

Abstract
XPA is a unique and essential protein required for the nucleotide excision DNA repair pathway and represents a therapeutic target in oncology. Herein, we are the first to develop novel inhibitors of the XPA–DNA interaction through structure-guided drug design efforts. Ester derivatives of the compounds 1 (X80), 22, and 24 displayed excellent inhibitory activity (IC50 of 0.82 ± 0.18 μM and 1.3 ± 0.22 μM, respectively) but poor solubility. We have synthesized novel amide derivatives that retain potency and have much improved solubility. Furthermore, compound 1 analogs exhibited good specificity for XPA over RPA (replication protein A), another DNA-binding protein that participates in the nucleotide excision repair (NER) pathway. Importantly, there were no significant interactions observed by the X80 class of compounds directly with DNA. Molecular docking studies revealed a mechanistic model for the interaction, and these studies could serve as the basis for continued analysis of structure–activity relationships and drug development efforts of this novel target.
Graphical Abstract
INTRODUCTION
Targeting DNA repair and the DNA damage response for cancer therapy has gained increasing attention with the recent U.S. FDA (December 2014) approval of the poly-ADP ribose polymerase (PARP) inhibitor olaparib (Lynparza, AstraZeneca) as the first DNA repair targeting agent for cancer treatment.1,2 While olaparib is approved as a single agent, the full utility of DNA repair targeted inhibitors can be expanded by their use in combination treatment regimens with DNA damaging chemotherapeutics including the platinum (Pt)-based agents cisplatin, carboplatin, and oxaliplatin.3 However, this utility requires knowledge of the relevant repair pathways involved in repairing and tolerating platinum-induced DNA damage.
Approximately half of all cancer patients who receive anticancer chemotherapy are treated with a platinum drug at some point within their treatment regimen with widely varied outcomes. Most cancers display a good initial response, but unfortunately treatment failure ensues due to development of intrinsic or extrinsic drug resistance.4 There are multiple factors involved in platinum resistance; among them increased capacity of DNA damage repair is one of the major concerns.5 Therefore, we have focused on directly targeting the nucleotide excision repair (NER) pathway which is responsible for repairing the vast majority of platinum-induced DNA damage.1,6–8 Several studies revealed that the overexpression of NER proteins (mainly XPA, RPA, ERCC1, XPC, and XPF) and repair of DNA damage by these proteins are directly linked to platinum resistance which ultimately hampers the efficacy of platinum-based therapy.6,9–14 The suppression of NER activity has been potentially recognized as a highly effective adjuvant therapy with DNA damaging agents such as platinum drugs and radiotherapy toward maximizing efficacy, overcoming resistance, and reducing the toxicities associated with the current regimen. The DNA damage recognition process is the limiting step in NER pathway, and specifically the XPA (xeroderma pigmentosum group A) protein binds to the damaged duplex DNA in the DNA damage identification and verification process.15,16 XPA does not possess any enzymatic activity but is an integral component for which there is no redundant or compensatory protein.17,18 In addition, XPA has been shown to have a greater affinity for damaged DNA over undamaged DNA and is required for the removal of all types of DNA lesions repaired by NER. In fact, as there are no redundant proteins that can compensate for the loss of XPA activity, decreased expression of XPA has been observed in testicular cancers where 95% of patients are cured by a platinum-based therapy.16,19,20 Therefore, XPA is a novel therapeutic target and the chemical synthetic lethality approach can be exploited with XPA inhibitors to increase the effectiveness of platinum-based chemotherapy in cancers in which there is only modest clinical success (Figure 1).6 While NER is responsible for removal of Pt-DNA damage, the homologous recombination repair (HRR) pathway allows the tolerance of Pt-DNA damage. We have devised a chemical synthetic lethality approach that provides novel mechanisms to specifically target cancer cells while sparing normal noncancer cells and thereby reducing toxicity associated with treatment (Figure 1). This model is fundamentally different from that of the synthetic lethality described for PARP inhibitors, which relies on endogenous single strand DNA damage to be converted to DNA double strand breaks,21–23 while our approach is designed specifically for Pt-combination therapy.
Figure 1.

Chemical synthetic lethality in Pt-combination cancer therapy. A HRR (homologous recombination repair) deficient cancer (blue cells) is depicted in a HRR proficient patient (black cells). Treatment with an XPA inhibitor (or NER inhibitor) renders both cell types unable to repair the Pt-DNA adducts via NER. The HRR proficient normal cells are able to tolerate or repair the Pt-DNA damage, which is anticipated to reduce toxicity. The HRR deficient cancer cells are unable to repair or tolerate the Pt-damage and thus are sensitive to the combination therapy.
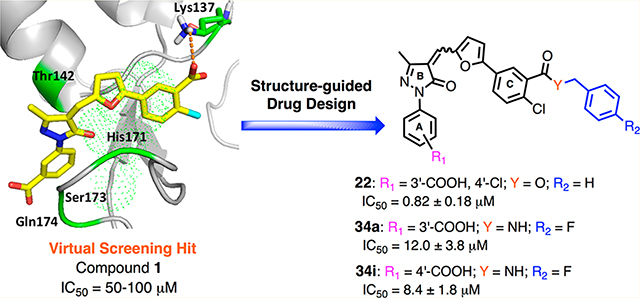
Human XPA is a relatively small 273-residue protein (39 kDa) that contains multiple domains and interaction motifs that support binding to DNA and other DNA repair proteins (Figure 2).17,18,24,25 The recently refined structural analysis of human XPA revealed that DNA binding activity resides in a 142 amino acid (XPA98–239) minimal DNA binding domain (MBD/DBD) spanning from the C4 zinc finger through the α-helix basic motif.26,27 The zinc containing globular core of XPA is responsible for binding both the ssDNA to dsDNA junction (Y junction) and the RPA70 domain.28,29 More recently, Koch et al. reported the first high-resolution X-ray crystal structures of the MBD of the yeast XPA homolog Rad14 bound to damage containing duplex DNA with either a cisplatin lesion (1,2-GG) or an acetylaminofluorene adduct (AAF-dG).30 The interaction of XPA homolog Rad14 with the ss-dsDNA junction is consistent with previous studies indicating that human XPA also preferentially binds to the DNA junction.31 Inhibitors targeting XPA–DNA interfaces hold great potential to enhance the efficiency of treatment with DNA damaging agents and reverse the platinum drug resistance by reducing NER activity. Surprisingly, despite the potential physiological significance and extensive scientific progress on XPA protein, very little progress has been made to date to develop small molecule inhibitors targeting XPA. In our previous studies,8 a 3D structure of the XPA MBD revealed a cleft that includes a number of conserved basic amino acids which has direct contact with the DNA in conjunction with surrounding residues and it also has an impact on binding to kinked DNA substrates, presumably similar to those formed by bulky DNA adducts that are repaired by NER.32,33 With further structure-based in silico screening of a virtual small molecule library targeting this cleft, we identified 5-(5-((1-(3-carboxyphenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)methyl)furan-2-yl)-2-chlorobenzoic acid 1 (X80 or TDRL-X80, Figure 3A)8 as an XPA–DNA interaction inhibitor by using fluorescence polarization and ELISA assays. In this continuation of our research, we are the first to report the structure-guided development of the X80 class of XPA small molecule inhibitors with the identification of more potent inhibitors of the XPA–DNA interaction.
Figure 2.

Schematic representation of human XPA protein and XPA interaction partners, mainly NER proteins.
Figure 3.

(A) Structure of compound 1. (B) Schematic representation of SAR exploration rationale: a pocket surrounding ring C of compound 1 for further structural optimization. Potential hydrogen bond contacts are shown as dashed magenta lines.
Results and Discussion
Initial Screening of XPA–DNA Interaction Inhibitors
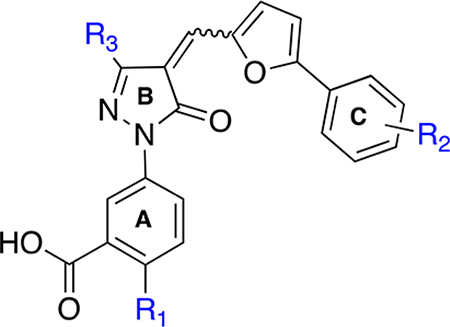
By use of the 3D structure of XPA determined by solution NMR (PDB code 1XPA), the coordinates of the C-terminal subdomain (residues 131–210) were found to have direct contact with a DNA ligand. Therefore, during our previous studies8 and also in these studies, we have targeted the cleft consisting of amino acid residues 138–142, 165–171, 174, and 177–181 for small molecule docking. Initial molecular docking studies with the compound 1 revealed that the interaction of compound 1 carboxylic acid (ring C) with the cleft contacting Lys137 is critical for inhibitory activity, and there is a large space-filling pocket around aromatic ring C that can be exploited for further structural optimization (Figure 3B). To further investigate the feasibility of targeting druglike binding pockets and identify inhibitors with improved potency, we first searched the virtual ChemDiv (San Diego, CA, USA) and AKos GmbH (Steinen, Germany) library for compound 1 analogs with a criterion of 85–95% structural similarity. Approximately 30 commercially available analogs (Table 1) were acquired and tested for their activity in inhibiting the XPA–DNA interaction. Purified full length XPA protein was used to screen a series of compound 1 analogs in an electrophoretic mobility shift assay (EMSA)29,34,35 that involved binding to a duplex DNA substrate containing a site specific cisplatin-DNA adduct centrally located on the duplex. In this study, we have expanded the in vitro assays to include EMSA to avoid the spectrophotometric interference by the colored nature of the XPA compounds. This allowed us to validate our findings and further refine the structure–activity relationships necessary for XPA inhibition. The results presented in Figure 4 (see Supporting Information Figure S1 for % DNA binding data) reveal a wide array of activities ranging from no inhibition to 100% inhibition at the relatively high concentration of 50 μM tested. Compounds that displayed greater than 80% inhibition at 50 μM were analyzed in titration experiments to determine IC50 values (Table 1).
Table 1.
Percentage Inhibition and IC50 Values of Compound 1 Analogsa
 | |||||
|---|---|---|---|---|---|
| compdb | R1 | R2 | R3 | % inhibition at 50 μMc | IC50 (μM)c,d |
| 1 | H | 3′-COOH, 4′-Cl | CH3 | 40 | >50 |
| 2 | H | 3′-COOH | CH3 | 31.2 | >50 |
| 3 | H | 2′-COOH | CH3 | 12.6 | >50 |
| 4 | H | 4′-COOH | CH3 | 21.8 | >50 |
| 5 | Cl | 2′-Br, 4′-NO2 | CH3 | 97.2 | 17.80 ± 0.75 |
| 6 | H | 2′-Br, 4′-NO2 | CH3 | 100 | 13.1 ± 2.9 |
| 7 | H | 2′-OMe, 4′-NO2 | CH3 | 90 | 12.0 ± 0.6 |
| 8 | H | 2′–OH, 5′-NO2 | CH3 | 60 | ND |
| 9 | H | 2′-CH3, 5′-NO2 | CH3 | 65 | ND |
| 10 | H | 3′-COMe | CH3 | 52 | ND |
| 11 | H | 3′-CO2Me | CH3 | 68.4 | ND |
| 12 | H | 3′-CO2Me | CF3 | 100 | 11.5 ± 1.4 |
| 13 | H | 3′-CO2Me, 4′-Cl | CH3 | 70.3 | ND |
| 14 | H | 3′-CO2Et, 4′-Cl | CH3 | 81.7 | 4.82 ± 0.55 |
| 15 | Cl | 3′-CO2Et | CH3 | 100 | 5.23 ± 0.1 |
| 16 | H | 3′-CO2nPr, 4′-Cl | CH3 | 87.7 | 12.6 ± 1.6 |
| 17 | Cl | 3′-CO2allyl | CH3 | 100 | ND |
| 18 | Cl | 3′-CO2allyl, 4′-Cl | CH3 | 100 | 6.8 ± 0.13 |
| 19 | Cl | 3′-CO2iPr | CH3 | 100 | 1.6 ± 0.8 |
| 20 | H | 3′-CO2nBu, 4′-Cl | CH3 | 98.6 | 9.1 ± 0.2 |
| 21 | Cl | 3′-CO2nBu, 4′-Cl | CH3 | 100 | 9.6 ± 1.1 |
| 22 | Cl | 3′-CO2CH2Ph, 4′-Cl | CH3 | 100 | 0.82 ± 0.18 |
| 23 | H | 3′-CO2CH2(2-Cl-Ph), 4′-Cl | CH3 | 100 | 1.93 ± 0.86 |
| 24 | Cl | 3′-CO2CH2(2-Cl-Ph), 4′-Cl | CH3 | 100 | 1.3 ± 0.22 |
Determined using EMSA, binding of full length human XPA to a cisplatin-modified DNA was assessed.
Compounds were obtained from ChemDiv and AKos GmbH library.
The assay was performed using full length XPA.
Compounds that displayed greater than 80% inhibition at 50 μM were analyzed in titration experiments. IC50 values are the mean of a minimum of triplicate independent experiments, and data are presented as the mean ± SD.
ND = not determined.
Figure 4.

EMSA analysis of XPA–DNA binding: Purified XPA (lanes 2–18) was mixed with DMSO control (lane 2, C) or the indicated compound at a concentration of 50 μM (lanes 3–18). 32P labeled ds-platinum damaged DNA was added and incubation continued for 30 min on ice. The products were separated on a 6% native gel by electrophoresis at 4 °C. Products were detected and quantified by PhosphorImager analysis. The free DNA is indicated by the red diamond and the XPA–DNA complex by the blue asterisk.
Optimization of Ring C and SAR Analysis
Quantification of the EMSA data and additional concentration dependent analysis of compound 1 analogs provided potential insight into the structure–activity relationships. The IC50 values, reported in Table 1, showed that the most efficient inhibitors of XPA–DNA binding were compounds 19 (IC50 = 1.6 ± 0.8 μM), 22 (IC50 = 0.82 ± 0.18 μM), 23 (IC50 = 1.93 ± 0.86 μM), and 24 (IC50 = 1.3 ± 0.22 μM). In agreement with the postulated molecular docking binding mode, the in vitro data indicate that the improved inhibitory activity of the compound 1 analogs is strongly related to the increasing hydrophobic interactions (by masking ionic carboxylic acid at ring C) in the binding pocket of XPA. Nitro and halogen substitutions on ring C (compounds 5 and 6) exhibited a considerable increase in XPA inhibitory activity in comparison with carboxylic acid substitution on ring C (compounds 1–4). The replacement of 2′-Br substitution (6) with 2′-OMe (7) on ring C in nitro containing compound retained XPA inhibitory activity. However, the inclusion of 2′-OH (8) and 2′-CH3 (9) at position 2 on ring C resulted in a decrease in inhibitory activity.
A decreased inhibitory activity observed for 3′-substituted acetyl containing compound 10 as compared to ester derivative (11) shows a clear agreement with our initial docking studies that carboxylic acid or ester substitution on ring C is very important to maintain the interaction with Lys137 and to retain inhibitory activity.
A series of 3′-substituted esters (compounds 11–24) resulted in increased inhibitory activity. A trend in inhibition was observed in the analysis of methyl, ethyl, and propyl esters where the addition of each methyl group resulted in a 10% increase in XPA inhibitory activity. There was an increase in the activity when replacing a bioisosteric methyl group (compound 11) with a trifluoromethyl group (compound 12) on the pyrazolone ring (ring B). An addition of a chloro group on ring A exhibited a slight increase in the XPA inhibitory activity (compound 24 vs compound 23).
All binding analyses thus far have employed using the full length human XPA; however while doing these studies, we have also performed binding studies with the minimal DNA-binding domain (DBD) consisting of amino acids 98–239 (XPA98–239). To ensure the inhibitor effects of the compounds via binding to this domain of XPA, we have overexpressed and purified the XPA DBD as an N-terminal tagged construct. We have evaluated the activity of compound 1 and one of our most potent inhibitor, 22, on purified minimal DNA-binding region XPA98–239 using EMSA. Figure 5 shows inhibition of full length XPA and minimal DBD XPA98–239 with increasing concentrations of compound 1 and compound 22. Interestingly, the compound 1 displayed greater potency toward full length XPA compared to the DNA-binding region XPA98–239, and this difference was dramatically reduced when assessing compound 22. These data suggest that there may be other interactions beyond the minimal DBD that stabilize the interactions of compound 1 with XPA but are not required for the higher affinity of compound 22.
Figure 5.

(A) EMSA with increasing concentration of compound 1 (12.5–100 μM) and 22 (1.6–25 μM) relative to DMSO control on full-length XPA (FL XPA) and XPA98–239 DBD. (B) Quantification and concentration-dependent analysis of DNA-binding activity of compounds 1 and 22 relative to DMSO control on full-length XPA and XPA98–239 DBD.
Compounds 19, 22, 23, and 24 displayed excellent inhibitory activity (IC50 = 1.6 ± 0.8 μM, 0.82 ± 0.18 μM, 1.93 ± 0.86 μM, and 1.3 ± 0.22 μM, respectively); however each of the commercially available compounds showed poor solubility. Therefore, with the aim to improve solubility, to establish a structure–activity relationship, and to define the structural determinants required to inhibit the XPA–DNA interactions, we synthesized and evaluated several novel compound 1 analogs.
Chemistry
We have developed a six- to seven-step synthesis for novel XPA inhibitors with an overall yield of ~15–20% using a highly efficient and operationally convenient protocol. The synthesis of final target compounds 34a–k is depicted in Scheme 1. We have obtained precursors 31a,b (Scheme 1) or 37a,b (Scheme 2) from starting materials 25a/25b by modifying existing synthetic approaches.36–39 Generally, the substituted dihydro-1H-pyrazol-1-yl 26a,b were prepared by refluxing corresponding substituted phenylhydrazines 25a,b with ethyl acetoacetate in acetic acid. The esterification of carboxylic acids 26a,b using H2SO4 in ethanol furnished 27a,b in good yields. Aldehyde 30 was prepared in high yield by optimizing Pd-catalyzed Suzuki coupling of 5-bromofuran-2-carbaldehyde 28 with boronic acid 29. Precursors 31a,b were prepared in moderate to good yields as a mixture of E/Z isomers by condensing corresponding pyrazolones 27a,b with aldehyde 30 in acetic acid under Knoevenagel reaction condition. To confirm the activity of compound 1, we have synthesized compound 1 and its 4′-substituted analog 32 by hydrolysis of corresponding esters (31a and 31b) using 2 N NaOH at room temperature. Alkyl- and arylamides 33a–j were prepared from 31a/31b in moderate yields by utilizing EDCI/HOBt amide synthetic protocol. Final target compounds 34a–d and 34f–k were obtained from compounds 33a–j by hydrolysis of corresponding esters using 10 N LiOH at room temperature. Target compound 34e was obtained from 34d and cyclopropylmethylamine using EDCI/HOBt amide synthesis strategy.
Scheme 1. Synthesis of Analogs 34a–ka.

aReagents and conditions: (a) ethyl acetoacetate, AcOH, reflux for 12 h, 72–76%; (b) H2SO4, EtOH, reflux for 12 h, 77–80%; (c) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O (1:1:0.3), 90 °C for 15 h, 92%; (d) AcOH, reflux for 3 h, 76–87%; (e) 2 N NaOH, THF/MeOH (2:1), rt for 6 h, 82–85%; (f) alkyl- or arylamine, EDCI, HOBt, DIPEA, DMF, rt for 18 h, 62–81%; (g) LiOH, THF/EtOH/H2O (4:2:1), rt for 12 h, 60–76% (after recrystallization).
Scheme 2. Synthesis of Analogs 39a–da.

aReagents and conditions: (a) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O (1:1:0.3), 90 °C for 15 h, 90%; (b) AcOH, reflux for 3 h, 78–85%; (c) alkylor arylamine, EDCI, HOBt, DIPEA, DMF, rt for 18 h, 74–80%; (d) LiOH, THF/EtOH/H2O (4:2:1), rt for 12 h, 64–77% (after recrystallization).
To explore the effects of amide substitution on para-position at ring C, we have synthesized target compounds 39a,b as depicted in Scheme 2 Aldehyde 36 was prepared from 5-bromofuran-2-carbaldehyde 28 and boronic acid 35 using in house optimized Suzuki coupling reaction by an above-described synthesis of aldehyde 30. Precursors 37a,b were prepared in moderate to good yields by condensing corresponding pyrazolones 27a,b with aldehyde 36 by using an above-described Knoevenagel reaction condition. Final compounds 39a–d were prepared from 37a or 37b using EDCI/HOBt amide synthesis protocol, followed by ester hydrolysis of compounds 38a–d. All biologically important compounds are ≥95% pure as determined by HPLC coupled to electrospray ionization mass spectrometry (LC/ESI-MS) analysis. Importantly, this protocol is feasible for large scale production, and synthesis on a 2 g scale is easily achievable. Esters 33a–j and final compounds 34a–k, 39a–d exist as a mixture of E/Z isomers, whereas Z isomer is the major product (~70–80%) as determined by NOESY NMR analysis of a mixture of isomers (see the Supporting Information Figure S2 for details). While we performed separation of the E/Z isomers of our final compounds using preparative HPLC, each isolated isomer rapidly equilibrated to give back the original mixture of isomers, and therefore separation of two isomers is practically impossible.
Structural Modification of Compound 22 To Improve Solubility and Further SAR Analysis
The identification of compounds 22 and 24 as the most potent XPA inhibitors in the ester series limited by poor solubility led us to focus our synthetic efforts on improving the aqueous solubility profile. The ester-containing compounds exhibited a high cLogP, and therefore, we utilized small isosteric modifications to improve solubility and metabolic stability. This was accomplished by replacing the metabolically labile ester group with a more chemically and metabolically stable amide. The novel synthesized compounds’ in vitro data are presented in Table 2. Replacement of the ester in compound 22 (IC50 = 0.82 ± 0.18 μM) with an amide and addition of a p-fluoro group 34a (IC50 = 12 ± 3.8 μM) led to dramatically increased solubility and decreased in potency but at least retained low micromolar activity. The difference in activity might be due to their ability to interact with a critical amino acid, Lys137, to retain and improve XPA inhibitory activity or might be due to result of the balance between the differences in the strength of interactions with another important cleft containing XPA backbone amino acid residues.
Table 2.
IC50 Values of Novel Synthetic Compound 1 Analogsa
 | ||||
|---|---|---|---|---|
| Compound | Series | R1 | R2 | IC50 (μM)b |
| 33a | A | 3-COOEt |  |
>25 |
| 34a | A | 3-COOH |  |
12.0 ± 3.8 |
| 34b | A | 3-COOH | 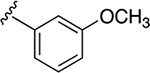 |
14.1 ± 3.0 |
| 34c | A | 3-COOH | 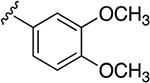 |
11.7 ± 1.2 |
| 34d | A | 3-COOH | 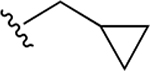 |
9.8 ± 2.6 |
| 34e | A | 3-CONHCH2C3H5 | 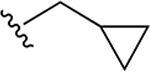 |
>50 |
| 34f | A | 3-COOH |  |
>25 (∼32) |
| 34g | A | 3-COOH | 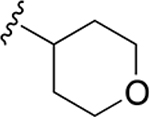 |
>25 (∼40) |
| 34h | A | 3-COOH | 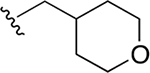 |
14.3 ± 2.1 |
| 34i | A | 4-COOH | 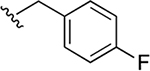 |
8.4 ± 1.8 |
| 34j | A | 4-COOH | 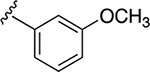 |
11.9 ± 3.15 |
| 34k | A | 4-COOH | 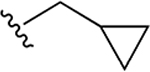 |
9.1 ± 2.4 |
| 39a | B | 3-COOH | 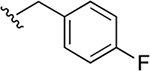 |
>50 |
| 39b | B | 3-COOH | 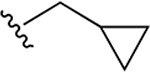 |
>50 |
| 39c | B | 4-COOH | 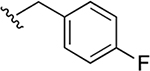 |
9.1 ± 4.3 |
| 39d | B | 4-COOH | 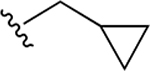 |
13.4 ± 3.8 |
Determined using EMSA, binding of full length human XPA to a cisplatin-modified DNA was assessed.
The assay was performed using full length XPA. IC50 values are the mean of a minimum of triplicate independent experiments, and data are presented as the mean ± SD.
Subsequently, several alkylamide- and arylamide-containing substituents at ring C were investigated as represented by 34b–h, with 4-fluorobenzyl (34a), dimethoxyphenyl (34c), and cyclopropylmethyl (34d) being the optimal fit in the space filling pocket. Cyclopropylmethyl containing amide 34d (IC50 = 9.8 ± 2.6 μM) at ring C exhibited almost 3.2-fold increased in activity compared to cyclopropylamide 34f (IC50 ≈ 32 μM), aligning with our previous inhibition trend observed for extended ester derivatives (13–21). Insertion of a heteroatom such as oxygen into the cyclohexane ring in the form of a pyran was tolerated, with more preference for the extended 4-methyltetrahydropyran 34h (IC50 = 14.3 ± 2.1 μM) over the 4-tetrahydropyran 34g (IC50 ≈ 40 μM). The amide substitution at 4-position of ring C with fluorobenzyl (39a) and cyclopropylmethyl (39b) showed no XPA inhibitory activity (IC50 ≥ 50 μM), which may be due to structural constraints imposed by such a modification to fit in the space-filling binding cleft. On the contrary, 4-substitued fluorobenzylamide (39c) and cyclopropylmethylamide (39d) at ring C along with para-substituted carboxylic acid on ring A showed potent XPA inhibitory activity in EMSA. Most likely, structural conformations of compounds 39c and 39d allow flexibility in accommodating these inhibitors within the XPA binding cavity.
Simultaneously, we have evaluated the influence of the carboxylic acid (ring A) of the arylpyrazolone moiety by replacing it with ester and amide groups. EMSA analysis revealed a dramatically decreased or almost no XPA inhibitory activity for ester (33a) and amide (34e) containing compounds, highlighting the importance of the carboxylic acid at ring A to maintain the interaction with XPA backbone residues and ultimately to retain inhibition activity. Interestingly, 4-substitued carboxylic acid at ring A exhibited promising activity with 4-fluorobenzyl 34i (IC50 = 8.4 ± 1.8 μM) and cyclopropylmethyl 34k (IC50 = 9.1 ± 2.4 μM) amide containing compounds.
Overall, on the basis of XPA inhibition data (Table 1 and 2) confirmed by EMSA analysis, among the compounds those with the alkyl/alkylaryl ester substitution at ring C were the most potent XPA inhibitors. In addition, alkyl/alkylaryl amide substitution at ring C along with carboxylic acid substitution on 3-position or 4-position at ring A displayed inhibition of XPA–DNA interaction at submicromolar concentrations. These results strongly correlate with the molecular docking data (Figure 6), showing compounds 22 and 34i interact strongly by positioning themselves in the space-filling pocket and cleft of the XPA binding site.
Figure 6.

Molecular docking studies (PDB code 1XPA). (A) Molecular interactions of compound 22 (yellow carbon) Z-isomer with hXPA (key amino acids are shown in green carbon, surface is shown in aquamarine, and cartoon is shown in light orange). (B) Molecular interactions of 34i (yellow carbon) Z-isomer with hXPA (key amino acids are shown in green carbon, surface is shown in aquamarine, and cartoon is shown in light orange). Interaction with amino acid side chains is indicated with the dashed magenta lines, π−π stacking interactions are shown in solid magenta dumbbell, cation–π interactions are shown in solid one sided magenta arrow, and salt-bridge interactions are shown in dashed two-sided magenta arrow. Distances are indicated in Å.
Molecular Docking
Initial molecular docking studies allowed us to expand our SAR and potentially correlate the results from the XPA inhibitory studies with the docking-based binding analysis of our compounds. To delineate the key interactions responsible for differences in binding affinity and to understand the SAR, the structures of XPA inhibitors were flexibly docked into the XPA minimal DNA binding domain (PDB code 1XPA). Docking studies with the most active XPA inhibitors revealed that only the Z isomer can be effectively docked in the XPA binding cavity. Binding modes for the Z isoform is predictable and consistent, while the E isoform does not adopt a consistent binding. This is likely an indication that E-isomer is inactive (see the molecular docking section in the Supporting Information for more details). Compounds with methyl substitution on pyrazolone ring have a higher ratio of the Z-isomer (~70–80%) as determined by NOE NMR analysis. As docking studies predicted, only the Z-isomer is likely contributing to the XPA inhibition activity; therefore we expect lower IC50 values (greater potency) with the compounds displayed in Tables 1 and 2. All Z-isomer compounds bind similarly to parent compound 1 by extending through the cleft and positioning ring C ester/amide in range for interactions with a critical amino acid, Lys137. As with the ester (11–24) and amide (34a–k and 39a–d) derivatives, alkyl or more bulky alkylaryl substituents at position 3 of aromatic ring C occupy the space filling binding pocket of the XPA binding site very efficiently.
Figure 6 shows the binding and molecular interactions of 22 and 34i Z-isomers within XPA MBD. The molecular interaction of 22 (Figure 6A) and 34i (Figure 6B) Z-isomer is largely ascribed to various electrostatic interactions, including the following. (i) Compound 22 ester carbonyl and compound 34i amide carbonyl make hydrogen bond contacts with the amine of Lys137. (ii) 3′-COOH (ring A) of compound 22 shows hydrogen bond contacts with the backbone amine of Gln174, while most interestingly, 4′-COOH of compound 34i does not interact with Gln174 but makes salt-bridge interactions with one of the most important cleft amino acid, Lys179 (Lys179 shown to impact binding to kinked DNA substrates).8 (iii) There are π−π stacking interactions between the furan moiety and the aromatic ring of His171 in both compounds. In addition, compound 22 phenyl moiety is also well positioned to make additional π−π stacking interactions with His171. (iv) Both substituted phenyl groups of ester (22) and amide (34i) are optimally positioned to make cation–π interactions with the cleft amino acid, Lys167. (v) Both compound’s N1–N2 of pyrazolone ring position reveal the potential for hydrogen bond contacts with the hydroxyl group of Ser173 and also with the cleft amino acid, Thr142. Docking studies also predicted a stronger affinity of the ester containing Z-isomer of compound 22 than amide containing Z-isomer of compounds 34a, 34i, and 39c (see Figures S3–S6 in the Supporting Information for details). This fact could be a result of the balance between the differences in the strength of the π−π stacking interactions between the furan as well as phenyl moieties and the aromatic ring of His171. Additionally, the increased and tighter interactions by the ester series of compounds with the XPA backbone amino acid residues Lys137, Gln174, and His171 might be one of the reasons that the ester containing compounds show higher affinities than the amide series of compounds. Particularly, the higher affinity of ester containing compound 22 than bioisoteric amide containing 34a might be due to it is potential to make hydrogen bond contacts with the backbone amine of the Gln174 while compound 34a lacks these interactions (see the Supporting Information Figures S3, S4, and S5 for details). On a particular note, these molecular docking studies represent models for potential interactions between our small molecule inhibitors (SMIs) and the XPA MBD region. The differences in inhibitory activity of our SMIs might be due to different structural conformation and their ability to interact with amino acid residues located within the XPA binding cleft.
DNA Intercalation Assay and Specificity
A competitive DNA intercalation assay was performed to determine if the compounds’ activity could be the result of binding to DNA. To access this as a potential mechanism of inhibition, compounds 22, 24, 34a, 34d, 34i, 34k, and 39c were analyzed in the fluorescence displacement assay along with using doxorubicin (Dox), a known noncovalent DNA binding chemotherapeutic, as a positive control. The results presented in Figure 7 demonstrate that no significant DNA binding activity was observed for recently synthesized novel XPA inhibitors (34a, 34d, 34i, 34k, and 39c). However, commercially available compounds 22 and 24 exhibited DNA binding activity above their IC50 value concentrations. These results are consistent with our XPA compounds that specifically inhibit protein–DNA interaction by binding to the XPA protein and not via binding to the DNA.
Figure 7.

Analysis of compound interactions with DNA. The indicated concentrations of compounds 22, 24, 34a, 34d, 34i, 34k, and 39c were analyzed for the ability to displace a fluorescent Sybr-green DNA intercalator as a measure of compound DNA interactions. The assay was performed and fluorescence measured as described in “Experimental Details”. The data represent the average and SD of three independent experimental determinations performed in duplicates.
Furthermore, to access specificity of our compounds, we have evaluated inhibitory activity against RPA (replication protein A) by using our previously described EMSA.7 RPA is a single-stranded DNA binding protein and forms a complex with XPA in duplex damaged DNA binding activity (DNA damage recognition process) in the NER pathway.17,18,40,41 RPA is also involved in other metabolic pathways, including DNA replication and recombination. Most importantly, there was no RPA inhibition observed at 10 μM with compounds 22, 24, 34i, and 34k in RPA-based EMSA assay.
Conclusions
Inhibition of macromolecular protein–DNA interactions by small molecules represents a relatively new class of important interactions that can be targeted for therapeutic intervention. Because of XPA’s interaction with Pt-damaged DNA, its absolute requirement in the initiation of Pt-DNA-damage repair and with refined structural analysis of the protein, XPA has emerged as an attractive drug target for combination therapy. A broad range of cancers such as lung, ovarian, bladder, and head and neck will receive a Pt therapy with modest efficacy. Effective inhibition of XPA and ultimately preventing repair of Pt-DNA damage has the potential to sensitize these cancers and reverse resistance to afford better clinical outcomes.
Therefore, on the basis of our previous molecular insights, we have designed and identified highly potent inhibitors of the XPA–DNA interaction. Compounds 19, 22, 23, and 24 exhibited inhibition of XPA–DNA interaction at submicromolar concentration; however, further analysis of ester derivatives was hampered due to poor solubility. Further systematic structure-guided drug design and synthetic SAR analysis of compound 1 derivatives led to the identification of 34a, 34c, 34d, 34i, 34k, and 39c as the most promising compounds in the series.
To our current knowledge, we are the first to develop highly potent and novel XPA inhibitors, and most importantly these compounds do not bind directly with DNA. In addition, these compounds do not interact with multifunctional RPA, a protein that forms a complex with XPA in duplex damaged DNA binding activity. These results offer new knowledge regarding the structural interaction of XPA inhibitors and its binding site that could be utilized in the design of future XPA inhibitors. Together the properties of these novel XPA inhibitors make them promising lead compounds for development into anticancer drugs that are to be used in combination therapy or as reagents for biochemical, cell and cancer biology studies. Further SAR analysis and biological evaluation of lead compounds are underway.
Experimental Details
General
All commercially available compounds 1–24 were purchased from ChemDiv (San Diego, CA, USA) and AKos GmbH (Steinen, Germany) library with highest purity (≥95%) and prepared at 10 mM stock solution in 100% DMSO. All chemicals used for synthesis were purchased from Aldrich, Alfa Aesar, Acros, Fisher Scientific, AK Scientific, and Combi-Blocks Chemical Co. (USA) and used without further purification. Anhydrous solvents were obtained from Fisher Scientific or Aldrich and used directly. All reactions involving air- or moisture-sensitive reagents were performed under a nitrogen atmosphere. 1H NMR spectra were recorded at 300 and 500 MHz using Bruker AV NMR spectrometer. 13C NMR spectra were recorded at 75 and 125 MHz using Bruker AV NMR spectrometer. The chemical shifts were reported as δ ppm relative to TMS, using the residual solvent peak as the reference unless otherwise noted. All coupling constants (J) are given in hertz. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), number of protons and coupling constants. Thin layer chromatography was performed using Merck silica gel 60 F-254 thin layer plates, which were developed using one of the following techniques: UV fluorescence (254 nm), alkaline potassium permanganate solution (0.5% w/v) or ninhydrin (0.2% w/v) and iodine vapors. Automated flash column chromatography was carried out on prepacked silica cartridges using the indicated solvent system on Biotage Isolera chromatography system. Target compounds 33a, 34a–k, and 39a–d were crystallized in ethanol, solid was collected, washed with EtOAc and then hot solutions of 20–30% EtOAc in hexanes to afford red to orange solids. If necessary, the products were purified with automated flash column chromatography. The chemical purity of target compounds was ≥95% determined by HPLC coupled to electrospray ionization mass spectrometry (LC/ESI-MS) analysis. LC–MS analyses and purity data of compounds were obtained using an Agilent 6130 quadrupole LC–MS instrument connected to an Agilent 1200 HPLC system, and both instruments were connected to an Agilent diode array detector. A C-18 reversed phase column (Vydac monomeric/Phenomenex/Kinetex 2.6 μm XB-C18, 50 mm × 4.6 mm) was used as stationary phase, and water and methanol/acetonitrile (both containing 0.1–0.25% TFA) were used as mobile phase (gradient 0–100% methanol, flow 0.8 mL/min, run time 15 min). UV absorbance at the fixed wavelength of 254 nm and positive and negative ESI-MS data were recorded. The retention time and corresponding ESI-MS data were used to identify molecules. HRMS data were obtained using Waters/Macromass LCT electrospray ionization (ESI) on a time-of-flight (TOF) mass spectrometer at the Mass Spectrometry Facility at Indiana University Chemistry Department (http://msf.chem.indiana.edu).
Synthesis of 26a and 26b
3-(3-Methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (26a)
Ethyl acetoacetate (2.01 mL, 1.2 equiv) was added to a solution of 3-hydrazinobenzoic acid 25a (2 g, 1 equiv) in glacial acetic acid (30 mL) under an argon atmosphere. After addition, the reaction mixture was heated at reflux with stirring for 12 h. Once the reaction was allowed to cool to room temperature, the reaction mixture was concentrated in vacuo resulting in the formation of a precipitate. The solid was filtered and washed with 5% MeOH in DCM (two times) and then two times with DCM to obtain 26a as an off-white solid (2.06 g, 72% yield, requires no further purification). TLC: 4% MeOH in DCM, Rf = 0.42; visualized with UV. 1H NMR (500 MHz, DMSO): δ 13.22 (brs, 1H, COOH), 8.36 (s, 1H), 8.05 (d, 1H, J = 8.5 Hz), 7.94 (d, 1H, J = 8.0 Hz), 7.70 (t, 1H, J = 8.0 and 16 Hz), 5.97 (s, 1H), 2.45 (s, 3H, CH3). 13C NMR (125 MHz, DMSO): δ 166.48, 158.79, 154.09, 150.10, 144.90, 136.81, 132.15, 129.96, 127.57, 124.16, 120.66, 104.64, 102.19, 19.12, 14.25. HRMS (ESI): calcd for C11H11N2O3 [M + H]+ m/z = 219.0770, found 219.0764.
4-(3-Methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (26b)
26b was prepared by an above-described procedure using 4-hydrazinobenzoic acid hydrochloride 25b (2 g) as a starting material. Off-white solid (1.76 g, 76% yield, requires no further purification). TLC: 4% MeOH in DCM, Rf = 0.42; visualized with UV. 1H NMR (300 MHz, DMSO): δ 12.87 (brs, 1H, COOH), 7.98 (d, 2H, J = 8.8 Hz), 7.88 (d, 2H, J = 8.4 Hz), 5.38 (s, 1H), 2.12 (s, 3H, CH3). 13C NMR (125 MHz, DMSO): δ 172.17, 170.56, 167.32, 159.86, 150.25, 142.66, 142.06, 130.80, 126.86, 126.46, 119.33, 117.49, 117.34, 43.61, 14.45. HRMS (ESI): calcd for C11H9N2O3 [M − H]− m/z = 217.0613, found 217.0619.
Synthesis of 27a and 27b
Ethyl 3-(3-Methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (27a)
To a stirred suspension of 3-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic acid 26a (1.95 g) in anhydrous ethanol (30 mL) was added a catalytic amount of concentrated sulfuric acid (1.5 mL) slowly under an argon atmosphere. The reaction mixture was refluxed for 12 h, and then it was allowed to cool to room temperature. The solvent was removed under vacuum, and the obtained residue was dissolved in ethyl acetate and washed successively with saturated NaHCO3 (2 × 10 mL), water, and brine solution. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude residue was purified by Biotage automated flash column chromatography using 0–50% EtOAc in hexanes as the eluent to furnish ethyl 3-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate 27a as a red oil (1.69 g, 77% yield). TLC: 45% EtOAc in hexanes, Rf = 0.44; visualized with UV. 1H NMR (300 MHz, CDCl3): δ 8.41 (s, 1H), 8.05 (d, 1H, J = 8.2 Hz), 7.78 (d, 1H, J = 8.0 Hz), 7.38 (t, 1H, J = 7.95 and 15.99 Hz), 4.35−4.28 (q, 2H, OCH2), 3.37 (s, 2H, CH2), 2.11 (s, 3H, CH3), 1.33 (t, 3H, J = 7.11 and 14.25 Hz, CH3). 13C NMR (75 MHz, CDCl3): δ 170.70, 166.14, 156.84, 138.18, 131.22, 128.82, 125.77, 122.69, 119.46, 61.10, 43.02, 16.93, 14.29. MS (ESI) m/z = 247.1 [M + H]+.
Ethyl 4-(3-Methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (27b)
27b was prepared by an above-described procedure using 26b (1.60 g) as a starting material. White solid (1.44 g, 80% yield). TLC: 40% EtOAc in hexanes, Rf = 0.44; visualized with UV. 1H NMR (300 MHz, CDCl3): δ 8.05 (d, 2H, J = 8.97 Hz), 8.01 (d, 2H, J = 8.94 Hz), 4.40−4.33 (q, 2H, OCH2), 3.46 (s, 2H, CH2), 2.22 (s, 3H, CH3), 1.39 (t, 3H, J = 7.11 and 14.25 Hz, CH3). 13C NMR (75 MHz, CDCl3): δ 170.78, 166.18, 156.89, 141.69, 130.54, 126.42, 117.61, 60.90, 43.17, 17.09, 14.36. MS (ESI) m/z = 247.1 [M + H]+.
Synthesis of 2-Chloro-5-(5-formylfuran-2-yl)benzoic Acid (30)
A solution of K2CO3 (2.37 g, 3 equiv) in water (10 mL) was added to a mixture of 4-chloro-3-carboxyphenylboronic acid 29 (1.37 g, 1.2 equiv) and 5-bromo-2-furaldehyde 28 (1 g, 1 equiv) in toluene/ethanol (1:1, v/v, 60 mL). The mixture was degassed with argon for 5 min, and then Pd(PPh3)4 (330 mg, 0.05 equiv) was added. The reaction mixture was stirred at 90 °C for 15 h. The reaction mixture was cooled to room temperature, filtered through Celite, and washed with water (2 × 10 mL). The pH of the solution was adjusted to 1–2 by addition of 6 N HCl solution. The precipitated reaction mixture was extracted with dichloromethane (3 × 100 mL); the combined organic fractions were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was triturated with 20–30% EtOAc in hexanes (2 times), and solid was filtered to afford 2-chloro-5-(5-formylfuran-2-yl)benzoic acid 30 (1.24 g, 87% yield) as an off-white solid. TLC: 60% EtOAc in hexanes, Rf = 0.40; visualized with UV and KMnO4 solution. 1H NMR (300 MHz, DMSO): δ 13.74 (brs, 1H, COOH), 9.63 (s, 1H, CHO), 8.23 (d, 1H, J = 2.22 Hz), 8.01 (dd, 1H, J = 2.28 and 8.43 Hz), 7.70 (d, 1H, J = 8.34 Hz), 7.67 (d, 1H, J = 2.85 Hz), 7.45 (d, 1H, J = 3.75 Hz). 13C NMR (75 MHz, DMSO): δ 178.64, 166.63, 156.44, 152.47, 132.93, 132.78, 132.13, 129.00, 128.10, 127.23, 110.56. MS (ESI) m/z = 249.0 [M − H]−.
Synthesis of 31a and 31b
(Z)-2-Chloro-5-(5-((1-(3-(ethoxycarbonyl)phenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)benzoic Acid (31a)
Ethyl 3-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate 27a (1 g, 1 equiv) and 2-chloro-5-(5-formylfuran-2-yl)benzoic acid 30 (1.01 g, 1 equiv) were dissolved in glacial acetic acid (50 mL). The reaction mixture was heated at reflux with stirring for 3 h. Solvent was removed in vacuo, and solid was suspended in EtOH, filtered, washed with EtOH, EtOAc, and DCM (2 times each) to obtain 31a as a red solid (1.48 g, 76% yield, requires no further purification). TLC: 5% MeOH in DCM, Rf = 0.45; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.75 (brs, 1H, COOH), 8.63 (d, 1H, J = 3.87 Hz), 8.48 (t, 1H, J = 1.86 and 3.69 Hz), 8.27 (d, 1H, J = 2.22 Hz), 8.19 (d, 1H, J = 7.08 Hz), 8.01 (dd, 1H, J = 2.22 and 8.43 Hz), 7.76−7.64 (m, 3H), 7.56−7.51 (m, 2H), 4.36−4.29 (q, 2H, OCH2), 2.64 (s, 0.29H, minor isomer, CH3), 2.32 (s, 2.71H, major isomer, CH3), 1.33 (t, 3H, J = 7.08 and 14.16 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 166.59, 165.88, 162.11, 157.65, 151.64, 150.82, 138.96, 133.15, 132.67, 131.11, 130.91, 130.08, 129.77, 128.96, 127.84, 127.26, 125.17, 122.41, 121.60, 118.43, 112.91, 61.38, 14.65, 13.29. MS (ESI) m/z = 477.1 [M − H]−.
(Z)-2-Chloro-5-(5-((1-(4-(ethoxycarbonyl)phenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)benzoic Acid (31b)
31b was prepared by an above-described procedure using 27b (1 g, 1 equiv) and 30 (1.01 g, 1 equiv) as starting materials. Red solid (1.69 g, 87% yield). TLC: 5% MeOH in DCM, Rf = 0.48; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.72 (brs, 1H, COOH), 8.62 (d, 1H, J = 3.84 Hz), 8.33 (d, 1H, J = 2.19 Hz), 8.15−7.90 (m, 5H), 7.79 (s, 1H), 7.71 (d, 1H, J = 8.49 Hz), 7.58 (d, 1H, J = 3.84 Hz), 4.33−4.26 (q, 2H, OCH2), 2.68 (s, 0.51H, minor isomer, CH3), 2.34 (s, 2.49H, major isomer, CH3), 1.32 (t, 3H, J = 7.11 and 14.19 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 166.60, 165.69, 162.41, 157.81, 152.26, 150.83, 142.42, 133.23, 132.70, 132.18, 130.71, 129.03, 127.87, 127.35, 125.42, 121.39, 117.39, 112.97, 61.01, 14.67, 13.35. MS (ESI) m/z = 477.1 [M − H]−.
Synthesis of Compounds 1 and 32
(Z)-5-(5-((1-(3-Carboxyphenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)-2-chlorobenzoic Acid (1)
To a stirred suspension of compound 31a (150 mg) in THF/MeOH (2:1, v/v, 10 mL) was added 2 N NaOH (1 mL) solution. The reaction mixture was stirred at room temperature for 6 h. Solvent was removed in vacuo, and residue was acidified to pH 2−3 using 20% citric acid solution. The product was extracted with EtOAc (3 × 15 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The product was crystallized in EtOAc and triturated with 30% EtOAc in hexanes to afford compound 1 (120 mg, 85% yield) as an orange solid. Isomer data: 1H NMR (300 MHz, DMSO) δ 13.27 (brs, 1H, COOH), 12.88 (brs, 1H, COOH), 8.61 (d, 1H, J = 3.7 Hz), 8.50 (t, 1H, J = 1.95 and 3.85 Hz), 8.23 (d, 1H, J = 1.95 Hz), 8.20 (d, 1H, J = 7.35 Hz), 8.01 (m, 1H), 7.76−7.66 (m, 3H), 7.58−7.53 (m, 2H), 2.65 (s, 0.77H; minor isomer, CH3), 2.37 (s, 2.23H; major isomer, CH3). MS (ESI) m/z = 473.1 [M + Na]+. HRMS (ESI): calcd for C23H13N2O6Cl [M − 2H]− m/z = 448.0462, found 448.0469. HPLC purity: 95.36%.
(Z)-5-(5-((1-(4-Carboxyphenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)-2-chlorobenzoic Acid (32)
32 was prepared by an above-described procedure using 31b (200 mg) as starting material. Orange solid (154 mg, 82% yield). 1H NMR (300 MHz, DMSO): δ 13.19 (brs, 1H, COOH), 12.84 (brs, 1H, COOH), 8.67 (d, 1H, J = 3.84 Hz), 8.10−7.91 (m, 6H), 7.80−7.77 (m, 1H), 7.70 (d, 1H, J = 8.64 Hz), 7.59−7.53 (m, 1H), 2.68 (s, 0.58H; minor isomer, CH3), 2.34 (s, 2.43H; major isomer, CH3). MS (ESI) m/z = 448.1 [M − 2H]−. HRMS (ESI): calcd for C23H14N2O6Cl [M − H]− m/z = 449.0540, found 449.0547. HPLC purity: 95.13%.
General Synthesis of Amides 33a–j
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-((4-fluorobenzyl)carbamoyl)phenyl)furan-2-yl)-methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33a)
To a solution of compound 31a (300 mg, 1 equiv) in dry DMF (6 mL) were added EDCI·HCl (180 mg, 1.5 equiv), HOBt (127 mg 1.5 equiv), and DIPEA (0.16 mL, 1.5 equiv), and the mixture was stirred for 30 min at room temperature under an argon atmosphere. 4-Fluorobenzylamine (75 μL, 1.05 equiv) and DIPEA (0.16 mL, 1.5 equiv) were added to the reaction mixture. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was poured into water and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with saturated NaHCO3 (2 × 10 mL), brine, dried over Na2SO4, and concentrated under reduced pressure. The product was triturated with mixture of EtOAc in hexanes (2–3 times) to afford 33a (279 mg, 76% yield) as a red solid. TLC: 3% MeOH in DCM, Rf = 0.45; visualized with UV. Isomer data: 1H NMR (300 MHz, DMSO) δ 9.16 (t, 1H, J = 5.52 and 11.28 Hz), 8.66 (d, 1H, J = 3.72 Hz), 8.53 (t, 1H, J = 1.5 and 3.6 Hz, major), 8.23 (d, 1H, J = 8.34 Hz), 8.03−7.92 (m, 2H), 7.81−7.74 (m, 2H), 7.72−7.67 (q, 1H), 7.62−7.54 (m, 2H), 7.43 (t, 2H, J = 8.43 and 14.07 Hz), 7.24−7.16 (m, 2H), 4.49 (d, 2H, J = 5.73 Hz, NHCH2), 4.36−4.30 (q, 2H, OCH2), 2.64 (s, 1.56H, CH3), 2.35 (s, 1.44H, CH3), 1.34 (t, 3H, J = 7.08 and 14.16 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 166.24, 165.92, 162.23, 158.06, 151.81, 150.81, 139.01, 138.22, 135.56, 131.55, 131.33, 131.17, 130.25, 129.84, 129.74, 128.04, 127.85, 127.34, 125.51, 125.33, 122.67, 121.57, 118.62, 115.69, 115.41, 112.91, 61.44, 42.34, 14.67, 13.34. MS (ESI) m/z = 586.1 [M + H]+. HRMS (ESI): calcd for C32H26N3O5ClF [M + H]+ m/z = 586.1545, found 586.1548. HPLC purity: 98.73%.
Compounds 33b–j were synthesized by an above synthetic procedure described for the preparation of amide 33a using appropriate starting materials. Each compound was triturated with the mixture of EtOAc in hexanes (2–3 times) to afford desired compound.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-((3-methoxyphenyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33b)
Red solid (226 mg, 62% yield). TLC: 3% MeOH in EtOAc, Rf = 0.47; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 10.64 (s, 1H, NH), 8.65 (d, 1H, J = 3.81 Hz), 8.52 (t, 1H, J = 1.83 and 3.63 Hz), 8.22−8.14 (m, 2H), 8.06 (dd, 1H, J = 2.16 and 8.46 Hz), 7.81−7.70 (m, 3H), 7.64−7.53 (m, 2H), 7.43 (s, 1H), 7.29−7.27 (m, 2H), 6.74−6.70 (m, 1H), 4.37−4.30 (q, 2H, OCH2), 3.75 (s, 3H, OCH3), 2.70 (s, 0.58H; minor isomer, CH3), 2.33 (s, 2.42H; major isomer, CH3), 1.33 (t, 3H, J = 7.11 and 14.19 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 165.91, 164.69, 162.20, 160.01, 157.96, 151.75, 150.82, 140.40, 139.00, 138.22, 131.78, 131.52, 131.21, 130.66, 127.96, 125.69, 125.28, 121.63, 118.58, 112.32, 109.89, 105.83, 61.42, 55.51, 14.66, 13.30. MS (ESI) m/z = 584.1 [M + H]+. HRMS (ESI): calcd for C32H26N3O6Cl [M]+ m/z = 583.1510, found 583.1524.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-((3,4-dimethoxyphenyl)-carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33c)
Red solid (261 mg, 68% yield). TLC: 3% MeOH in EtOAc, Rf = 0.54; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 10.52 (s, 1H, NH), 8.67 (d, 1H, J = 3.72 Hz), 8.56−8.50 (m, 1H), 8.27−8.14 (m, 2H), 8.06− 7.94 (m, 1H), 7.82−7.73 (m, 3H), 7.64−7.54 (m, 2H), 7.48−7.44 (m, 1H), 7.29−7.26 (dd, 1H, J = 2.16 and 8.67 Hz), 6.97−6.94 (d, 1H, J = 8.82 Hz), 4.38−4.31 (q, 2H, OCH2), 3.75 (s, 6H, diOCH3), 2.72 (s, 0.59H; minor isomer, CH3), 2.34 (s, 2.41H; major isomer, CH3), 1.34 (t, 3H, J = 7.08 and 14.16 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 165.92, 164.21, 162.21, 158.01, 151.75, 150.81, 150.35, 149.00, 148.71, 145.79, 138.01, 138.83, 138.36, 138.22, 132.85, 131.84, 131.59, 131.19, 131.01, 130.18, 129.88, 128.01, 127.92, 127.38, 127.05, 125.72, 125.28, 122.59, 121.60, 118.58, 118.32, 112.93, 112.44, 111.97, 104.97, 61.43, 56.17, 55.84, 55.38, 14.66, 13.30. MS (ESI) m/z = 614.1 [M + H]+. HRMS (ESI): calcd for C33H29N3O7Cl [M + H]+ m/z = 614.1694, found 614.1696.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-((cyclopropylmethyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33d)
Red solid (249 mg, 75% yield). TLC: 3% MeOH in DCM, Rf = 0.43; visualized with UV. Major Z-isomer data: 1H NMR (500 MHz, DMSO) δ 8.68−8.64 (m, 2H), 8.54 (t, 1H, J = 1.8 and 3.55 Hz), 8.23−8.20 (m, 1H), 8.01−7.92 (m, 2H), 7.78−7.74 (m, 2H), 7.68 (d, 1H, J = 8.3 Hz), 7.60−7.57 (m, 2H), 4.38−4.33 (q, 2H, OCH2), 3.18 (m, 2H, NHCH2), 2.72 (s, 0.64H; minor isomer, CH3), 2.35 (s, 2.36H; major isomer, CH3), 1.35 (t, 3H, J = 7.1 and 14.2 Hz, CH3), 1.06−1.0 (m, 1H, CH), 0.49−0.45 (m, 2H, CH2), 0.28−0.25 (m, 2H, CH2). 13C NMR (125 MHz, DMSO): δ 165.51, 165.44, 161.74, 157.65, 151.27, 150.29, 138.54, 138.14, 131.07, 130.56, 129.68, 129.37, 127.54, 127.29, 126.66, 124.96, 124.80, 112.16, 121.05, 118.16, 112.28, 60.92, 43.24, 14.17, 12.81, 10.65. MS (ESI) m/z = 532.1 [M + H]+. HRMS (ESI): calcd for C29H27N3O5Cl [M + H]+ m/z = 532.1639, found 532.1640.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-(cyclopropylcarbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33e)
Red solid (223 mg, 69% yield). TLC: 3% MeOH in DCM, Rf = 0.46; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.68 (d, 1H, J = 4.26 Hz), 8.63 (d, 1H, J = 3.6 Hz), 8.52 (s, 1H), 8.22 (d, 1H, J = 7.41 Hz), 7.97−7.88 (m, 2H), 7.79−7.73 (m, 2H), 7.67−7.52 (m, 3H), 4.37−4.30 (q, 2H, OCH2), 2−88−2.82 (m, 1H, CH) 2.68 (s, 0.46H; minor isomer, CH3), 2.35 (s, 2.54H; major isomer, CH3), 1.33 (t, 3H, J = 7.08 and 14.13 Hz, CH3), 0.75−0.69 (m, 2H, CH2), 0.58−0.53 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.19, 165.92, 162.20, 158.09, 151.76, 150.75, 139.02, 138.83, 138.34, 138.06, 131.04, 130.17, 129.88, 128.05, 127.75, 127.16, 125.51, 125.27, 122.59, 121.50, 118.58, 112.83, 61.42, 23.23, 14.67, 13.33, 6.16. MS (ESI) m/z = 518.1 [M + H]+. HRMS (ESI): calcd for C28H25N3O5Cl [M + H]+ m/z = 518.1483, found 518.1488.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-((tetrahydro-2H-pyran-4-yl)-carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33f)
Red solid (246 mg, 70% yield). TLC: 3% MeOH in DCM, Rf = 0.45; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.69 (m, 2H), 8.53 (t, 1H, J = 1.71 and 3.51 Hz), 8.26−8.20 (m, 1H), 8.02−7.87 (m, 2H), 7.79 (s, 1H), 7.75 (d, 1H, J = 8.1 Hz), 7.69−7.55 (m, 3H), 4.38−4.31 (q, 2H, OCH2), 4.04−3.92 (m, 1H, CH), 3.90−3.84 (m, 2H, CH2), 3.45−3.39 (m, 2H, CH2), 2.71 (s, 0.58H; minor isomer, CH3), 2.34 (s, 2.42H; major isomer, CH3), 1.85−1.75 (m, 2H, CH2), 1.60−1.45 (m, 2H, CH2), 1.33 (t, 3H, J = 7.08 and 14.01 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 166.29, 165.92, 165.37, 162.21, 158.13, 156.81, 152.40, 151.81, 150.77, 146.86, 141.35, 139.03, 138.55, 138.40, 131.41, 131.02, 130.59, 130.24, 127.93, 127.78, 127.16, 125.33, 124.64, 122.65, 121.49, 118.62, 110.44, 66.29, 61.44, 46.06, 32.62, 14.67, 13.36. MS (ESI) m/z = 562.1 [M + H]+. HRMS (ESI): calcd for C30H29N3O6Cl [M + H]+ m/z = 562.1745, found 562.1753.
(Z)-Ethyl 3-(4-((5-(4-Chloro-3-(((tetrahydro-2H-pyran-4-yl)-methyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33g)
Red solid (267 mg, 74% yield). TLC: 3% MeOH in DCM, Rf = 0.45; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.69−8.63 (m, 2H), 8.53 (s, 1H), 8.22 (d, 1H, J = 9.06 Hz), 7.99−7.90 (m, 2H), 7.81−7.74 (m, 1H), 7.77 (s, 1H), 7.67−7.53 (m, 3H), 4.38−4.28 (q, 2H, OCH2), 3.90−3.81 (m, 2H, CH2), 3.32−3.22 (m, 2H, CH2), 3.19−3.13 (m, 2H, NHCH2), 2.72 (s, 0.58H; minor isomer, CH3), 2.34 (s, 2.42H; major isomer, CH3), 1.84−1.71 (m, 1H, CH), 1.69−1.60 (m, 2H, CH2), 1.33 (t, 3H, J = 7.08 and 13.95 Hz, CH3), 1.26−1.15 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 178.58, 166.65, 166.24, 165.92, 162.22, 158.13, 156.80, 152.40, 151.79, 150.77, 146.85, 141.35, 139.02, 138.68, 138.52, 131.72, 131.02, 130.60, 127.95, 127.88, 127.79, 125.30, 124.65, 122.65, 121.51, 119.60, 112.86, 110.42, 108.52, 67.22, 61.43, 45.27, 36.25, 31.22, 14.69, 13.43. MS (ESI) m/z = 576.2 [M + H]+. HRMS (ESI): calcd for C31H31N3O6Cl [M + H]+ m/z = 576.1901, found 576.1913.
(Z)-Ethyl 4-(4-((5-(4-Chloro-3-((4-fluorobenzyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33h)
Red solid (300 mg, 79% yield). TLC: 3% MeOH in DCM, Rf = 0.47; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 9.17 (t, 1H, J = 5.88 and 11.76 Hz), 8.62 (s, 1H), 8.16−7.97 (m, 6H), 7.75 (s, 1H), 7.69 (d, 1H, J = 8.4 Hz), 7.60 (d, 1H, J = 3.33 Hz), 7.45−7.41 (m, 2H), 7.22−7.18 (m, 2H), 4.49 (d, 2H, J = 5.76 Hz, NHCH2), 4.32−4.25 (q, 2H, OCH2), 2.62 (s, 0.74H; minor isomer, CH3), 2.33 (s, 2.26H; major isomer, CH3), 1.31 (t, 3H, J = 7.08 and 14.1 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 166.23, 166.07, 165.23, 162.53, 158.66, 158.19, 154.90, 151.00, 150.74, 150.37, 149.06, 144.85, 142.46, 138.22, 138.04, 135.57, 131.61, 131.34, 130.78, 130.35, 129.73, 127.99, 127.83, 126.52, 125.53, 124.10, 121.33, 118.28, 117.52, 115.91, 115.69, 115.41, 112.89, 61.05, 44.84, 14.69, 13.47. MS (ESI) m/z = 586.1 [M + H]+. HRMS (ESI): calcd for C32H26N3O5ClF [M + H]+ m/z = 586.1545, found 586.1549.
(Z)-Ethyl 4-(4-((5-(4-Chloro-3-((3-methoxyphenyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33i)
Dark red solid (272 mg, 67% yield). TLC: 3% MeOH in EtOAc, Rf = 0.49; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 10.66 (s, 1H, NH), 8.60 (d, 1H, J = 3.0 Hz), 8.20 (d, 1H, J = 1.5 Hz), 8.14−7.94 (m, 5H), 7.82−7.73 (m, 2H), 7.63−7.54 (m, 1H), 7.45−7.39 (m/brs, 1H), 7.29−7.23 (m, 2H), 6.74−6.69 (m, 1H), 4.323−4.26 (q, 2H, OCH2), 3.75 (s, 3H, OCH3), 2.70 (s, 072.H; minor isomer, CH3), 2.32 (s, 2.31H; major isomer, CH3), 1.32 (t, 3H, J = 7.05 and 14.04 Hz, CH3). 13C NMR (75 MHz, DMSO): δ 165.70, 164.69, 162.43, 160.01, 158.07, 152.30, 150.79, 150.44, 142.44, 140.41, 138.20, 131.58, 131.22, 130.74, 130.16, 129.06, 127.92, 125.76, 125.46, 121.36, 117.42, 112.97, 112.31, 109.89, 105.82, 61.04, 55.51, 55.38, 54.95, 14.69, 13.34. MS (ESI) m/z = 584.1 [M + H]+. HRMS (ESI): calcd for C30H21N3O6Cl [M − H]− m/z = 584.1588, found 584.1596.
(Z)-Ethyl 4-(4-((5-(4-Chloro-3-((cyclopropylmethyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (33j)
Red solid (364 mg, 82% yield). TLC: 3% MeOH in DCM, Rf = 0.44; visualized with UV. Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.74 (t, 1H, J = 5.31 and 10.5 Hz), 8.59 (s, 1H), 8.07 (t, 2H, J = 8.73 and 17.7 Hz), 8.0 (d, 4H, J = 7.83 Hz), 7.75 (s, 1H), 7.66 (d, 1H, J = 8.85 Hz), 7.58 (d, 1H, J = 3.96 Hz), 4.32−4.25 (q, 2H, OCH2), 3.16 (t, 2H, J = 6.09 and 12.42 Hz, NHCH2), 2.68 (s, 0.66H; minor isomer, CH3), 2.32 (s, 2.34H; major isomer, CH3), 1.31 (t, 3H, J = 7.11 and 14.16 Hz, CH3), 1.09−0.98 (m, 1H, CH), 0.48−0.42 (m, 2H, CH2), 0.28−0.23 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 166.00, 166.07, 165.92, 162.49, 158.74, 157.92, 152.39, 150.77, 150.34, 148.97, 142.47, 138.63, 138.42, 131.62, 131.12, 130.77, 127.95, 127.73, 127.20, 125.50, 121.07, 119.60, 117.51, 112.81, 61.06, 43.68, 14.70, 13.39, 11.17, 3.36. MS (ESI) m/z = 532.1 [M + H]+. HRMS (ESI): calcd for C29H27N3O5Cl [M + H]+ m/z = 532.1639, found 532.1642.
General Synthesis of Target Compounds 34a−d and 34f−k
(Z)-3-(4-((5-(4-Chloro-3-((4-fluorobenzyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-benzoic Acid (34a)
To a stirred suspension of ester 33a (80 mg, 1 equiv) in THF/EtOH/H2O (4:2:1, 7 mL) was added LiOH (32 mg, 10 equiv). The reaction mixture was stirred at room temperature for 12 h. Solvent was removed in vacuo, and residue was acidified to pH 2−3 using 20% citric acid solution. The product was extracted with EtOAc (3 × 15 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The product was crystallized in EtOH, and solid was collected, washed with EtOAc and then hot solutions of 20−30% EtOAc in hexanes to afford target compound 34a (47 mg, 62% yield) as a red solid. Major Z-isomer data: 1H NMR (500 MHz, DMSO) δ 9.15 (t, 1H, J = 5.8 and 11.7 Hz), 8.68 (d, 1H, J = 3.05 Hz), 8.56 (t, 1H, J = 1.7 and 3.85 Hz), 8.20 (d, 1H, J = 7.45 Hz), 8.04−7.94 (m, 2H), 7.82−7.69 (m, 2H), 7.71 (t, 1H, J = 8.15 and 16.45 Hz), 7.62−7.55 (m, 2H), 7.45−7.42 (m, 2H), 7.22−7.17 (m, 2H), 4.49 (d, 2H, J = 5.85 Hz, NHCH2), 2.68 (s, 0.76H; minor isomer, CH3), 2.36 (s, 2.24H; major isomer, CH3). MS (ESI) m/z = 556.1 [M − H]−. HRMS (ESI): calcd for C30H20N3O5ClF [M − H]− m/z = 556.1076, found 556.1079. HPLC purity: 97.24%.
Target compounds 34b–d and 34f–k were synthesized by an above synthetic procedure described for the preparation of compound 34a using appropriate starting materials. Each compound was crystallized in EtOH, and solid was collected, washed with EtOAc and then hot solutions of 20–30% EtOAc in hexanes to afford desired final compound. If necessary, the products were purified using 2–5% MeOH in DCM (1% AcOH in DCM) solvent system on automated flash column chromatography.
(Z)-3-(4-((5-(4-Chloro-3-((3-methoxyphenyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34b)
Red solid (111 mg, 69% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.04 (brs, 1H, COOH), 10.65 (s, 1H, NH), 8.69 (d, 1H, J = 3.16 Hz), 8.55 (t, 1H, J = 1.95 and 3.5 Hz), 8.31−8.19 (m, 2H), 8.08−7.97 (m, 1H), 7.80−7.70 (m, 3H), 7.65−7.55 (m, 2H), 7.43 (s, 1H), 7.29−7.28 (m, 2H), 6.74−6.69 (m, 1H), 3.76 (s, 3H, OCH3), 2.73 (s, 0.51H; minor isomer, CH3), 2.34 (s, 2.49H; major isomer, CH3). 13C NMR (75 MHz, DMSO): δ 172.50, 167.51, 167.28, 165.08, 164.69, 162.22, 160.01, 157.94, 151.59, 140.41, 138.93, 132.03, 131.93, 131.22, 130.72, 129.72, 129.47, 125.48, 124.69, 122.30, 121.74, 112.32, 109.89, 105.83, 55.52, 13.30. MS (ESI) m/z = 554.1 [M − H]−. HRMS (ESI): calcd for C30H21N3O6Cl [M − H]− m/z = 554.1119, found 554.1124. HPLC purity: 98.63%.
(Z)-3-(4-((5-(4-Chloro-3-((3,4-dimethoxyphenyl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34c)
Red solid (60 mg, 63% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.14 (brs, 1H, COOH), 10.52 (s, 1H, NH), 8.70 (d, 1H, J = 3.75 Hz), 8.56−8.51 (m, 1H), 8.28−8.15 (m, 2H), 8.08−7.95 (m, 1H), 7.80−7.74 (m, 3H), 7.65−7.55 (m, 2H), 7.47−7.43 (m, 1H), 7.30−7.27 (m, 1H), 6.97−6.90 (m, 1H), 3.75 and 3.74 (s, 6H, diOCH3), 2.74 (s, 0.49H; minor isomer, CH3), 2.35 (s, 2.49H; major isomer, CH3). 13C NMR (75 MHz, DMSO): δ 167.51, 164.22, 164.69, 162.22, 157.99, 151.70, 150.84, 149.00, 145.79, 138.93, 138.37, 131.93, 131.58, 130.46, 129.74, 127.94, 127.39, 125.73, 125.43, 122.30, 121.71, 118.96, 112.44, 111.98, 104.97, 105.83, 56.17, 55.84, 13.30. MS (ESI) m/z = 584.1 [M − H]−. HRMS (ESI): calcd for C31H23N3O7Cl [M − H]− m/z = 584.1225, found 584.1229. HPLC purity: 95.07%.
(Z)-3-(4-((5-(4-Chloro-3-((cyclopropylmethyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34d)
Red solid (56 mg, 60% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.10 (brs, 1H, COOH), 8.71−8.65 (m, 2H), 8.55 (s, 1H), 8.20 (d, 1H, J = 7.74 Hz), 8.02−7.94 (m, 2H), 7.83−7.66 (m, 3H), 7.62−7.53 (m, 2H), 3.19−3.13 (m, 2H, NHCH2), 2.73 (s, 0.71H; minor isomer, CH3), 2.35 (s, 2.29H; major isomer, CH3), 1.07−0.95 (m, 1H, CH), 0.48−0.42 (m, 2H, CH2), 0.28−0.23 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.50, 166.00, 162.22, 158.11, 151.71, 150.80, 138.94, 138.63, 131.93, 131.54, 130.07, 129.73, 128.01, 127.78, 127.15, 125.49, 122.31, 121.61, 118.97, 112.81, 43.71, 13.32, 11.16, 3.73. MS (ESI) m/z = 502.1 [M − H]−. HRMS (ESI): calcd for C27H21N3O5Cl [M − H]− m/z = 502.1170, found 502.1172. HPLC purity: 97.11%.
(Z)-3-(4-((5-(4-Chloro-3-(cyclopropylcarbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34f)
Red solid (64 mg, 68% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.12 (brs, 1H, COOH), 8.66−8.63 (m, 2H), 8.54 (t, 1H, J = 1.77 and 3.54 Hz), 8.20 (d, 1H, J = 8.16 Hz), 8.00−7.94 (m, 2H), 7.82−7.73 (m, 2H), 7.69−7.64 (m, 1H), 7.60−7.53 (m, 2H), 2.89−2.80 (m, 1H, CH), 2.71 (s, 1.10H; minor isomer, CH3), 2.34 (s, 1.90H; major isomer, CH3), 0.77−0.69 (m, 2H, CH2), 0.58−0.53 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.50, 167.19, 162.21, 158.06, 151.69, 150.78, 138.94, 138.38, 131.93, 131.57, 131.06, 130.15, 129.73, 127.99, 127.78, 127.15, 126.49, 122.29, 121.62, 118.95, 112.81, 23.22, 13.32, 6.18. MS (ESI) m/z = 488.1 [M − H]−. HRMS (ESI): calcd for C26H19N3O5Cl [M − H]− m/z = 488.1013, found 488.1017. HPLC purity: 95.74%.
(Z)-3-(4-((5-(4-Chloro-3-((tetrahydro-2H-pyran-4-yl)carbamoyl)-phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34g)
Red solid (72 mg, 76% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.11 (brs, 1H, COOH), 8.65−8.59 (m, 2H), 8.54 (t, 1H, J = 1.71 and 3.42 Hz), 8.20 (d, 1H, J = 8.16 Hz), 8.01−7.93 (m, 2H), 7.82−7.73 (m, 2H), 7.70−7.65 (m, 1H), 7.61−7.53 (m, 2H), 4.05−3.95 (m, 1H, CH), 3.91−3.84 (m, 2H, CH2), 3.46−3.41 (m, 2H, CH2), 2.72 (s, 0.77H; minor isomer, CH3), 2.34 (s, 2.23H; major isomer, CH3), 1.86−1.79 (m, 2H, CH2), 1.59−1.46 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.50, 165.37, 162.19, 158.07, 151.68, 150.79, 138.94, 138.57, 131.92, 131.56, 131.03, 130.13, 129.71, 128.02, 127.80, 127.14, 125.34, 122.29, 121.60, 118.96, 112.81, 66.28, 46.05, 32.65, 13.32. MS (ESI) m/z = 532.1 [M − H]−. HRMS (ESI): calcd for C28H23N3O6Cl [M − H]− m/z = 532.1275, found 532.1281. HPLC purity: 96.33%.
(Z)-3-(4-((5-(4-Chloro-3-(((tetrahydro-2H-pyran-4-yl)methyl)-carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34h)
Red solid (66 mg, 70% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.12 (brs, 1H, COOH), 8.65−8.59 (m, 2H), 8.54 (s, 1H), 8.19 (d, 1H, J = 7.77 Hz), 8.01−7.92 (m, 2H), 7.82−7.73 (m, 2H), 7.69−7.65 (m, 1H), 7.60−7.54 (m, 2H), 3.90−3.81 (m, 2H, CH2), 3.28−3.22 (m, 2H, CH2), 3.19−3.13 (m, 2H, NHCH2), 2.70 (s, 0.67H; minor isomer, CH3), 2.34 (s, 2.33H; major isomer, CH3), 1.84−1.72 (m, 1H, CH), 1.69−1.61 (m, 2H, CH2), 1.30−1.14 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.00, 165.75, 161.70, 157.56, 151.18, 150.29, 138.42, 138.18, 131.41, 130.93, 130.54, 129.66, 129.22, 127.47, 127.31, 126.60, 124.98, 121.79, 121.10, 118.45, 112.32, 66.72, 44.78, 34.77, 30.40, 12.80. MS (ESI) m/z = 546.1 [M − H]−. HRMS (ESI): calcd for C29H25N3O6Cl [M − H]− m/z = 546.1432, found 546.1434. HPLC purity: 97.82%.
(Z)-4-(4-((5-(4-Chloro-3-((4-fluorobenzyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34i).
Red solid (66 mg, 70% yield). Isomer data: 1H NMR (300 MHz, DMSO) δ 12.85 (brs, 1H, COOH), 9.17 (t, 1H, J = 5.88 and 11.79 Hz), 8.64 (d, 1H, J = 3.66 Hz), 8.10−7.92 (m, 6H), 7.83−7.79 (m, 1H), 7.71 (dd, 1H, J = 2.67 and 8.4 Hz), 7.62−7.56 (m, 1H), 7.45−7.40 (m, 2H), 7.23−7.15 (m, 2H), 4.48 (d, 2H, J = 5.88 Hz, NHCH2), 2.66 (s, 1.65H, CH3), 2.34 (s, 1.35H, CH3). 13C NMR (75 MHz, DMSO): δ 167.31, 166.23, 166.11, 165.28, 162.44, 160.10, 159.56, 158.15, 152.22, 150.81, 150.33, 149.13, 142.20, 142.00, 138.22, 138.04, 135.56, 131.83, 131.59, 131.47, 131.17, 131.03, 130.93, 127.83, 127.73, 126.52, 125.90, 125.54, 121.41, 119.71, 117.47, 117.25, 115.69, 115.42, 112.93, 42.32, 13.37. MS (ESI) m/z = 556.1 [M − H]−. HRMS (ESI): calcd for C30H20N3O5ClF [M − H]− m/z = 556.1076, found 556.1077. HPLC purity: 98.93%.
(Z)-4-(4-((5-(4-Chloro-3-((3-methoxyphenyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34j)
Red solid (60 mg, 63% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 12.86 (brs, 1H, COOH), 10.55 (s, 1H, NH), 8.14−7.96 (m, 4H), 7.95−7.85 (m, 3H), 7.83−7.75 (m, 1H), 7.73−7.66 (m, 1H), 7.61−7.54 (m, 1H), 7.47−7.40 (m, 1H), 7.32−7.23 (m, 2H), 6.75−6.67 (m, 1H), 3.74 (s, 3H, OCH3), 2.74 (s, 0.24H; minor isomer, CH3), 2.35 (s, 2.87H; major isomer, CH3). 13C NMR (75 MHz, DMSO): δ 167.21, 165.10, 164.66, 162.20, 159.98, 157.91, 150.39, 140.46, 137.86, 132.01, 131.58, 130.86, 130.07, 129.92, 128.42, 127.51, 125.52, 123.55, 122.26, 119.96, 119.68, 112.32, 109.79, 105.80, 55.47, 13.36. MS (ESI) m/z = 554.1 [M − H]−. HRMS (ESI): calcd for C30H21N3O6Cl [M − H]− m/z = 554.1119, found 554.1122. HPLC purity: 96.34%.
(Z)-4-(4-((5-(4-Chloro-3-((cyclopropylmethyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (34k)
Red solid (89 mg, 63% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 12.83 (brs, 1H, COOH), 8.67 (t, 1H, J = 5.1 and 10.2 Hz), 8.60 (s, 1H), 8.06 (t, 2H, J = 8.01 and 14.19 Hz), 8.0 (d, 4H, J = 8.55 Hz), 7.76 (s, 1H), 7.67 (d, 1H, J = 8.64 Hz), 7.58 (d, 1H, J = 3.9 Hz), 3.17 (t, 2H, J = 5.7 and 11.4 Hz, NHCH2), 2.70 (s, 0.81H; minor isomer, CH3), 2.33 (s, 2.19H; major isomer, CH3), 1.08−0.94 (m, 1H, CH), 0.49−0.40 (m, 2H, CH2), 0.28−0.22 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.31, 166.01, 165.91, 162.41, 159.64, 158.22, 152.18, 150.76, 150.29, 149.09, 142.21, 138.60, 138.39, 131.83, 131.05, 130.21, 128.18, 127.74, 127.16, 126.44, 125.45, 121.33, 119.62, 117.542, 112.81, 43.71, 13.35, 11.17, 3.73. MS (ESI) m/z = 502.1 [M − H]−. HRMS (ESI): calcd for C27H21N3O5Cl [M − H]− m/z = 502.1170, found 502.1171. HPLC purity: 95.18%.
Synthesis of Target Compound 34e.
(Z)-2-Chloro-N-(cyclopropylmethyl)-5-(5-((1-(3-((cyclopropylmethyl)carbamoyl)phenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)-benzamide (34e)
34e was synthesized using an above synthetic procedure described for the preparation of compound 33a using 34d as a starting material. Product was crystallized in EtOH, and solid was collected, washed with EtOAc and then hot solutions of 20−30% EtOAc in hexanes to afford 34e as a red solid (51 mg, 78% yield). TLC: 4% MeOH in DCM, Rf = 0.42; visualized with UV. Major Zisomer data: 1H NMR (300 MHz, DMSO) δ 8.70−8.65 (m, 3H), 8.36 (s, 1H), 8.11 (d, 1H, J = 7.83 Hz), 7.99−7.91 (m, 2H), 7.77 (s, 1H), 7.68−7.65 (m, 2H), 7.58 (d, 1H, J = 3.9 Hz), 7.54−7.48 (m, 1H), 3.19−3.13 (q, 4H, 2NHCH2), 2.72 (s, 0.56H; minor isomer, CH3), 2.34 (s, 2.44H; major isomer, CH3), 1.07−0.95 (m, 2H, 2CH), 0.49− 0.41 (m, 4H, 2CH2), 0.27−0.23 (m, 4H, 2CH2). 13C NMR (75 MHz, DMSO): δ 166.30, 166.01, 162.14, 159.49, 158.05, 151.50, 150.80, 138.76, 138.61, 136.01, 131.51, 131.23, 131.07, 130.05, 129.23, 127.91, 127.78, 125.41, 123.34, 121.66, 120.89, 117.69, 112.76, 44.08, 43.71 13.31, 11.49, 11.16, 3.73 (t). MS (ESI) m/z = 557.1 [M + H]+. HRMS (ESI): calcd for C31H30N4O4Cl [M + H]+ m/z = 557.1956, found 557.1957.
Synthesis of 4-(5-Formylfuran-2-yl)benzoic Acid (36)
Aldehyde 36 was synthesized using the Suzuki coupling reaction described for the preparation of compound 30 using 4-carboxyphenylboronic acid 35 as a starting material. The workup of this reaction was different from previous reaction as the product was insoluble in organic solvents after acidification. The reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The pH of the suspension was adjusted to 1−2 by the addition of 6 N HCl solution. The precipitated product was filtered, washed successively with water (3 × 15 mL), EtOAc (2 × 10 mL), DCM, and dried under high vacuum overnight to get 4-(5-formylfuran-2-yl)benzoic acid 36 (1.11 g, 90% yield) as a white solid. 1H NMR (500 MHz, DMSO): δ 13.16 (s, 1H, COOH), 9.66 (s, 1H, CHO), 8.05 (dd, 2H, J = 1.5 and 6.5 Hz), 8.00 (dd, 2H, J = 2.0 and 7.0 Hz), 7.70 (d, 1H, J = 3.5 Hz), 7.46 (d, 1H, J = 4.0 Hz). 13C NMR (125 MHz, DMSO): δ 178.70, 167.17, 157.36, 152.66, 132.78, 131.73, 130.61, 125.47, 111.05. MS (ESI) m/z = 215.1 [M − H]−. HRMS (ESI): calcd for C12H7O4 [M − H]− m/z = 215.0344, found 215.0351.
Synthesis of Compounds 37a and 37b
37a and 37b were prepared using an above Knoevenagel condensation reaction procedure described for the preparation of compound 31a using 4-(5-formylfuran-2-yl)benzoic acid 36 and 27a (600 mg) and 27b (600 mg), respectively, as starting materials.
(Z)-4-(5-((1-(3-(Ethoxycarbonyl)phenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)benzoic Acid (37a)
Red solid (844 mg, 78% yield). Major Z-isomer data: 1H NMR (500 MHz, DMSO) δ 13.14 (s, 1H, COOH), 8.63 (d, 1H, J = 3.5 Hz), 8.49 (t, 1H, J = 1.5 and 3.5 Hz), 8.20 (d, 1H, J = 8.5 Hz), 8.03−7.90 (m, 4H), 7.76−7.71 (m, 1H), 7.65 (s, 1H), 7.56−7.51 (m, 2H), 4.35−4.29 (q, 2H, OCH2), 2.67 (s, 0.68H; minor isomer, CH3), 2.32 (s, 2.32H; major isomer, CH3), 1.34 (t, 3H, J = 7.5 and 14.5 Hz, CH3). 13C NMR (125 MHz, DMSO): δ 166.62, 165.40, 161.60, 158.05, 151.10, 150.51, 138.48, 133.15, 131.98, 131.25, 130.09, 129.45, 129.29, 127.32, 124.91, 124.70, 121.27, 118.02, 112.91, 60.88, 14.15, 12.78. MS (ESI) m/z = 443.1 [M − H]−. HRMS (ESI): calcd for C25H19N2O6 [M − H]− m/z = 443.1243, found 443.1254.
(Z)-4-(5-((1-(4-(Ethoxycarbonyl)phenyl)-3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)methyl)furan-2-yl)benzoic Acid (37b)
Red solid (920 mg, 85% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.08 (brs, 1H, COOH), 8.56 (s, 1H), 7.98−7.81 (m, 7H), 7.62−7.39 (m, 3H), 4.27−4.20 (q, 2H, OCH2), 2.59 (s, 0.68H; minor isomer, CH3), 2.28 (s, 2.32H; major isomer, CH3), 1.28 (t, 3H, J = 6.63 and 12.57 Hz, CH3). MS (ESI) m/z = 443.1 [M − H]−. HRMS (ESI): calcd for C25H19N2O6 [M − H]− m/z = 443.1243, found 443.1249.
Synthesis of Amides 38a–d
Compounds 38a,b and 38c,d were prepared using an above synthetic procedure described for the preparation of compound 33a using 37a (300 mg) and 37b (300 mg), respectively, and corresponding amines as a starting material.
(Z)-Ethyl 3-(4-((5-(4-((4-Fluorobenzyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-benzoate (38a).
Red solid (294 mg, 79% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 9.29 (t, 1H, J = 5.7 and 11.7 Hz), 8.64 (d, 1H, J = 3.72 Hz), 8.56−8.47 (m, 1H), 8.21 (d, 1H, J = 8.1 Hz), 8.06−7.86 (m, 4H), 7.76−7.61 (m, 2H), 7.57−7.44 (m, 2H), 7.42−7.32 (m, 2H), 7.20−7.12 (m, 2H), 4.45 (d, 2H, J = 5.67 Hz, NHCH2), 4.36−4.27 (q, 2H, OCH2), 2.67 (s, 0.49H; minor isomer, CH3), 2.32 (s, 2.51H; major isomer, CH3), 1.35−1.29 (m, 3H, CH3). 13C NMR (75 MHz, DMSO): δ 166.59, 166.05, 162.49, 159.25, 158.43, 151.15, 150.67, 147.18, 139.31, 136.55, 135.55, 132.60, 131.50, 131.28, 130.89, 130.04 129.94, 129.01, 128.69, 128.34, 125.75, 125.61, 122.86, 121.76, 119.92, 118.85, 115.84, 115.75, 113.36, 61.70 (d), 42.74, 14.99, 13.76 (d). MS (ESI) m/z = 552.1 [M + H]+. HRMS (ESI): calcd for C32H27N3O5F [M + H]+ m/z = 552.1935, found 552.1939.
(Z)-Ethyl 3-(4-((5-(4-((Cyclopropylmethyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (38b)
Red solid (248 mg, 74% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.71−8.64 (m, 2H), 8.52 (t, 1H, J = 1.8 and 3.6 Hz), 8.21 (d, 1H, J = 8.22 Hz), 8.03−7.91 (m, 4H), 7.78−7.70 (m, 2H), 7.59−7.50 (m, 2H), 4.37−4.28 (q, 2H, OCH2), 3.16 (t, 2H, J = 6.21 and 12.36 Hz, NHCH2), 2.70 (s, 0.71H; minor isomer, CH3), 2.34 (s, 2.29H; major isomer, CH3), 1.36−1.30 (m, 3H, CH3), 1.08−0.98 (m, 1H, CH), 0.47−0.41 (m, 2H, CH2), 0.26−0.21 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 165.91, 165.64, 165.01, 162.18, 159.00, 151.69, 150.82, 139.02, 138.84, 135.64, 131.01, 130.97, 130.09, 129.83, 128.56, 128.04, 127.00, 125.25, 122.53, 121.43, 118.53, 112.96, 61.40, 44.11, 14.66, 13.31, 11.46, 3.83. MS (ESI) m/z = 498.1 [M + H]+. HRMS (ESI): calcd for C29H28N3O5 [M + H]+ m/z = 498.2029, found 498.2030.
(Z)-Ethyl 4-(4-((5-(4-((4-Fluorobenzyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-benzoate (38c)
Red solid (297 mg, 80% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 9.20 (m, 1H), 8.64 (d, 1H, J = 3.84 Hz), 8.11−7.92 (m, 8H), 7.73 (d, 1H, J = 11.7 Hz), 7.58−7.49 (m, 1H), 7.40−7.31 (m, 2H), 7.19−7.09 (m, 2H), 4.48−4.42 (q, 2H, NHCH2), 4.32−4.21 (m, 2H, OCH2), 2.66 (s, 0.56H; minor isomer, CH3), 2.34 (s, 2.44H; major isomer, CH3), 1.33−1.25 (m, 3H, CH3). MS (ESI) m/z = 552.1 [M + H]+. HRMS (ESI): calcd for C32H27N3O5F [M + H]+ m/z = 552.1935, found 552.1943.
(Z)-Ethyl 4-(4-((5-(4-((Cyclopropylmethyl)carbamoyl)phenyl)-furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoate (38d)
Red solid (251 mg, 75% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 8.69 (t, 1H, J = 5.58 and 11.55 Hz), 8.63 (d, 1H, J = 3.66 Hz), 8.09−7.90 (m, 8H), 7.70 (s, 1H), 7.56−7.50 (m, 1H), 4.31−4.22 (q, 2H, OCH2), 3.15 (t, 2H, J = 5.7 and 12.0 Hz, NHCH2), 2.68 (s, 0.70H; minor isomer, CH3), 2.33 (s, 2.30H; major isomer, CH3), 1.33−127 (m, 3H, CH3), 1.08−0.99 (m, 1H, CH), 0.47−0.41 (m, 2H, CH2), 0.26−0.21 (m, 2H, CH2). MS (ESI) m/z = 498.1 [M + H]+. HRMS (ESI): calcd for C29H28N3O5 [M + H]+ m/z = 498.2029, found 498.2034.
Synthesis of Target Compounds 39a–d
Target compounds 39a–d were synthesized by an above synthetic procedure described for the preparation of compound 34a using appropriate starting materials. Each compound was crystallized in EtOH, and solid was collected, washed with EtOAc and then hot solutions of 20–30% EtOAc in hexanes to afford the desired final compound. If necessary, the products were purified using 2–5% MeOH in DCM (1% AcOH in DCM) solvent system on automated flash column chromatography.
(Z)-3-(4-((5-(4-((4-Fluorobenzyl)carbamoyl)phenyl)furan-2-yl)-methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (39a)
Red solid (103 mg, 73% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 13.12 (s, 1H, COOH), 9.20−9.16 (m, 1H), 8.69 (d, 1H, J = 3.84 Hz), 8.55 (t, 1H, J = 1.83 and 3.6 Hz, major isomer), 8.21 (d, 1H, J = 8.16 Hz), 8.08−7.98 (m, 4H), 7.83−7.73 (m, 2H), 7.60−7.53 (m, 2H), 7.40−7.35 (m, 2H), 7.16 (t, 2H, J = 8.94 and 17.82 Hz), 4.48 (d, 2H, J = 5.79 Hz, NHCH2), 2.75 (s, 1.06H; minor isomer, CH3), 2.35 (s, 1.94H; major isomer, CH3). 13C NMR (75 MHz, DMSO): δ 167.03, 165.34, 162.76, 161.74, 159.56, 158.43, 149.97, 148.23, 138.46, 135.68, 134.79, 131.44, 130.79, 129.32, 129.22, 128.19, 127.51, 125.04, 124.87, 121.82, 121.13, 119.46, 118.47, 115.15, 114.87, 112.60, 42.01, 12.84. MS (ESI) m/z = 522.1 [M − H]−. HRMS (ESI): calcd for C30H21N3O5F [M − H]− m/z = 522.1465, found 522.1467. HPLC purity: 97.73%.
(Z)-3-(4-((5-(4-((Cyclopropylmethyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (39b)
Red solid (90 mg, 64% yield). Isomer data: 1H NMR (300 MHz, DMSO) δ 13.11 (s, 1H, COOH), 8.72−8.65 (m, 2H), 8.55 (d, 1H, J = 14.52 Hz), 8.19 (d, 1H, J = 6.15 Hz), 8.05−7.92 (m, 4H), 7.82−7.70 (m, 2H), 7.59−7.51 (m, 2H), 3.16 (t, 2H, J = 6.35 and 12.28 Hz, NHCH2), 2.73 (s, 1.65H, CH3), 2.34 (s, 1.35H, CH3), 1.09− 0.97 (m, 1H, CH), 0.48−0.40 (m, 2H, CH2), 0.27−0.20 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 167.07, 165.22, 161.75, 159.93, 158.54, 151.20, 150.41, 138.49, 138.33, 135.20, 131.45, 130.77, 130.60, 129.65, 129.25, 128.14, 127.56, 124.99, 124.82, 121.82, 121.09, 118.48, 112.52, 112.29, 43.67, 12.87, 11.02, 3.39. MS (ESI) m/z = 568.1 [M − H]−. HRMS (ESI): calcd for C27H22N3O5 [M − H]− m/z = 468.1559, found 468.1561. HPLC purity: 96.48%.
(Z)-4-(4-((5-(4-((4-Fluorobenzyl)carbamoyl)phenyl)furan-2-yl)-methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (39c)
Red solid (109 mg, 77% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 12.82 (s, 1H, COOH), 9.18 (t, 1H, J = 5.7 and 11.64 Hz), 8.65 (d, 1H, J = 3.45 Hz), 8.10−7.94 (m, 8H), 7.74 (s, 1H), 7.59−7.54 (m, 1H), 7.40−7.32 (m, 2H), 7.16 (t, 2H, J = 8.85 and 17.67 Hz), 4.48 (d, 2H, J = 5.55, NHCH2), 2.73 (s, 0.64H; minor isomer, CH3), 2.34 (s, 2.36H; major isomer, CH3). 13C NMR (75 MHz, DMSO): δ 166.65, 165.14, 161.76, 158.36, 151.51, 150.21, 141.54, 135.53, 135.49, 130.56, 130.26, 129.14, 129.03, 128.00, 125.78, 124.70, 120.68, 116.77, 114.97, 114.68, 112.44, 41.83, 12.69. MS (ESI) m/z = 522.1 [M − H]−. HRMS (ESI): calcd for C30H21N3O5F [M − H]− m/z = 522.1465, found 522.1465. HPLC purity: 98.52%.
(Z)-4-(4-((5-(4-((Cyclopropylmethyl)carbamoyl)phenyl)furan-2-yl)methylene)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)benzoic Acid (39d).
Red solid (94 mg, 67% yield). Major Z-isomer data: 1H NMR (300 MHz, DMSO) δ 12.82 (s, 1H, COOH), 8.70−8.64 (m, 2H,), 8.09−7.79 (m, 8H), 7.73 (s, 1H), 7.57−7.53 (m, 1H), 3.15 (t, 2H, J = 6.0 and 12.0 Hz, NHCH2), 2.72 (s, 0.45H; minor isomer, CH3), 2.33 (s, 2.55H; major isomer, CH3), 1.09−0.97 (m, 1H, CH), 0.46−0.40 (m, 2H, CH2), 0.26−0.21 (m, 2H, CH2). 13C NMR (75 MHz, DMSO): δ 166.76, 165.09, 161.86, 158.55, 151.59, 150.27, 141.65, 135.21, 130.85, 130.44, 128.03, 125.87, 124.73, 120.71, 116.86, 112.45, 43.56, 12.79, 10.90, 3.27 MS (ESI) m/z = 568.1 [M − H]−. HRMS (ESI): calcd for C27H22N3O5 [M − H]− m/z = 468.1559, found 468.1557. HPLC purity: 95.37%.
Overexpression and Purification of Full Length Human XPA
The full length human XPA protein was purified according to previously described protocol.8,34 Briefly, Sf9 cells were infected with XPA virus, and the cellular pellet was lysed by dounce homogenization in buffer A containing 50 mM Tris, 100 mM NaCl, 0.1% (v/v) Triton X-100, 10% (v/v) glycerol, and 10 mM BME, along with a protease inhibitor cocktail. Following sonication, imidazole was added at 1 mM to the cellular extract, which was then loaded onto a 2 mL nickel-NTA agarose column. Bound protein was eluted in buffer A with 80 mM imidazole, and protein containing fractions were identified using Bradford analysis. Protein containing fractions were then pooled and loaded directly onto a 2 mL heparin-Sepharose column. Protein was eluted using a gradient from 100 mM to 1 M NaCl in heparin buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 10% (v/v) glycerol, and 1 mM DTT with protease inhibitor mix). Fractions containing XPA were identified using Bradford and SDS–PAGE analysis, pooled, and dialyzed overnight in heparin buffer and stored at −80 °C.
Overexpression and Purification of XPA98–239 MBD (DBD)
The XPA minimal binding domain was expressed in E. coli. Bl-21(DE3) from a plasmid driving expression of an N-terminal 6-histidine tagged fragment corresponding to amino acids 98−239.26,42 The recombinant protein was purified by a two-step column chromatography procedure similar to the methodology used for full length XPA.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was carried out using previously described procedure with the following modifications.7,29,43 Reactions were performed in volume of 20 μL containing a p32 labeled 30bp duplex DNA containing a single 1,2 dGpG cisplatin adduct.29 Compounds were preincubated on ice with the indicated amount of XPA and products separated by electrophoresis on a nondenaturing 6% polyacrylamide gel. Gels were cooled and loaded, and electrophoresis was performed at 4 °C. Gels were used to expose a PhosphorImager screen, band intensity was determined, and % binding and inhibition were calculated as we have previously described.7,43
DNA Intercalation Fluorescence Displacement Assay
The analysis of DNA binding or intercalation of XPA inhibitory compounds was performed as we have described previously.7 A competitive DNA intercalation assay was performed using SYBR-Green (Sigma) and salmon sperm DNA (Fisher). Reactions were carried out in 25 mM MOPS (pH 6.5) containing sonicated salmon sperm DNA (8.29 ng/μL), SYBR-Green, and varying concentrations of XPA inhibitors. Reactions were performed in a black 96-well plate in a final volume of 110 μL. Doxorubicin, a known noncovalent DNA binding chemotherapeutic, was used as a positive control. Fluorescence was measured using a BioTek Synergy H1 hybrid multimode microplate reader with an excitation wavelength of 485 nm, emission wavelength of 528 nm, and a read height of 7 mm. Data were collected using BioTek Gen5 reader software. Reactions were incubated a maximum of 5 min before measurements were collected.
Molecular Docking
During our previous studies8 we identified compound 1 interactions with XPA protein by examining the two solution NMR structures of the central domain of XPA (PDB codes 1XPA and 1D4U).24,25,44 Comparison of these two NMR structures showed that the putative DNA binding groove in the 1XPA structure (PDB code 1XPA) is narrower and deeper with a more closed conformation. The difference between these two structures may explain the flexibility of XPA in recognizing a wide variety of DNA lesions. The more closed and deep groove of 1XPA makes it more suitable for performing molecular docking experiments.
We have performed docking studies using solution NMR structure and PDB code 1XPA obtained from the Protein Data Bank (PDB)45 and prepared them using the Protein Preparation Wizard.46 In this step, force field atom types and bond orders are assigned, missing atoms are added, tautomer/ionization states are assigned, water orientations are sampled, Asn, Gln, and His residues are flipped to optimize the hydrogen bond network, and a constrained energy minimization is performed. XPA inhibitors were drawn in ChemDraw as MDL molfiles and prepared for docking using LigPrep47 including a minimization with the OPLS3 force field.48,49 All chiral centers were retained as specified in the literature. One low energy ring conformation per compound was generated. Ionization states and tautomer forms were enumerated at pH 7.0 ± 2.0 with Epik.50–52
XPA inhibitors were flexibly docked into the cleft defined by residues 138–142, 165–171, 174, and 177–181 using the Glide SP protocol53–55 with default settings. Docking poses were evaluated based on visual interrogation and calculated docking score. Potential amino acid interactions were determined based on proximity to each compound as revealed by docking analysis. XPA interactions with small molecules were viewed using Pymol using cartoon, surface, and compounds interaction views. All the molecular modeling within this study was performed using Maestro software, version 11 (Schrödinger),56 operating in a Linux environment.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to Dr. Lifan Zeng, Dr. Tax Georgiadis, and Jiyoon Li for technical assistance with HPLC and mass spectrometry (LCMS and HRMS). We are also grateful to Dr. Noeris K. Salam (Schrödinger LLC) for his assistance in docking studies. This work is supported by NIH Grants R01-CA180710 and R41-CA162648 and the Tom and Julie Wood Family Foundation.
Abbreviations Used
- XPA
xeroderma pigmentosum group A
- Pt
platinum
- NER
nucleotide excision repair
- HRR
homologous recombination repair
- MBD
minimal binding domain
- DBD
DNA binding domain
- EMSA
electrophoretic mobility shift assay
- SAR
structure–activity relationship
- Dox
doxorubicin
- RPA
replication protein A
Footnotes
Notes
The authors declare the following competing financial interest(s): Dr. Turchi is a co-founder and the Chief Scientific Officer of NERx Biosciences.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00780.
Molecular formula strings and biological data (CSV) Initial XPA inhibitory screening data, structures of commercially available compound 1 analogs, 1H NMR spectra of synthesized final compounds, E/Z isomers determination using NOESY NMR and molecular docking, overlays and docking poses of Z and E isomer of potent XPA inhibitors, and 2D interactions of XPA inhibitors with XPA protein (PDF)
REFERENCES
- (1).Gavande NS; VanderVere-Carozza PS; Hinshaw HD; Jalal SI; Sears CR; Pawelczak KS; Turchi JJ DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wang Y-Q; Wang P-Y; Wang Y-T; Yang G-F; Zhang A; Miao Z-H An update on poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors: Opportunities and challenges in cancer therapy. J. Med. Chem. 2016, 59, 9575–9598. [DOI] [PubMed] [Google Scholar]
- (3).Lee J-M; Ledermann JA; Kohn EC PARP Inhibitors for BRCA½ mutation-associated and BRCA-like malignancies. Ann. Oncol 2014, 25, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Galluzzi L; Senovilla L; Vitale I; Michels J; Martins I; Kepp O; Castedo M; Kroemer G Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- (5).Martin LP; Hamilton TC; Schilder RJ Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [DOI] [PubMed] [Google Scholar]
- (6).Gavande NS; VanderVere-Carozza PS; Pawelczak KS; Turchi JJ Targeting the nucleotide excision repair pathway for therapeutic applications In DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications; Kelley M, Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2016; pp 135–150. [Google Scholar]
- (7).Mishra AK; Dormi SS; Turchi AM; Woods DS; Turchi JJ Chemical inhibitor targeting the Replication Protein A–DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 2015, 93, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Neher TM; Shuck SC; Liu JY; Zhang JT; Turchi JJ Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010, 5, 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fu X; Hu J; Han HY; Hua YJ; Zhou L; Shuai WD; Du WY; Kuang CM; Chen S; Huang W; Liu RY High expression of XPA confers poor prognosis for nasopharyngeal carcinoma patients treated with platinum-based chemoradiotherapy. Oncotarget 2015, 6, 28478–28490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liu Y; Bernauer AM; Yingling CM; Belinsky SA HIF1 regulated expression of XPA contributes to cisplatin resistance in lung cancer. Carcinogenesis 2012, 33, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Weaver DA; Crawford EL; Warner KA; Elkhairi F; Khuder SA; Willey JC ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with cisplatin chemoresistance in non-small cell lung cancer cell lines. Mol. Cancer 2005, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lai TC; Chow KC; Fang HY; Cho HC; Chen CY; Lin TY; Chiang IP; Ho SP Expression of xeroderma pigmentosum complementation group C protein predicts cisplatin resistance in lung adenocarcinoma patients. Oncol. Rep. 2011, 25, 1243–1251. [DOI] [PubMed] [Google Scholar]
- (13).Jun HJ; Ahn MJ; Kim HS; Yi SY; Han J; Lee SK; Ahn YC; Jeong HS; Son YI; Baek JH; Park K ERCC1 expression as a predictive marker of squamous cell carcinoma of the head and neck treated with cisplatin-based concurrent chemoradiation. Br. J. Cancer 2008, 99, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jekimovs C; Bolderson E; Suraweera A; Adams M; O’Byrne KJ; Richard DJ Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Front. Oncol 2014, 4, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Luijsterburg MS; von Bornstaedt G; Gourdin AM; Politi AZ; Mone MJ; Warmerdam DO; Goedhart J; Vermeulen W; van Driel R; Hofer T Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell Biol. 2010, 189, 445–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koberle B; Roginskaya V; Wood RD XPA protein as a limiting factor for nucleotide excision repair and UV sensitivity in human cells. DNA Repair 2006, 5, 641–648. [DOI] [PubMed] [Google Scholar]
- (17).Sugitani N; Sivley RM; Perry KE; Capra JA; Chazin WJ XPA: A key scaffold for human nucleotide excision repair. DNA Repair 2016, 44, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fadda E Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput. Struct. Biotechnol. J 2016, 14, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Welsh C; Day R; McGurk C; Masters JRW; Wood RD; Koberle B Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [DOI] [PubMed] [Google Scholar]
- (20).Einhorn LH Curing metastatic testicular cancer. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 4592–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [DOI] [PubMed] [Google Scholar]
- (22).Chan DA; Giaccia AJ Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discovery 2011, 10, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ashworth AA Synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [DOI] [PubMed] [Google Scholar]
- (24).Ikegami T; Kuraoka I; Saijo M; et al. Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat. Struct. Mol. Biol. 1998, 5, 701–706. [DOI] [PubMed] [Google Scholar]
- (25).Buchko GW; Ni S; Thrall BD; Kennedy MA Structural features of the minimal DNA binding domain (M98-F219) of human nucleotide excision repair protein XPA. Nucleic Acids Res. 1998, 26, 2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sugitani N; Shell SM; Soss SE; Chazin WJ Redefining the DNA-binding domain of human XPA. J. Am. Chem. Soc. 2014, 136, 10830–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hilton B; Shkriabai N; Musich PR; Kvaratskhelia M; Shell S; Zou Y A new structural insight into XPA-DNA interactions. Biosci. Rep. 2014, 34, 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Saijo M; Takedachi A; Tanaka K Nucleotide excision repair by mutant Xeroderma Pigmentosum Group A (XPA) proteins with deficiency in interaction with RPA. J. Biol. Chem. 2011, 286, 5476–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Patrick SM; Turchi JJ Xeroderma pigmentosum complementation group A protein (XPA) modulates RPA-DNA interactions via enhanced complex stability and inhibition of strand separation activity. J. Biol. Chem. 2002, 277, 16096–16101. [DOI] [PubMed] [Google Scholar]
- (30).Koch SC; Kuper J; Gasteiger KL; Simon N; Strasser R; Eisen D; Geiger S; Schneider S; Kisker C; Carell T Structural insights into the recognition of cisplatin and AAF-dG lesion by Rad14 (XPA). Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8272–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yang Z; Roginskaya M; Colis LC; Basu AK; Shell SM; Liu Y; Musich PR; Harris CM; Harris TM; Zou Y Specific and efficient binding of xeroderma pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3′- and/or 5′-ssDNA branches. Biochemistry 2006, 45, 15921–15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Buchko GW; Tung CS; McAteer K; Isern NG; Spicer LD; Kennedy MA DNA-XPA interactions: a (31)P NMR and molecular modeling study of dCCAATAACC association with the minimal DNA-binding domain (M98-F219) of the nucleotide excision repair protein XPA. Nucleic Acids Res. 2001, 29, 2635–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Camenisch U; Dip R; Schumacher SB; Schuler B; Naegeli H Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat. Struct. Mol. Biol. 2006, 13, 278–284. [DOI] [PubMed] [Google Scholar]
- (34).Hermanson IL; Turchi JJ Overexpression and purification of human XPA using a Baculovirus expression system. Protein Expression Purif. 2000, 19, 1–11. [DOI] [PubMed] [Google Scholar]
- (35).Hellman LM; Fried MG Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kumar V; Chang C-K; Tan K-P; Jung Y-S; Chen S-H; Cheng Y-SE; Liang P-H Identification, synthesis, and evaluation of new neuraminidase inhibitors. Org. Lett. 2014, 16, 5060–5063. [DOI] [PubMed] [Google Scholar]
- (37).Liang Z; Ding X; Ai J; Kong X; Chen L; Chen L; Luo C; Geng M; Liu H; Chen K; Jiang H Discovering potent inhibitors against c-Met kinase: molecular design, organic synthesis and bioassay. Org. Biomol. Chem. 2012, 10, 421–430. [DOI] [PubMed] [Google Scholar]
- (38).Bowers EM; Yan G; Mukherjee C; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Moreau F; Desroy N; Genevard JM; et al. Discovery of new gram-negative antivirulence drugs: Structure and properties of novel E. coli WaaC inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4022–4026. [DOI] [PubMed] [Google Scholar]
- (40).Wang M; Mahrenholz A; Lee S-H RPA stabilizes the XPA-damaged DNA complex through protein-protein interaction. Biochemistry 2000, 39, 6433–6439. [DOI] [PubMed] [Google Scholar]
- (41).Lee S-H; Kim DK; Drissi R Human Xeroderma Pigmentosum Group A protein interacts with human Replication Protein A and inhibits DNA replication. J. Biol. Chem. 1995, 270, 21800–21805. [DOI] [PubMed] [Google Scholar]
- (42).Jones CJ; Wood RD Preferential binding of the Xeroderma Pigmentosum Group A complementing protein to damaged DNA. Biochemistry 1993, 32, 12096–12104. [DOI] [PubMed] [Google Scholar]
- (43).Shuck SC; Turchi JJ Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Buchko GW; Daughdrill GW; de Lorimier R; Rao BK; Isern NG; Lingbeck JM; Taylor J-S; Wold MS; Gochin M; Spicer LD; Lowry DF; Kennedy MA Interactions of human nucleotide excision repair protein XPA with DNA and RPA70 Delta C327: chemical shift mapping and 15N NMR relaxation studies. Biochemistry 1999, 38, 15116–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (47).Schrod̈inger release 2015–2: LigPrep, version 3.4; Schrödinger LLC: New York, NY, 2015. [Google Scholar]
- (48).Shivakumar D; Harder E; Damm W; Friesner RA; Sherman W Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J. Chem. Theory Comput. 2012, 8, 2553–8. [DOI] [PubMed] [Google Scholar]
- (49).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (50).Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya M Epik: A software program for Pk(a) prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–91. [DOI] [PubMed] [Google Scholar]
- (51).Greenwood JR; Calkins D; Sullivan AP; Shelley JC Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [DOI] [PubMed] [Google Scholar]
- (52).Schrod̈inger release 2015–2: Epik, version 3.2; Schrödinger LLC: New York, NY, 2015. [Google Scholar]
- (53).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–49. [DOI] [PubMed] [Google Scholar]
- (54).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–59. [DOI] [PubMed] [Google Scholar]
- (55).Small-molecule drug discovery suite 2015–2: Glide, version 6.7; Schrödinger LLC: New York, NY, 2015. [Google Scholar]
- (56).Maestro, version 11; Schrödinger, LLC, New York, NY, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.