et al., 2017; Xu et al., 2014). Several studies also implicate the involvement of PGAM in human disease. Although PGAM protein and activity are upregulated in many cancerous tissues (Durany et al., 1997), patients with PGAM deficiencies are also reported (Naini et al., 2009). However, the precise regulatory role of PGAM in glycolysis remains unclear.
Here, we report a previously unappreciated role for PGAM in cancerous glycolytic regulation. We observed that PGAM significantly affected the global profiles of glycolysis in cancerous cells. PGAM cooperated with Chk1, previously known as a checkpoint kinase for p53, to boost glycolysis under oncogenic conditions, but not in standard cells. The significance of the PGAM-Chk1 interaction in cancerous glycolysis was validated by several lines of evidence with genetic or chemical ablation of PGAM-Chk1 binding, especially under oncogenic Ras expressing conditions. Thus, PGAM and Chk1 cooperated to regulate cancerous glycolysis.
Results
PGAM Overexpression In Vivo Promotes Chemically Induced Tumorigenesis with Global Increase in Glycolytic mRNAs
We previously reported that heart-specific Pgam2-transgenic (Tg) mice displayed almost normal glycolytic features in the heart (Okuda et al., 2013). However, the impact of global PGAM overexpression in vivo remains unclear. As it has been demonstrated that the overexpression of either PGAM isoform confers similar physiological impact (Kondoh et al., 2005; Mikawa et al., 2014; Zhang et al., 2001), we utilized Pgam2-Tg mice, which exhibited a significant increase of PGAM protein in the whole body by the CAG-promoter driven Pgam2-FLAG transgene (Figure S1A) (Mikawa et al., 2014). Global overexpression of PGAM in vivo did not affect the profiles for glycolytic mRNAs among the various tissues (skin, liver, kidney, muscle, WAT, lung, or heart) under physiological conditions or in MEFs under standard culture conditions (Figures S1B and S1C). Although both isoforms of PGAM were expressed in skin of wild-type mice (Mikawa et al., 2014), however, we noticed that skin in Pgam2-Tg mice was more vulnerable to inflammation caused by a proinflammatory agent, 12-O-tetradecanoylphorbol-13-acetate (TPA) (Figures S2A and S2B). These findings prompted further investigation into the possibility that PGAM overexpression in vivo may affect glycolysis under TPA stress. For this purpose, we applied a protocol of chemically induced carcinogenesis utilizing 7,12-dimethylbenz[a]anthracene (DMBA) initiation followed by TPA promotion in mice skin from control and Pgam2-Tg mice (Figure S2C). We observed that the total number of tumors in Pgam2-Tg mice was slightly increased (Figure S2D), whereas the tumor sizes were strikingly larger than those of control mice throughout the protocol. At the early stage of the experimental process (8–12 weeks) small tumors in Pgam2-Tg mice were already observed (Figure 1A, top panel). Subsequently (12–24 weeks), some tumors in the Pgam2-Tg mice grew to over 6 mm in their diameters, which was not observed in control mice (Figure 1A, middle and bottom panels). Notably, some of the largest tumors in the Pgam2-Tg mice were accompanied by moderate-to-severe ulceration as shown in Figure 1B. At the end of the protocol (24 weeks), pathologic analysis was performed for all tumors in mice. Histologically, tumors in both groups showed acanthosis and hyperkeratosis. All tumors in the control mice were pathologically benign (squamous cell papillomas), whereas 3 of 25 tumors that developed in Pgam2-Tg mice showed marked dyskeratosis, numerous mitoses, and severe disturbances of stratification, which are characteristic of squamous cell carcinoma (SCC) (Figures 1C and 1D).
Figure 1.

PGAM Overexpression In Vivo Promotes Chemically Induced Tumorigenesis with Significant Increment in Glycolytic mRNAs
Chemical-induced skin tumorigenesis was performed. Control (n = 7) and Pgam2-Tg mice (n = 7) were first treated with DMBA followed by TPA treatment twice a week.
(A) Comparison of tumor numbers between control (wild-type) and Pgam2-Tg mice. Tumors were classified into three subgroups according to their diameters (top panel for tumor size <3 mm; middle panel, 3–6 mm; and bottom panel, >6 mm). The average number of tumors per mouse was compared at the indicated time points.
(B) Representative tumors in control or Pgam2-Tg mice at 24 weeks are indicated.
(C and D) Histopathological analysis of skin tumors. (C) Skin papillomas in control (left panel) and SCC in Pgam2-Tg (right panel) with hematoxylin and eosin staining. Scale bar indicates 200 μm. (D) Summary of pathologic diagnoses of skin tumors in control and Pgam2-Tg mice.
(E) Comparison of mRNA levels for glycolytic enzymes between control and Pgam2-Tg mice. The results in non-tumor forming skin, skin papillomas, and skin SCCs are shown. Data for the indicated genes are shown as relative values against the counterparts in non-tumor-forming skin from control mice. Abbreviations for glycolytic enzymes were shown in Figure S1. ∗p < 0.05 and ∗∗p < 0.005, Dunnett's multiple comparison test. Data represent the mean ± SEM.
See also Figures S1 and S2.
Notably, we identified clear differences in the mRNA profiles of glycolytic enzymes in the skins of this setting. The levels of glycolytic mRNAs were markedly increased both in papillomas and in skins without tumors from Pgam2-Tg mice compared with those in control mice (Figure 1E). In addition, the expression of these mRNAs was further enhanced in malignant tumors from Pgam2-Tg skin than in benign papillomas of the same genetic background (Figure 1E). Thus, the overexpression of PGAM promotes the expression of glycolytic mRNAs in vivo in chemically induced tumorigenesis.
Abrogation of PGAM Decreases Glycolytic mRNAs in Cancer Cells but Not in Standard Cells In Vitro
We next examined the impact of PGAM inactivation on glycolytic profiles in vitro. First, PGAM1 siRNA was transfected into cancer cell types in which PGAM1 is dominantly expressed: non-small cell lung carcinoma (H1299) and squamous cell carcinoma of human skin (HSC-1) cell lines (Figures S3A–S3C). We observed that PGAM1 knockdown in these cells significantly decreased their proliferative capacities (Figures 2A and S3D), accompanied by a marked reduction in glycolytic measurements such as glucose consumption, glycolytic flux, and lactate production (Figures 2B–2D and S3E–S3G). We observed that mRNA, protein levels, and enzymatic activities of other glycolytic enzymes were also reduced in both cell lines (Figures 2E–2G and S3H–S3J). Notably, the decline in overall glycolytic mRNAs after PGAM1 knockdown was restored by ectopic expression of either mouse Pgam1 or Pgam2 (Figures S4A and S4B), probably due to the high similarity in their sequences and enzymatic activities.
Figure 2.

Ablation of PGAM Downregulates Glycolysis In Vitro
The impact of PGAM1 knockdown on glycolysis in cancer cells in vitro. H1299 cells were transfected with siPGAM1 (n = 3) or scrambled RNA (n = 3) (A–G).
(A) PGAM1-knockdown H1299 cells were passaged according to 3T3 cell culture protocol. The proliferation curves show population doublings. The glycolytic profiles were evaluated in PGAM1-knockdown H1299 cells (B–G).
(B–D) Glucose consumption was evaluated by measuring glucose concentration in medium (B), whereas glycolytic flux was evaluated using 3H-labeled glucose (C). Lactate production was determined by the measurements of lactate concentration in medium (D).
(E) The mRNA levels for glycolytic enzymes were assessed. Data are relative to those in scrambled RNA-transfected H1299 cells. ∗p < 0.05 and ∗∗p < 0.005, Student's t test.
(F) The protein levels of the indicated glycolytic enzymes were analyzed by immunoblot using anti-aldolase (ALDO), anti-GAPDH, anti-PGK1, anti-PGAM, anti-enolase1 (ENO1), and anti-actin antibodies. Intensity of immunoblotting bands was normalized to that of actin. Relative values were shown, compared with scrambled RNA-transfected H1299 cells.
(G) Enzymatic activity of aldolase, GAPDH, PGK, PGAM, and enolase was measured by spectrometric assay.
(H and I) Pgam1 was ablated in primary MEFs from Pgam1flox/flox mice harboring Cre-ER by treatment with 4-hydroxytamoxifen (4-OHT) for 4 days. (H) PGAM protein levels in the indicated MEFs were analyzed by immunoblotting. (I) Comparison of glycolytic mRNAs among the indicated MEFs after 4-OHT treatment (n = 3). Cre(−); Pgam1flox/flox MEFs, Cre(+); Pgam1flox/flox + Cre-ER MEFs.
Data are relative to those in control. ∗p < 0.05 and ∗∗p < 0.005, Dunnett's multiple comparison test. Data represent the mean ± SEM.
See also Figures S3 and S4.
Next, Pgam1flox/flox MEFs expressing Cre-ER were generated by crossing CAG-Cre-ER-Tg mice and Pgam1flox/flox mice, which we established (Figure S4C). Pgam1 mRNA and protein were efficiently ablated after 4-hydroxytamoxifen (4-OHT) treatment of these MEFs (Figures 2H and 2I), followed by a premature senescent growth arrest (Figures S4D and S4E). However, glycolytic mRNA expression in these cells was similar to Pgam1-intact cells (Figure 2I). Similar results were obtained following transfection with Pgam1 siRNA in primary MEFs and human fibroblast WI-38 cells, which predominantly express PGAM1 (Figures S4F and S4G). Collectively, the abrogation of PGAM in standard cells did not affect the levels of other glycolytic mRNAs, in sharp contrast to our findings in cancer cells.
PGAM Attenuates p53 Phosphorylation and Interacts with Chk1
In addition to the susceptibility of globally PGAM-overexpressing mice to skin carcinogenesis (Figure 1), we previously reported that PGAM was ubiquitinated via the Mdm2/p53 axis under senescence-inducing stress (Mikawa et al., 2014). We also noticed that, in prematurely senescent MEFs by Pgam1 ablation, the levels of several p53 targets (p21Cip, Bax, and Fas) and p16ink4 were significantly increased (Figure 3A). To gain insights into the molecular mechanism of glycolytic regulation by PGAM, we further investigated the possible link between PGAM and the tumor suppressor p53. As p53 activity and protein stability are regulated largely by post-transcriptional modification including its phosphorylation, we examined the p53 profile under oncogenic stress both in Pgam2-Tg and control MEFs expressing Ras-G12V. The protein levels of p21 and Mdm2, downstream effectors of p53, were upregulated in Ras-G12V-expressing control MEFs but were moderately impaired in Ras-G12V-expressing Pgam2-Tg MEFs (Figure 3B). Oncogene-induced stress also upregulated p53 protein levels in control MEFs, accompanied by the enhanced phosphorylation of serine residues 18 and 23 (Di Micco et al., 2006). In contrast, in Ras-G12V-expressing Pgam2-Tg MEFs, p53 serine 23 phosphorylation was largely impaired compared with control, whereas serine 18 phosphorylation was moderately downregulated (Figure 3B). We also observed that ectopic expression of PGAM1 also suppresses p53 serine 23 phosphorylation in MEFs under oncogene-induced stress (Figure S5A). We also examined the DNA damage response in H1299 cell line with transfected WT p53. Similarly, p53 phosphorylation at serine 20, corresponding to serine 23 in mouse p53, was attenuated by ectopic PGAM expression during etoposide-induced DNA damage in H1299 cells (Figure S5B). These data indicate that PGAM partially interferes with p53 function by impacting phosphorylation at serine 23.
Figure 3.

PGAM Interacts with and Modulates p53 Phosphorylation by Chk1
(A) The mRNA levels for p16Ink4 and p53 targets (p21Cip, Bax, and Fas) were analyzed by quantitative PCR in the indicated MEFs. Indicated MEFs were treated with or without 4-OHT for 4 days, as in Figure 2H.
(B) The assessment for the effect of PGAM overexpression on the profiles of p53, Mdm2, p21, Chk1, and Chk2. Oncogenic Ras (Ras-G12V) was ectopically expressed in control (wild-type) and Pgam2-Tg MEFs via retroviral infection.
(C) Immunoprecipitation assay was performed using p53−/− MEFs expressing Chk1-myc-his with or without PGAM-FLAG. MEFs expressing PGAM1-FLAG (upper) or PGAM2-FLAG (lower) were examined. After treatment with 20 μM MG132 for 3 h, cells were lysed and immunoprecipitated with an anti-FLAG antibody and immunoblotted for Chk1 and FLAG.
(D) Schematic diagram of various fragments of PGAM2: full-length (T1), N-terminal (TN), middle-region (T4), and C-terminal (TC) fragments (upper panel). The binding of each fragment to Chk1 was assessed by co-immunoprecipitation (lower panels). Extracts from p53−/− MEFs transfected with the indicated plasmids were immunoprecipitated with an anti-FLAG antibody.
(E) Interaction between endogenous Chk1 and PGAM protein was evaluated with immunoprecipitation in H1299 and WI-38 cells.
(F) In vitro phosphatase activity against various peptides, phospho-p53 (Ser15), phospho-p53 (Ser20), and phospho-p53 (Ser15/Ser20) was determined using recombinant PGAM1, PGAM2, or λPPase. The peptide sequences of phospho-p53 are shown in Figure S5F. The amount of released phosphate from phosphopeptides was monitored.
∗p < 0.05 and ∗∗p < 0.005, Dunnett's multiple comparison test. Data represent the mean ± SEM.
See also Figure S5.
p53 phosphorylation at serine 23 after DNA damage and oncogenic stress is mediated either by Chk1 or Chk2, checkpoint serine/threonine kinases, followed by p53 stabilization and activation (Shieh et al., 2000). In cells ectopically expressing PGAM, total protein levels and phosphorylation status of Chk1 and Chk2 (serine 345 and threonine 68, respectively) (Ahn et al., 2000; Zhao and Piwnica-Worms, 2001) were comparable with control cells (Figure 3B). However, we observed that both PGAM1 and PGAM2 physically interacted with Chk1, but not with Chk2, in p53−/− MEFs, as shown with an immunoprecipitation assay (Figures 3C and S5C). To identify the Chk1-interacting domain in PGAM, a series of deletions in PGAM1- and PGAM2-FLAG were assessed for their ability to bind Chk1 (Figures S5D and 3D). Chk1-myc-his efficiently co-immunoprecipitated with full-length PGAM1- and 2-FLAG (T1) and the N-terminal one-third of PGAM (TN) but not with the other central (T4) or C-terminal fragments (TC). Consistently, endogenous PGAM protein co-immunoprecipitated with endogenous Chk1 protein in H1299 cancer cells but not in human standard cells WI-38 (Figure 3E).
Although PGAM has been shown to function as a phosphatase for metabolite (White and Fothergill-Gilmore, 1992), it is unclear whether PGAM possesses activity as a protein phosphatase. We assessed the potential of PGAM to display protein phosphatase activity. Recombinant PGAM1 or PGAM2 proteins with intact PGAM activity were prepared from Escherichia coli (Figure S5E). None of the recombinant PGAM proteins displayed any phosphatase activity against phospho-Ser/Thr peptides as a substrate, at any concentrations (125, 250, or 500 ng) or in any pH conditions tested (pH 6.2, 7.2, 8.2, and 9.2), whereas λPPase exhibited clear phosphatase activity (Figures S5F–S5H). In addition, recombinant PGAM proteins exhibited no phosphatase activity in vitro against phosphopeptides that mimicked phospho-p53 (Ser15, Ser20, or Ser15/Ser20; Figures 3F and S5F). Taken together, these data imply that PGAM interacts with and impairs p53 phosphorylation by Chk1, although PGAM is less likely to function as a protein phosphatase.
PGAM and Chk1 Cooperate to Regulate Glycolytic mRNAs
We noticed that the interaction of PGAM with Chk1 in p53−/− MEFs was enhanced under TPA stress (Figures 4A and S6A), which upregulates glycolytic mRNAs during in vivo chemical tumorigenesis protocol (Figure 1E). To investigate the physiological significance of the interaction between PGAM and Chk1, we evaluated the impact of Chk1 on glycolytic mRNA profiles in vitro. We observed that Chk1 knockdown in the p53-null cell line H1299 was followed by a significant reduction in glucose consumption, glycolytic flux, and lactate production (Figures 4B–4D). Significant reductions in glycolytic mRNAs and proteins were also observed in these cells (Figures 4E and 4F). Treatment with the Chk1 kinase inhibitor UCN01, but not that with the Chk2 kinase inhibitor (Chk2-inhibitor II), induced the downregulation of glycolytic flux and glycolytic mRNAs in H1299 cells (Figures S6B and S6C). Interestingly, although a low concentration of UCN01 (50 nM) did not affect glycolytic mRNA levels in wild-type MEFs, the downregulation of glycolytic mRNAs was observed in Pgam1+/− MEFs (Figure 4G), which displayed the same glycolytic profiles and proliferative capacity as those in wild-type MEFs under standard culture condition (Figures S6D–S6G). These data support the notion that PGAM and Chk1 cooperatively affect glycolytic mRNA profiles.
Figure 4.

Cooperation between PGAM and Chk1 Regulates Glycolytic mRNAs
(A) The impact of TPA treatment on the interaction between PGAM1 and Chk1. p53−/− MEFs with expression of Chk1-myc-his and PGAM1-FLAG were treated with or without 0.1 μg/mL TPA for 48 h, followed by treatment with MG132 for 3 h. The cell lysates were immunoprecipitated with an anti-FLAG antibody.
(B–F) Impact of Chk1 inactivation on glycolytic profile in Chk1 siRNA-transfected H1299 cells (n = 3). Glucose consumption, glycolytic flux, and lactate production were shown (B–D). The mRNA and protein levels of glycolytic enzymes were evaluated in indicated cells (E and F). Intensity of immunoblotting bands was normalized to that of actin. Relative values were shown, compared with scrambled RNA-transfected H1299 cells.
(G) Comparison of glycolytic mRNAs between WT (n = 3) and Pgam1+/− MEFs (n = 3) with or without mild treatment of UCN01, a Chk1 kinase inhibitor at a very low UCN01 concentration (50 nM) that exhibits minimal effect in WT MEFs. Data are relative to WT MEFs, respectively.
∗p < 0.05 and ∗∗p < 0.005, Student's t test.
See also Figure S6.
Oncogenic Ras Pathway Is Required for the PGAM-Chk1 Interaction
To evaluate the clinical relevance of PGAM-Chk1 cooperation, the impact of PGAM/Chk1 axis on prognosis of patients with cancer was evaluated. According to the database of non-small cell lung cancer (NSCLC; jacob-00182-MSK) (Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma, et al., 2008) in PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) (Mizuno et al., 2009), we divided 104 patients with NSCLC into four groups in terms of the abundance of PGAM1 and Chk1 in cancers; Low-PGAM1 + Low-Chk1 (n = 23), Low-PGAM1 + High-Chk1 (n = 12), High-PGAM1 + Low-Chk1 (n = 29), and High-PGAM1 + High-Chk1 (n = 40) (Figure S7A). We noticed that the prognostic values were most significantly declined in High-PGAM1 + High-Chk1 group, compared with the others (Figure 5A).
Figure 5.

Oncogenic Ras Enhances PGAM-Chk1 Interaction
(A) Association between the survival and PGAM1/Chk1 levels was analyzed in patients with non-small cell lung cancer (NSCLC) by PrognoScan database. The survivals of 104 patients with NSCLC (Dataset Jacob-00182-MSK) were plotted with Kaplan-Meier curve. Patients with NSCLC were divided into four groups: Low-PGAM1+Low-Chk1 (black; n = 23), Low-PGAM1+High-Chk1 (blue; n = 12), High-PGAM1+Low-Chk1 (green; n = 29), and High-PGAM1+High-Chk1 (orange; n = 40). ∗p < 0.05 and ∗∗p < 0.005, Log rank test.
(B) Summary of glycolytic mRNA downregulation by PGAM1-knockdown in 15 cancer cell lines tested. Original data of glycolytic mRNA expression were shown in Figure S7B. (−), <20% reduction; (+), reduction from 20 to 40%; (++), >40% reduction. ∗p < 0.05 and ∗∗p < 0.005, Student's t test.
(C) Summary of correlation coefficients between Chk1 and glycolytic genes, which were evaluated in dataset of 26 NSCLC cell lines. All data were obtained from DBKERO. NSCLC cell lines were classified into two subgroups according to gene aberrations of p53 and Ras pathways as shown in Table S2.
(D) The impact of Ras-G12V expression on PGAM-Chk1 binding. His-tagged protein pull-down assay was performed in indicated cells. PGAM1- or PGAM2-FLAG proteins were detected in left or right panel, respectively.
(E–H) The impact of MEK and RSK inhibition against PGAM-Chk1 interaction. H1299 cells were treated with either U126 (MEK inhibitor) or BI-D1870 (RSK inhibitor). Immunoprecipitation assay was performed to evaluate the interaction between PGAM and Chk1 (E and F). Phosphorylation of Thr202/Tyr204 on ERK1/2 and Ser112 on Bad, which is downstream target of MEK/RSK pathway, was analyzed for assessment of U126 and BI-D1870, respectively. Glycolytic flux was measured in indicated cells (G and H).
See also Figures S7 and S8.
Then, we addressed the question of whether the PGAM/Chk1 axis is involved in any cancer-related genetic events. In addition to H1299 and HSC-1 (Figures 2E and S3H), we evaluated the impact of PGAM1 siRNA among thirteen other cancer cells, including skin, breast, colon, bone, and liver cancer cell lines. Among these 15 in total, four cancer cell lines (H1299, HSC-1, SW480, and DLD-1) displayed the most prominent changes in glycolytic mRNAs (ALDO1, TPI, GAPDH, and ENO1) following PGAM1 knockdown (Figures 5B and S7B). For the clinical practice of precision medicine, subsets of oncogene and tumor suppressors are tested to select tailor-made therapy for individuals; for example, 50 cancer-related genes are listed according to Tsongalis et al. (2014) (Table S1). We surveyed 15 cancer cell lines we tested, regarding the mutations in these 50 cancer-related genes according to previous studies (Cancer Genome Atlas Research, 2008; Fujii et al., 1995; Hori et al., 2009; Klijn et al., 2015; Oliner et al., 1992). We noticed that aberrations both in the p53 and Ras pathways were the most common genetic features, shared in these four cell lines (H1299, HSC-1, SW480, and DLD-1) displaying significant glycolytic decline by PGAM1 knockdown (Table S1). As patients with NSCLC with High-PGAM1 + High-Chk1 were classified in low-prognosis subgroup (Figure 5A), we also analyzed the PGAM/Chk1 axis in 26 human lung adenocarcinoma cells. Based on the database of Kashiwa Encyclopedia for human genome mutations in Regulatory regions and their Omics contexts (DBKERO; http://kero.hgc.jp/) (Suzuki et al., 2015), these 26 cells are classified into two subgroups; cells with genetic aberrations either in the p53 or Ras pathway (n = 14) and those with aberrations in both (n = 12) (Table S2). Then, we analyzed Pearson correlation between the levels of Chk1 and glycolytic enzymes. Correlation coefficient between Chk1 and most glycolytic enzymes (HK2, GPI, ALDOA, TPI, GAPDH, PGK1, PGAM1, ENO1, and PKM1) is high (over 0.4) in the subgroup with aberrations in both, whereas only that between Chk1 and TPI is high in the former subgroup (Figures 5C, S7C and S7D).
Consistent with these implications on the significance of Ras pathway against PGAM/Chk1 axis, we noticed that PGAM-Chk1 binding is enhanced by the expression of oncogenic Ras-G12V in p53−/− MEFs (Figure 5D). To gain the molecular insight on the impact of Ras pathway against PGAM-Chk1 interaction, we applied several inhibitors for the downstream kinases of canonical Ras pathway in H1299; U126 (MEK inhibitor), BI-D1870 (inhibitor for ribosomal S6 kinase [RSK], a downstream kinase of MEK), and Triciribine (Akt kinase inhibitor). Although Akt inhibition unlikely affected the PGAM-Chk1 interaction (Figure S8A), the inactivation of MEK/RSK interferes the binding between PGAM-Chk1 (Figures 5E and 5F), followed by significant reduction in glycolytic flux (Figures 5G and 5H). Thus, oncogenic Ras signals via MEK/RSK pathway are much involved in PGAM-Chk1 interaction.
Nutlin-3a Interferes with the PGAM-Chk1 Interaction
Next, we addressed the question of whether interference with the PGAM-Chk1 interaction affects glycolysis. Nutlin-3a is a compound originally designed to inhibit the physical binding between p53 and Mdm2 (Figure S8B) (Vassilev et al., 2004). We examined the effect of Nutlin-3a on PGAM protein, as PGAM also binds Mdm2 under senescence-inducing stress (Mikawa et al., 2014). Nutlin-3a did not affect the enzymatic activity of recombinant PGAM in vitro, even with the increased concentrations tested (Figure S8C). Although PGAM-Mdm2 binding was not affected by Nutlin-3a (Figures 6A and S8D), Nutlin-3a treatment strikingly abolished PGAM-Chk1 binding in p53−/− MEFs (Figures 6B and S8E). We also treated p53-null H1299 cells with Nutlin-3a to disrupt PGAM-Chk1 binding to evaluate the effect on glycolytic regulation (Figure S8F). Indeed, glycolytic flux and glycolytic mRNAs in H1299 cells were downregulated by Nutlin-3a in a dose-dependent manner (Figures 6C and 6D).
Figure 6.

Cooperation of PGAM and Chk1 for Glycolytic Regulation Is Abolished by Interfering with Their Binding
(A and B) Immunoprecipitation assay or His-tagged protein pull-down assay was performed to evaluate the effect of Nutlin-3a on the interaction between PGAM and Mdm2 (A) or PGAM-Chk1 binding (B). p53−/− MEFs expressing indicated vectors were prepared. Cells were exposed to 5 or 10 μM Nutlin-3a for 48 h. After treatment with 20 μM MG132 for 3 h, cell lysates were collected. PGAM2-FLAG proteins were immunoprecipitated by anti-FLAG antibody (A) or Chk1-myc-his proteins were precipitated by Ni-NTA beads (B).
(C and D) The effect of Nutlin-3a on glycolytic profile. Comparison of glycolytic flux (C) and glycolytic mRNAs (D) among H1299 cells treated with indicated concentration of Nutlin-3a for 48 h (n = 3). Data are relative to control cells. ∗p < 0.05 and ∗∗p < 0.005, Dunnett's multiple comparison test.
(E) Diagram of the consensus motif of Nutlin-3-target proteins in the amino terminus of PGAM and p53. Consensus motifs, [L/I/V/M]-[W/Y/F]-x-x-[L/I/V/W], are indicated in blue and green. The alignments of the conserved consensus sequence in the amino terminus of PGAM1, 2, and p53 (human and mouse) are also presented. Orange bar, a site for Chk1 binding identified in Figure 3D. Purple bar, site for Mdm2 binding.
(F) PGAM enzymatic activity was measured in Chk1-expressing p53−/− MEFs retrovirally infected with the indicated vectors (empty vector, PGAM2-WT-FLAG, PGAM2-W68A-FLAG, and PGAM2-W78A-FLAG) (n = 3 each; upper panel). Protein levels for various versions of PGAM2-FLAG were shown by western blotting (lower panels). ∗∗p < 0.005, Dunnett's multiple comparison test.
(G) The Chk1-binding activity of PGAM2 mutants (W68A and W78A) compared with that of PGAM2-WT was analyzed with a His-tagged protein pull-down assay. Data represent the mean ± SEM.
See also Figure S8.
Recently, the consensus sequence ([L/I/V/M]-[W/Y/F]-x-x-[L/I/V/W]) for Nutlin-3a responsive proteins, including p53, was determined by proteomic analysis (Nicholson et al., 2014). We noticed that two repeats of this consensus motif are located and conserved in the N terminus of human and mouse PGAM, which overlap with the Chk1-interacting domains (Figures 3D and 6E). PGAM mutants (W68A and W78A) with amino acid substitutions in these two repeats of the consensus sequence were generated by PCR mutagenesis (Figure 6E). Although the enzymatic activity and protein levels were maintained in both W68A and W78A mutants (Figures 6F and S8G), PGAM-Chk1 binding was largely impaired (Figures 6G and S8H). Thus, Nutlin-3a interferes with the PGAM-Chk1 interaction and the maintenance of glycolytic features but not with the enzymatic activity of PGAM.
Physical Interaction between PGAM and Chk1 Is Essential for Both Maintenance of Glycolysis and Proliferative Potential in Cancerous Cells
As PGAM mutants for Nutlin consensus motif (W68A and W78A) attenuate the interaction with Chk1 in p53−/− MEFs with or without Ras-G12V expression (Figures S8I and S8J), we evaluated the physiological impact of these mutants (W68A and W78A) in the conditions with or without Ras-G12V. For this purpose, Chk1-expressing p53−/− MEFs with or without Ras-G12V were prepared, followed by retroviral infection with various versions of PGAM. In the absence of Ras-G12V, co-expression of Chk1 with WT-, W68A-, or W78A-PGAM resulted in similar profiles for overall glycolytic mRNAs and proliferative capacity (Figures S8K, S9D, 7F, S8L, S9F, and S9G). In sharp contrast, in the presence of Ras-G12V, ectopic expression of PGAM-WT in Chk1-expressing p53−/− MEFs increased the glycolytic parameters (glucose consumption, glycolytic flux, and lactate production), compared with the vector control (Figures 7A–7C and S9A–S9C). Consistently, the expression of glycolytic mRNAs and proteins were upregulated in these cells (Figures 7D, 7E, S9D, and S9E). In the same genetic background, however, PGAM-W68A and W78A mutants displayed no such enhancement in glycolytic profiles (Figures 7A–7E and S9A–S9E).
Figure 7.

W68A and W78A in PGAM, Binding-Deficient Mutations with Intact Enzyme Activity, Abolished the Enhancement of Glycolytic Profiles and Proliferative Capacity in the Presence of Ras-G12V.
(A–E) Comparison of glycolytic profiles among the cells expressing PGAM2 variants (WT, W68A, or W78A) in the presence of Ras-G12V. Glucose consumption (A), glucose flux (B), and lactate production (C) were analyzed among indicated cells on the common genetic background of p53−/− with Ras-G12V and Chk1 (n = 3). The mRNA and protein levels of glycolytic enzymes were also evaluated in indicated cells (D and E). Intensity of immunoblotting bands was normalized to that of actin. Data are relative to controls expressing the empty vector.
(F) The effect of indicated PGAM2 variants on cell growth was assessed by crystal-violet staining in Chk1-expressing p53−/− MEFs with or without Ras-G12V.
(G and H) In vivo tumor growth assay in nude mice. p53−/− MEFs, expressing Ras-G12V, Chk1, and the indicated versions of PGAM2, were injected subcutaneously into nude mice (n = 6). (G) Representative images of tumors. (H) Measurement of tumor volumes are shown. ∗p < 0.05 and ∗∗p < 0.005, Dunnett's multiple comparison test. Data represent the mean ± SEM.
(I) Schematic model on the significance of PGAM-Chk1 interaction in cancerous glycolysis. Oncogenic Ras augmented PGAM-Chk1 interaction, followed by enhanced glycolysis in cancer. Genetic or chemical ablation of the interaction abrogated such enhancement.
See also Figures S8–S11.
Strikingly, in the presence of Ras-G12V, both the in vitro proliferative capacity and in vivo tumor growth of PGAM-WT overexpressing cells were much enhanced than control (Figures 7F–7H, S8L, and S9F–S9I). In the same genetic background, however, the cells with binding-deficient mutations (W68A and W78A) showed the similar proliferative potentials in vitro and in vivo to those in control (Figures 7F–7H, S8L, and S9F–S9I). Next, we addressed the question whether W68A or W78A mutation abolished PGAM-Chk1 binding through modulating PGAM dimerization. For this purpose, NanoLuc Binary Technology (NanoBiT) system (Promega) is applied, which enables us to quantify protein-protein interaction between the proteins with large and small NanoBiT tag (Figure S10A) (Dixon et al., 2016). We purified recombinant PGAM1, 2-WT, W68A, and W78A proteins with NanoBiT tags from the extract of bacteria (Figure S10B). We observed these recombinant PGAM proteins displayed similar intact enzymatic activity (Figure S10C). Consistently, the dimerization of either PGAM versions displayed the similar values of Kd (dissociation constant), shown by the saturation curves of NanoBiT detection; 62.5 to 67.1 nM for various versions of PGAM1 and 60.6 to 65.9 nM for those of PGAM2 (Figure S10D). Thus, PGAM-W68 and W78A did not affect dimerization and enzymatic activity of PGAM. However, we noticed that monomer forms of PGAM were much abundant in H1299 cancer cells, compared with those in standard WI-38 cells (Figure S10E). These findings support the possibility that nonenzymatic PGAM is more accumulated in cancer cells.
Finally, we evaluated the impact of enzymatic PGAM mutants in the same setting. As the patients with PGAM deficiency were previously reported (Naini et al., 2009), the point mutation R90W in the patients abolished PGAM enzymatic activity (Tsujino et al., 1995). We generated cDNA clone of PGAM1 and 2 with R90W mutation. We observed that such mutation in both PGAM isoforms abolished PGAM activity (Figures S11A and S11B) but still retains proliferative advantage compared with vector control, in Chk1 and Ras-G12V-expressing p53−/− MEFs (Figures S11C and S11D).
Thus, in accordance with the observation of chemically interfering by Nutlin, the genetical interference of PGAM-Chk1 binding with intact PGAM activity (W68A, W78A mutations) abolished cancerous proliferation and enhanced glycolytic profiles, in the presence of oncogenic Ras (Figure 7I).
Discussion
Here we demonstrated a previously unappreciated regulatory mechanism for cancerous glycolysis via the PGAM-Chk1 interaction. Genetic and chemical dissection for its interaction disclosed a “non-enzymatic” role for PGAM, especially under oncogenic Ras expressing conditions. Thus, we unveiled the noteworthy impact of interfering with the PGAM/Chk1 axis in the cancerous glycolysis.
First, our data indicate the evident genetic link between PGAM and the global enhancement of glycolytic profiles frequently observed in cancerous cells. We found a molecular interaction between PGAM and Chk1 as notable glycolytic regulator, which is operating in cancer cells, not in standard cells. Moreover, in addition to the data on the prognosis of patients with cancer, several close correlations between Chk1/PGAM and glycolytic enzymes identified oncogenic Ras mutations, which are highly prevalent both in human cancers and DMBA-treated tissues (Karnoub and Weinberg, 2008), as enhancer of PGAM-Chk1 interaction. Although the other research has also emphasized the distinct impact of PGAM in cancer metabolism (Evans et al., 2005; Vander Heiden et al., 2010), we identified the essential effect of PGAM on the boost of glycolysis in the cancerous conditions.
Second, our study also disclosed the key metabolic role of Chk1 for glycolytic regulation in cancer. Chk1, initially identified as a checkpoint kinase, was known to induce cell-cycle arrest after its activation during DNA damage or senescence-inducing stress (Sancar et al., 2004). Other studies, however, have suggested the opposing notions that Chk1 would be the critical driver of cellular proliferation in ES cells and cancer cells in vitro (Liu et al., 2000; Shimada et al., 2008) and in carcinogenic events in vivo (Tho et al., 2012). Interestingly, Chk1 was reported to be regulated by RSK, the downstream kinase of Ras pathway, during cell cycle progression (Li et al., 2012). RSK is upregulated in many types of cancer including lung cancer and promotes cancerous proliferation (Houles and Roux, 2018), whose impacts on the Warburg effect were scarcely reported. As our findings suggest that oncogenic Ras/RSK activation are closely involved in PGAM-Chk1 binding, the opposing roles of Chk1 with respect to cell proliferation would be cellular-context dependent, including the modulation of Ras/RSK pathway.
Third, our findings in the binding-deficient mutants of PGAM implicate that regardless of its enzymatic activity, PGAM plays a critical role in the regulation of glycolytic upregulation. Although neither the phosphatase nor enzymatic activity of PGAM was found to be required for the functional cooperation with Chk1, the physical interaction between PGAM and Chk1 is essential for glycolytic upregulation. These findings are rather consistent with the previous notion that the multifaceted roles of some enzymes are independent of their catalytic activity (Mdm2 for Poyurovsky et al. [2003]; PP2A for Takemoto et al. [2009]). It is possible that in cancerous cells with accumulation of PGAM proteins, “nonenzymatic” PGAM cooperates with the Chk1 pathway to boost glycolysis, whose activation is required for the survival of cancers bearing oncogenic Ras mutation (Dietlein et al., 2015). A plausible explanation for the Warburg effect has been proposed as follows: besides hypoxic adaptation, it might not only enable cancer cells to meet demands both for energy and biosynthesis (Lunt and Vander Heiden, 2011) but also protect them from oxidative damages (Kondoh et al., 2007). Interestingly, the recent studies suggested that PGAM interacts with actin proteins as nonenzymatic role, to facilitate cancer motility (Huang et al., 2019; Zhang et al., 2017). In addition to the above, the identification of nonenzymatic role for PGAM suggested that enhanced glycolysis might support proliferative capacity of rapidly dividing cells through the cooperation with Chk1.
Finally, our analysis also supported the clinical significance of PGAM-Chk1 interaction. Nutlin-3a, an anticancer drug as inhibitor of p53-Mdm2 binding (Burgess et al., 2016), interfered with the PGAM-Chk1 interaction in a p53-independent manner but not with PGAM enzymatic activity. Thus, Nutlin-3a likely also functions as a glycolytic modulator through interfering with the PGAM-Chk1 interaction. Since the discovery of the Warburg effect, cancer metabolism has long been assumed to be a potential anticancer therapeutic target. However, enzymatic inhibition of glycolysis has failed in clinical trials as anticancer drugs owing to their profound adverse effects (Granchi and Minutolo, 2012). Therefore, targeting the binding between PGAM and Chk1 would represent a potential candidate strategy for anti-cancer therapy.
Limitation of the Study
Skin carcinogenesis protocol was performed in Pgam2-Tg mice, whereas in vivo tumor formation assay in nude mice was performed in PGAM1- or PGAM2-expressing cells. In the future, it would be worthy to explore the PGAM/Chk1 axis in human clinical samples.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr.Hiroshi Kondoh, hkondoh@kuhp.kyoto-u.ac.jp.
Materials Availability
All unique reagents generated in this study are available on reasonable request with a completed Materials Transfer Agreement.
Data and Code Availability
All data are available in the main text or the Supplemental Information.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank all the staff of Geriatric unit and of the Department of Diabetes, Endocrinology and Nutrition at Kyoto University for their cooperation; Dr. Kayoko Maehara for the pBabe-puro-Ras-G12V vector; Dr. Toshio Kitamura for the PLAT-A cells. This work was supported in part by grants from the Japan Society for the Promotion of Science, Japan (Grants No. 26310103 and No. 15K19283) and by the Japan Agency for Medical Research and Development (AMED), Japan, Core Research for Evolutional Science and Technology, Japan (CREST JP17gm0610002h0306). The animal experiments and radio-isotope experiments were performed at the Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, and in the Kyoto University Hospital Radioisotopes Research Laboratory, respectively.
Author Contributions
T.M., E.S., M.S., K.I., T.I., H. Kanda, K.T., and M.E.L. researched and analyzed the data. N.I. and M.Y. provided technical support and contributed to discussions. H. Kondoh wrote the paper and supervised this study. All the authors read and approved the manuscript.
Declaration of Interests
T.M. and H. Kondoh have submitted relevant patent on the impact of PGAM-Chk1 for glycolysis.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101306.
Supplemental Information
References
- Ahn J.Y., Schwarz J.K., Piwnica-Worms H., Canman C.E. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- Bensaad K., Vousden K.H. p53: new roles in metabolism. Trends Cell Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Burgess A., Chia K.M., Haupt S., Thomas D., Haupt Y., Lim E. Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimenti M.S., Triggianese P., Conigliaro P., Candi E., Melino G., Perricone R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. 2015;6:e1887. doi: 10.1038/cddis.2015.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C.V., Semenza G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre M., Nuciforo P.G., Bensimon A. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Dietlein F., Kalb B., Jokic M., Noll E.M., Strong A., Tharun L., Ozretic L., Kunstlinger H., Kambartel K., Randerath W.J. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell. 2015;162:146–159. doi: 10.1016/j.cell.2015.05.053. [DOI] [PubMed] [Google Scholar]
- Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. Shedden K., Taylor J.M., Enkemann S.A., Tsao M.S., Yeatman T.J., Gerald W.L., Eschrich S., Jurisica I., Giordano T.J. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat. Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A.S., Schwinn M.K., Hall M.P., Zimmerman K., Otto P., Lubben T.H., Butler B.L., Binkowski B.F., Machleidt T., Kirkland T.A. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016;11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- Durany N., Joseph J., Campo E., Molina R., Carreras J. Phosphoglycerate mutase, 2,3-bisphosphoglycerate phosphatase and enolase activity and isoenzymes in lung, colon and liver carcinomas. Br. J. Cancer. 1997;75:969–977. doi: 10.1038/bjc.1997.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.J., Saghatelian A., Sorensen E.J., Cravatt B.F. Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat. Biotechnol. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]
- Fujii K., Dousaka-Nakajima N., Imamura S. Epidermal growth factor enhancement of HSC-1 human cutaneous squamous carcinoma cell adhesion and migration on type I collagen involves selective up-regulation of alpha 2 beta 1 integrin expression. Exp. Cell Res. 1995;216:261–272. doi: 10.1006/excr.1995.1032. [DOI] [PubMed] [Google Scholar]
- Goyal M.S., Vlassenko A.G., Blazey T.M., Su Y., Couture L.E., Durbin T.J., Bateman R.J., Benzinger T.L., Morris J.C., Raichle M.E. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017;26:353–360.e3. doi: 10.1016/j.cmet.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granchi C., Minutolo F. Anticancer agents that counteract tumor glycolysis. ChemMedChem. 2012;7:1318–1350. doi: 10.1002/cmdc.201200176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T., Zhou L., Elf S., Fan J., Kang H.B., Seo J.H., Shan C., Dai Q., Zhang L., Xie J. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori M., Suzuki K., Udono M.U., Yamauchi M., Mine M., Watanabe M., Kondo S., Hozumi Y. Establishment of ponasterone A-inducible the wild-type p53 protein-expressing clones from HSC-1 cells, cell growth suppression by p53 expression and the suppression mechanism. Arch. Dermatol. Res. 2009;301:631–646. doi: 10.1007/s00403-008-0915-5. [DOI] [PubMed] [Google Scholar]
- Houles T., Roux P.P. Defining the role of the RSK isoforms in cancer. Semin. Cancer Biol. 2018;48:53–61. doi: 10.1016/j.semcancer.2017.04.016. [DOI] [PubMed] [Google Scholar]
- Huang K., Liang Q., Zhou Y., Jiang L.L., Gu W.M., Luo M.Y., Tang Y.B., Wang Y., Lu W., Huang M. A novel allosteric inhibitor of phosphoglycerate mutase 1 suppresses growth and metastasis of non-small-cell lung cancer. Cell Metab. 2019;30:1107–1119.e8. doi: 10.1016/j.cmet.2019.09.014. [DOI] [PubMed] [Google Scholar]
- Karnoub A.E., Weinberg R.A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn C., Durinck S., Stawiski E.W., Haverty P.M., Jiang Z., Liu H., Degenhardt J., Mayba O., Gnad F., Liu J. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015;33:306–312. doi: 10.1038/nbt.3080. [DOI] [PubMed] [Google Scholar]
- Kondoh H., Lleonart M.E., Gil J., Wang J., Degan P., Peters G., Martinez D., Carnero A., Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Kondoh H., Lleonart M.E., Nakashima Y., Yokode M., Tanaka M., Bernard D., Gil J., Beach D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid. Redox Signal. 2007;9:293–299. doi: 10.1089/ars.2006.1467. [DOI] [PubMed] [Google Scholar]
- Koppenol W.H., Bounds P.L., Dang C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Li P., Goto H., Kasahara K., Matsuyama M., Wang Z., Yatabe Y., Kiyono T., Inagaki M. P90 RSK arranges Chk1 in the nucleus for monitoring of genomic integrity during cell proliferation. Mol. Biol. Cell. 2012;23:1582–1592. doi: 10.1091/mbc.E11-10-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Guntuku S., Cui X.S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Mikawa T., Maruyama T., Okamoto K., Nakagama H., Lleonart M.E., Tsusaka T., Hori K., Murakami I., Izumi T., Takaori-Kondo A. Senescence-inducing stress promotes proteolysis of phosphoglycerate mutase via ubiquitin ligase Mdm2. J. Cell Biol. 2014;204:729–745. doi: 10.1083/jcb.201306149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikawa T., ME L.L., Takaori-Kondo A., Inagaki N., Yokode M., Kondoh H. Dysregulated glycolysis as an oncogenic event. Cell Mol. Life Sci. 2015;72:1881–1892. doi: 10.1007/s00018-015-1840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H., Kitada K., Nakai K., Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med. Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naini A., Toscano A., Musumeci O., Vissing J., Akman H.O., DiMauro S. Muscle phosphoglycerate mutase deficiency revisited. Arch. Neurol. 2009;66:394–398. doi: 10.1001/archneurol.2008.584. [DOI] [PubMed] [Google Scholar]
- Nicholson J., Scherl A., Way L., Blackburn E.A., Walkinshaw M.D., Ball K.L., Hupp T.R. A systems wide mass spectrometric based linear motif screen to identify dominant in-vivo interacting proteins for the ubiquitin ligase MDM2. Cell Signal. 2014;26:1243–1257. doi: 10.1016/j.cellsig.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Okuda J., Niizuma S., Shioi T., Kato T., Inuzuka Y., Kawashima T., Tamaki Y., Kawamoto A., Tanada Y., Iwanaga Y. Persistent overexpression of phosphoglycerate mutase, a glycolytic enzyme, modifies energy metabolism and reduces stress resistance of heart in mice. PLoS One. 2013;8:e72173. doi: 10.1371/journal.pone.0072173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner J.D., Kinzler K.W., Meltzer P.S., George D.L., Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- Poyurovsky M.V., Jacq X., Ma C., Karni-Schmidt O., Parker P.J., Chalfie M., Manley J.L., Prives C. Nucleotide binding by the Mdm2 RING domain facilitates Arf-independent Mdm2 nucleolar localization. Mol. Cell. 2003;12:875–887. doi: 10.1016/s1097-2765(03)00400-3. [DOI] [PubMed] [Google Scholar]
- Rodwell V.W., Towne J.C., Grisolia S. The kinetic properties of yeast and muscle phosphoglyceric acid mutase. J. Biol. Chem. 1957;228:875–890. [PubMed] [Google Scholar]
- Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn J., Tamai K., Taya Y., Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Shimada M., Niida H., Zineldeen D.H., Tagami H., Tanaka M., Saito H., Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Wakaguri H., Yamashita R., Kawano S., Tsuchihara K., Sugano S., Suzuki Y., Nakai K. DBTSS as an integrative platform for transcriptome, epigenome and genome sequence variation data. Nucleic Acids Res. 2015;43:D87–D91. doi: 10.1093/nar/gku1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto A., Maeshima K., Ikehara T., Yamaguchi K., Murayama A., Imamura S., Imamoto N., Yokoyama S., Hirano T., Watanabe Y. The chromosomal association of condensin II is regulated by a noncatalytic function of PP2A. Nat. Struct. Mol. Biol. 2009;16:1302–1308. doi: 10.1038/nsmb.1708. [DOI] [PubMed] [Google Scholar]
- Taylor M., Wallhaus T.R., Degrado T.R., Russell D.C., Stanko P., Nickles R.J., Stone C.K. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in Patients with Congestive Heart Failure. J. Nucl. Med. 2001;42:55–62. [PubMed] [Google Scholar]
- Tho L.M., Libertini S., Rampling R., Sansom O., Gillespie D.A. Chk1 is essential for chemical carcinogen-induced mouse skin tumorigenesis. Oncogene. 2012;31:1366–1375. doi: 10.1038/onc.2011.326. [DOI] [PubMed] [Google Scholar]
- Tsongalis G.J., Peterson J.D., de Abreu F.B., Tunkey C.D., Gallagher T.L., Strausbaugh L.D., Wells W.A., Amos C.I. Routine use of the Ion Torrent AmpliSeq cancer Hotspot panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2014;52:707–714. doi: 10.1515/cclm-2013-0883. [DOI] [PubMed] [Google Scholar]
- Tsujino S., Shanske S., Sakoda S., Toscano A., DiMauro S. Molecular genetic studies in muscle phosphoglycerate mutase (PGAM-M) deficiency. Muscle Nerve Suppl. 1995;3:S50–S53. doi: 10.1002/mus.880181412. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Locasale J.W., Swanson K.D., Sharfi H., Heffron G.J., Amador-Noguez D., Christofk H.R., Wagner G., Rabinowitz J.D., Asara J.M. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Wang Y., Cai W.S., Chen L., Wang G. Molecular dynamics simulation reveals how phosphorylation of tyrosine 26 of phosphoglycerate mutase 1 upregulates glycolysis and promotes tumor growth. Oncotarget. 2017;8:12093–12107. doi: 10.18632/oncotarget.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- White M.F., Fothergill-Gilmore L.A. Development of a mutagenesis, expression and purification system for yeast phosphoglycerate mutase. Investigation of the role of active-site His181. Eur. J. Biochem. 1992;207:709–714. doi: 10.1111/j.1432-1033.1992.tb17099.x. [DOI] [PubMed] [Google Scholar]
- Xu Y., Li F., Lv L., Li T., Zhou X., Deng C.X., Guan K.L., Lei Q.Y., Xiong Y. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res. 2014;74:3630–3642. doi: 10.1158/0008-5472.CAN-13-3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Jin N., Sun W., Li X., Liu B., Xie Z., Qu J., Xu J., Yang X., Su Y. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene. 2017;36:2900–2909. doi: 10.1038/onc.2016.446. [DOI] [PubMed] [Google Scholar]
- Zhang J., Yu L., Fu Q., Gao J., Xie Y., Chen J., Zhang P., Liu Q., Zhao S. Mouse phosphoglycerate mutase M and B isozymes: cDNA cloning, enzyme activity assay and mapping. Gene. 2001;264:273–279. doi: 10.1016/s0378-1119(00)00597-7. [DOI] [PubMed] [Google Scholar]
- Zhao H., Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the Supplemental Information.