

Graphical abstract

Keywords: CRISPR-Cas9, Substitution, DNA repair, Non-homologous end joining, High-throughput sequencing
Abstract
CRISPR-Cas9 induces DNA cleavages at desired target sites in a guide RNA-dependent manner; DNA editing occurs through the resulting activity of DNA repair processes including non-homologous end joining (NHEJ), which is dominant in mammalian cells. NHEJ repair frequently causes small insertions and deletions (indels) near DNA cleavage sites but only rarely causes nucleotide substitutions. High-throughput sequencing is the primary means of assessing indel and substitution frequencies in bulk populations of cells in the gene editing field. However, it is difficult to detect bona fide substitutions, which are embedded among experimentally-induced substitution errors, in high-throughput sequencing data. Here, we developed a novel analysis method, named CRISPR-Sub, to statistically detect Cas9-mediated substitutions in high-throughput sequencing data by comparing Mock- and CRISPR-treated samples. We first pinpointed ‘hotspot positions’ in target sequences at which substitution mutations were quantitatively observed much more often (p > 0.001) in CRISPR- versus Mock-treated samples. We refer to the substitution mutations in defined hotspot positions as ‘apparent substitutions’ and ultimately calculated ‘apparent substitution frequencies’ for each target. By examining 51 endogenous target sites in HeLa cells, we found that the average apparent substitution frequency was 0.8% in all queries, that apparent substitutions frequently occur near CRISPR-Cas9 cleavage sites, and that nucleotide conversion showed no meaningful nucleotide preference patterns. Furthermore, we generated NHEJ-inhibited cell lines (LIG4−/−) by knockout of the gene encoding ligase IV and found that the apparent substitution frequencies were significantly decreased in LIG4−/− cells, strongly suggesting that DNA substitutions are generated by the NHEJ pathway.
1. Introduction
The CRISPR (clustered regularly interspaced short palindromic repeat)-Cas (CRISPR associated) system [1], [2], an adaptive immune response in bacteria and archaea, facilitates RNA-guided site-specific DNA cleavage in various organisms [3], [4]. DNA double-strand breaks (DSBs) induced by CRISPR endonucleases are typically repaired by a cell’s own repair processes [5], such as the homology-directed repair (HDR) pathway, the non-homologous end joining (NHEJ) pathway, or an alternative KU-independent process such as the microhomology-mediated end joining (MMEJ) pathway [6], [7], [8], [9]. Among these, the error-free HDR pathway, in the presence of an inserting donor DNA, is useful for generating targeted gene knock-ins or gene corrections [10], [11], [12], whereas the error-prone NHEJ pathway is frequently accompanied with small insertions and deletions (indels) and MMEJ causes DNA sequence-dependent deletions, both resulting in gene disruption at desired target sites [13], [14], [15], [16]. Among various CRISPR effectors [17], type II Cas9 [18] derived from Streptococcus pyogenes (SpCas9) is the most widely used due to its high efficacy and simple DNA recognition sequences (5′-NGG-3′), which are also called protospacer adjacent motifs (PAMs) [19], [20]. CRISPR-Cas9 has been utilized in many research areas to achieve goals such as improving plants [21], [22], [23], detecting and curing diseases [24], [25], [26], and revealing genes’ functions [27], [28].
Until now, most gene editing studies performed in the absence of an HDR donor have focused on the measurement of indel mutation frequencies at target sites and utilized the mutations for gene disruption or rescue [29], [30], [31]. But a few studies have reported the presence of single nucleotide substitutions near DNA cleavage sites after CRISPR-Cas9 treatment [15], [32]. For example, Wang et al. reported that expression of Cas9 with single guide RNAs (sgRNAs) in human KBM-7 cells ultimately induced various types of DNA mutations; Sanger sequencing of single colony transformants revealed an average of 91% deletions, 6% insertions, and 3% substitutions. However, these substitution mutations caused by CRISPR-Cas9 have not attracted serious attention and are typically thought to be an exception. Furthermore, it is difficult to measure the frequency of substitution mutations accurately, especially in a bulk population of cells. When gene editing is assessed in such populations in high-throughput manner using next generation sequencing (NGS) technologies, DNA substitutions are normally tainted by substitution errors, which are mainly derived from the DNA amplification process [33] and DNA sequencers [34], [35]. Therefore, although substitution mutations have been reported and are expected to exist after CRISPR-Cas9 treatment, most researchers have focused solely on the analysis of indel patterns. Previously, computational tools such as CRISPResso2 [36] and Amplican [37] were developed to calculate substitution frequency in addition to insertion and deletion frequencies, but they are not substitution-dedicated tools for systematically evaluation of substitution patterns and frequencies from the high-throughput sequencing data. In this study, we developed a novel analysis method, CRISPR-Sub, to measure the frequency of Cas9-mediated substitutions by comparing high-throughput sequencing data from Mock- and CRISPR-treated samples. For a massive evaluation of DNA substitutions, we employed high-throughput sequencing using an Illumina Miniseq platform.
2. Materials and methods
2.1. Generation of sgRNA-encoding plasmids
Each oligo including sgRNA was purchased from Macrogen (South Korea). Oligos were heated and cooled down to make double-strand oligos. pRG2 sgRNA expression vector was cleaved with Bsa1 and then ligated with double-strand oligos. Target sequences were selected using Cas-OFFinder [38] to have one on-target and no potential off-targets having mismatches up to 2 in the genome for each sgRNA. The list of oligomers for target sequences are in Supplementary Table S1.
2.2. Cell culture and transfection
HeLa (ATCC®, CCL-2™) and HEK293T (ATCC®, CCL-3216™) cells were cultivated in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 units/mL streptomycin. Prior to transfection, 1 × 105 cells from each cell line were cultivated overnight in 24-well plates. For CRISPR-treated cells, 750 ng of SpCas9 expression plasmid and 250 ng of sgRNA expression plasmid were mixed with 100 μl of Opti-MEM medium and 2 μl of Lipofectamine 2000 and incubated for 20 min at room temperature. For Mock-treated cells, the mixture contained 750 ng of SpCas9 expression plasmid only without sgRNA expression plasmid. The mixtures were then gently added to the wells containing cells. After 3 days, transfected cells were detached using 0.05% trypsin-EDTA and genomic DNA was extracted using NucleoSpin Tissue (MARCHEREY-NAGEL & Co. KG).
2.3. LIG4 knockout cell lines
1 × 105 HeLa cells were transfected with Cas9 expression plasmid (750 ng) and sgRNA expression plasmid (250 ng) expressing an sgRNA targeting exon 1 in the DNA ligase IV gene LIG4 using Lipofectamine 2000. After 3 days, the transfected cells were detached from the wells and the SpCas9 efficiency was determined in a portion of the cells by deep sequencing. The remaining cells were distributed in 96-well plates, at a density of one cell per well. The presence of LIG4 mutations in each cell line was determined; cell lines with confirmed mutations were used in later experiments.
2.4. Targeted deep sequencing
The targeted region (200–270 nucleotides in length) of genomic DNA was amplified using Phusion polymerase in three separate reactions. The PCR products were subjected to paired-end read sequencing using Illumina Mini-seq. The number of total reads is required >20,000 for verification (Supplementary Fig. 1). The unjoined NGS results were joined using Fastq-join tool [39] (https://github.com/brwnj/fastq-join). Insertion and deletion frequencies were analyzed by Cas-Analyzer [40] (http://www.rgenome.net/cas-analyzer/) to confirm the activity of each Cas9/sgRNA.
2.5. Off-line tool for detecting CRISPR-induced substitutions
The off-line version of CRISPR-Sub was developed using Python 3.6. The program extracts the relevant sequence information from the NGS results and removes the sequences with insertion or deletion mutations by comparing their length to that of the wild-type sequence. From the NGS outcomes, we calculated a substitution fold (Sub-fold) value and a frequency of count at each position. When Sub-score of Mock-treated data and/or CRISPR-treated data is 0 at one position, the case will not be included to the dataset for calculating Sub-folds and the cut-off threshold. We used a Gaussian function for a curve fitting to the frequency count data and calculated the threshold for Sub-fold values with P-value under 0.001, using the function “scipy.optimize.curve_fit” and “scipy.stats.norm” in SciPy module (https://www.scipy.org/). After counting the number of CRISPR-induced substitution mutations, the program creates a results file using the xlsxwriter module (https://xlsxwriter.readthedocs.io/).
2.6. Web-based tool for detecting CRISPR-induced substitutions
CRISPR-Sub, a web-based tool for detecting CRISPR-induced substitutions, was developed using the backend program Django2.2 (https://www.djangoproject.com/) and Bootstrap library (https://getbootstrap.com/). Gzipped files are decompressed by the JavaScript library pako (http://nodeca.github.io/pako). Almost all sequence analysis occurs at the user-client site by JavaScript, whereas curve fitting by the SciPy module (https://www.scipy.org/) occurs at the server. Resulting graphs are visualized using Plotly.js (https://plot.ly/javascript/). All sequences are aligned by EMBOSS needle [41].
3. Results
3.1. Strategy for calculating the frequency of apparent CRISPR-induced substitutions in a bulk population of cells
In high-throughput sequencing data from cell populations, bona fide substitution mutations caused by CRISPR-Cas9 are usually embedded in a set of false-positive substitution errors derived from NGS-related processes. We hypothesized that the false-positive substitutions would occur uniformly within sequenced regions, whereas the bona fide CRISPR-mediated substitutions would be abundant near DNA cleavage sites. Therefore, we expected to measure the total frequency of bona fide substitutions statistically by comparing CRISPR-treated sample data against Mock-treated sample data as a negative control, although it is almost impossible to distinguish whether any given substitution is CRISPR-derived or a false positive.
To concretize our initial idea, we developed a novel analysis platform, named CRISPR-Sub, for NGS data as follows (Fig. 1). (i) For NGS outcomes, we first aligned all sequencing queries to a reference sequence (wild type; WT) and classified them into three different groups: WT-length, insertion, or deletion according to their lengths. It is of note that the WT-length group includes DNA sequences with substitutions in addition to WT sequences. (ii) For queries in the WT-length group only, we counted the number of mismatched nucleotides at each position in the entire DNA sequence. Then, the substitution frequency (Sub-score) at each position was determined by dividing the total number of substitutions at each position by the total number of WT-length queries. (iii) We repeated the above steps for CRISPR-treated and Mock-treated samples, and ultimately obtained Sub-scores at each position for both. In this step, bona fide and false-positive substitutions are sometimes hard to distinguish because of substitution noise. However, dividing the Sub-scores of the CRISPR-treated data by those of the Mock-treated data to obtain their fold difference (Sub-fold value) should reveal positions containing bona fide substitutions, because they would have higher Sub-fold values than other positions. If there is a natural single-nucleotide variation (SNV) in the DNA queries, it would also fade out in this step because both CRISPR-treated and Mock-treated sequencing data would contain the SNV in common. Thus, CRISPR-Sub analysis of data from a bulk population of cells allows us to pinpoint hotspot positions at which apparent CRISPR-induced substitutions (hereafter, apparent substitutions) are frequently observed, as well as to identify apparent substitutions at hotspot positions.
Fig. 1.
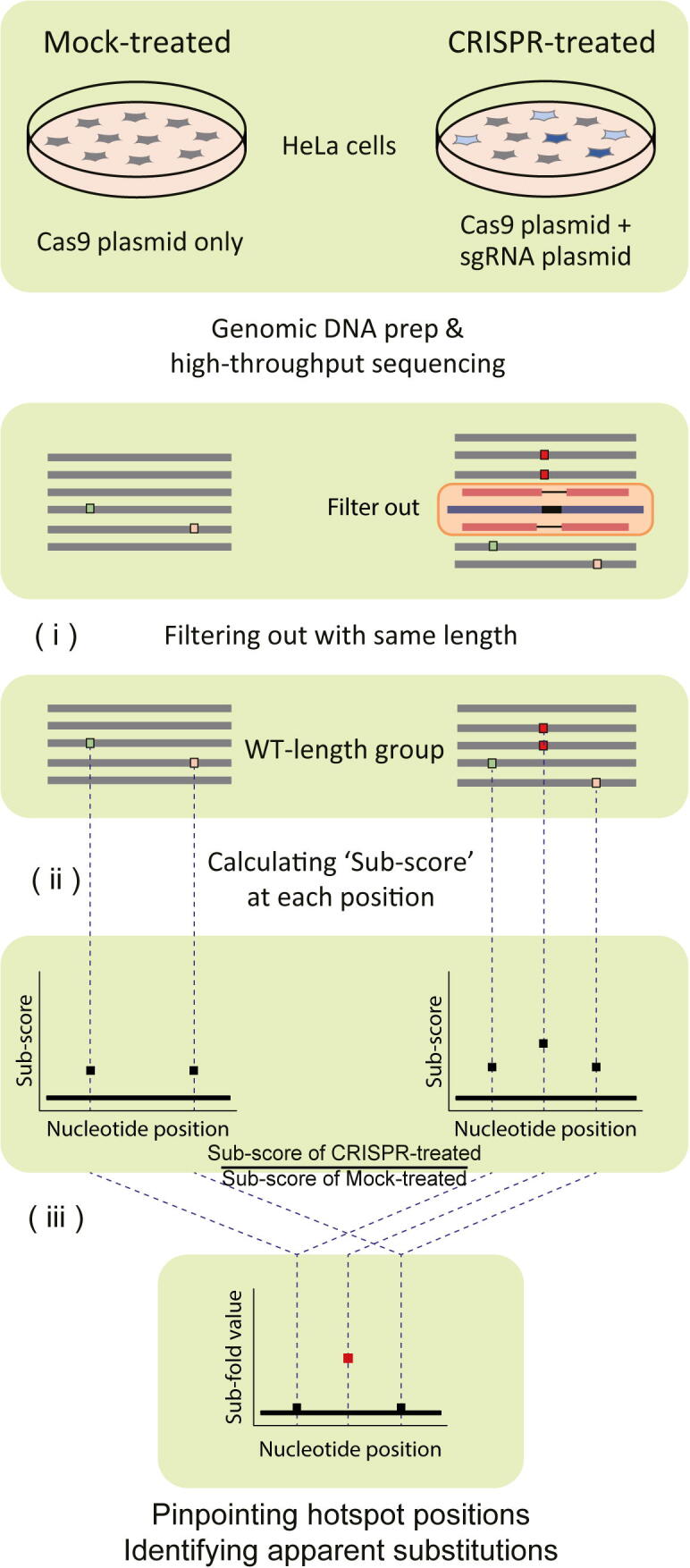
Scheme of the CRISPR-Sub analysis platform. Mock-treated Hela cells are treated only with a plasmid encoding SpCas9, whereas CRISPR-treated Hela cells are treated with plasmids encoding SpCas9 and sgRNA. Genomic DNA is extracted from both Mock- and CRISPR-treated cells, amplified by targeted deep-sequencing, and analyzed by NGS. Sequences in the WT-length group are collected by filtering out the reads having a length different from that of WT. Sub-scores at each position are calculated by dividing the total number of substitutions at each position by the total number of WT-length queries. To exclude NGS and PCR error signals, the Sub-fold value is calculated by dividing the Sub-score of the CRISPR-treated data by that of the Mock-treated data.
3.2. Analysis of apparent substitutions in the PYK2 gene as a proof-of-concept
To demonstrate our strategy experimentally, as a proof-of-concept we arbitrarily selected one target site in the PYK2 gene and transfected plasmids that encoded CRISPR-Cas9 and an sgRNA targeting the gene into human HeLa cells. For the Mock-transfected, negative control, we transfected the CRISPR-Cas9-encoding plasmid alone. For both CRISPR-treated and Mock-treated samples, genomic DNAs were prepared, the DNA target sites were amplified by PCR, and the PCR amplicons were subjected to paired-end read sequencing using an Illumina MiniSeq platform. Then, both sets of high-throughput sequencing data were analyzed by CRISPR-Sub. For the CRISPR-treated samples, the WT-length, insertion, and deletion groups respectively accounted for 11,116 (17.1%), 8944 (13.7%), and 45,042 (69.2%) of the total of 65,102 reads (i.e., the indel frequency was 82.9%), whereas for the Mock-treated samples, the three groups respectively accounted for 53,097 (99.5%), 38 (0.1%), and 243 (0.5%) among the total of 53,378 reads (i.e., the indel frequency was 0.5%) (Supplementary Fig. 2).
For queries in the WT-length group, we calculated Sub-scores at each position for both Mock-treated (Fig. 2a) and CRISPR-treated samples (Fig. 2b). When the results were plotted, no points with values meaningfully above zero were observed for the Mock-treated data at any of the target positions, whereas a few prominent points near the cleavage site (zero on the X-axis) were observed for the CRISPR-treated data. Then, we divided the Sub-scores of the CRISPR-treated sample by those of the Mock-treated sample at each position to obtain Sub-fold values for all DNA positions. As shown in Fig. 2c, the Sub-scores of certain prominent points were much amplified, indicating their identity as apparent substitutions, but random errors caused by the NGS procedure were depleted. We also confirmed that no points above the threshold were observed when the Sub-scores of the Mock-treated sample were divided by the Sub-scores from another set of Mock-treated data, further supporting the utility of CRISPR-Sub (Supplementary Fig. 3).
Fig. 2.

Analysis of apparent substitutions in PYK2 with CRISPR-Sub. (a), (b) Scatter plots of Sub-scores at each nucleotide position in WT-length sequences from the Mock-treated and SpCas9-treated samples. (c) Scatter plot of Sub-fold values, with a graph on the right showing the count frequency fitted with a Gaussian curve. The blue dashed line indicates the threshold value. The red points have Sub-fold values above the threshold, indicating that they represent hotspot positions. (d) (Top) The WT sequence surrounding the target region, with the PAM nucleotides indicated in white on a gray background. (Bottom) The five most abundant mutant sequences in the WT-length group contain apparent substitutions at hotspot positions. The heat map shows Sub-fold values at the hotspot positions. (e) Apparent substitution frequencies in PYK2 in all queries and in the WT-length group. (f) One pattern of apparent substitutions that was detected in colonies derived from single cells. One allele contains a 7-bp deletion and another contains a C > T substitution. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To pinpoint hotspot positions at which apparent substitutions are frequently observed, we further made a frequency histogram of Sub-fold values fitted it with a Gaussian curve (Fig. 2c right panel and Supplementary Fig. 4). We confirmed that Sub-fold values have normality using a Shapiro-wilk test in two Mock-treated samples (Supplementary Fig. 5). From the Gaussian fitting, we calculated a standard Sub-fold value (p-value > 0.001), which serves as a threshold for determining hotspot positions statistically. In this case, the threshold value was 4.56; a total of 12 positions with Sub-fold values higher than 4.56 (indicated as red squares in Fig. 2c and d) were determined to be hotspot positions; i.e., substitution mutations at these positions are statistically likely to be apparent substitutions. We found that the five most abundant mutant sequences in the WT-length group had substitutions exactly at the estimated hotspot positions (Fig. 2d), supporting that CRISPR-Sub is relevant for measuring apparent substitutions. In addition, we counted the number of reads having substitution mutations at the 12 hotspot positions, and calculated apparent substitution frequencies by dividing by the total number of all queries (0.43%) and the total number of queries in the WT-length group (2.55%) (Fig. 2e).
To further confirm that our predicted apparent substitutions really exist in cells, we diluted CRISPR-treated cells and obtained about 960 colonies derived from single cells. We performed high-throughput sequencing for each colony and found that 6 colonies (0.63%) had substitution patterns. The allele frequency of apparent substitutions was 0.31% (6/1920), which is similar with the calculated apparent frequency in all queries (0.43%) in Fig. 2e. One representative colony showed heterozygous mutation patterns, with a C > T substitution in one allele and a 7-nt deletion in the other (Fig. 2f).
3.3. Comprehensive evaluation of apparent substitutions in 50 additional endogenous human targets
We next chose 50 additional targets in endogenous sites in the human genome and applied the CRISPR-Sub method at all targets. Similar to the experiments described above, we treated HeLa cells with a plasmid encoding CRISPR-Cas9 in the presence or absence of a plasmid encoding the appropriate sgRNA, after which we performed high-throughput sequencing at all targets. We found that indel frequencies in the CRISPR-treated samples ranged from 7.12% to 99.92% (Fig. 3a), whereas the average indel frequencies in the Mock-treated samples were insignificant (0.33 ± 0.31%), indicating that the selected sgRNAs worked efficiently at all tested sites. Then, we applied the CRISPR-Sub method to pinpoint hotspot positions for each target (Supplementary Fig. 6). and found that the hotspot positions were positioned near cleavage sites (Fig. 3b and Supplementary Fig. 7). Note that hotspot positions distal to the expected cleavage site have low probability of bona-fide substitutions.
Fig. 3.

Frequencies and patterns of indel mutations and apparent substitutions at endogenous sites in HeLa cells. (a) Bar graph showing the frequencies of insertions (blue) and deletions (red) at 51 targets (n = 1). (b) Frequency of hotspot positions at each nucleotide position. (c) Box plot showing the apparent substitution frequencies in all queries and in WT-length group (n = 51). (d) Heat map of mutation patterns in Mock-treated samples. (e) Heat map of mutation patterns at hotspots in CRISPR-treated samples. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We next calculated apparent substitution frequencies for each target both in all queries (average 0.80 ± 0.1.52%) and in the WT-length group (average 2.64 ± 3.88%) (Fig. 3c). Notably, the apparent substitution frequencies in WT-length groups have a positive correlation with indel frequencies (i.e. the Pearson’s coefficient is 0.58), whereas the apparent substitution frequencies in all queries have a lower correlation with indel frequencies (the Pearson’s coefficient is −0.22) (Supplementary Fig. 8). For example, a target in the ARG gene showed a 97.48% indel frequency. In this case, the apparent substitution frequency is very high (12.62%) in the WT-length group but very low (0.32%) in the whole set of sequences because most sequences have indel mutations and a few sequences only have substitution mutations. Therefore, it is necessary to measure the apparent substitution frequencies in the WT-length group, which reflects the tendency and characteristics of substitution mutations more directly, as well as in the whole set of sequences for substitution analysis to investigate hotspot positions and substitution patterns.
We further investigated the nucleotide conversion patterns at hotspot positions in all targets. Initially, we hypothesized that substitution errors in NGS data would mainly be the result of errors by DNA polymerase and NGS sequencers. It was previously reported that Phusion DNA polymerase causes transition (mainly A > G and T > C) rather than transversion (i.e., purine to pyrimidine or pyrimidine to purine) mutations during PCR [42]. On the other hand, it is known that Illumina Mi-seq and Mini-seq sequencers on average have a substitution error rate of 0.1%, primarily causing A > T or T > A conversions, at levels that are affected by the GC content [43], [44]. We first measured all nucleotide conversion patterns for the Mock-treated samples (Fig. 3d) and found that G > A or C > T transitions were dominant; presumably these mutations were induced during the PCR process. To our surprise, however, the substitution patterns at hotspot positions in the CRISPR-treated samples showed no dominant conversion pattern(s) (Fig. 3e), indicating that CRISPR-mediated substitutions lack sequence dependence and conversion preferences.
3.4. The NHEJ pathway is a major cause of the observed nucleotide substitutions
We next investigated which cellular repair pathway is responsible for the apparent substitution mutations. In contrast to MMEJ, which induces deletions according to short homology sequences, NHEJ induces indels regardless of sequence context. Therefore, we strongly postulated that NHEJ would be the dominant cause of base substitutions after CRISPR-Cas9 treatment. It is well known that Ku70, Ku80, DNA ligase IV, and XRCC4 are essential components of the NHEJ repair pathway in human cells [45]. To demonstrate a correlation between NHEJ and the substitutions we observed, we sought to inhibit the NHEJ pathway by disrupting one component, DNA ligase IV. Toward this end, we used CRISPR-Cas9 to target the LIG4 gene and generate DNA ligase IV-deficient HeLa cell lines. We next selected eight endogenous targets that exhibited high rates of indel formation in the experiment in Fig. 3a, targeted those sites in the LIG4−/− cells using CRISPR-Cas9, and applied the CRISPR-Sub method of analysis (Fig. 4a). Interestingly, NGS data showed that insertion frequencies in the LIG4−/− cells were significantly reduced compared to frequencies in WT cells, whereas deletion frequencies in the LIG4−/− cells were increased (Fig. 4b and c). We further calculated the frequency of MMEJ-mediated deletion, which have microhomology sequences of at least 2 bases, and found that MMEJ-mediated deletion frequencies in the LIG4−/− cells were significantly increased, suggesting that disruption of NHEJ may promote the alternative MMEJ pathway [46], [47]. On the other hand, the apparent substitution frequencies were decreased dramatically at most targets (six of the eight sites) in the LIG4−/− versus the WT HeLa cells (Fig. 4d), strongly suggesting that the NHEJ repair process is primarily responsible for the nucleotide substitutions.
Fig. 4.

Frequencies of indels and apparent substitutions in WT and LIG4−/− HeLa cells. (a) Experimental scheme. (b), (c) Bar graphs showing the frequencies of insertion (blue), deletion (red), and MMEJ-mediated deletion (orange) at eight endogenous targets in WT and LIG4−/− cells (n = 1). (d) Apparent substitution frequencies in the WT-length groups. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. CRISPR-induced substitutions in human HEK293T cells
One question of interest is whether CRISPR-induced substitutions occur in other than HeLa cells. To address this issue, we selected nine representative targets that exhibited high rates (average 60%) of indel formation in the experiment in Fig. 3a, targeted those sites in the human HEK293T cell line using CRISPR-Cas9, and applied the CRISPR-Sub method. We found that the indel frequencies ranged from 10.93% to 72.63% in the CRISPR-treated samples, whereas average indel frequencies were insignificant (0.16 ± 0.12%) in the Mock-treated samples, indicating that the selected sgRNAs worked efficiently at all tested sites (Supplementary Fig. 9). The average apparent substitution frequencies in the WT-length group were significantly higher for the CRISPR-treated (1.03 ± 0.78%) versus the Mock-treated samples (0.38 ± 0.24%), indicating that CRISPR-mediated substitutions occur similarly in HEK293T and HeLa cells.
3.6. Construction of a web-based CRISPR-Sub tool
The CRISPR-Sub method requires the step-by-step use of several statistical and computational analysis programs, which are deposited at github (https://github.com/Gue-ho/CRISPR-Sub). Users who do not have a computational background may not be comfortable using these command-line programs. Furthermore, CRISPR-Sub requires a relevant computational environment, including the LINUX operating system. Therefore, we constructed an online version of the CRISPR-Sub program ((http://www.rgenome.net/crispr-sub), through which users can easily analyze CRISPR-mediated substitutions by simply clicking on a few buttons. Because the CRISPR-Sub web tool was developed using JavaScript, it is almost completely used at a client-side web browser on-the-fly and there is no need to upload large NGS datasets to a server, which reduces running time. The CRISPR-Sub web tool receives NGS raw files (unjoined fastq files, gziped unjoined fastq files, or fastqjoin files) and displays the results in a web browser. The CRISPR-Sub web tool can analyze a 1.1 GB fastqjoin file and a 1.2 GB control fastqjoin file via Ryzen3 3850x and 32 GB RAM in 75 s. The results are provided with useful information including the Sub-fold value at each position, sequence reads containing substitutions, apparent substitution frequencies, and apparent substitution sequences aligned to the reference sequence (Fig. 5).
Fig. 5.

A sample CRISPR-Sub web tool results page. In Mock-treated data, apparent substitution frequencies are indicated as N.A. The heat map for hotspot positions is calculated by aligning 5′–3′ by target sequence. The aligned results are classified in each group at CRISPR- and Mock-treated data.
4. Discussion
In this study, we developed a novel analysis method, named CRISPR-Sub, to statistically detect apparent substitutions in high-throughput sequencing data by comparing data from Mock- and CRISPR-treated samples. CRISPR-Sub distinguishes apparent substitutions from false-positive substitution errors that are mainly introduced by the PCR process and determines apparent substitution frequencies in the WT-length group of sequences or in all queries.
To test the reproducibility of the CRISPR-Sub method, we prepared identical genomic DNA sample and performed CRISPR-Sub repeatedly. The results showed that there were no substantial differences between replicates in terms of apparent substitution frequencies, verifying the reproducibility of CRISPR-Sub (Supplementary Fig. 10). In addition, we examined the DNA polymerase dependence of CRISPR-Sub. To this end, we tested three different DNA polymerases (SUN Tag, KOD Taq, and Phusion polymerase) that have been known to have different fidelities. As expected, the Sub-scores in Mock-treated sample as well as CRISPR-treated sample varied according to the type of DNA polymerases but the Sub-fold showed less variable values (Supplementary Fig. 11). This result suggests that DNA polymerase-mediated errors can be destructive by dividing the CRISPR-treated with Mock-treated Sub-scores to determine the Sub-fold values.
In summary, we detected apparent CRISPR-induced substitution mutations and measured apparent substitution frequencies in bulk cell populations via CRISPR-Sub. Recently, DNA base editors have been developed to induce C-to-T or A-to-G substitution without generating DNA cleavages [48], [49], but they are also limited in the types of targeted base conversions and unintended promiscuous substitutions with side effects in genomic DNA or RNA levels [50], [51], [52]. Therefore, going forward, use of CRISPR-Sub will enable consideration of CRISPR-mediated substitutions in gene editing experiments. Both off-line (https://github.com/Gue-ho/crispr-sub) and online (http://rgenome.net/crispr-sub) versions of CRISPR-Sub are available for users’ convenience.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Dr. Heather McDonald for editing of this manuscript. This research was supported by grants from the National Research Foundation of Korea (NRF) (no. 2018M3A9H3022412), the Next Generation BioGreen 21 Program (PJ01319301), and the Technology Innovation Program (no. 20000158) to S.B.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.06.026.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 3.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 4.Garneau J.E., Dupuis M.E., Villion M., Romero D.A., Barrangou R. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 5.Cong L., Ran F.A., Cox D., Lin S., Barretto R. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieber M.R., Ma Y., Pannicke U., Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 7.Mao Z., Bozzella M., Seluanov A., Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothkamm K., Krüger I., Thompson L.H., Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang L., Deng L., Nguyen S.C., Zhao X., Maulion C.D. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–3310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan Q., Wang Y., Li J., Zhang Y., Chen K. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol. 2013;31:686–688. doi: 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 11.Wu Y., Liang D., Wang Y., Bai M., Tang W. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimi K., Kunihiro Y., Kaneko T., Nagahora H., Voigt B., Mashino T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 2016;7:10431. doi: 10.1038/ncomms10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakuma T., Nakade S., Sakane Y., Suzuki K.T., Yamamoto T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc. 2015;11:118–133. doi: 10.1038/nprot.2015.140. [DOI] [PubMed] [Google Scholar]
- 14.Auer T.O., Bene D. CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods. 2014;69:142–150. doi: 10.1016/j.ymeth.2014.03.027. [DOI] [PubMed] [Google Scholar]
- 15.Bassett A.R., Tibbit C., Ponting C.P., Liu J.L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013;4(1):220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koonin E.V., Makarova K.S., Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chylinski K., Makarova K.S., Charpentier E., Koonin E.V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:6091–6105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esvelt K.M., Mali P., Braff J.L., Moosburner M., Yaung S.J., Church G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao J., Guo D., Zhang J., Huang Q., Qin G. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 2013;23:1233–1236. doi: 10.1038/cr.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu R., Yang Y., Qin R., Li H., Qiu C. Rapid improvement of grain weight via highly efficient CRISPR/Cas9-mediated multiplex genome editing in rice. J Genet Genomics. 2016;43:529–532. doi: 10.1016/j.jgg.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Sánchez-León S., Gil-Humanes J., Ozuna C.V., Giménez M.J., Sousa C. Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol J. 2018;16:902–910. doi: 10.1111/pbi.12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S.H., Yu J., Hwang G.H., Kim S., Kim H.S. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene. 2017;36:6823–6829. doi: 10.1038/onc.2017.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park C.Y., Kim D.H., Son J.S., Sung J.J., Lee J. Functional correction of large factor VIII gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Wang L., Smith J., Breton C., Clark P., Zhang J. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat Biotechnol. 2018;36:717–725. doi: 10.1038/nbt.4182. [DOI] [PubMed] [Google Scholar]
- 27.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R. High-resolution CRISPR Screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Wang D., Mou H., Li S., Li Y., Hough S. Adenovirus-mediated somatic genome editing of pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther. 2015;26:432–442. doi: 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mou H., Smith J.L., Peng L., Yin H., Moore J. CRISPR/Cas9-mediated genome editing induces exon skipping by alternative splicing or exon deletion. Genome Biol. 2017;18:108. doi: 10.1186/s13059-017-1237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramakrishna S., Kwaku Dad A.B., Beloor J., Gopalappa R., Lee S.K., Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNa. Genome Res. 2014;24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke LA, REbelo CS, Goncalves J, Boavida MG, Jordan P. PCR amplification introduces errors into mononucleotide and dinucleotide repeat sequences. Mol Pathol. 2001;54:351-353. [DOI] [PMC free article] [PubMed]
- 34.Fox E.J., Reid-Bayliss K.S., Emond M.J., Loeb L.A. Accuracy of next generation sequencing platforms. Next Gener Seq Appl. 2014;1:1000106. doi: 10.4172/jngsa.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glen T.C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 36.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabun K., Guo X., Chavez A., Chruch G., Gagnon J.A., Valen E. Accurate analysis of genuine CRISPR editing events with ampliCan. Genome Res. 2019;29:843–847. doi: 10.1101/gr.244293.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aronesty E. Comparison of sequencing utility programs. Open Bioinform J. 2013:7–8. [Google Scholar]
- 40.Park J., Lim K., Kim J.S., Bae S. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 2017;33:286–288. doi: 10.1093/bioinformatics/btw561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice P., Longden I., Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 42.Mclnerney P., Adams P., Hadi M.Z. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol Biol Int. 2014 doi: 10.1155/2014/287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schirmer M., D’Amore R., Ijaz U.Z., Gall N., Quince C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinf. 2016;17:125. doi: 10.1186/s12859-016-0976-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma X., Shao Y., Tian L., Flasch D.A., Mulder H.L. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019;20:50. doi: 10.1186/s13059-019-1659-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang K., Guo R., Xu D. Non-homologous end joining: advances and frontiers. Acta Biochim Biophys Sin (Shanghai) 2016;48:632–640. doi: 10.1093/abbs/gmw046. [DOI] [PubMed] [Google Scholar]
- 46.Sishc B.J., Davis A.J. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers. 2017;9:81. doi: 10.3390/cancers9070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiruvella K.K., Liang Z., Wilson T.E. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;553:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nautre. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zuo E., Sun Y., Wei W., Yuan T. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou C., Sun Y., Yan R., Lui Y. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–285. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 52.Kim H.S., Jeong Y.K., Hur J.K., Kim J.S., Bae S. Adenine base editors catalyze cytosine conversions in human cells. Nat biotechnol. 2019;37:1145–1148. doi: 10.1038/s41587-019-0254-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.