

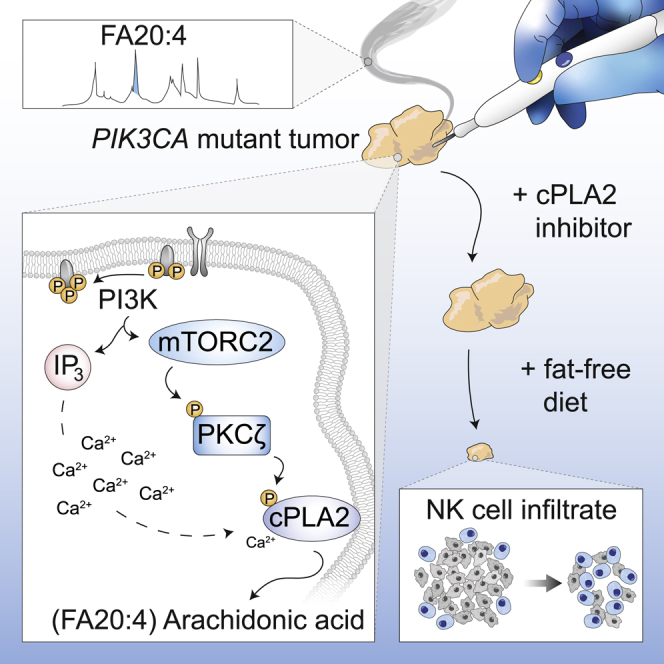
Summary
Oncogenic transformation is associated with profound changes in cellular metabolism, but whether tracking these can improve disease stratification or influence therapy decision-making is largely unknown. Using the iKnife to sample the aerosol of cauterized specimens, we demonstrate a new mode of real-time diagnosis, coupling metabolic phenotype to mutant PIK3CA genotype. Oncogenic PIK3CA results in an increase in arachidonic acid and a concomitant overproduction of eicosanoids, acting to promote cell proliferation beyond a cell-autonomous manner. Mechanistically, mutant PIK3CA drives a multimodal signaling network involving mTORC2-PKCζ-mediated activation of the calcium-dependent phospholipase A2 (cPLA2). Notably, inhibiting cPLA2 synergizes with fatty acid-free diet to restore immunogenicity and selectively reduce mutant PIK3CA-induced tumorigenicity. Besides highlighting the potential for metabolic phenotyping in stratified medicine, this study reveals an important role for activated PI3K signaling in regulating arachidonic acid metabolism, uncovering a targetable metabolic vulnerability that largely depends on dietary fat restriction.
Video Abstract
Keywords: PIK3CA, mTORC2, PKCζ, cPLA2, arachidonic acid, eicosanoids, iKnife, fat restriction, diet, cancer metabolism
Graphical Abstract
Highlights
-
•
The iKnife offers near real-time diagnosis of PIK3CA mutant breast cancers
-
•
Oncogenic PIK3CA promotes enhanced arachidonic acid via mTORC2-PKCζ-cPLA2 signaling
-
•
Mutant PIK3CA regulates proliferation beyond a cell autonomous manner
-
•
cPLA2 inhibition and dietary fat restriction suppress PIK3CA-induced tumorigenicity
Metabolic fingerprinting using the iKnife offers near real-time diagnosis of PIK3CA mutant breast cancers and connects oncogenic PIK3CA with enhanced arachidonic acid metabolism. cPLA2 inhibition shows remarkable synergy with dietary fat restriction to restore tumoral immune cell infiltration and inhibit growth of mutant PIK3CA-bearing breast tumors.
Introduction
In addition to the routine use of histological subtyping in cancer diagnosis and therapy decision-making, a renewed interest has emerged in the identification of novel predictive factors to improve patient stratification and therapeutic response. Malignant transformation and disease progression involve multiple changes in biosynthetic and energy production pathways, offering opportunities to exploit the clinical utility of metabolic tracking in diagnosis and disease monitoring (Vander Heiden and DeBerardinis, 2017). Cells can also reprogram pathways of nutrient acquisition and processing as a result of oncogenic activation of commonly perturbed signaling pathways (Tarrado-Castellarnau et al., 2016). A characteristic example is the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway, which regulates a wide range of transcriptional and post-translational programs to support the anabolic and catabolic requirements of proliferating cells (Carracedo and Pandolfi, 2008, Dibble and Manning, 2013, Fruman et al., 2017, Lien et al., 2016).
These observations raise some fundamental questions: whether we can use metabolic tracking for more effective screening of the molecular features underlying tumor pathogenesis and, ultimately, whether this information can be translated into better and more efficacious treatment strategies for each patient. Indeed, the concept of metabotyping has been widely applicable in characterizing functionally distinct traits that have the power to influence clinical decision-making (Gavaghan et al., 2000, Holmes et al., 2008, Nicholson et al., 2002, Nicholson et al., 2012). Here, we used rapid evaporative ionization mass spectrometry (REIMS), which coupled to the intelligent surgical device, also known as iKnife, allows for instantaneous chemical analysis of the aerosol generated during electrosurgical tissue ablation and cauterization, in the form of gas-phase ionic species. Unlike other technologies that are commonly used for metabolite profiling, such as liquid chromatography-mass spectrometry (LC-MS), REIMS analysis requires no sample preparation and allows for near real-time (1–2 s) lipidomic analysis and tissue recognition, based on multivariate classification analysis of spectral libraries of reference mass spectra.
The iKnife/REIMS can be used both in the intraoperative and biopsy collection settings to differentiate cancerous from non-cancerous tissues with very high precision, based on their lipidomic composition (Alexander et al., 2017, Balog et al., 2013, St John et al., 2017). However, the potential of using this technology beyond the pattern-level identification of tissues, to reveal the biological mechanisms underlying unique metabolic signatures, or identify which patients will likely benefit from a given treatment, has not yet been explored.
Results
Metabolic Phenotyping Using REIMS Predicts Molecular Markers Including Oncogenic Mutations in PIK3CA
We first examined whether REIMS-detected lipid signatures correlate with any established molecular markers of breast cancer of known prognostic and therapeutic value (Figure 1A). For this, we selected a panel of 43 breast cancer cell lines, 18 patient-derived xenograft (PDX), and 12 primary breast tumors that are well characterized for their estrogen (ER), progesterone (PR), and HER2 receptor status. REIMS profiling of cell lines consistently classified ER, HER2, and triple negative status (TN) with area under the curve (AUC) accuracies between 0.8–0.9, and 0.6–0.7 for PR (Figure 1B).
Figure 1.

REIMS Analysis Predicts Breast Cancer Molecular Markers Including Oncogenic Mutations in PIK3CA
(A) Schematic overview of sample preparation for REIMS analysis.
(B) Area under the curve (AUC) classification accuracies for ER, PR, HER2 receptor, and triple negative status of 43 breast cancer (BC) cell lines (median intensity of n = 3 biological replicates) following feature selection for phospholipids in the m/z range 600–900, and leave-one-out cross validation.
(C) Immunoblot analysis of estrogen inducible protein pS2 and predicted ESR1 expression in ER+ve MCF7 cells following treatment with 0.1% DMSO or indicated concentrations of 4-OHT for 72 h.
(D) Unsupervised hierarchical clustering of 872 lipid species detected by REIMS across 43 BC cell lines.
(E) Dendrogram of BC cell lines and isogenic MCF10A cells harboring either WT or MUT (E545K or H1047R) PIK3CA.
(F) Immunoblot analysis of mature SREBP1 transcription factor expression in nuclear extracts of the MCF10A PIK3CA isogenic panel.
(G) Relative exogenous fatty acid uptake in MCF10A PIK3CA WT and MUT cells following serum starvation for 1 h and supplementation with fluorescently labeled dodecanoic acid (n = 5 replicates).
(H and I) Unsupervised hierarchical clustering of 9 PIK3CA WT and 9 MUT breast PDX tumors (H) and (I) 5 WT and 7 MUT primary breast tumors. Individual rows in the heatmaps in (D), (H) and (I) correspond to scaled Z score phospholipid intensities (n = 3 biological replicates). Error bars represent ± SEM. n.s., not significant; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. p values in (C, bottom panel) and (G) were calculated with one-way ANOVA, followed by unpaired, two-tailed Student’s t test with Bonferroni correction.
Consistent with previous studies (Hilvo et al., 2011), the most striking differences in lipid profiles were observed between ER-positive (+ve) and -negative (−ve) breast cancer cell lines (Figures 1B and S1A; Table S1) and tumor specimens (Figure S1B). A surrogate marker for ER positivity, aside from its routine determination by immunohistochemistry (IHC), is expression of the estrogen receptor 1 (ESR1) gene. We built a regression model to predict ESR1 expression based on the spectral profiles obtained by REIMS and tested this in representative ER+ve cell lines treated with or without 4-hydroxy-tamoxifen (4-OHT). Of note, the predicted ESR1 expression was significantly reduced following 4-OHT treatment as compared to untreated controls (Figures 1C and S1C), suggesting that the modulation of ER signaling induces distinct lipidomic alterations, which are detectable by REIMS and are reversible by ER inhibition.
Figure S1.

Related to Figure 1
(A) Volcano plots of significantly altered phospholipids between receptor positive and negative cell lines. Black dots: not significantly altered; Red dots: significantly upregulated; Green dots: significantly downregulated phospholipids. (B) Area under the curve (AUC) classification accuracies for estrogen (ER), progesterone (PR), HER2 receptor and triple negative status of 30 primary and PDX breast tumors (median intensity of n = 3 separate sections per tumor) following feature selection for phospholipids in the m/z range 600-900 and leave-one-out cross validation. (C) Immunoblot analysis of estrogen inducible protein pS2 (top) and prediction of ESR1 expression (bottom) in ER+ve T47D cells following treatment with 0.1% DMSO or indicated concentrations of 4-OHT for 72 hours using REIMS. (D) NMF consensus maps summarizing the clustering of cell lines used in Figure 1D. The color map represents the correlation between cell lines in the same cluster when samples are divided into 2-6 groups. The highest cophenetic score was obtained for two clusters. (E) REIMS analysis of MCF10A PIK3CA WT and MUT cells cultured as 3D spheroids for 10 days. Clustering was performed as in Figure 1D using the median lipid intensities of 3 biological replicates. (F) Overall, precision and recall classification accuracies for PIK3CA mutation status in primary and PDX breast tumors (n = 30 in total), using all detectable lipid features (n = 1147) following 3-fold cross validation repeated 100 times with random forest as a classifier. n.s., not significant; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. P values in (C, bottom panel) were calculated with one-way ANOVA, followed by unpaired, two-tailed Student’s t test with Bonferroni correction.
With a robust lipidomic profile obtained using REIMS, we next performed unsupervised hierarchical clustering to partition all breast cancer cell lines on the basis of their spectral similarities measured over 872 lipid species. This analysis revealed two subtypes with distinctive signatures, in which REIMS-detected lipid species were significantly enriched (black) or depleted (gray) (Figure 1D). The observed clusters were also confirmed using a consensus non-negative matrix factorization (NMF) (Figure S1D).
To shed light on the mechanism that is driving this unique metabolic classification, we examined mutational enrichment of the cells between the two subtypes. Out of the top 150 genes that are frequently (>20%) mutated in these cell lines, oncogenic mutation in PIK3CA was the only one to be significantly (Fisher’s test, p value = 0.019) overrepresented in the lipid-enriched cluster (Figure 1E; Table S2). In accordance with this finding, analysis of isogenic MCF10A PIK3CA wild-type (WT) and mutant (MUT) (E545K and H1047R) cell lines also revealed clustering of the latter in the lipid-enriched group, both when cells were cultured in 2D, or 3D as spheroids (Figures 1E and S1E). Consistent with this stratification, performing gene and functional pathway enrichment analyses revealed KEGG-pathway ontologies relating to metabolic pathways that were significantly associated with the lipid-enriched subtype (Figures S2A and S2B). Specific overexpressed genes included FASN and ELOVL6, which are involved in de novo lipogenesis, and LDLRAP1, which facilitates exogenous lipid uptake (Figures S2C–S2E). Indeed, PIK3CA MUT cells displayed elevated induction of the de novo lipogenesis transcriptional regulator SREBP1 (Figure 1F) and higher exogenous FA uptake capacity (Figure 1G), suggesting that both could contribute to the lipid-enriched metabotype.
Figure S2.

(A) Pathway enrichment analysis of genes corresponding to the three most significantly enriched KEGG pathways determined from the 512 significantly upregulated genes in the lipid-enriched cluster from Figure 1D. (B) Gene interaction networks corresponding to the 55 genes encompassing the KEGG pathways in (A). (C) FASN, (D) ELOVL6, and (E) LDLRAP1 mRNA expression between cell lines in the lipid-enriched (n = 19) and depleted (n = 15) clusters. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. P values in (C), (D), and (E) were calculated with unpaired, two-tailed Student’s t test.
Most importantly, the observed metabolic stratification was also evident among PIK3CA WT and MUT breast cancer PDXs (Figure 1H) and primary tumors (Figure 1I; Table S3). Among the PDX tumors assessed (n = 18), only one was misclassified (BR5017), and this harbored a rare I391M mutation that has activity reminiscent of WT PIK3CA (Dan et al., 2010) (Figure 1H). Overall, PIK3CA mutation status in both PDX and primary tumors could be classified with an accuracy of 90% using all measurable lipid species (Figure S1F), suggesting that the iKnife/REIMS could be used for near real-time diagnosis of PIK3CA MUT breast cancers by MS analysis of aerosolized tissue material.
mTORC2 Signaling Downstream of Oncogenic PIK3CA Drives the Lipid-Enriched Phenotype
The effects of PI3K/Akt/mTOR signaling on lipid metabolism have been observed on numerous levels (Dibble and Manning, 2013, Lien et al., 2016, Saxton and Sabatini, 2017). To elucidate the specific mechanisms underlying the observed phenotype, we treated PIK3CA MUT cells with inhibitors targeting the activity of key nodes in the PI3K pathway (Figure 2A), albeit at concentrations that do not affect cell viability (Figure S3A). PI3K (BYL719, BKM120) and mTOR (rapamycin, torin 1) inhibition dramatically reduced relative phospholipid levels, but surprisingly, Akt inhibition with either MK2206 or GSK690693 did not (Figure 2B). Similar results were observed in a panel of 5 PIK3CA MUT breast cancer cell lines, with the exception of MCF7 cells that also responded to MK2206 (Figure S3B).
Figure 2.

Oncogenic PIK3CA Drives the Lipid-Enriched Phenotype via mTORC2 Signaling
(A) MCF10A PIK3CA MUT cells were treated with BYL719 or BKM120 (100 nM), MK2206, or GSK690693 (150 nM) for 72 h, or rapamycin (20 nM) for 4 h, or rapamycin or torin 1 (20 nM) for 72 h.
(B) Unsupervised hierarchical clustering of MCF10A E545K and H1047R MUT cells treated with PI3K, AKT, and mTOR inhibitors.
(C and D) Immunoblot analysis (C) and unsupervised hierarchical clustering (D) of MCF10A E545K and H1047R cells transfected with RAPTOR, RICTOR, or mTOR siRNA. Individual rows in the heatmaps in (B) and (D) correspond to scaled Z score phospholipid intensities (n = 3 biological replicates).
Figure S3.

Related to Figure 2
(A) Cell viability of MCF10A PIK3CA MUT cells following treatment with increasing concentrations of rapamycin, torin 1, BYL-719, BKM120, MK2206 or GSK690693 for 72 hours. (B) Unsupervised hierarchical clustering of the median phospholipid intensities of 5 PIK3CA MUT breast cancer cell lines (MCF7, T47D, MDAMB361, MDAMB453 and BT474) treated with 20 nM rapamycin, 100 nM BYL-719 and 150 nM MK2006 for 72 hours. (C) Immunoblot analysis of mTORC1 and mTORC2 signaling in the PIK3CA MUT isogenic panel. Cells were serum and growth-factor starved for 16 hours and subsequently stimulated with 5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone and 10 μg/ml insulin for 30 min. Data in (A) are presented as the mean ± SEM of n = 4 biological replicates and are representative of at least two independent experiments. n.s., not significant; ∗p ≤ 0.05; ∗∗p ≤ 0.01. P values in (A) were calculated with unpaired, two-tailed Student’s t test.
Interestingly, we did not observe an effect on relative phospholipid abundances following acute exposure to rapamycin for 4 h, despite inhibition of mTORC1 (Figures 2A, bottom left panel, and 2B). Because extended rapamycin treatment inhibits mTORC1 and mTORC2 (Figure 2A, bottom right panel) (Sarbassov et al., 2006), and both impinge upon lipogenesis (Düvel et al., 2010, Griffiths et al., 2013, Guri et al., 2017, Lee et al., 2017, Ricoult et al., 2016), we sought to investigate which of these complexes might contribute to the regulation of the observed phenotype. Knockdown of RICTOR or mTOR, but not RAPTOR, led to a significant reduction in relative phospholipid abundances (Figures 2C and 2D), pointing to a PIK3CA- and mTORC2-dependent metabolic phenotype that is largely independent of mTORC1 or Akt inhibition.
Oncogenic PIK3CA Drives Enhanced Arachidonic Acid Metabolism, thereby Promoting Cell Proliferation beyond a Cell-Autonomous Manner
Given that global lipidomic profiles could stratify breast cancer cell lines and tumors based on PIK3CA mutation status, we next aimed to characterize specific lipid alterations that are associated with oncogenic PIK3CA. Fatty acids (FAs), which are the main constituents of phospholipids and have additional effector functions in cancer pathogenesis, were profiled in the PIK3CA isogenic panel using REIMS. Among the most abundant FAs in PIK3CA MUT compared to WT cells were palmitoleate (FA16:1), palmitic acid (FA16:0), and oleic acid (FA18:1), all of which are established products of elevated lipogenesis and in line with the observed lipid enriched phenotype (Figure 1D; Table S4). Interestingly, the second most significantly elevated FA after palmitoleate was arachidonic acid (AA) (FA20:4), an omega-6 FA which is predominantly found in animal fats and is of particular relevance as a major regulator of pro-inflammatory responses in cancer, through the production of bio-active lipids known as eicosanoids (Wang and Dubois, 2010) (Figure 3A; Table S4). Importantly, in addition to the cell lines, significant elevations in AA were also observed in all the PIK3CA MUT breast PDX and primary tumors (Figures 3B and 3C) and across tumors of other tissue types including ovarian, pancreatic, and sarcomas (Figure 3D). In agreement with our REIMS findings, AA and downstream eicosanoids were also found to be significantly elevated in both PIK3CA MUT cells using LC-MS (Figures 3E, S4A, and S4B).
Figure 3.

Oncogenic PIK3CA Drives Enhanced Arachidonic Acid Metabolism
(A and B) Arachidonic acid (AA) levels measured by REIMS in MCF10A PIK3CA WT and MUT cells cultured under full 5% horse serum or fatty acid-free (FAF) conditions for 72 h (n = 3 biological replicates) (A). AA levels of 18 breast PDX tumors (n = 9 PIK3CA WT and n = 9 MUT) (left) (B). Three sections corresponding to different tumor regions were analyzed with REIMS. Data are summarized in the boxplot to the right.
(C) 12 primary breast tumors (n = 5 PIK3CA WT and n = 7 MUT) (left). Data are summarized in the boxplot to the right.
(D) Breast, ovarian, pancreatic, sarcoma, and colorectal PDX tumors (n = 5 PIK3CA WT and MUT tumors for breast, pancreatic, sarcoma, and colorectal tissues, and n = 4 PIK3CA WT and MUT ovarian PDX tumors).
(E) Heatmap and Venn diagram summarizing the intracellular eicosanoids that were significantly different between MCF10A PIK3CA WT and MUT cells. Rows correspond to the Z score scaled eicosanoid intensities detected by LC-MS (n = 3 biological replicates).
(F) Heatmap and Venn diagram summarizing the eicosanoids of the conditioned media (CM) that were significantly different between MCF10A PIK3CA WT and MUT cells.
(G) Cell proliferation assays of MCF10A PIK3CA WT cells cultured in CM derived from WT or H1047R MUT cells before or after lipid depletion (LD), with or without the supplementation of 25 μM AA, palmitate, or palmitoleate.
(H) Cell proliferation assays of MCF10A PIK3CA H1047R MUT cells before or after LD, with or without the supplementation of 25 μM AA, palmitate, or palmitoleate. Sulforhodamine B (SRB) protein staining was used in (G) and (H) to measure cell proliferation over 5 days (replicates from n = 3 wells). Error bars in (G) and (H) represent mean ± SEM for each time point. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. p values in (A)–(D) were calculated with unpaired, two tailed Student’s t test. Two-way ANOVA was used for (G) and (H).
Figure S4.

Intracellular levels of (A) Arachidonic acid (AA) and (B) PGE2 levels as measured by LC-MS profiling. (C) AA levels from the conditioned media (CM) of indicated cells before and after lipid depletion (LD). (D) Cell proliferation assays of MCF10A PIK3CA WT cells cultured in CM derived from WT or E545K MUT cells before or after LD, with or without the supplementation of 25 μM AA, palmitate or palmitoleate. (E) Cell proliferation assays of MCF10A PIK3CA E545K MUT cells before or after LD, with or without the supplementation of 25 μM AA, palmitate or palmitoleate. (F) Diacylglycerol (DAG) levels in MCF10A PIK3CA WT and MUT cells. (G) Diagram summarizing DAG contribution to AA production. Sulforhodamine B (SRB) protein staining was used in (D) and (E) to measure cell proliferation over 5 days. Data are presented as the mean ± SEM of n = 3-4 biological replicates and are representative of at least two independent experiments. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. P values in (A), (B), (C) and (F) were calculated using one-way ANOVA followed by unpaired, two-tailed Student’s t test with Bonferroni correction, and in (D) and (E) with two-way ANOVA.
To measure FAs that are secreted from cells, as opposed to those that might already exist in serum-supplemented media, cells were grown under FA-deprived conditions. Pro-inflammatory derivatives of AA were significantly increased in the media of PIK3CA MUT cells (Figure 3F), signifying a potential role for these bio-active lipids in tumor microenvironment (TME) interactions.
Next, to ascertain the functional consequences of elevated eicosanoid metabolism, the effects of PIK3CA MUT-derived conditioned media (CM) were assessed. PIK3CA WT cells displayed dramatically increased proliferative rate following incubation with CM obtained from MUT cells, and this was effectively rescued by depleting the lipids from the media (Figures 3G, S4C, and S4D). Moreover, the proliferation of both PIK3CA MUT cell lines was significantly reduced following incubation with their respective lipid-deprived CM (Figures 3H and S4E), whereas supplementation of lipid-deprived CM with AA, but not palmitate or palmitoleate, restored proliferation in both WT and MUT cells (Figures 3G, 3H, S4D, and S4E). Together, these data support not only a critical role for oncogenic PIK3CA in influencing autocrine- and paracrine-mediated cell proliferation, but also point to AA as an easily measured metabolic biomarker that could help with the diagnosis and treatment of PIK3CA MUT tumors.
Oncogenic PIK3CA Promotes Enhanced Arachidonic Acid Production via mTORC2-PKCζ-cPLA2 Signaling
To better understand the mechanism by which PIK3CA MUT cells have elevated AA, we assessed various pathways which contribute to its cellular pool, including: direct exogenous uptake, synthesis from linoleic acid, hydrolysis from diacylglycerol (DAG), or endogenous release from membrane phospholipid through phospholipase (PLAs) activity. Curiously, we noted a persistent increase in AA in PIK3CA MUT isogenic panel even when cells were cultured with FA-free media (Figure 3A). Additionally, DAG levels—that can be in part generated from the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2)—were significantly reduced in PIK3CA MUT cells (Figures S4F and S4G).
These results pointed to a potential role for phospholipases (PLAs), of which three main classes predominate: cytosolic/calcium-dependent (cPLA2), calcium-independent (iPLA2), and secretory (sPLA2) phospholipase A2 (Burke and Dennis, 2009). Among these, only cPLA2 displayed significantly higher enzymatic activity (Figure 4A), as well as elevated total protein levels and stability (Figures S5A and S5B) in the presence of oncogenic PIK3CA, whereas expression of PLA2G4A—the gene encoding cPLA2α—remained unchanged (Figure S5C).
Figure 4.

Oncogenic PIK3CA Signaling Triggers cPLA2-Induced Arachidonic Acid Production
(A) Enzymatic activity of cPLA2, iPLA2, and sPLA2 in the MCF10A PIK3CA isogenic panel.
(B–D) cPLA2 activity (B) and AA levels (C and D) measured by REIMS in MCF10A H1047R PIK3CA MUT cells following RAPTOR or RICTOR siRNA-mediated knockdown (C), or treatment with 100 nM ASB14780, 1 μM each of PKCα, β, ε, or ζ peptide inhibitors, 250 μM GSK650394, or 150 nM MK2206 for 72 h (D). Cells were grown under exogenous FAF conditions.
(E) cPLA2 activity following PKCζ inhibition with 1 μM peptide inhibitor for 72 h.
(F and G) Immunoblot analysis of the MCF10A PIK3CA isogenic panel following growth factor deprivation for 16 h and 30 min stimulation with serum and growth factors (F) or PKCζ inhibition with 1 μM peptide inhibitor for 72 h (G).
(H) Immunoblot analysis of activated Rac-1 and p38 MAPK in the MCF10A PIK3CA isogenic panel following PKCζ inhibition with 1 μM peptide inhibitor for 72 h.
(I) In vitro kinase assay of 100 ng and 0.5 μg/μL purified PKCζ and cPLA2 proteins, respectively.
(J) Immunoblot analysis of anti-HA immunoprecipitates derived from HA-tagged cPLA2 transfected MCF10A PIK3CA WT and MUT cells.
(K) Immunoblot analysis of anti-HA immunoprecipitates derived from HA-tagged cPLA2 transfected MCF10A PIK3CA WT and MUT cells treated where indicated with 1 μM PKCζ peptide inhibitor for 48 h.
(L) AA levels across H1047R MUT cells with CRISPR knockout of PLA2G4A reconstituted with WT or phosphoresistant cPLA2 isoforms.
(M) Diagram summarizing the proposed model for PI3K-mTORC2-PKCζ and calcium-dependent activation of cPLA2, leading to a concomitant increase in AA and downstream eicosanoids. Data are presented as the mean ± SEM of n = 3–6 biological replicates and are representative of at least two independent experiments. n.s., not significant; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. p values in (A) were calculated with unpaired, two tailed Student’s t test, and in (B)–(E), (I), and (L) with one-way ANOVA, followed by unpaired, two-tailed Student’s t test with Bonferroni correction.
Figure S5.

Related to Figure 4
(A) Immunoblot of phospholipases in MCF10A PIK3CA WT and MUT cells following serum and growth-factor deprivation for 16 hours and stimulation with serum and growth factors for 30 min. (B) Immunoblot analysis of cPLA2 protein decay following treatment with 50 μM cycloheximide (CHX) for the indicated times. Image and quantification is from one experiment. (C) Real-time quantitative PCR of PLA2G4A expression in the MCF10A PIK3CA isogenic panel. (D) AA levels measured by REIMS in MCF10A E545K PIK3CA MUT cells following RAPTOR and RICTOR siRNA-mediated knockdown 48 hours post transfection under exogenous FAF conditions. ELISA analysis of (E) AA and (F) PGE2 in the MCF10A PIK3CA isogenic panel 48 hours post RICTOR siRNA-mediated knockdown. (G) Immunoblot analysis of cPLA2 protein decay (top) and quantification (bottom) following RICTOR siRNA-mediated knockdown and treatment with 50 μM cycloheximide for the indicated times. Image and quantification is from one experiment. (H) Immunoblot analysis of substrates of conventional PKCα/β (p-IκBα Ser32 and p-RB Thr821/826), novel PKCε (p-STAT3 Ser727 and p-PKD Ser744/748) and atypical PKCζ (p-RKIP Ser153 and p-cPLA2 T376) isoforms following treatment of MCF10A E545K and H1047R MUT cells with 1 μM of each PKCα, β, ε, and ζ peptide inhibitors for 72 hours. (I) Enzymatic activity of cPLA2, iPLA2, and sPLA2 in the MCF10A PIK3CA isogenic panel following treatment with 100 nM ASB14780 for 72 hours. (J) AA levels measured by REIMS in MCF10A E545K MUT cells treated with 100 nM ASB14780, 1 μM each of PKCα, β, ε, and ζ peptide inhibitors, 250 μM GSK650394, or 150 nM MK2206 for 72 hours under exogenous FAF conditions. (K) Immunoblot (right) of total PKCζ and phospho-S505 and T376 cPLA2 of MCF10A PIK3CA WT and MUT cells following PKCζ siRNA-mediated knockdown, and cPLA2 activity (left) following 48 hours post-transfection. (L) Representative phospho-PKCζ Thr560 immunoreactivity images (left) of 9 PIK3CA MUT (blue) and 9 WT (red) breast PDX tumors. Scale bar = 250 μm. Quantification of percent positive regions (right) was performed using the IHC profiler plug-in for ImageJ. Data are presented as the mean ± SEM of n = 3-5 biological replicates and are representative of at least two independent experiments. n.s., not significant, ∗p ≤ 0.05; ∗∗p ≤ 0.01. P values in (C), (D), (E), (F), (I), (J) and (K, right) with one-way ANOVA followed by unpaired, two-tailed Student’s t test with Bonferroni correction, and in (L) with unpaired, two-tailed Student’s t test.
Consistent with the predominant role of mTORC2 in driving the lipid enriched phenotype in PIK3CA MUT cells (Figures 2C and 2D), RICTOR, but not RAPTOR, silencing rescued cPLA2 activity (Figure 4B), and led to a concomitant reduction in AA and prostaglandin E2 (PGE2) levels (Figures 4C and S5D–S5F). Importantly, mTORC2-specific inhibition was also accompanied by decreased cPLA2 stability (Figure S5G). To elucidate the mechanism underlying this observation, known substrates of mTORC2 including serum/glucocorticoid-regulated kinase 1 (SGK-1) and the protein kinase C (PKC) isoforms were inhibited in PIK3CA MUT cells. ASB14780—an indole-based compound that inhibits both cPLA2 translocation to membrane compartments and the interaction between phospholipid substrates with the enzyme active site (McKew et al., 2008, Tomoo et al., 2014)—was used as a positive control. Importantly, a significant reduction in AA and cPLA2’s enzymatic activity, reminiscent of that observed upon ASB14780 inhibition, was only observed following pharmacological and small interfering RNA (siRNA)-mediated inhibition of PKCζ (Figures 4D, 4E, and S5H–S5K).
Previous studies have shown that phosphorylation of cPLA2 on S505 regulates its activity and stability and that this is mediated, at least in part, by the p38 MAPK/ERK signaling pathway (Kramer et al., 1996, Lin et al., 1993). The contribution of PI3K-mTORC2 signaling to this process is unknown, as is the role of PKCζ in the regulation of cPLA2. Consistent with PKCζ being a direct substrate of PDK-1 (Chou et al., 1998) and mTORC2 phosphorylation (Li and Gao, 2014), it was found to be hyperphosphorylated in PIK3CA MUT cells (Figure 4F) and breast PDX tumors (Figure S5L). Moreover, its inhibition led to a marked reduction in MAPK/ERK signaling (Figure 4G), as well as p38 MAPK phosphorylation and active GTP-bound Rac-1 (Figure 4H), culminating in reduced cPLA2 phosphorylation at the S505 site (Figures 4G and S5L).
In addition to S505 phosphorylation, an increase in intracellular calcium levels is essential for sustained phospholipase activity and liberation of AA by cPLA2 (Ambs et al., 1995, Clark et al., 1995). Given that elevated PIP3 levels, induced by oncogenic PIK3CA, promote the activation of phospholipase C gamma 1 (PLCγ1), leading to an increase in cytosolic calcium via generation of inositol-1,4,5-trisphosphate (IP3) (Rameh et al., 1998), we hypothesized that this signaling node could also play a role in regulating cPLA2 activity and AA release downstream of active PI3Kα. In line with this premise, we observed higher phosphorylation of PLCγ1 (Figure S6A) and significantly elevated intracellular calcium levels in PIK3CA MUT cells (Figure S6B). Although genetic and pharmacological (U73122) inhibition of PLCγ1 led to a significant reduction in calcium flux in both PIK3CA WT and MUT cells (Figures S6C–S6E), an inhibitory effect on cPLA2 activity and AA levels was only observed in the MUT cells (Figures S6F–S6H), highlighting the importance of PLCγ1-mediated Ca2+ flux in sustaining elevated cPLA2 activity in the context of oncogenic PIK3CA.
Figure S6.

(A) Immunoblot analysis of phospho-Tyr783 PLCγ1 in MCF10A PIK3CA isogenics following serum and growth factor deprivation for 16 hours, and stimulation with serum and growth factors for 30 min. Densitometry values are either scaled to unstimulated or stimulated (bold) WT samples. (B) Measurement of intracellular calcium flux in MCF10A PIK3CA isogenics following serum and growth factor deprivation and stimulation for 30 min. (C) Immunoblot of MCF10A PIK3CA WT and MUT cells following siRNA-mediated knockdown of PLCγ1. (D) Intracellular calcium flux of MCF10A PIK3CA WT (top), E545K (middle) and H1047R (bottom) cells 48 hours post transfection with siPLCγ1, or (E) treatment with 2 μM U73122 for 24 hours. For the final 18 hours of the treatments, cells were serum and growth factor deprived, and stimulated with full media immediately prior to the assay. cPLA2 activity in MCF10A PIK3CA WT and MUT following (F) siRNA-mediated knockdown of PLCγ1 for 48 hours, or (G) treatment with 2 μM U73122 for 24 hours. (H) AA levels measured by REIMS in MCF10A PIK3CA isogenics following treatment with 2 μM U73122 for 24 hours. (I) Representative confocal images and (J) quantification of in situ proximity ligation assay (PLA) between cPLA2 and phospho-Thr560 PKCζ in MCF10A PIK3CA WT and MUT cells. (K) Immunoblot analysis of phospho-cPLA2 (T376) custom antibody in the MCF10A isogenic panel following serum and growth factor deprivation for 18 hours and subsequent stimulation for 30 min (left), treatment with 1 μM PKCζ peptide inhibitor for 72 hours (middle), and in MCF10A H1047R cPLA2 CRISPR knockout cells overexpressing a phosphoresistant mutant (T376A) cPLA2 (right). (L) Activity of cPLA2 in MCF10A PIK3CA WT or H1047R cPLA2 CRISPR knockout cells transfected with 9 μg of either WT-cPLA2, or S505A/T376A phosphoresistant mutant cPLA2 constructs. Activity was measured 48 hours post-transfection. Cell proliferation of MCF10A (M) PIK3CA WT and (N) E545K MUT cells expressing control shGFP, cPLA2-sh1 or sh5 under exogenous FAF conditions. Sulforhodamine B (SRB) protein staining was used to measure cell proliferation over 5 days. Data in (B), (D), (E), (F), (G), (H), (L), (M) and (N) are presented as the mean ± SEM of n = 3-6 biological replicates and are representative of at least two independent experiments. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; P values in (B), (D), (E), (M) and (N) were calculated using two-way ANOVA. One-way ANOVA followed by Student’s t test with Bonferroni correction was used for (F), (G), (H), (J)and (L).
Finally, because cPLA2 has a predicted PKCζ phosphorylation site (T376), we tested the possibility that it could serve as a direct substrate for PKCζ. In vitro kinase assays using purified PKCζ and cPLA2 suggested a direct interaction and phosphorylation (Figure 4I), and this was confirmed following immunoprecipitation (Figure 4J) and proximity ligation activity (PLA) assays (Figures S6I and S6J). To further evaluate cPLA2 as a candidate substrate for PKCζ, we developed a custom antibody recognizing the cPLA2 T376 phosphorylation site. Specificity was validated in both serum starved/stimulated samples (Figure S6K, left panel), following PKCζ inhibition (Figures S5K and S6K, middle), and in cPLA2 CRISPR knockout cells overexpressing a phosphoresistant MUT (T376A) cPLA2 (Figure S6K, right panel). Importantly, increased T376 phosphorylation was observed in the presence of oncogenic PIK3CA and this was reduced to levels equivalent to PIK3CA WT cells upon PKCζ inhibition (Figure 4K).
To ascertain the functional significance of these two phosphorylation sites (S505 and T376), endogenous cPLA2 in PIK3CA WT and H1047R MUT cells was reconstituted with WT or phosphoresistant MUT of cPLA2 (S505A or T376A) (Figure 4L). Knockout of cPLA2 in H1047R cells reduced AA to levels equivalent to the PIK3CA WT background, and this could be rescued with ectopic expression of the WT, but not MUT (S505A or T376A) cPLA2 (Figure 4L). Interestingly, the activity of exogenously expressed WT or MUT cPLA2 largely mirrored the trends in AA that were previously detected by REIMS (Figure S6L), suggesting that cPLA2 activity is highly dependent on the phosphorylation of S505 and T376 in the context of oncogenic PIK3CA. Overall, our model provides a unifying framework for several previously unconnected components of PI3K signaling, which converge on cPLA2 activation and enhanced AA metabolism (Figure 4M).
cPLA2 Inhibition and Dietary Fat Restriction Suppress PIK3CA-Induced Tumorigenicity and Restore Anti-cancer Immune Responses
If cPLA2 is required for PIK3CA oncogenicity, then targeting this enzyme could represent an attractive therapeutic strategy. For this, we used the inhibitor ASB14780, which displays excellent oral bioavailability and higher specificity for the cPLA2 isoform than other commonly used compounds such as Efipladib and Ecopladib (Lee et al., 2007, Tomoo et al., 2014). Furthermore, ASB14780 has been shown to ameliorate inflammatory pathologies including non-alcoholic fatty liver disease (NAFLD) and chronic obstructive pulmonary disease (COPD) through suppression of AA and prostaglandin synthesis (Kanai et al., 2016, Tomoo et al., 2014). However, the potential anti-neoplastic properties of this inhibitor remain obscure. Of note, PIK3CA mutation sensitized cells to pharmacological inhibition of cPLA2 with ASB14780 (Figures 5A and 5B), and this effect was more prominent under exogenous FA-free conditions (Figures 5C and 5D), alleviating any compensatory mechanisms to obtain AA. In addition to viability, cPLA2 inhibition significantly reduced the clonogenicity of PIK3CA MUT cells, and, importantly, both could be rescued by exogenous supplementation of AA (Figures 5C, 5D, and 5E). Near-identical results were obtained following genetic knockdown of cPLA2 using two constitutive short-hairpin RNAs (shRNAs, denoted as cPLA2-sh1 and cPLA2-sh5) (Figures 5F, 5G, and S6M), while WT cells were unaffected, suggesting that cPLA2 is dispensable in this setting (Figure S6N). To further confirm the importance of cPLA2-induced AA metabolism in PIK3CA MUT cancers, we suppressed cPLA2 in the isogenic panel and assessed their ability to form colonies under FA-free conditions. Although no significant difference in colony formation was detected in PIK3CA WT cells, there was a marked reduction in the number of colonies following cPLA2 knockdown in the MUT cells that was restored in the presence of AA (Figure 5H). Furthermore, using an inducible shRNA against cPLA2, we demonstrated that PIK3CA MUT, but not WT cells, rely on cPLA2 to form epithelial acini in 3D culture and sustain their proliferation (Figures 6A–6C).
Figure 5.

Genetic and Pharmacological Inhibition of cPLA2 Selectively Reduces Oncogenic PIK3CA-Mediated Tumorigenicity
(A–D) Cell viability of (A) PIK3CA WT and MUT MCF10A cells, and (B) breast cancer cell lines (PIK3CA WT: MDAMB134, Hs578T, AU565; PIK3CA MUT: MCF-7, CAL-51, MDAMB453) following treatment with increasing concentrations (20 nM–10 μM) of ASB14780 under full serum conditions for 72 h. The same treatments were also performed under fatty acid-free conditions in (C) and (D), in the presence or absence of exogenous supplementation of 25 μM AA.
(E) Clonogenic assays of MCF10A PIK3CA WT and MUT cells treated with increasing concentrations of ASB14780 as in (A)–(D). Treatments were performed under fatty acid-free conditions, with or without the supplementation of 25 μM AA.
(F) Immunoblot analysis confirming specific knockdown of cPLA2 using two independent constitutive shRNAs (sh1 and sh5) (left) and reduction in AA levels in MCF10A E545K/H1047R MUT cells using REIMS.
(G) Proliferation of MCF10A H1047R MUT cells expressing shGFP, cPLA2-sh1, or cPLA2-sh5 under exogenous FAF conditions. Sulforhodamine B (SRB) protein staining was used to measure cell proliferation over 5 days.
(H) Clonogenic assays of MCF10A PIK3CA WT and MUT cells expressing shGFP, cPLA2-sh1, or cPLA2-sh5 under FAF conditions, supplemented with or without 25 μM AA. Data in (A)–(H) are presented as the mean ± SEM of n = 3–4 biological replicates and are representative of at least two independent experiments. Data in (D) are presented as the mean viability of three PIK3CA MUT (MCF-7, CAL-51, MDAMB453) and WT (MDAMB134, Hs578T, AU565) measured in triplicate wells. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; p values in (A)–(D) and (G) were calculated using two-way ANOVA. For (E, right), (F, right), and (H, right), one-way ANOVA followed by unpaired, two-tailed Student’s t test with Bonferroni correction was applied.
Figure 6.

Oncogenic PIK3CA Serves as a Defining Biomarker for Sensitivity of Pre-clinical Models to cPLA2 Inhibition
(A) Immunoblot analysis confirming inducible knockdown of cPLA2 following induction with 2 μg/mL doxycycline.
(B) 3D acini formation of MCF10A PIK3CA WT and MUT cells following doxycycline-induced cPLA2-sh1 or shGFP expression. Cells were stained for Ki-67 (pink, Alexa Fluor 546), F-actin (red, Phalloidin 633), and DAPI (blue).
(C) Quantification of Ki-67 staining from treatments in (B).
(D) Schematic of in vivo experimental design and tumor profiling with REIMS.
(E and F) Tumor weights of (E) PIK3CA WT (BR1458) and (F) C420R MUT (BR1282) breast PDX tumors treated with 100 mg/kg of the cPLA2α pharmacological inhibitor ASB14780 under FAF diet (n = 8 mice for the BR1458 model, and n = 7 mice for the BR1282 for both the vehicle- and ASB14780-treated groups).
(G) Representative images of H&E staining from resected tumors in (E) and (F). The black masks in (G) represent viable tumor area, while unshaded regions correspond to necrotic tissue.
(H and I) Quantification of viable tumor area from (H) PIK3CA WT (BR1458) and (I) PIK3CA MUT (BR1282) tumor sections based on the analysis depicted in (G).
(J and K) AA levels measured by REIMS in (J) PIK3CA WT (BR1458) and (K) MUT (BR1282) tumors excised and snap frozen 2 h after the final dosing. Error bars in (C), (J), and (K) represent mean ± SEM, with data in (J) and (K) corresponding to tumor REIMS measurements from n = 7–8 mice. n.s., not significant;∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; p values in (E), (F), and (H)–(K) were calculated using one-way ANOVA followed by unpaired, two-tailed Student’s t test with Bonferroni correction.
To further evaluate the therapeutic effect of cPLA2 inhibition in primary breast cancers, we treated triple negative breast cancer (TNBC) (Figure 6D) PIK3CA WT (Figure 6E) and MUT (Figure 6F) PDX-bearing mice with the ASB14780 inhibitor in conjunction with a normal or near-isocaloric fat-free diet. A significant reduction in tumor weight was only observed in the PIK3CA MUT PDX model when both the inhibitor and a fat-free diet were administered in combination (Figure 6F). Corroborating these observations, histological analysis did not reveal any changes in tumor area for the PIK3CA WT BR1458 model (Figures 6G, left panels, and 6H), while a striking reduction in viable tumor regions was observed in PIK3CA MUT PDX-bearing mice treated with ASB14780 in fat-free diet (Figures 6G, right panels, and 6I). The concomitant increase in necrotic regions (as indicated by areas of pale eosinophilic cytoplasms, in addition to loss of nuclei and karyolysis), evidently contributed to a substantial proportion of the overall weight of the PIK3CA MUT-bearing tumor that was left after treatment of ASB14780 in fat-free diet (Figure 6G, bottom right panel). In the interest of measuring AA levels at the end of the treatment regime (Figure 6D), resected tumors were analyzed directly with REIMS. Although a fat-free diet alone led to a modest, yet significant reduction in the AA levels of the PDX tumors, the decrease was significantly more pronounced when accompanied with cPLA2 inhibition (Figures 6J and 6K), highlighting the role of dietary AA restriction in therapy response (Figures 6E–6I).
The observation that cPLA2 inhibition and fat-free diet selectively reduces the tumorigenicity of PIK3CA MUT cells raises the interesting prospect of modulating this response by altering dietary fat content. Indeed, it is becoming increasingly appreciated that the diet of Western populations commonly contains an excess of pro-inflammatory omega-6 to omega-3 FAs by up to 50 times (popularly referred to as the “Western” diet), and this may be implicated in the progression of breast and colorectal cancers (Patterson et al., 2012, Simopoulos, 2008). To further explore this premise, we injected triple negative CAL51 (PIK3CA MUT) and Hs578T (PIK3CA WT) cell lines stably expressing either control shGFP, or two independent shRNAs targeting cPLA2 (cPLA2-sh1 or sh5) into the mammary fat pad of BALB/c nude mice that had been preconditioned on either fat-free, balanced (omega6:omega3 = 1:1) or “Western” (omega6:omega3 = 50:1) diets (Figure 7A). Consistent with pharmacological inhibition, knockdown of cPLA2 significantly impaired the growth of PIK3CA MUT tumor xenografts under fat-free diet conditions (Figure 7B), and this therapeutic effect was completely reversed when animals were fed the AA-enriched “Western” diet (Figures 7C and 7D). Although there was a trend toward a reduction in overall tumor weights under a balanced diet, this did not reach statistical significance (Figures S7A and S7B). In accordance with our model, PIK3CA WT tumors were unaffected by these treatments (Figures 7E–7G, S7C, and S7D). Further corroborating our findings, genetic inhibition of cPLA2 only reduced viable tumor area in animals with PIK3CA MUT tumors when those were fed a fat-free diet, while no anti-neoplastic benefit was conferred under a “Western” diet (Figures 7H–7J).
Figure 7.

Dietary Supplementation of Arachidonic Acid Reverses the Sensitivity of PIK3CA Mutant Tumors to cPLA2 Inhibition
(A) Schematic of in vivo experimental design and profiling of breast cancer cell line xenografts with REIMS.
(B and C) Relative tumor growth of CAL-51 (PIK3CA MUT)-derived xenografts stably expressing control shGFP or two independent shRNAs targeting cPLA2 (cPLA2-sh1 and cPLA2-sh5) under (B) fat-free or (C) “Western” diets.
(D) Weights of tumors excised at the end of the experiments (B) and (C).
(E and F) Relative tumor growth of Hs578T (PIK3CA WT)-derived xenografts stably expressing control shGFP or two independent shRNAs targeting cPLA2 under (E) fat-free or (F) “Western” diets.
(G) Weights of tumors excised at the end of the experiments (E) and (F).
(H) Representative images of H&E staining from resected tumors in (D) and (G). The black masks in (H) represent viable tumor area, while unshaded regions correspond to necrotic tissue.
(I and J) Quantification of viable tumor area from (I) PIK3CA MUT (CAL-51) and (J) PIK3CA WT (Hs578T) tumor sections based on the analysis depicted in (H).
(K and L) AA levels measured by REIMS in (K) PIK3CA MUT (CAL-51) and (L) PIK3CA WT (Hs578T) snap frozen excised tumors. AA intensities are reported as scaled values to the appropriate shGFP-fat-free diet condition. Data in (B), (C), (E), (F), (K), and (L) represent the mean ± SEM of relative tumor growth or tumor REIMS measurements from n = 3–5 mice. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; p values in (B), (C), (E), and (F) were calculated using two-way ANOVA, and one-way ANOVA followed by unpaired, two-tailed Student’s t test with Bonferroni correction was used in (D), (G), and (I)–(L).
Figure S7.

(A) Relative tumor growth and (B) tumor weights of CAL51 (PIK3CA MUT)-derived xenografts stably expressing control shGFP or two independent shRNAs targeting cPLA2 (cPLA2-sh1 and cPLA2-sh5) in mice fed a balanced omega3:omega6 diet. (C) Relative tumor growth and (D) tumor weights of Hs578T (PIK3CA WT)-derived xenografts stably expressing control shGFP or cPLA2-sh1 or cPLA2-sh5 in mice fed a balanced omega3:omega6 diet. AA levels measured by REIMS in (E) PIK3CA MUT (CAL51) and (F) PIK3CA WT (Hs578T) snap frozen excised tumors. AA intensities are reported as scaled values to the appropriate shGFP-fat free diet control. Quantification of CCL5 from excised tumors derived from (G) PDX and (H) cell line-derived xenograft studies. Quantification of CX3CL1 from excised tumors derived from (I) PDX and (J) cell line-derived xenograft studies. Concentrations of chemokines in (G-J) were determined from whole tumor lysates using ELISA, and normalized to protein content. (K) Representative immunohistochemical staining of the activated NK cell marker NKp46 in BR1282 (PIK3CA MUT) and BR1458 (PIK3CA WT) PDX tumors. (L) and (M) Quantification of positively immunostained areas from (K). (N) Representative immunohistochemical staining of NKp46 in shGFP, cPLA2-sh1 and cPLA2-sh5 expressing CAL51 (PIK3CA MUT) and Hs578T (PIK3CA WT)-derived xenograft tumors under fat free or ‘Western’ diets. (O) and (P) Quantification of positively immunostained areas from (N). Data in (A), (C), and (E) to (J) are presented as the mean ± SEM of n = 3–5 mice for cell line xenograft or n = 7–8 mice for PDX studies. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; P values in (A) and (C) were calculated using two-way ANOVA, and one-way ANOVA followed by unpaired, two tailed Student’s t test with Bonferroni correction was used in (B), (D), (E), (F), (G), (H), (I), (J), (L), (M), (O), (P).
REIMS profiling of excised tumors revealed that AA levels were significantly altered in concordance with targeting cPLA2 and dietary fat intake (Figures 7K and S7E). Interestingly, a more substantial AA reduction was observed in PIK3CA MUT xenograft tumors following cPLA2-knockdown and administration of fat-free diet (40%–50% decrease), as compared to WT tumors (20% decrease) (Figures 7K and 7L, S7E, and S7F). In line with our previous in vivo study (Figure 6), these findings demonstrate that the modulation of dietary fat content, and supplementation of omega-6 FAs either in a balanced ratio with omega-3 FAs, or to a much larger extent in the “Western” diet, completely abolishes the therapeutic benefit of cPLA2 inhibition in PIK3CA MUT tumors.
In addition to promoting growth and proliferation through both autocrine and paracrine mechanisms, AA and its downstream metabolites have also been implicated in the metabolic remodeling of the tumor microenvironment. One of their major consequences is inhibition of the anti-cancer immune responses, ultimately leading to immune evasion and tumor progression (Böttcher et al., 2018, Zelenay et al., 2015). Although the BALB/c nude mice used in this study lack adaptive immunity in the form of T cells, these animals mount robust innate immune responses predominantly mediated by natural killer (NK) cells (Lee et al., 2015, Okada et al., 2019). We therefore sought to investigate how the various therapeutic and dietary regimes that were used in this study impact NK cell responses. To do this, the levels of type I interferon-induced chemokines (CCL5 and CX3CL1) and expression of a major NK cell-activating receptor (NKp46) were measured in tumors from different treatment groups.
Coinciding with the largest reduction in AA levels (Figure 6K), a marked increase in CCL5 and CX3CL1 was only observed in the BR1282 (PIK3CA MUT) PDX tumor following co-administration of ASB14780 and fat-free diet (Figures S7G and S7I). Similar results were obtained in the CAL51 (PIK3CA MUT)-derived xenograft tumor model, where the increase in chemokine levels was rescued when mice were fed the AA-enriched “Western” diet (Figures S7H and S7J).
In agreement with the chemokine analysis, tumor infiltration of NK cells was significantly increased in PIK3CA MUT tumors by dietary and therapeutic interventions, with dual inhibition (either with ASB14780 or shRNA) of cPLA2 and fat-free diet leading to the largest increase in NKp46 staining (Figures S7K, S7L, S7N, and S7O). It is also noteworthy that PIK3CA WT PDX and cell line-derived xenograft tumors contained relatively higher baseline levels of CCL5, CX3CL5, and NKp46 expression, as compared to MUT tumors, and these were not significantly altered by cPLA2 inhibition and/or changes in the diet (Figures S7G–S7J, S7M, and S7P). Considered together, these data suggest that oncogenic PIK3CA might suppress BC immunogenicity, at least in part, through regulation of AA metabolism, and this can be reversed through co-administration of cPLA2 inhibition and dietary fat restriction.
Discussion
Unraveling the interplay between genotype and metabolic phenotype, as well as their complex interactions with nutrient availability, unequivocally plays a major role in understanding disease pathogenesis and identifying novel therapeutic interventions. Here, we demonstrate that the iKnife/REIMS enables close to real-time prediction of clinically relevant tumor features based on their metabolic fingerprints, offering a novel repertoire for cancer diagnosis and therapy decision-making. Among these, is oncogenic PIK3CA, which triggers almost “the perfect storm” of signaling events, culminating in the overproduction of AA and downstream eicosanoids via the activation of cPLA2.
We have evidence of the central role of AA and eicosanoids in a wide range of disorders including cancer, obesity, diabetes, asthma, and autoimmune disorders (Dennis and Norris, 2015, Sonnweber et al., 2018, Wang and Dubois, 2010). However, the signal transduction pathways behind their activation, as well as the molecular cues that link these bio-active lipids with growth factor-independent cell proliferation have remained largely obscure. Our results demonstrate that mTORC2 downstream of oncogenic PIK3CA acts as a pivotal signaling hub for driving enhanced AA metabolism to sustain cell proliferation beyond a cell autonomous manner. This is particularly interesting in light of evidence obtained from in situ single-cell analysis of primary breast tumors, showing often that only a small fraction of cancer cells within a tumor carry PIK3CA mutations, while many of the neighboring cancer and stromal cells are WT (Janiszewska et al., 2015). Given that AA has been shown to induce both PI3K (Hughes-Fulford et al., 2006) and MAPK (Alexander et al., 2006) signaling, PIK3CA MUT cells could trigger a snowball effect, through overproduction of AA, affecting not only their own signaling and proliferation, but also that of their adjacent PIK3CA WT cells. Moreover, in light of the role of prostaglandins in lymphangiogenesis (Lala et al., 2018), the paracrine effects of AA could be of further relevance to the activity of PI3Kα in endothelial cells (Okkenhaug et al., 2016, Wang and Dubois, 2010).
Eicosanoids no longer represent the missing link between inflammation and cancer (Greene et al., 2011). Elevated tumor-derived PGE2 contributes to immune evasion by preventing the interferon gamma (IFNγ)-dependent upregulation of ICAM-1 that is pertinent for complete CD8(+) T cells activation (Basingab et al., 2016). In addition, autocrine PGE2 impairs NK cell viability and chemokine production and leads to downregulation of the chemokine receptors of cDC1 that promote their recruitment into tumors (Böttcher et al., 2018). Importantly, there is a strong positive correlation between the gene signature of cDC1 and NK cells and better overall survival in melanoma and breast cancers (Böttcher et al., 2018), suggesting that monitoring the immunomodulatory functions of prostaglandins via PI3K/Akt pathway inhibition could have important clinical implications. Indeed, we have shown that modulation of AA levels in PIK3CA MUT tumors through cPLA2 inhibition in combination with dietary fat restriction increases intra-tumor infiltration of NK cells and their associated chemokines, while this can be reversed by the “Western” diet, which contains an excess of omega6-FAs. NK cell markers were largely unaffected in PIK3CA WT tumors, and this could reflect their lower intra-tumor AA levels that are likely attributable to reduced cPLA2 activity and FA uptake, as compared to PIK3CA MUT cells. In light of recent evidence showing that blocking PI3K signaling with the pan-PI3K inhibitor BKM120 increases tumor-immune infiltrate and renders PIK3CA MUT mouse bladder tumors more susceptible to PD-1 blockade (Borcoman et al., 2019), our model raises important considerations for how immunotherapies may be successfully applied to oncogenic PIK3CA MUT tumors that may be inherently less immunogenic, at least in part, due to enhanced AA production.
Another way to relieve the immunosuppressive effects of tumor cells is by inhibiting COX activity via the use of non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin. Notably, oncogenic PIK3CA has been shown to sensitize cancer cells to aspirin (Henry et al., 2017, Liao et al., 2012) and, based on our findings, it is tempting to speculate that this connection could be true, in part because of the heightened capacity of PIK3CA MUT cells for high AA production. However, further studies are needed to ascertain this connection, because evidence suggests that the growth inhibitory effect of aspirin in PIK3CA MUT cells is likely to be COX-2-independent (Henry et al., 2017).
Although PI3K pathway inhibitors have shown some efficacy in treating advanced solid tumors, the majority has been associated with only partial tumor remission and they are often accompanied by severe side effects (Fruman et al., 2017, Li et al., 2018). Recent evidence suggests that one way to enhance their efficacy is by suppressing their insulin feedback through adoption of a ketogenic diet (Hopkins et al., 2018). Indeed, diet could play a much more significant role in therapy response than previously anticipated. Our data suggest that a diet rich in FAs limits the efficacy of the cPLA2 inhibitor, as PIK3CA MUT tumors likely depend on their high flux of extracellular FA intake to compensate for the loss of AA. This observation raises the possibility that adopting a diet without meat and dairy products (major sources of AA) could dramatically improve the sensitivity of the cPLA2 inhibitor and help restore tumor immunogenicity, suggesting a novel path for future clinical trials where nutrition will play a major role in disease management and treatment.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-beta-actin | Cell Signaling Technology | Cat# 4967; RRID: AB_10695744 |
| Rabbit polyclonal anti-Akt antibody | Cell Signaling Technology | Cat# 9272; RRID: AB_329827 |
| Rabbit monoclonal anti-phospho-Akt (Ser473) | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Rabbit polyclonal anti-PRAS40 | Cell Signaling Technology | Cat# 2610; RRID: AB_916206 |
| Rabbit monoclonal anti-phospho-PRAS40 (Thr246) | Cell Signaling Technology | Cat# 2997; RRID: AB_2258110 |
| Mouse monoclonal anti-S6 Ribosomal Protein | Cell Signaling Technology | Cat# 2317; RRID: AB_2238583 |
| Rabbit polyclonal anti-phospho-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technology | Cat# 2211; RRID: AB_331679 |
| Rabbit polyclonal anti-NDRG1 | Cell Signaling Technology | Cat# 5196; RRID: AB_10626626 |
| Rabbit monoclonal anti-phospho-NDRG1 (Thr376) | Cell Signaling Technology | Cat# 5482; RRID: AB_10693451 |
| Rabbit polyclonal anti-Raptor | Cell Signaling Technology | Cat# 2280; RRID: AB_561245 |
| Rabbit polyclonal anti-Rictor | Cell Signaling Technology | Cat# 2140; RRID: AB_2179961 |
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | Cat# 2983; RRID: AB_2105622 |
| Rabbit polyclonal anti-P70 S6 Kinase | Cell Signaling Technology | Cat# 9202; RRID: AB_331676 |
| Rabbit polyclonal anti-phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat# 9205; RRID: AB_330944 |
| Rabbit polyclonal anti-4E-BP1 (53H11) | Cell Signaling Technology | Cat# 9644; RRID: AB_2097841 |
| Rabbit polyclonal anti-phospho-4E-BP1 (Ser65) | Cell Signaling Technology | Cat# 9451; RRID: AB_330947 |
| Rabbit monoclonal anti-Lamin B1 (D4Q4Z) | Cell Signaling Technology | Cat# 12586; RRID: AB_2650517 |
| Rabbit polyclonal anti-phospho-PKC (pan) (βII Ser660) | Cell Signaling Technology | Cat# 9371; RRID: AB_2168219 |
| Rabbit polyclonal anti-PKCdelta | Cell Signaling Technology | Cat# 2058; RRID: AB_10694655 |
| Rabbit polyclonal anti-phospho-PKCdelta/theta (Ser643/676) | Cell Signaling Technology | Cat# 9376; RRID: AB_2168834 |
| Rabbit polyclonal anti-RKIP (G38) | Cell Signaling Technology | Cat# 5060; RRID: AB_1904081 |
| Rabbit polyclonal anti-c-Raf | Cell Signaling Technology | Cat# 9422; RRID: AB_390808 |
| Rabbit monoclonal anti-phospho-c-Raf (Ser338) | Cell Signaling Technology | Cat# 9427; RRID: AB_2067317 |
| Rabbit polyclonal anti-MEK1/2 | Cell Signaling Technology | Cat# 9122; RRID: AB_823567 |
| Rabbit polyclonal anti-phospho-MEK1/2 (Ser217/Ser221) | Cell Signaling Technology | Cat# 9121; RRID: AB_331648 |
| Rabbit polyclonal anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat# 9102; RRID: AB_330744 |
| Rabbit polyclonal anti-phospho-p44/42 MAPK (Thr202/Tyr204) | Cell Signaling Technology | Cat# 9101; RRID: AB_331646 |
| Rabbit polyclonal anti-p38 MAPK | Cell Signaling Technology | Cat# 9212; RRID: AB_330713 |
| Rabbit polyclonal anti-phospho-p38 MAPK (Thr180/Tyr182) | Cell Signaling Technology | Cat# 9211; RRID: AB_331641 |
| Rabbit polyclonal anti-cPLA2 | Cell Signaling Technology | Cat# 2832; RRID: AB_2164442 |
| Rabbit polyclonal anti-phospho-cPLA2 (Ser505) | Cell Signaling Technology | Cat# 2831; RRID: AB_2164445 |
| Rabbit monoclonal anti-HA-Tag | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 2118; RRID: AB_561053 |
| Rabbit polyclonal anti-PLCgamma1 | Cell Signaling Technology | Cat# 2822; RRID: AB_2163702 |
| Rabbit polyclonal anti-phospho-PLCgamma1 (Tyr783) | Cell Signaling Technology | Cat# 2821; RRID: AB_330855 |
| Rabbit polyclonal anti-phospho-threonine | Cell Signaling Technology | Cat# 9381; RRID: AB_330301 |
| Rabbit polyclonal anti-IkBalpha | Cell Signaling Technology | Cat# 9242; RRID: AB_331623 |
| Rabbit monoclonal anti-phospho-IkBalpha (Ser32) | Cell Signaling Technology | Cat# 2859; RRID: AB_561111 |
| Rabbit monoclonal anti-Stat3 | Cell Signaling Technology | Cat# 4904; RRID: AB_331269 |
| Rabbit polyclonal anti-phospho-Stat3 (Ser727) | Cell Signaling Technology | Cat# 9134; RRID: AB_331589 |
| Rabbit monoclonal anti-PKD/PKCμ | Cell Signaling Technology | Cat# 90039; RRID: AB_2800149 |
| Rabbit polyclonal anti-phospho-PKD/PKCμ (Ser744/748) | Cell Signaling Technology | Cat# 2054; RRID: AB_2172539 |
| Mouse monoclonal anti-Rb (4H1) | Cell Signaling Technology | Cat# 9309; RRID: AB_823629 |
| Rabbit monoclonal anti-estrogen inducible protein pS2 | Abcam | Cat# ab92377; RRID: AB_10562122 |
| Rabbit polyclonal anti-PKCzeta | Abcam | Cat# ab59364; RRID: AB_944858 |
| Rabbit monoclonal anti-phospho-PKCzeta (Thr560) | Abcam | Cat# ab62372; RRID: AB_946309 |
| Rabbit polyclonal anti-PKCepsilon | Abcam | Cat# ab63638; RRID: AB_1142276 |
| Rabbit polyclonal anti-phospho-PKCepsilon (Ser729) | Abcam | Cat# ab63387; RRID: AB_1142277 |
| Rabbit monoclonal anti-secretory phospholipase A2 | Abcam | Cat# ab139692 |
| Mouse monoclonal anti-PKCbeta II (F-7) | Santa Cruz Biotechnology | Cat# sc-13149; RRID: AB_628144 |
| Mouse monoclonal anti-PKCzeta (B-7) | Santa Cruz Biotechnology | Cat# sc-393218 |
| Mouse monoclonal anti-phospho-RKIP (Ser153) | Santa Cruz Biotechnology | Cat# sc-135779; RRID: AB_2163163 |
| Normal rabbit IgG | Santa Cruz Biotechnology | Cat# sc-2027; RRID: AB_737197 |
| Mouse monoclonal anti-phospho-Rb (Thr821/826) | Santa Cruz Biotechnology | Cat# sc-271930; RRID: AB_670923 |
| Rabbit polyclonal anti-phospholipase A2 (iPLA2) | Sigma-Aldrich | Cat# SAB4200129; RRID: AB_11129638 |
| Rabbit monoclonal anti-phospho-PKCzeta (Thr410) | Thermo Fisher Scientific | Cat# MA5-15060; RRID: AB_10983263 |
| Mouse monoclonal anti-SREBP1 | BD Biosciences | Cat#557036; RRID: AB_396559 |
| Goat anti-rabbit IgG (H+L)-HRP conjugate | Bio-Rad | Cat#170-6515; RRID: AB_11125142 |
| Goat anti-mouse IgG (H+L)-HRP conjugate | Bio-Rad | Cat#170-6516; RRID: AB_11125547 |
| Mouse polyclonal anti-Nkp46/ncr1 | R and D systems | Cat#AF2225; RRID: AB_355192 |
| Rabbit monoclonal anti-Ki-67 (D3B5) | Cell Signaling Technology | Cat# 9129; RRID: AB_2687446 |
| Goat anti-Mouse IgG (H+L) secondary antibody, Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11003; RRID: AB_2534071 |
| Phalloidin-iFluor 633 | Abcam | Cat# ab176758 |
| Rabbit polyclonal anti-phospho-cPLA2 (Thr376) (Peptide name: PLA2G4A-369:383-pT376) | This paper (produced by Thermo Fisher Scientific) | Cat# UE1820P-T-AB1792 |
| Bacterial and Virus Strains | ||
| MAX Efficiency DH5α Competent Cells | Thermo Fisher Scientific | Cat#18258012 |
| Biological Samples | ||
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB180 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB83 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB092 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB519 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB512 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB367 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB541 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB383 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB323 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB210 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB334 |
| Primary human breast tissue | Imperial College Tissue Bank | Cat# IKB283 |
| Breast PDX model | CrownBioscience | Cat# BR6695 |
| Breast PDX model | CrownBioscience | Cat# BR5020 |
| Breast PDX model | CrownBioscience | Cat# BR5009 |
| Breast PDX model | CrownBioscience | Cat# BR5022 |
| Breast PDX model | CrownBioscience | Cat# BR1282 |
| Breast PDX model | CrownBioscience | Cat# BR5011 |
| Breast PDX model | CrownBioscience | Cat# BR5014 |
| Breast PDX model | CrownBioscience | Cat# BR5012 |
| Breast PDX model | CrownBioscience | Cat# BR5017 |
| Breast PDX model | CrownBioscience | Cat# BR5013 |
| Breast PDX model | CrownBioscience | Cat# BR1474 |
| Breast PDX model | CrownBioscience | Cat# BR3267 |
| Breast PDX model | CrownBioscience | Cat# BR5010 |
| Breast PDX model | CrownBioscience | Cat# BR5015 |
| Breast PDX model | CrownBioscience | Cat# BR5337 |
| Breast PDX model | CrownBioscience | Cat# BR1115 |
| Breast PDX model | CrownBioscience | Cat# BR1283 |
| Breast PDX model | CrownBioscience | Cat# BR1458 |
| Breast PDX model | Champions Oncology | Cat# CTG1059 |
| Breast PDX model | Champions Oncology | Cat# CTG1350 |
| Breast PDX model | Champions Oncology | Cat# CTG1941 |
| Breast PDX model | Champions Oncology | Cat# CTG2308 |
| Breast PDX model | Champions Oncology | Cat# CTG0033 |
| Breast PDX model | Champions Oncology | Cat# CTG0012 |
| Breast PDX model | Champions Oncology | Cat# CTG0017 |
| Breast PDX model | Champions Oncology | Cat# CTG0018 |
| Breast PDX model | Champions Oncology | Cat# CTG0437 |
| Breast PDX model | Champions Oncology | Cat# CTG0473 |
| Ovarian PDX model | Champions Oncology | Cat# CTG0253 |
| Ovarian PDX model | Champions Oncology | Cat# CTG1423 |
| Ovarian PDX model | Champions Oncology | Cat# CTG1602 |
| Ovarian PDX model | Champions Oncology | Cat# CTG1627 |
| Ovarian PDX model | Champions Oncology | Cat# CTG0252 |
| Ovarian PDX model | Champions Oncology | Cat# CTG0258 |
| Ovarian PDX model | Champions Oncology | Cat# CTG0259 |
| Ovarian PDX model | Champions Oncology | Cat# CTG0486 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0292 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0381 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0391 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG1485 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG2205 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0282 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0283 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0284 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0285 |
| Pancreatic PDX model | Champions Oncology | Cat# CTG0286 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0886 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG1084 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG1116 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG1255 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG1628 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0142 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0143 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0241 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0242 |
| Sarcoma PDX model | Champions Oncology | Cat# CTG0243 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0083 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0129 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0360 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0706 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0799 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0058 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0062 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0063 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0065 |
| Colorectal PDX model | Champions Oncology | Cat# CTG0066 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hydrocortisone | Sigma-Aldrich | Cat# H-0888 |
| Insulin-transferrin-selenium | GIBCO | Cat# 41400-045 |
| Epidermal growth factor (EGF) | Peprotech | Cat# AF-100-15 |
| Cholera toxin | Sigma-Aldrich | Cat# C-8052 |
| Insulin | Sigma-Aldrich | Cat# I-1882 |
| Fatty acid free bovine serum albumin | Sigma-Aldrich | Cat# A8806 |
| PKC zeta pseudosubstrate inhibitory peptide | Sigma-Aldrich | Cat# P1614 |
| PKC beta II peptide inhibitor | Sigma-Aldrich | Cat# P-0102 |
| FIPI hydrochloride hydrate | Sigma-Aldrich | Cat# F5808 |
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# T176 |
| Bromoenol lactone | Sigma-Aldrich | Cat# B1552 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| Arachidonic acid | Sigma-Aldrich | Cat# 10931 |
| Palmitoleate | Sigma-Aldrich | Cat# P9417 |
| Palmitate | Sigma-Aldrich | Cat# P0500 |
| PKC alpha (C2-4) inhibitor peptide | Santa Cruz Biotechnology | Cat# sc-304 |
| PKC epsilon inhibitor peptide | Cambridge Bioscience | Cat# CAY17476 |
| Rapamycin | Selleckchem | Cat# S1039 |
| Torin 1 | Selleckchem | Cat#S2827 |
| BYL719 | Selleckchem | Cat# S2814 |
| BKM120 | Selleckchem | Cat# 2247 |
| MK2206 | Selleckchem | Cat# S1078 |
| GSK690693 | Selleckchem | Cat# S1113 |
| GSK650394 | Tocris Bioscience | Cat# 3572 |
| U73122 | Tocris Bioscience | Cat# 1268 |
| ASB14780 | Axon Medchem | Cat# 2578 |
| DharmaFECT-1 transfection reagent | Dharmacon | Cat# T-2001-02 |
| FuGENE HD Transfection reagent | Promega | Cat# E2311 |
| Lipid Removal Adsorbent | Supelco | Cat# 13358 |
| Matrigel | Corning | Cat# 354230 |
| Paraformaldehyde | Sigma-Aldrich | Cat# 158127 |
| Triton X-100 | Sigma-Aldrich | Cat# X100 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat# D1306 |
| RIPA buffer | Thermo Fisher Scientific | Cat# 89900 |
| Leupeptin | Sigma-Aldrich | Cat# L2884 |
| Pepstatin | Sigma-Aldrich | Cat# P5318 |
| Na3VO4 | Sigma-Aldrich | Cat# 450243 |
| DL-Dithiothreitol | Sigma-Aldrich | Cat# 646653 |
| Calyculin A | Cell Signaling Technology | Cat# 9902 |
| Beta-glycerophosphate | Sigma-Aldrich | Cat# G9422 |
| PMSF protease inhibitor | Cell Signaling Technology | Cat# 8553 |
| Bradford reagent | Bio-Rad | Cat# 5000006 |
| ALLN protease inhibitor | Merck-Millipore | Cat# 208719 |
| 2x Laemmli sample buffer | Bio-Rad | Cat# 161-0737 |
| 4x Laemmli sample buffer | Bio-Rad | Cat# 161-0747 |
| 10X Cell lysis buffer | Cell Signaling Technology | Cat# 9803 |
| Recombinant human protein kinase C zeta | Insight Biotechnology | Cat# TP302472 |
| Recombinant human phospholipase A2, group IVA | Insight Biotechnology | Cat# TP320972 |
| Magnesium chloride | Sigma-Aldrich | Cat# M8266 |
| Bovine serum albumin | Sigma-Aldrich | Cat# A2153 |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| Blasticidin | Invivogen | Cat# ant-bl-1 |
| Critical Commercial Assays | ||
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat# 210518 |
| CellTiter96 Aqueous Non-radioactive (MTS) cell proliferation assay | Promega | Cat# G5430 |
| Fatty Acid Uptake Kit | Sigma-Aldrich | Cat# MAK156 |
| Cytosolic Phospholipase A2 Assay Kit | Abcam | Cat# ab133090 |
| Secretory Phospholipase A2 Assay Kit | Abcam | Cat# ab133089 |
| Diacylglycerol (DAG) Assay Kit | Cell Biolabs Inc | Cat# MET-5028 |
| Active Rac1 Detection Kit | Cell Signaling Technology | Cat# 8815 |
| Duolink In Situ Detection Reagents Red Kit | Sigma-Aldrich | Cat# DUO92008 |
| Minus and Plus PLA probes | Sigma-Aldrich | Cat# DUO92004 and DUO92002 |
| Fluo-4 Direct Calcium Assay Kit | Thermo Fisher Scientific | Cat# F10471 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74134 |
| QuantiTect Reverse Transcription Kit | QIAGEN | Cat#205311 |
| SYBR Select Master Mix | Thermo Fisher Scientific | Cat# 4472908 |
| QIAamp DNA mini kit | QIAGEN | Cat# 51304 |
| PNAClamp PIK3CA Mutation Detection Kit | Panagene | Cat# PNAC-4001 |
| ADP-Glo Kinase Assay | Promega | Cat# V6930 |
| Arachidonic Acid ELISA Kit | Generon | Cat# CEB098Ge |
| Prostaglandin E2 ELISA Kit | Enzo Life Sciences | Cat# ADI-900-001 |
| Mouse RANTES (CCL5) ELISA Kit | Abcam | Cat# ab100739 |
| Mouse Fractalkine (CX3CL1) ELISA Kit | Abcam | Cat# ab100683 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Deposited Data | ||
| Custom script for quantification of proximity ligation assay images | This manuscript | Github: https://github.com/adamltyson/foci2D |
| Custom script for quantification of acini images | This manuscript | Github: https://github.com/adamltyson/cell-coloc-3D |
| Custom script for quantification of calcium flux | This manuscript | Github: https://github.com/adamltyson/CalciumAnalysis |
| REIMS data for Figure 1D | This manuscript | Mendeley Data; https://doi.org/10.17632/xcgc5kpntm.1 |
| REIMS data for Figure 1E | This manuscript | Mendeley Data; https://doi.org/10.17632/xcgc5kpntm.1 |
| REIMS data for Figure 1H | This manuscript | Mendeley Data; https://doi.org/10.17632/xcgc5kpntm.1 |
| REIMS data for Figure 3B | This manuscript | Mendeley Data; https://doi.org/10.17632/xcgc5kpntm.1 |
| REIMS data for Figure 3D | This manuscript | Mendeley Data; https://doi.org/10.17632/xcgc5kpntm.1 |
| Significantly altered phospholipids across breast cancer cell lines of different receptor, or triple negative status | This manuscript | Table S1 |
| Significantly different fatty acids between MCF10A PIK3CA wild-type and E545K/H1047R mutant cells | This manuscript | Table S4 |
| Experimental Models: Cell Lines | ||
| Human PIK3CA (H1047R/+) MCF10A | Horizon Discovery | Cat# HD 101-011 |
| Human PIK3CA (E545K/+) MCF10A | Horizon Discovery | Cat# HD 101-002 |
| AU565 (human breast carcinoma) | ATCC | Cat# CRL-2351; RRID: CVCL_1074 |
| BT20 (human breast carcinoma) | ATCC | Cat# HTB-19; RRID: CVCL_0178 |
| BT474 (human breast carcinoma) | ATCC | Cat# HTB-20; RRID: CVCL_0179 |
| BT549 (human breast carcinoma) | ATCC | Cat# HTB-122; RRID: CVCL_1092 |
| CAL51 (human breast carcinoma) | DSMZ | Cat# ACC-302; RRID: CVCL_1110 |
| CAMA1 (human breast carcinoma) | ATCC | Cat# HTB-21; RRID: CVCL_1115 |
| EFM19 (human breast carcinoma) | DSMZ | Cat# ACC-231; RRID: CVCL_0253 |
| Hs578T (human breast carcinoma) | ATCC | Cat# HTB-126; RRID: CVCL_0332 |
| JIMT1 (human breast carcinoma) | DSMZ | Cat# ACC-589; RRID: CVCL_2077 |
| KPL1 (human breast carcinoma) | DSMZ | Cat# ACC-317; RRID: CVCL_2094 |
| MCF7 (human breast carcinoma) | ATCC | Cat# HTB-22; RRID: CVCL_0031 |
| MDAMB134 (human breast carcinoma) | ATCC | Cat# HTB-23; RRID: CVCL_0617 |
| MDAMB157 (human breast carcinoma) | ATCC | Cat# HTB-24; RRID: CVCL_0618 |
| MDAMB231 (human breast carcinoma) | ATCC | Cat# HTB-26; RRID: CVCL_0062 |
| MDAMB361 (human breast carcinoma) | ATCC | Cat# HTB-27; RRID: CVCL_0620 |
| MDAMB436 (human breast carcinoma) | ATCC | Cat# HTB-130; RRID: CVCL_0623 |
| MDAMB453 (human breast carcinoma) | ATCC | Cat# HTB-131; RRID: CVCL_0418 |
| MDAMB468 (human breast carcinoma) | ATCC | Cat# HTB-132; RRID: CVCL_0419 |
| MFM223 (human breast carcinoma) | DSMZ | Cat# ACC-422; RRID: CVCL_1408 |
| S68 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_5585 |
| SKBR3 (human breast carcinoma) | ATCC | Cat# HTB-30; RRID: CVCL_0033 |
| T47D (human breast carcinoma) | ATCC | Cat# HTB-133; RRID: CVCL_0553 |
| UACC812 (human breast carcinoma) | ATCC | Cat# CRL-1897; RRID: CVCL_1781 |
| VP229 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_2754 |
| BT483 (human breast carcinoma) | ATCC | Cat# HTB-121; RRID: CVCL_2319 |
| HCC1143 (human breast carcinoma) | ATCC | Cat# CRL-2321; RRID: CVCL_1245 |
| HCC1395 (human breast carcinoma) | ATCC | Cat# CRL-2324; RRID: CVCL_1249 |
| HCC1428 (human breast carcinoma) | ATCC | Cat# CRL-2327; RRID: CVCL_1252 |
| HCC1500 (human breast carcinoma) | ATCC | Cat# CRL-2329; RRID: CVCL_1254 |
| HCC1569 (human breast carcinoma) | ATCC | Cat# CRL-2330; RRID: CVCL_1255 |
| HCC1937 (human breast carcinoma) | ATCC | Cat# CRL-2336; RRID: CVCL_0290 |
| HCC1954 (human breast carcinoma) | ATCC | Cat# CRL-2338; RRID: CVCL_1259 |
| HCC202 (human breast carcinoma) | ATCC | Cat# CRL-2316; RRID: CVCL_2062 |
| HCC38 (human breast carcinoma) | ATCC | Cat# CRL-2314; RRID: CVCL_1267 |
| HCC70 (human breast carcinoma) | ATCC | Cat# CRL-2315; RRID: CVCL_1270 |
| SUM52 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_3425 |
| ZR751 (human breast carcinoma) | ATCC | Cat# CRL-1500; RRID: CVCL_0588 |
| ZR7530 (human breast carcinoma) | ATCC | Cat# CRL-1504; RRID: CVCL_1661 |
| SUM44 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL-3424 |
| SUM159 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_5423 |
| SUM149 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_3422 |
| SUM225 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_5593 |
| SUM229 (human breast carcinoma) | Breast Cancer Now (Institute of Cancer Research) | RRID: CVCL_5594 |
| HEK293T (human embryonic kidney) | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Human MCF10A PIK3CA WT CRISPR control cell line | This manuscript | N/A |
| Human MCF10A PIK3CA WT cPLA2 CRISPR cell line | This manuscript | N/A |
| Human MCF10A PIK3CA H1047R (+/) CRISPR control cell line | This manuscript | N/A |
| Human MCF10A PIK3CA H1047R (+/−) cPLA2 CRISPR cell line | This manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c nude (female, age: 6–8 weeks) | Beijing Anikeeper Biotech (Beijing, China) | N/A |
| Mouse: BALB/c nude (female, age: 7–9 weeks) | Envigo | N/A |
| Oligonucleotides | ||
| Primers for cPLA2 shRNA amplification (Forward 5′-GTGGAAAGGACGAAACACCGGT-3′, Reverse 5′-TTTGTCTCGAGGTCGAGAATTC-3′ | Sigma-Aldrich | N/A |
| Mutagenesis primers to generate cPLA2 S505A (Forward 5′-GCAAAGTCACTCAAAGGAGCCAGTG GATAAGATGTATTG-3′, Reverse 5′-CAATACATCTTATCCACTGGCTCCTTT GAGTGACTTTGC-3′ |
Sigma-Aldrich | N/A |
| Mutagenesis primers to generate cPLA2 T376A (Forward 5′-TTCTTC ATACTTCTTAACGACTGCTCCCATAAAAA ATTTGCTTCCAA-3′, Reverse 5′-TTGGAAGCAAATTT TTTATGGGAGCAGTCGTTAAG AAGTATGAAGAA-3′ |
Sigma-Aldrich | N/A |
| qPCR primers for human cPLA2 (PLA2G4A) (Forward 5′-GATGAAACTCTAGGGACAGCAAC-3′, Reverse 5′-CTGGGCATGAGCAAACTTCAA-3′ | Sigma-Aldrich | N/A |
| qPCR primers for human beta-actin (Forward 5′-GACCCAGATCATGTTTGAGACC-3′, Reverse 5′-CTTCATGAGGTAGTCAGTCAGG-3′) | Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| pLKO-Tet-On Vector | Wiederschain et al., 2009 | Addgene ID: 21915 |
| pCMV3-HA-PLA2G4A | Sino Biological | Cat# HG13126-NY |
| RICTOR ON-TARGETplus SMARTPool human siRNA | Dharmacon | Cat# L-016984-00-0005 |
| RAPTOR ON-TARGETplus SMARTPool human siRNA | Dharmacon | Cat# L-004107-00-0005 |
| FRAP1 ON-TARGETplus SMARTPool human siRNA | Dharmacon | Cat# L-003008-00-0005 |
| PLCγ1 ON-TARGETplus SMARTPool human siRNA | Dharmacon | Cat# L-003559-00-0005 |
| PRKCZ ON-TARGETplus SMARTPool human siRNA | Dharmacon | Cat# L-003526-00-0005 |
| Non-targeting siRNA control | Dharmacon | Cat# D-001810-01-05 |
| TRC Lentiviral eGFP shRNA positive control | Dharmacon | Cat# RHS4459 |
| TRC Lentiviral Human PLA2G4A shRNA 1 | Dharmacon | Cat# TRCN0000050263 |
| TRC Lentiviral Human PLAG2G4A shRNA 5 | Dharmacon | Cat# TRCN0000050267 |
| LentiCRISPR v2 | Sanjana et al., 2014 | Addgene ID: 52961 |
| PLA2G4A sgRNA CRISPR/Cas9 All-in-One Lentivector Target 2 | Applied Biological Materials | Cat# K1659207 |
| pCMV-HA-PLA2G4A-S505A | This manuscript | N/A |
| pCMV-HA-PLA2G4A-T376A | This manuscript | N/A |
| Inducible pLKO-Tet-On-TRC Lentiviral Human PLA2G4A shRNA 1 | This manuscript | N/A |
| Software and Algorithms | ||
| R statistical software (version 3.5.1) | The R Project | https://www.r-project.org/ |
| ProteoWizard MsConvert software (version 3.0.11781) | Chambers et al., 2012 | http://proteowizard.sourceforge.net/download.html |
| MALDIquant package | Gibb and Strimmer, 2012 | https://cran.r-project.org/web/packages/MALDIquant/index.html |
| ReactomePA package | Yu and He, 2016 | https://bioconductor.org/packages/release/bioc/html/ReactomePA.html |
| MATLAB (2014a, version 8.3.0.532) | Mathworks | https://www.mathworks.com/products/matlab.html |
| GraphPad Prism (version 8.0.1) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8) | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| TargetLynx software | Waters Corporation | https://www.waters.com/waters/home.htm |
| Image Lab Software (version 5.2.1) | Bio-Rad | https://www.bio-rad.com/en-uk/product/image-lab-software?ID=KRE6P5E8Z |
| ImageJ (version 1.51) | NIH | https://imagej.nih.gov/ij/download.html |
| Other | ||
| Protein G Sepharose beads | Sigma-Aldrich | Cat# P3296 |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | GIBCO | Cat# 41965-039 |
| Roswell Park Memorial Institute (RPMI) 1640 | GIBCO | Cat# 10220-106 |
| Ham’s F12 media | Thermo Fisher Scientific | Cat# 11765054 |
| DMEM/F-12 | GIBCO | Cat# 31330-038 |
| Horse Serum | Thermo Fisher Scientific | Cat# 16050-122 |
| 4–15% Criterion TGX Precast Midi Protein Gel | Bio-Rad | Cat# 5671084 |
| 4–15% Mini-PROTEAN TGX Precast Protein Gel | Bio-Rad | Cat# 4561083 |
| Electrosurgical bipolar forceps | Erbe Elektromedizin (Germany) | N/A |
| ForceTriad electrosurgical unit | Covidien (Ireland) | N/A |
| Thermo Exactive orbitrap instrument | Thermo Scientific | N/A |
| Waters HSS T3 UPLC column | Waters Corporation | Cat# 186005614 |
| Waters Xevo TQ-S triple quadrupole mass spectrometer | Waters Corporation | N/A |
| Normal diet (for PDX study) | Keaoxieli Feed (Beijing, China) | Cat# 2152 |
| Fat free diet (for PDX study) | Xietong Organism (Beijing, China) | Cat# RD17112401 |
| Western diet (omega-3/omega-6 = 1:50) (For cell line xenograft study) | Research Diets | Cat# D19032707 |
| Balanced diet (omega-3/omega-6 = 1:1) (For cell line xenograft study) | Research Diets | Cat# D19032708 |
| Fat free diet (For xenograft study) | Research Diets | Cat# D19032705 |
| Precellys Lysing Soft tissue homogenizing kit | Precellys | Cat# P000912-LYSK0 |
| Precellys24 homogenizer | Bertin Instruments | Cat# P000669-PR240-A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Poulogiannis (george.poulogiannis@icr.ac.uk).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The code generated during this study are available at GitHub using the following accessions: https://github.com/adamltyson/CalciumAnalysis, https://github.com/adamltyson/cell-coloc-3D, and https://github.com/adamltyson/foci2D. These accessions are also provided in the Key Resources Table. The published article includes all REIMS m/z values and putative annotations for significantly different lipids between various receptor subtypes and MCF10A PIK3CA isogenics in the Supplementary Information in Tables S1 and S4, respectively. Original/source data of REIMS profiles for Figures 1D, 1E, 1H, 3B, and 3D in the paper corresponding to breast cancer cell lines and tumors is available through Mendeley Data (https://doi.org/10.17632/xcgc5kpntm.1)
Experimental Model and Subject Details
Human samples
12 primary breast cancer samples from female patients (> 18 years of age) who consented to utilization of tissue for research were provided by the Imperial College Healthcare NHS Trust Tissue Bank. Other investigators may have received samples from these same tissues. The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Human samples used in this research project were obtained with evaluation and approval from the Wales Research Ethics Committee Reference 17/WA/0161 (Imperial College Healthcare Tissue Bank Human Tissue Authority license: 12275; Project number R18024), the East of England – Cambridge East Research Ethics Committee Reference 14/EE/0024, and the project was registered under the Imperial College Tissue Bank.
Animals
Mouse PDX experiments were performed by Crown Bioscience in accordance with approved Institutional Animal Care and Use Committee (IACUC) protocols and ethical guidelines, and in strict accordance with the Crown Bioscience Guidelines and Standard Operating Procedures. Two primary human triple-negative breast cancer PDX corresponding to PIK3CA WT (BR1458) or PIK3CA C420R MUT (BR1282) tumor fragments (2-3 mm in diameter) were inoculated subcutaneously into the breast pad of 7-9-week-old female immunodeficient BALB/c nude mice weighing 17-23 g and which had not received previous treatments or procedures. Once tumors reached a volume of 100-200 mm3, mice were randomized into four groups corresponding to either a normal or fatty acid free (FAF) diet and administered with vehicle (0.5% hydroxypropyl cellulose in sterile water) or 100 mg/kg cPLA2 inhibitor ASB14780 (2578, Axon Medchem) daily through oral gavage for 21 days. Animals were housed in a specific pathogen free facility in individually vented cages and provided with diets and distilled water ad libitum. Room temperature was monitored and maintained at 20-25°C with the light cycle set at 12 hours. All animals were checked daily for signs of ill health, as well as for any effects of tumor growth and treatments on behavior such as mobility, food and water consumption, and body weight gain/loss. Researchers were not blinded to treatment groups. Tumors were excised 2 hours after the final dosing and snap frozen in preparation for metabolomics/REIMS processing and histopathological assessment. The mean tumor area as a percent of the total tissue area was initially assessed by an independent histopathologist, and subsequently quantified using ImageJ version 1.51. Normal and FAF diets were purchased from Keaoxieli Feed (2152) and Xietong Organism (RD17112401), respectively, and their compositions are summarized in Table S6.
All animal work undertaken at the Institute of Cancer Research was carried out under UK Home Office Project Licenses P6AB1448A (Establishment License, X702B0E74 70/2902) and was approved by the Animal Welfare and Ethical Review Body at the ICR. For cell line-derived xenograft studies, 7-9 week old female immunodeficient BALB/c nude mice weighing 18-25 g and which had not received previous treatments or procedures were initially pre-conditioned on fat free, balanced (omega3/omega6 1:1) or Western (omega3/omega6 1:50) diets for 2 weeks to assess tolerability. In order for the animal feed to be controlled, cages were randomized to treatment groups rather than individual mice, and this occurred prior to orthotopic injections. Animals were subsequently injected with 2.5x106 triple negative CAL51 (PIK3CA mutant) or Hs578T (PIK3CA WT) cells expressing either control shGFP or two independent shRNAs targeting cPLA2 (cPLA2-sh1 or cPLA2-sh5) in 100 μL PBS:matrigel (50:50) into the right mammary fat pad. Animals were housed in a specific pathogen free facility in individually vented cages (no more than 4 mice per cage) and provided with diets and distilled water ad libitum. Room temperature was monitored and maintained at 20-25°C with the light cycle set at 12 hours. All animals were checked daily for signs of ill health, as well as for any effects of tumor growth and treatments on behavior such as mobility, food and water consumption, and body weight gain/loss. Owing to the nature of the diets, blinding was not possible. Tumor measurements were taken twice weekly in three dimensions (width, length and depth), and presented as relative tumor growth normalized to first measurement day. Mice were excluded from the analysis if the primary tumor engrafted subcutaneously or into the peritoneum instead of the mammary fat pad. On the final day of the experiment, tumors were excised and immediately snap frozen in preparation for metabolomics/REIMS processing and histopathological assessment. The mean tumor area was quantified as a percent of the total tissue area using ImageJ version 1.51. Fat free, balanced and Western diets were purchased from Research Diets Inc. (D19032705, D19032708, D19032707, respectively). The composition of the diets used for the cell line xenograft study are summarized in Table S6
Fresh frozen breast patient-derived xenograft (PDX) tumors were obtained from Crown Bioscience, and additional breast, ovarian, pancreatic, sarcoma and colorectal PDX tumors were kindly provided by Champions Oncology. These are described in the Key Resources Table. The PIK3CA mutational status for all 66 PDX tumor samples is summarized in Table S5.
Cell culture
Human female breast carcinoma cell lines AU565 BT20, BT474, BT549, CAL51, CAMA1, EFM19, Hs578T, JIMT1, KPL1, MCF7, MDAMB134, MDAMB157, MDAMB231, MDAMB361, MDAMB436, MDAMB453, MDAMB468, MFM223, S68, SKBR3, T47D, UACC812 and VP229 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (GIBCO, 41965-039) and BT483, HCC1143, HCC1395, HCC1428, HCC1500, HCC1569, HCC1937, HCC1954, HCC202, HCC38, HCC70, SUM52, ZR751 and ZR7530 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 media (Sigma-Aldrich, R8758), both supplemented with 10% fetal bovine serum (FBS) (GIBCO, 10220-106). SUM44 (human, female), SUM159 (human, female), SUM149 (human, female), SUM225 (human, female) and SUM229 (human, female) were cultured in Ham’s F12 media (Thermo Fisher Scientific, 11765054) supplemented with 5% FBS, 0.5 μg/ml hydrocortisone (Sigma-Aldrich, H-0888) and 0.4% insulin-transferrin-selenium (GIBCO, 41400-045) and MCF10A (human, female) cells (including the PIK3CA MUT isogenic panel) were cultured in DMEM/F-12 (GIBCO, 31330-038) supplemented with 5% horse serum (Thermo Fisher Scientific, 16050-122), 20 ng/ml epidermal growth factor (EGF) (Peprotech, AF-100-15), 100 ng/ml cholera toxin (Sigma-Aldrich, C-8052), 0.5 mg/ml hydrocortisone (Sigma-Aldrich, H-0888) and 10 μg/ml insulin (Sigma-Aldrich, I1882). For cell culture conditions free of exogenous sources of fatty acids, 10% FBS or 5% horse serum was replaced with 1% fatty acid bovine serum albumin (BSA) (Sigma-Aldrich, A8806). All cell lines were maintained at 37°C, 5% CO2. All cell lines were authenticated by short tandem repeat analysis (Eurofins Scientific) and were tested and confirmed to be negative for mycoplasma infection.
Method Details
Experimental design
Experiments were repeated multiple times across different cell line and tumor models with similar results as indicated in the figure legends. Key findings from in vivo experiments were reproduced using orthogonal approaches including cell line xenograft models and genetic inhibitions. Animals were randomized into treatment groups either individually following PDX engraftment and growth to 100-200 mm3, or in cages following diet preconditioning. Throughout the study researchers were not blinded as data analysis required prior knowledge of the sample annotation. For in vitro and in vivo experiments, sample size was chosen based on preliminary experiments and previous experience with protocols. No completed data were excluded from the analysis performed in this manuscript.
Mass spectrometry analysis
Cell lines were cultured in 10 cm2 plates and harvested in biological triplicate for REIMS analysis when 80% confluent. After replacing the media one hour prior to harvesting, the plates were washed twice with ice-cold PBS, and immediately flash frozen in liquid nitrogen. 1 mL PBS was added to the cells and cell extracts were scraped and collected in eppendorf tubes and centrifuged for 5 min at 5000rpm. Pellets were flash frozen and stored at −80°C. For fresh frozen PDX and primary breast tumors, three separate regions were sectioned for analysis.
REIMS analysis was performed with commercially available electrosurgical bipolar forceps (Erbe Elektromedizin, Germany) connected to a ForceTriad electrosurgical unit (Covidien, Ireland) programmed in Macro bipolar setting using 4 W or 30 W power for cell lines and tumors, respectively. Bipolar forceps were connected to the inlet capillary of a Thermo Exactive orbitrap instrument (Thermo Scientific) using PTFE tubing, allowing for the direct suction of aerosol generated from rapid biomass heating to the mass spectrometer (set up is shown in Figure 1A). The mass spectrometer settings used for phospholipid and fatty acid profiling are summarized in Table S7.
Metabolomics data pre-processing and analysis
Raw mass spectrometric files were converted to mzXML format using the ProteoWizard MSConvert software (Version 3.0.11781) (Chambers et al., 2012) and imported into RStudio (Version 3.4.4). Data were pre-processed using the MALDIquant package (Gibb and Strimmer, 2012). Prior to pre-processing, high quality spectra corresponding to the maximum total ion (TIC) count for each measurement were selected from each individual sample runs. Spectra were normalized using median scaling determined from the non-zero intensities across the full scan range (m/z 150-2000), and square root transformed. Peaks were aligned using the locally weighted scatterplot smoothing (LOWESS) function and detected using median absolute deviation (MAD) with a signal to noise ratio (SNR) equal to 3. Peak matching was subject to a maximum peak shift of 5 ppm. For further analysis, the median intensity of all biological and technical replicates was calculated, such that each sample is represented by a single spectrum. The mass ranges of m/z 600-900 and m/z 150-500 were recorded for the detection of phospholipids and fatty acids, respectively. m/z values of interest which discriminated significantly between experimental conditions were annotated based on previously performed tandem mass spectrometry (MS/MS) fragmentation analysis and data available from Lipid Maps (https://www.lipidmaps.org). To assess instrument reproducibility, the MDAMB468 cells were included in every REIMS run as a quality control. The spectra obtained were compared among different batches using unsupervised or guided principal component analysis (gPCA) and no significant batch effect was observed. Classification and feature-selection was performed using leave-one-out or 3-fold cross validation with random forest as the classifier, applying the ‘caret’ and ‘randomForest’ packages, while unsupervised analysis was performed using hierarchical clustering (Euclidean distance, complete linkage), or non-negative matrix factorization (NMF) using the ‘NMF’ package. Prediction of continuous variables such as ESR1 gene expression was performed using random forest regression analysis with the TreeBagger function in MATLAB (2014a, version 8.3.0.532).
Transfections and site directed mutagenesis
Sub-cloning of cPLA2 sh1 into the pLKO-Tet-On inducible vector was done by first amplifying the shRNA sequence of interest with the following primers: Forward 5′-GTGGAAAGGACGAAACACCGGT-3′ and Reverse 5′-TTTGTCTCGAGGTCGAGAATTC-3′, and subsequently introducing compatible AgeI and EcoRI restriction sites in the shRNA oligonucleotides and pLKO-Tet-On vector (TET-pLKO puro was a gift from D. Wiederschain, Addgene plasmid 21915). For lentiviral gene knockdown, PLA2G4A or GFP shRNAs were transfected in 293T cells using FuGENE® HD Transfection reagent (Promega, E2311), according to the manufacturer’s instructions and infected cells were selected in the presence of 1 μg/ml puromycin. For siRNA knockdown, MCF10A PIK3CA WT and E545K/H1047R MUT cells were transfected with 25 nM ON-TARGETplus SMARTpool human siRNAs targeting RAPTOR, RICTOR, FRAP1, PLCγ1 or PRKCZ using DharmaFECT-1 Transfection Reagent for 48 hours. HA-tagged cPLA2 was transiently overexpressed using 9 μg pCMV3-HA-PLA2G4A vector DNA and 18 μL FuGENE® HD Transfection reagent, and experiments were performed 48 hours post transfection.
For generating CRISPR knockouts, MCF10A PIK3CA WT and H1047R cells were transfected with PLA2G4A sgRNA CRISPR/Cas9 All-in-One Lentivector Target 2 (Applied Biological Materials, K1659207) or control LentiCRISPR v2 (lentiCRISPR v2 was a gift from Feng Zhang, Addgene plasmid #52961) according to the manufacturer’s instructions. Immunoblotting and DNA sequencing were used to validate the isolation of PLA2G4A deleted clones.
Phosphoresistant isoforms of cPLA2 were generated using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, 210518) according to the manufacturer’s instructions. Briefly, 100 ng of pCMV3-HA-PLA2G4A vector DNA was incubated with 125 ng of the appropriate mutagenesis primers:
S505A Forward 5′-GCAAAGTCACTCAAAGGAGCCAGTGGATAAGATGTATTG-3′, Reverse 5′-CAATACATCTTATCCACTGGCTCCTTTGAGTGACTTTGC-3′;
T376A
Forward 5′-TTCTTCATACTTCTTAACGACTGCTCCCATAAAAAATTTGCTTCCAA-3′ Reverse 5′-TTGGAAGCAAATTTTTTATGGGAGCAGTCGTTAAGAAGTATGAAGAA-3′;
PCR cycling parameters were as follows: initial denaturing at 95°C for 2 min, followed by 18 cycles of 20 s denaturing (95°C), 10 s annealing (60°C) and 4.5 min elongation (68°C). A final elongation step occurred for 5 min (68°C). To assess the effects of these phosphomutants on cPLA2 activity or arachidonic acid (AA) levels, cells were transiently transfected with 9 μg pCMV3-HA-PLA2G4A, pCMV3-HA-PLA2G4A-S505A or pCMV3-HA-PLA2G4A-T376A vector DNA and 18 μL FuGENE® HD Transfection reagent, and experiments were performed 48 hours post transfection.
Cell based assays
For colony formation assays, single cell suspensions containing 5x102 MCF10A PIK3CA WT or E545K/H1047R MUT cells were seeded in 6 well plates and allowed to adhere for 18 hours. Wells were subsequently washed twice with PBS and DMEM-F/12 media containing growth factors and 1% FAF BSA (herein called ‘FAF media’), supplemented either with 0.1% DMSO or 25 μM AA. After 14 days, colonies were stained with crystal violet, and quantified using ImageJ. For 3D spheroid culture, 5x103 cells were seeded in round bottom, low attachment 96 well plates (Corning, 7007) and incubated for 14 days. Media was replaced every 3 days. For ASB14780 viability assays, 5x103 PIK3CA WT (MCF10A, MDAMB134, AU565, Hs578T) or PIK3CA MUT (MCF10A E545K/H1047R, CAL51, MCF7, MDAMB453) were seeded in 96 well plates in full media and allowed to adhere for 18 hours. Wells were washed twice with PBS, and FAF media containing either 0.1% DMSO, 20 nM-10 μM ASB14780 or the inhibitor supplemented with 25 μM AA was added. Viability was assessed after 72 hours using the CellTiter 96 Aqueous Non-Radioactive (MTS) cell proliferation assay (Promega, G5430).
To measure cell proliferation, 5x103 MCF10A isogenic cells expressing GFP control or cPLA2 shRNAs (sh1 and sh5) were seeded in 96 well plates, left to adhere for 18 hours, washed twice with PBS, and incubated with FAF media. Cell number was determined on Days 0, 1, 3 and 5 using the Sulforhodamine B (SRB) assay.
To assess eicosanoid-mediated autocrine and paracrine effects on cell proliferation, conditioned media were derived from MCF10A PIK3CA WT or MUT isogenics. Cells were grown to 80% confluency in full media, after which residual media were washed off twice with PBS, and cells were cultured with FAF media for 48 hours. The conditioned media were centrifuged at 2000 g for 20 min to remove cell debris and incubated with Lipid Removal Adsorbent (LRA, Supelco, 13358) at 0.4 g of LRA per 10 mL media to deplete secreted lipids. Following overnight incubation with gentle shaking, lipid-deprived media were centrifuged at 2000 g for 20 min and supernatants were collected. Proliferation was assessed over 5 days using the SRB assay.
To assess fatty acid uptake, 5x104 MCF10A PIK3CA WT or MUT cells were seeded in 100 μL full medium containing 5% horse serum in a 96-well plate and incubated for 24 hours at 37°C, 5% CO2. Cells were washed twice with PBS and serum deprived for 1 hour before adding 100 μL TF2-C12 Fatty Acid Stock Solution (Sigma-Aldrich, MAK156). After incubating the cells for 1 hour at 37°C, the fluorescence signal was measured at Ex/Em = 485/515 nm.
Three-dimensional cell culture
For 3D acini formation, MCF10A cells were grown in Matrigel (Corning, 354230) as described previously (Debnath et al., 2003). Briefly, PIK3CA isogenic MCF10A cells expressing pLKO-Tet-On inducible shRNA against cPLA2 or GFP were induced with 2 μg/ml doxycycline for 48 hours prior to seeding. A single cell suspension (3x103 cells per well) was plated in assay medium containing 1% FAF BSA onto eight-well chamber slides coated with a layer of pure growth factor–reduced Matrigel. Cells were then overlaid with a 300 μL of assay medium containing 2% Matrigel and cultured for 10 days. Growth media was replaced every 3 days.
Confocal microscopy
For immunofluorescence, cells were fixed with 4% PFA at room temperature, permeabilized with pre-chilled 0.5% Triton X-100 for 10 min prior to blocking with 2% BSA/PBS for 1 hour. After blocking, cells were incubated overnight at 4°C with primary Ki-67 antibody (Cell Signaling Technology, 9129) in a humidified chamber. The following day cells were incubated with fluorescently labeled secondary antibodies Alexa Fluor 546-conjugated secondary antibody (Thermo Fischer Scientific, A-11003) and Phalloidin 633 (Abcam, ab176758) to visualize Ki-67 and F-actin, respectively, 1% BSA/PBS for 1 hour. Slides were mounted using Prolong Gold anti-fade reagent with DAPI (Invitrogen, D1306). Images were captured using a Zeiss AxioObserver microscope equipped with a Yokogawa CSU-W1 spinning disk unit (Intelligent Imaging Innovations) and a 40x oil objective. Serial z stacks of the acini structures were acquired at 5 um intervals (usually 10-15 sections per field), and then analyzed with a custom MATLAB script (2017b, The Mathworks Inc.). Images were resampled to isotropic resolution and each spheroid was manually segmented. The DAPI signal was thresholded using Otsu’s method (Otsu, 1979) following intensity depth correction and smoothing. Holes were then filled and small, non-cellular objects were removed. The resulting binary nuclei image was used as a mask to measure cellular proliferation.
Enzymatic assays
cPLA2, iPLA2 and sPLA2 activities were measured using commercially available assays (Abcam, ab133089 or ab133090) according to the manufacturers instructions. Briefly, total cell lysates were obtained using 1x Cell Lysis Buffer (Cell Signaling Technology, 9803) under non-denaturing conditions. For cPLA2 activity, 10 μL lysate, 5 μL Assay buffer and 200 μL substrate solution containing arachidonoyl Thio-PC were incubated at room temperature for one hour. For iPLA2 activity, cell lysates were either untreated (measuring both cPLA2 and iPLA2 activity) or treated with 5 μM of the iPLA2 specific inhibitor bromoenol lactone (BEL), and activity was determined as follows: iPLA2 activity = (Activity without BEL) – (Activity with BEL). For sPLA2 activity, conditioned media was obtained from MCF10A PIK3CA WT and MUT cells and concentrated using a centrifugal vacuum evaporator (“SpeedVac”). Dried samples were resuspended in 100 μL Assay Buffer, and 10 μL of this was used for the assay. Cellular diacylglycerol (DAG) levels were measured using a DAG assay kit according to the manufacturer’s instructions (Cell Biolabs Inc., MET-5028). Briefly, ∼1x107 cells were harvested by scraping with 1 mL cold PBS, and pellets were obtained after centrifugation at 1500 g for 10 min. Lipids were extracted following sonication and incubation with methanol, sodium chloride and chloroform. The lower chloroform phase was washed twice with pre-equilibrated upper phase (PEU) and dried under a stream of nitrogen. 50 μL of assay buffer was used to resuspend the dried sample, 20 μL of which was used for the assay.
Immunoblot analysis
Cells were washed with ice-cold PBS and lysed on ice for 30 min with cell lysis buffer containing RIPA buffer (Thermo Scientific, 89900) supplemented with 4 μg each of leupeptin (Sigma-Aldrich, L2884) and pepstatin (Sigma-Aldrich, P5318), 2 mM Na3VO4 (Sigma-Aldrich, 450243), 1 mM DL-Dithiothreitol (Sigma-Aldrich, 646653), 10 μM Calyculin A (Cell Signaling Technology, 9902), 250 μM β-glycerophosphate (Sigma-Aldrich, G9422) and 400 μM PMSF protease inhibitor (Cell Signaling Technology, 8553). Lysates were subjected to centrifugation at 12,000 g for 30 min at 4°C, and protein concentrations were determined using the Bradford assay (Bio-Rad, 5000006). Nuclear isolation for mature SREBP probing was performed using a Nuclear Extract Kit (Active Motif, 40010) with 10 μg/ml of the protease inhibitor ALLN (Millipore, 208719). Protein lysates were boiled for 10 min and subjected to SDS-PAGE electrophoresis using 4%–15% precast gels (Bio-Rad, 567-1084). Densitometry was calculated using the Image Lab Software 5.2.1 (Bio-Rad). Affinity purified custom antibodies for phosphorylated cPLA2 on Thr376 were developed by Thermo Fisher Scientific using the PLA2G4A-369:383 peptide antigen and used at a dilution of 1:500 in 5% BSA/TBST. All other primary antibodies were used at a dilution of 1:1000 in 5% BSA/TBST solution, and secondary antibodies at 1:5000 in 5% milk/TBST.
Immunoprecipitation analysis
MCF10A PIK3CA WT and MUT isogenics were transiently transfected with HA-tagged cPLA2 (pCMV3-HA-PLA2G4A, Sino Biological, HG13126-NY) and lysed with 1X Cell Lysis Buffer (Cell Signaling Technologies, 9803) 48 hours post transfection. Equal volume of diluted cell lysates containing 4 mg of soluble protein were incubated with 2 μg rabbit anti-HA (Cell Signaling Technology, 3724) or 2 μg normal rabbit IgG (Santa-Cruz Biotechnology, sc-2027) as a negative control, and were pre-coupled with protein G Sepharose beads (Sigma-Aldrich, P3296) following incubation at 4°C for 4 hours rotating. 1X Cell Lysis Buffer was used to wash the beads four times, after which samples were resuspended in 30 μL 2x Laemmli sample buffer (Bio-Rad, 161-0737) and boiled for 10 min to release bound proteins.
To detect GTP-bound Rac1, the Active Rac1 Detection Kit (Cell Signaling Technology, 8815) was used, following the manufacturer’s instructions. Briefly, cell lysates were harvested under non-denaturing conditions using 1X Cell Lysis Buffer. To affinity precipitate activated G-protein, spin cups were incubated with 100 μL of glutathione resin and subsequently washed with 1X Cell Lysis Buffer. 20 μg of GST-PAK1-PBD was added to spin cups containing glutathione resin, after which 700 μL of the cell lysate containing 1 mg total protein was added, and the mix was incubated at 4°C for 1 hour with gentle shaking. Following three washes with 400 μL 1X Cell Lysis Buffer, samples were eluted by adding 2X reducing sample buffer containing 200 mM DTT in 2X SDS sample buffer, and subsequently heated for 5 min at 95°C. 25 μL of eluted samples were loaded onto an SDS-PAGE gel for subsequent immunoblot analysis using the provided Rac1 mouse monoclonal antibody (1:1000 dilution).
Immunohistochemistry analysis
10 μM thick sections were obtained from formalin-fixed and paraffin-embedded tumors and stained using an anti-phospho PKCζ Thr560 antibody (Abcam, ab62372) at a dilution of 1:100. For breast PDX and cell line-derived xenograft tumors, fresh frozen tumor pieces were mounted on Optimal Cutting Temperature (OCT) compound and sectioned to 10 μM thickness. To assess the presence of activated natural killer cells, slides were stained using goat anti-mouse NKp46/ncr1 polyclonal antibody (R and D systems, 170-6516) at a final concentration of 3 μg/ml. Images were captured on a Hamamatsu NanoZoomer, and staining was quantified using the IHC Profiler plug-in for ImageJ.
Proximity ligation assay
For the proximity ligation assay (PLA), all incubations were performed in a humidity chamber as per the manufacturer’s instructions using a Duolink In Situ Detection Reagents Red kit (Sigma-Aldrich, DUO92008). Briefly, MCF10A PIK3CA isogenic cells were seeded on glass coverslips, serum-starved overnight, and stimulated with full serum and growth factors for 30 min; control cells were untreated. They were then fixed with 4% paraformaldehyde (PFA), blocked and incubated with primary antibodies (anti-cPLA2 and anti-phospho PKCζ Thr560) for 1 hour at room temperature. Samples were then incubated with Minus and Plus PLA probes (Sigma-Aldrich, DUO92004 and DUO92002) for 1 hour at 37°C, followed by a ligation step for 30 min at 37°C, and an amplification step for 100 min at 37°C. Finally, the coverslips were mounted with the Duolink PLA mounting media and DAPI, and fluorescence was visualized with a Zeiss 710 Confocal microscope with a 40x oil objective. Images were analyzed using a custom MATLAB (2017b) script. Briefly, the DAPI signal was segmented by taking a maximum intensity projection, smoothing, and then thresholding (Otsu, 1979). Borders between cells were estimated by finding the midpoints between nuclei using a watershed approach. A summed projection was calculated for the probe signal, this was smoothed (Gaussian kernel sigma of 1 pixel), and thresholded. The threshold calculated from a positive control image was applied to all images. The segmented image of the probe was used to calculate the number of foci on a per cell basis.
Fluorescent calcium assay
Calcium flux was measured using the Fluo-4 Direct™ Calcium Assay Kit (Thermo Fisher Scientific, F10471). Full MCF10A growth medium was removed and replaced with FBS-free medium for 18 hours. Control cells were treated with equal volume of the culture media of the 2x Fluo-4 Direct™ calcium assay reagent solution for 30 min in 37°C, 5% CO2, without removing the assay media. The induced cells were stimulated with full media for 30 min, while incubated with the calcium assay reagent solution. Cells were then analyzed by monitoring the fluorescence of the Fluo-4 dye using an ImageXpress® Micro Confocal High-Content Imaging System (Molecular Devices) and a 20x objective with 488 nm excitation. The fluorescence per field before and after induction was calculated using a MetaXpress Software Custom Module.
To assess the effects of PLCγ1 inhibition on calcium flux, cells were treated with either 2 μM U73122 for 24 hours, or transiently transfected with 25 nM ON-TARGETplus SMARTpool human siRNA targeting PLCγ1 for 48 hours. For the final 18 hours, full MCF10A growth medium was removed and replaced with FBS-free medium. Immediately prior to the assay, cells were treated with equal volume culture media and 2x Fluo-4 Direct™ calcium assay reagent, and baseline fluorescence was monitored using an ImageXpress® Micro Confocal High-Content Imaging System (Molecular Devices). After 300 ms, cells were induced by adding 1 mL full MCF10A growth medium, and fluorescence of the Fluo-4 dye was monitored using a 20x objective with 488 nm excitation. Calcium flux was calculated by first normalizing fluorescence readings to baseline measurements using the formula: dF = F(t) – F(0)/F(0), where F = fluorescence at 488 nm, t = time, and F(0) represents the average baseline readings from 1-299 ms. Fluorescence values were subsequently normalized to a unit interval between 0 and 1 and presented as the time required post stimulation to reach a maximal calcium intensity.
Quantitative RT-PCR and PIK3CA mutation analysis
RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN, 74134) and 1 μg of total RNA was used to synthesize complementary DNA (cDNA) using the QuantiTect Reverse Transcription Kit (QIAGEN, 205311). Primer sequences used are as follows: Human cPLA2, forward: 5′-GATGAAACTCTAGGGACAGCAAC-3′,
reverse: 5′-CTGGGCATGAGCAAACTTCAA-3′, Human β-actin: forward: 5′-GACCCAGATCATGTTTGAGACC-3′,
reverse: 5′-CTTCATGAGGTAGTCAGTCAGG. Reactions were performed with SYBR Select Master Mix (ThermoFisher, 4472908) using the TProfessional Thermocycler from Biometra and analyzed with the qPCRsoft version 3.1 (Thistle Scientific/Analytik Jena). The following cycle reactions were used: pre-denaturation for 3 min at 95°C, followed by 45 cycles of 5 s at 95°C (denaturation), 5 s at 55.8°C (annealing) and 15 s at 72°C (elongation).
To detect PIK3CA mutations in primary breast tumor samples, DNA was extracted from 10 mg of tissue using the QIAamp DNA mini kit (QIAGEN, 51304). Mutations in exon 9 (helical domain) and exon 20 (kinase domain) of PIK3CA were assessed using the PNAClamp PIK3CA Mutation Detection Kit (Panagene, PNAC-4001), according to the manufacturer’s instructions. Briefly, reactions were performed using 10 ng DNA with a SYBR Green PCR reaction premix and primer premixes detecting E542, E545, Q546 and H1047 mutations using the TProfessional Thermocycler. The following cycle reactions were used: pre-denaturation for 5 min at 94°C, followed by 40 cycles of 30 s at 94°C (denaturation), 20 s at 70°C (peptide nucleic acid clamping), 30 s at 63°C (annealing) and 30 s at 72°C (extension). PIK3CA mutations were assessed based on manufacturer’s instructions.
In vitro kinase assay
In vitro kinase assays were performed using the ADP-Glo Kinase Assay (Promega, V6930), according to the manufacturer’s instructions. Briefly, 100 ng recombinant human protein kinase C zeta (PKCζ, Insight Biotechnology, TP302472) and 0.5 μg/ml phospholipase A2, group IVA (cPLA2, Insight Biotechnology, TP320972) were incubated in kinase buffer comprising 40 mM TRIS buffer pH 7.5, 20 mM MgCl2 (Sigma-Aldrich, M8266), 0.1 mg/ml BSA (Sigma-Aldrich, A2153) and 50 μM DTT (Sigma-Aldrich, 646563), and supplemented with 20 μM ATP. Reactions were incubated at 30°C for 3 hours, after which luminescence was measured following an integration time of 1 s.
Chemokine assays
Tumor samples were placed in 2 mL lysing tubes prefilled with 1.4 mm ceramic (zirconium oxide) beads and 1 mL of chilled PBS. Samples were homogenized with a Precellys24 homogenizer programmed with three 30 s cycles at 6,500 Hz and 4-min pause times. At the end of the homogenization cycle, samples were centrifuged at 5,000rpm for 10 min at 4°C, and supernatants were transferred to fresh Eppendorf tubes
To measure CCL5 (RANTES) chemokine levels, the CCL5 Mouse ELISA kit (Abcam, ab100739) was used according to the manufacturer’s instructions. Briefly, 100 μL of standards prepared in Assay Diluent A and sample supernatants were added to appropriate wells and incubated at room temperature for 2.5 hours. Wells were subsequently washed 4 times with 1X Wash Solution, and 100 μL of 1X Biotinylated CCL5 (RANTES) Detection Antibody was added and incubated for 1 hour at room temperature with gentle shaking. Following the incubation with the detection antibody, wells were washed 4 times and incubated with 100 μL HRP-Streptavidin solution for 45 min at room temperature with gentle shaking. Following a final set of 4 washes, 100 μL of TMB One-Step Substrate Reagent was added to each well and incubated for 30 min at room temperature in the dark with gentle shaking. 50 μL of stop solution was subsequently added, and measurements were taken immediately at 450 nm.
To measure CX3CL1 chemokine levels, the CX3CL1 Mouse ELISA kit (Abcam, ab100683) was used according to the manufacturer’s instructions. Briefly, 100 μL of standards prepared in Assay Diluent C and sample supernatants were added to appropriate wells and incubated at room temperature for 2.5 hours. Wells were subsequently washed 4 times with 1X Wash Solution, and 100 μL of 1X Biotinylated CX3CL1 Detection Antibody was added and incubated for 1 hour at room temperature with gentle shaking. Following the incubation with the detection antibody, wells were washed 4 times and incubated with 100 μL HRP-Streptavidin solution for 45 min at room temperature with gentle shaking. Following a final set of 4 washes, 100 μL of TMB One-Step Substrate Reagent was added to each well and incubated for 30 min at room temperature in the dark with gentle shaking. 50 μL of stop solution was subsequently added, and measurements were taken immediately at 450 nm. All measurements were normalized to total protein content as determined using the BCA protein assay (Thermo Fisher Scientific, 23225).
Lipid extraction and eicosanoid profiling
Total lipids were extracted based on the Bligh and Dyer method (Bligh and Dyer, 1959) and analyzed as described previously (Wolfer et al., 2015). Briefly, cell lines were harvested when 80% confluent, and media was replaced one hour prior to lipid extraction. Cell pellets were resuspended in 1ml water, after which 3.75 mL chloroform/methanol mixture (1:2 v/v) was added and samples were vortexed and incubated for at least 30 min on ice. 1.25 mL chloroform was added, followed by 1.25 mL mili-Q water and after vortexing and centrifugation at 1000rpm for 10 min at 4°C, the organic bottom phase was separated and transferred to a 15 mL amber vial to get dried under a nitrogen flow. For conditioned media profiling, cells were grown to 80% confluency in full media, after which residual media was washed off twice with PBS, and cells were cultured with FAF media for 48 hours. The conditioned media was centrifuged at 2000 g for 20 min to remove cell debris. Samples were concentrated using a centrifugal vacuum evaporator. The lipid extraction was then performed as above.
Eicosanoid levels were determined using liquid chromatography with a Waters HSS T3 UPLC column connected to a Waters Xevo TQ-S triple quadrupole mass spectrometer with an electrospray ionization (ESI) source. TargetLynx (Waters, Manchester, UK) was used for peak detection and integration. Data was normalized to protein concentration.
To measure PGE2 (Enzo Life Sciences, ADI-900-001) and AA (Generon, CEB098Ge) concentrations, enzyme-linked immunosorbent assays were used, following the manufacturer’s instructions.
Bioinformatic analysis
Gene-centric RMA-normalized expression data and mutation status for available cell lines were obtained from the Cancer Cell Line Encyclopedia (CCLE). 150 frequently mutated genes were identified based on a mutation frequency of ≥ 20% or higher in n = 35 available cell lines, and significant enrichment in observed clusters was assessed using Fisher’s exact test. A functional annotation analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8) (Huang et al., 2009) was used to identify biologically relevant pathways represented by a set of 512 genes, which were significantly upregulated in the PIK3CA mutation- and lipid-enriched cluster. Interactions between genes involved in the identified pathways were visualized by functional gene networks using the ‘ReactomePA’ package in R (Yu and He, 2016).
Quantification and Statistical Analysis
Student t test, Fisher’s exact test, and one- or two-way ANOVA were used to evaluate statistical significance as indicated in the respective figure legends. The Shapiro-Wilk test was used to assess normality. The ‘N’ for each experiment can be found in the figure legends and represents independently generated samples for in vitro experiments, wells for cell-based assays, or mice for in vivo experiments. Bar graphs present the mean ± standard error of the mean (SEM). Significance was defined as p < 0.05, and denoted by asterisks throughout the figures as follows: n.s (not significant), ∗(p < 0.05), ∗∗(p < 0.01), ∗∗∗(p < 0.001). No statistical methods were used to determine sample size, and no completed data were excluded from the analysis performed in this manuscript. Statistical tests were performed using GraphPad Prism (version 8.0.1) and R statistical software (version 3.5.1).
Acknowledgments
We thank Naomi Guppy and Farzana Noor (Breast Cancer Now Histopathology, ICR, London, UK) and Elena Miranda and Adriana Resende Alves (Pathology Core Facility, University College London Cancer Institute) for support with immunohistochemistry, hematoxylin, and eosin analysis; and Champions Oncology (London, UK) for kindly providing breast, pancreatic, ovarian, sarcoma, and colorectal cancer PDX tumor specimens. We would also like to thank Edward St. John for enabling access to primary breast tumor samples, Verena M. Horneffer-van der Sluis for assistance with eicosanoid analysis, and the Biological Services Unit staff at the Institute of Cancer Research (Chelsea site) for their assistance with in vivo experiments. N.K. was supported by an ICR PhD studentship. The work described and the laboratory of G.P. was supported by the Institute of Cancer Research and a Cancer Research UK Grand Challenge award (C59824/A25044). Work in the Z.T. lab was supported by the European Research Council (MASSLIP Consolidator grant), Cancer Research UK Grand Challenge award (C59824/A25044), and the National Institute for Health Research (Imperial Biomedical Research Centre). A.V. was funded by the Ministry of Education, Culture and Sport under the Program for Promoting and Hiring of Talent and its Employability (Subprogram for Mobility “José Castillejo”) of the Spanish Government and by Comunitat Autònoma de les Illes Balears, Direcció General d’lnnovació i Recerca (AAEE003/2017) and Fons Europeu de Desenvolupament Regional de la Unió Europea (FEDER).
Author Contributions
N.K., Z.T., and G.P. designed the study with contributions from J.K.N. and R.C.G. Z.T. and G.P. directed the project and supervised data analysis. N.K. performed and analyzed most experiments. E.K. performed 3-D acini, proximity ligation assays, calcium flux analyses, the CRISPR knockdown generation, the site-directed mutagenesis constructs, and assisted with xenograft studies. A.T. performed in vitro kinase assays, ELISAs, and assisted with xenograft studies and REIMS analysis of tumor samples. A.V. assisted with estrogen receptor signaling experiments, the generation of dox-inducible cPLA2-knockdown cell lines, and lipid extractions for LC-MS analysis. P.I. developed essential platforms for pre-processing and interpretation of REIMS data. N.J.S.P. assisted with xenograft studies. D.J.M. assisted with chemokine assays. S.A.V. and G.A.E. cultured and analyzed cell lines grown under different microenvironment conditions. A.L.T. carried out image analysis. M.L.D. assisted with LC-MS profiling of eicosanoids. A.v.W. and C.M.I. helped with animal experiments. R.F.S. provided essential technical support for sample processing with REIMS. N.K. and G.P. wrote the manuscript. All authors edited the manuscript.
Declaration of Interests
N.K. and G.P. are inventors on a patent application covering new methods and compositions useful in the treatment of cancers with PIK3CA mutation (application number GB 2005874.9).
Published: June 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.05.053.
A video abstract is available at https://doi.org/10.1016/j.cell.2020.05.053#mmc8.
Contributor Information
Zoltan Takats, Email: z.takats@imperial.ac.uk.
George Poulogiannis, Email: george.poulogiannis@icr.ac.uk.
Supplemental Information
References
- Alexander L.D., Ding Y., Alagarsamy S., Cui X.L., Douglas J.G. Arachidonic acid induces ERK activation via Src SH2 domain association with the epidermal growth factor receptor. Kidney Int. 2006;69:1823–1832. doi: 10.1038/sj.ki.5000363. [DOI] [PubMed] [Google Scholar]
- Alexander J., Gildea L., Balog J., Speller A., McKenzie J., Muirhead L., Scott A., Kontovounisios C., Rasheed S., Teare J. A novel methodology for in vivo endoscopic phenotyping of colorectal cancer based on real-time analysis of the mucosal lipidome: a prospective observational study of the iKnife. Surg. Endosc. 2017;31:1361–1370. doi: 10.1007/s00464-016-5121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambs P., Baccarini M., Fitzke E., Dieter P. Role of cytosolic phospholipase A2 in arachidonic acid release of rat-liver macrophages: regulation by Ca2+ and phosphorylation. Biochem. J. 1995;311:189–195. doi: 10.1042/bj3110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balog J., Sasi-Szabó L., Kinross J., Lewis M.R., Muirhead L.J., Veselkov K., Mirnezami R., Dezső B., Damjanovich L., Darzi A. Intraoperative tissue identification using rapid evaporative ionization mass spectrometry. Sci. Transl. Med. 2013;5:194ra93. doi: 10.1126/scitranslmed.3005623. [DOI] [PubMed] [Google Scholar]
- Basingab F.S., Ahmadi M., Morgan D.J. IFNγ-Dependent Interactions between ICAM-1 and LFA-1 Counteract Prostaglandin E2-Mediated Inhibition of Antitumor CTL Responses. Cancer Immunol. Res. 2016;4:400–411. doi: 10.1158/2326-6066.CIR-15-0146. [DOI] [PubMed] [Google Scholar]
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Borcoman E., De La Rochere P., Richer W., Vacher S., Chemlali W., Krucker C., Sirab N., Radvanyi F., Allory Y., Pignot G. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. OncoImmunology. 2019;8:e1581556. doi: 10.1080/2162402X.2019.1581556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.C., Sahai E., Zelenay S., Reis E Sousa C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J.E., Dennis E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A., Pandolfi P.P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Chambers M.C., Maclean B., Burke R., Amodei D., Ruderman D.L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou M.M., Hou W., Johnson J., Graham L.K., Lee M.H., Chen C.S., Newton A.C., Schaffhausen B.S., Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr. Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Clark J.D., Schievella A.R., Nalefski E.A., Lin L.L. Cytosolic phospholipase A2. J. Lipid Mediat. Cell Signal. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- Dan S., Okamura M., Seki M., Yamazaki K., Sugita H., Okui M., Mukai Y., Nishimura H., Asaka R., Nomura K. Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010;70:4982–4994. doi: 10.1158/0008-5472.CAN-09-4172. [DOI] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S.K., Brugge J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Dennis E.A., Norris P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015;15:511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble C.C., Manning B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K., Yecies J.L., Menon S., Raman P., Lipovsky A.I., Souza A.L., Triantafellow E., Ma Q., Gorski R., Cleaver S. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Chiu H., Hopkins B.D., Bagrodia S., Cantley L.C., Abraham R.T. The PI3K Pathway in Human Disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavaghan C.L., Holmes E., Lenz E., Wilson I.D., Nicholson J.K. An NMR-based metabonomic approach to investigate the biochemical consequences of genetic strain differences: application to the C57BL10J and Alpk:ApfCD mouse. FEBS Lett. 2000;484:169–174. doi: 10.1016/s0014-5793(00)02147-5. [DOI] [PubMed] [Google Scholar]
- Gibb S., Strimmer K. MALDIquant: a versatile R package for the analysis of mass spectrometry data. Bioinformatics. 2012;28:2270–2271. doi: 10.1093/bioinformatics/bts447. [DOI] [PubMed] [Google Scholar]
- Greene E.R., Huang S., Serhan C.N., Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths B., Lewis C.A., Bensaad K., Ros S., Zhang Q., Ferber E.C., Konisti S., Peck B., Miess H., East P. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1:3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guri Y., Colombi M., Dazert E., Hindupur S.K., Roszik J., Moes S., Jenoe P., Heim M.H., Riezman I., Riezman H., Hall M.N. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell. 2017;32:807–823.e12. doi: 10.1016/j.ccell.2017.11.011. [DOI] [PubMed] [Google Scholar]
- Henry W.S., Laszewski T., Tsang T., Beca F., Beck A.H., McAllister S.S., Toker A. Aspirin Suppresses Growth in PI3K-Mutant Breast Cancer by Activating AMPK and Inhibiting mTORC1 Signaling. Cancer Res. 2017;77:790–801. doi: 10.1158/0008-5472.CAN-16-2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilvo M., Denkert C., Lehtinen L., Müller B., Brockmöller S., Seppänen-Laakso T., Budczies J., Bucher E., Yetukuri L., Castillo S. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;71:3236–3245. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- Holmes E., Wilson I.D., Nicholson J.K. Metabolic phenotyping in health and disease. Cell. 2008;134:714–717. doi: 10.1016/j.cell.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Hopkins B.D., Pauli C., Du X., Wang D.G., Li X., Wu D., Amadiume S.C., Goncalves M.D., Hodakoski C., Lundquist M.R. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560:499–503. doi: 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hughes-Fulford M., Li C.F., Boonyaratanakornkit J., Sayyah S. Arachidonic acid activates phosphatidylinositol 3-kinase signaling and induces gene expression in prostate cancer. Cancer Res. 2006;66:1427–1433. doi: 10.1158/0008-5472.CAN-05-0914. [DOI] [PubMed] [Google Scholar]
- Janiszewska M., Liu L., Almendro V., Kuang Y., Paweletz C., Sakr R.A., Weigelt B., Hanker A.B., Chandarlapaty S., King T.A. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015;47:1212–1219. doi: 10.1038/ng.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai S., Ishihara K., Kawashita E., Tomoo T., Nagahira K., Hayashi Y., Akiba S. ASB14780, an Orally Active Inhibitor of Group IVA Phospholipase A2, Is a Pharmacotherapeutic Candidate for Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2016;356:604–614. doi: 10.1124/jpet.115.229906. [DOI] [PubMed] [Google Scholar]
- Kramer R.M., Roberts E.F., Um S.L., Börsch-Haubold A.G., Watson S.P., Fisher M.J., Jakubowski J.A. p38 Mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J. Biol. Chem. 1996;271:27723–27729. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- Lala P.K., Nandi P., Majumder M. Roles of prostaglandins in tumor-associated lymphangiogenesis with special reference to breast cancer. Cancer Metastasis Rev. 2018;37:369–384. doi: 10.1007/s10555-018-9734-0. [DOI] [PubMed] [Google Scholar]
- Lee K.L., Foley M.A., Chen L., Behnke M.L., Lovering F.E., Kirincich S.J., Wang W., Shim J., Tam S., Shen M.W. Discovery of Ecopladib, an indole inhibitor of cytosolic phospholipase A2alpha. J. Med. Chem. 2007;50:1380–1400. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]
- Lee S.J., Kang W.Y., Yoon Y., Jin J.Y., Song H.J., Her J.H., Kang S.M., Hwang Y.K., Kang K.J., Joo K.M., Nam D.H. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011. doi: 10.1186/s12885-015-2034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Zheng Y., Cho S., Jang C., England C., Dempsey J.M., Yu Y., Liu X., He L., Cavaliere P.M. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell. 2017;171:1545–1558. doi: 10.1016/j.cell.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Gao T. mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 2014;15:191–198. doi: 10.1002/embr.201338119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Dai D., Chen B., Tang H., Xie X., Wei W. Efficacy of PI3K/AKT/mTOR pathway inhibitors for the treatment of advanced solid cancers: A literature-based meta-analysis of 46 randomised control trials. PLoS ONE. 2018;13:e0192464. doi: 10.1371/journal.pone.0192464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X., Lochhead P., Nishihara R., Morikawa T., Kuchiba A., Yamauchi M., Imamura Y., Qian Z.R., Baba Y., Shima K. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 2012;367:1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E.C., Lyssiotis C.A., Cantley L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016;207:39–72. doi: 10.1007/978-3-319-42118-6_3. [DOI] [PubMed] [Google Scholar]
- Lin L.L., Wartmann M., Lin A.Y., Knopf J.L., Seth A., Davis R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- McKew J.C., Lee K.L., Shen M.W., Thakker P., Foley M.A., Behnke M.L., Hu B., Sum F.W., Tam S., Hu Y. Indole cytosolic phospholipase A2 alpha inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J. Med. Chem. 2008;51:3388–3413. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- Nicholson J.K., Connelly J., Lindon J.C., Holmes E. Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002;1:153–161. doi: 10.1038/nrd728. [DOI] [PubMed] [Google Scholar]
- Nicholson J.K., Holmes E., Kinross J.M., Darzi A.W., Takats Z., Lindon J.C. Metabolic phenotyping in clinical and surgical environments. Nature. 2012;491:384–392. doi: 10.1038/nature11708. [DOI] [PubMed] [Google Scholar]
- Okada S., Vaeteewoottacharn K., Kariya R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells. 2019;8:889. doi: 10.3390/cells8080889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkenhaug K., Graupera M., Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016;6:1090–1105. doi: 10.1158/2159-8290.CD-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. [Google Scholar]
- Patterson E., Wall R., Fitzgerald G.F., Ross R.P., Stanton C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012;2012:539426. doi: 10.1155/2012/539426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh L.E., Rhee S.G., Spokes K., Kazlauskas A., Cantley L.C., Cantley L.G. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J. Biol. Chem. 1998;273:23750–23757. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- Ricoult S.J.H., Yecies J.L., Ben-Sahra I., Manning B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35:1250–1260. doi: 10.1038/onc.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D.D., Ali S.M., Sengupta S., Sheen J.H., Hsu P.P., Bagley A.F., Markhard A.L., Sabatini D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Saxton R.A., Sabatini D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simopoulos A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. (Maywood) 2008;233:674–688. doi: 10.3181/0711-MR-311. [DOI] [PubMed] [Google Scholar]
- Sonnweber T., Pizzini A., Nairz M., Weiss G., Tancevski I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018;19:3285. doi: 10.3390/ijms19113285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John E.R., Balog J., McKenzie J.S., Rossi M., Covington A., Muirhead L., Bodai Z., Rosini F., Speller A., Shousha S. Rapid evaporative ionisation mass spectrometry of electrosurgical vapours for the identification of breast pathology: towards an intelligent knife for breast cancer surgery. Breast Cancer Res. 2017;19 doi: 10.1186/s13058-017-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrado-Castellarnau M., de Atauri P., Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget. 2016;7:62726–62753. doi: 10.18632/oncotarget.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoo T., Nakatsuka T., Katayama T., Hayashi Y., Fujieda Y., Terakawa M., Nagahira K. Design, synthesis, and biological evaluation of 3-(1-Aryl-1H-indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2α inhibitors. J. Med. Chem. 2014;57:7244–7262. doi: 10.1021/jm500494y. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., DeBerardinis R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Dubois R.N. Eicosanoids and cancer. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederschain D., Wee S., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y.M., Lengauer C. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- Wolfer A.M., Gaudin M., Taylor-Robinson S.D., Holmes E., Nicholson J.K. Development and Validation of a High-Throughput Ultrahigh-Performance Liquid Chromatography-Mass Spectrometry Approach for Screening of Oxylipins and Their Precursors. Anal. Chem. 2015;87:11721–11731. doi: 10.1021/acs.analchem.5b02794. [DOI] [PubMed] [Google Scholar]
- Yu G., He Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- Zelenay S., van der Veen A.G., Böttcher J.P., Snelgrove K.J., Rogers N., Acton S.E., Chakravarty P., Girotti M.R., Marais R., Quezada S.A. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated during this study are available at GitHub using the following accessions: https://github.com/adamltyson/CalciumAnalysis, https://github.com/adamltyson/cell-coloc-3D, and https://github.com/adamltyson/foci2D. These accessions are also provided in the Key Resources Table. The published article includes all REIMS m/z values and putative annotations for significantly different lipids between various receptor subtypes and MCF10A PIK3CA isogenics in the Supplementary Information in Tables S1 and S4, respectively. Original/source data of REIMS profiles for Figures 1D, 1E, 1H, 3B, and 3D in the paper corresponding to breast cancer cell lines and tumors is available through Mendeley Data (https://doi.org/10.17632/xcgc5kpntm.1)