

Abstract
Epidermal growth factor (EGF) has many physiological roles. However, its effects on stem and progenitor Leydig cell development remain unclear. Rat stem and progenitor Leydig cells were cultured with different concentrations of EGF alone or in combination with EGF antagonist, erlotinib or cetuximab. EGF (1 and 10 ng/mL) stimulated the proliferation of stem Leydig cells on the surface of seminiferous tubules and isolated CD90+ stem Leydig cells and progenitor Leydig cells but it blocked their differentiation. EGF also exerted anti‐apoptotic effects of progenitor Leydig cells. Erlotinib and cetuximab are able to reverse EGF‐mediated action. Gene microarray and qPCR of EGF‐treated progenitor Leydig cells revealed that the down‐regulation of steroidogenesis‐related proteins (Star and Hsd3b1) and antioxidative genes. It was found that EGF acted as a proliferative agent via increasing phosphorylation of AKT1. In conclusion, EGF stimulates the proliferation of rat stem and progenitor Leydig cells but blocks their differentiation.
Keywords: EGF, StAR, stem/progenitor Leydig cells, steroidogenesis, steroids
1. INTRODUCTION
The androgen deficiency could be treated by transplanting stem (SLC)/progenitor (PLC) Leydig cells (LCs) into the host testis. 1 Androgen production not only depends upon LC number but also on its maturity. 2 The development of LCs starts from SLCs. 1 A set of testicular SLCs in the neonatal and adult rat, 3 , 4 mouse 5 , 6 , 7 and human 8 testes were identified. During puberty in rats, the SLC commits into a spindle‐shaped PLC around days 12‐14 postpartum. 9 , 10 PLCs begin to express some steroidogenic enzymes, including cytochrome P450 cholesterol side‐chain cleavage enzyme (CYP11A1), 3β‐hydroxysteroid dehydrogenase isoform 1 (HSD3B1) and cytochrome 17α‐hydroxylase/17,20‐lyase (CYP17A1). 9 However, PLCs do not express the last‐step androgen synthetic enzyme, 17β‐hydroxysteroid dehydrogenase isoform 3 (HSD17B3). 9 PLCs also express high levels of androgen metabolizing enzymes, including steroid 5α‐reductase 1 (SRD5A1) and 3α‐hydroxysteroid dehydrogenase. 9 Androstenedione made by PLCs after CYP17A1 catalysis was metabolized into androstanedione by SRD5A1 via 5α‐reduction and further into androsterone (AO) via 3α‐reduction, thus being the primary androgen secreted by PLCs. 9 PLCs undergo transitions into oval lipid‐droplet‐rich immature Leydig cells (ILCs) around days 28 postpartum, when ILCs appear to express HSD17B3 to synthesize testosterone (T). 9 ILCs still have higher levels of androgen metabolizing enzymes, and they metabolize T into dihydrotestosterone via 5α‐reduction by SRD5A1 and further into 5α‐androstane‐3α,17‐diol (DIOL) via 3α‐reduction. 9 After PLCs enter the ILC stage, an important glucocorticoid‐metabolizing 11β‐hydroxysteroid dehydrogenase 1 (HSD11B1) is expressed and is exclusively located in ILCs and adult Leydig cells (ALCs). 11 , 12 ILCs differentiate into ALCs around days 56 postpartum. 9 , 13 ALCs mainly secrete T because of the disappearance of SRD5A1 protein. 9
Leydig cell development during puberty mainly relies on the regulation of luteinizing hormone (LH), 14 , 15 because PLCs, ILCs and ALCs all contain its receptor (LHCGR). 2 , 16 Indeed, LH can stimulate PLC proliferation and differentiation. 9 , 14 , 15 , 17 However, SLCs do not express LHCGR and do not need LH for the regulation. 3 , 4 SLC proliferation and differentiation depend on signalling pathways acting between somatic and stem cells in rodents and humans during the perinatal and prepubertal period. 18 , 19 Many growth factors may have an impact on the development of LCs during puberty. 1 One of these growth factors could be epidermal growth factor (EGF). Evidence points to the regulation of EGF for the development of steroid‐producing gland. EGF has been found to promote functional maturation of the foetal primate adrenal glands. 20 EGF plays a role in the development of testis because EGF knockout mice are sterile. 21 Despite the physiological importance of EGF in testicular function, its mechanism of action on LC development remains unknown.
Epidermal growth factor is a growth factor that was first purified from mouse salivary glands. 22 It exerts numerous roles in different cell types. EGF exerts its action through binding to its transmembrane receptors, EGF receptors, thus stimulating proliferation, growth and differentiation of many types of cells. 22 Previous studies have demonstrated the presence of EGF receptors in human, bovine, rat and mouse LCs, suggesting that EGF has a role in LCs. 23 , 24 , 25 , 26
However, the effects of EGF on the development of SLCs/PLCs into the LC lineage are largely unclear. In our previous studies, we established a model for studying SLC development using an in vitro culture of LC‐depleted seminiferous tubules (STs). 4 , 27 ALCs not SLCs can be eliminated after intraperitoneal injection of 75 mg/kg ethane dimethane sulfonate (EDS) into rats. 28 , 29 LC‐depleted ST is isolated for the serum‐free culture. In this system, SLCs on the surface of STs can be induced to differentiate into the LC lineage under the LC differentiation medium (LDM) containing LH and insulin‐transferrin‐selenium supplement (ITS). 4 , 27 , 30 In the current study, we examined the effects of EGF on the development of SLCs/PLCs.
2. MATERIALS AND METHODS
2.1. Chemicals
Bovine serum albumin (BSA), ITS, dimethyl sulfoxide (DMSO), EGF, Percoll, M199, DMEM, F12 and HBSS medium were obtained from Sigma‐Aldrich. Click‐it EdU Alexa Fluor was obtained from Life Technologies. Erlotinib HCl (E), an EGFR kinase inhibitor, 31 was obtained from Selleck. LH was a gift of NIDDK (US). Cetuximab (Cet) was obtained from MCE (Cat No: HY‐P9905). EDS was purchased from Pterosaur Biotech. [3H]‐Thymidine, [3H]‐androsterone ([3H]‐AO) and [3H]‐testosterone ([3H]‐T) tracers were obtained from DuPont‐New England Nuclear. Hyamine hydroxide was purchased from ICN Radiochemicals. Primer information was listed in Table S1. Antibody information was listed in Table S2.
2.2. Animals
Male Sprague‐Dawley rats were purchased from Shanghai Laboratory Animal Co. Ltd. Forty male rats at age of 7 days were used for the isolation of CD90+ SLCs each time. Forty male rats at age of 21 days were used for the isolation of PLCs each time. Male rats at age of 90 days were used for ST isolation and culture of SLCs. The animal procedure was approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University and was performed in accordance with the Guide for the Care and Use of Laboratory Animals.
2.3. Purification and culture of CD90+ SLCs
Testicular CD90+ cells were thought to be the SLCs. 30 Purification and culture of CD90+ SLCs from 7‐day‐old male rats were performed. Peritubular testicular cells were obtained from collagenase (0.1 mg/mL)‐treated isolated STs and stained using CD90 antibody, and purified using BD IMag™ bead. Cells were incubated with CD90 antibody (1:100) in BD IMag™ Buffer for 20 minutes on ice. Beads were incubated with CD90‐conjugated cells for 30 minutes. After washing, the cells were separated by BD IMagnet™ for 10 minutes. Cells were suspended in M199 medium. The purity of CD90+ cells (SLCs) was over 99%. To study the proliferation of SLCs, CD90+ cells (1 × 104 cells/well) in LDM medium were seeded in 12‐well plate and incubated with control (LDM), EGF (10 ng/mL), Cet (an EGF antagonist, 5 µg/mL) and EGF (10 ng/mL)+Cet (5 µg/mL) for 24 hours. Then, cells were washed using PBS, and EdU incubation was performed as following section. Our previous study has demonstrated that the SLCs are cultured during the first week of culture, and the number of SLCs is greatly amplified. 30 Then, SLCs were switched to LDM for additional 14 days, the amplified SLCs could be differentiated into ALCs, and thus, the increased number of ALCs could contribute into the robust increase of T level in the medium. 30 Using this approach, EGF (0, 10 ng/mL) with or without EGF antagonist (Cet, 5 μg/mL) was added to SLCs (1 × 104 cells/well) in M199 and cultured at 34°C and 5% CO2 for 7 days, and then, SLCs were switched into LDM for additional 14 days. Media were collected for the measurement of T level.
To study the differentiation of SLCs, CD90+ cells (1 × 104 cells/well) in LDM medium were seeded in 12‐well plate and incubated with control (LDM), EGF (10 ng/mL), Cet (an EGF antagonist, 5 µg/mL) and EGF (10 ng/mL)+Cet (5 µg/mL) for 14 days. Media were collected for the measurement of T level.
2.4. ST isolation and culture of SLCs on the surface of STs
The procedure for ST isolation and culture was performed as previously described. 27 , 30 Briefly, EDS was dissolved in a 1:3 solution mixture of DMSO and water. One 90‐day‐old rat was selected and injected intraperitoneally with a single dose (75 mg/kg body weight) of EDS, which can effectively eliminate all LCs in the testis without damaging SLCs. 12 , 13 Seven days after EDS, LCs were all eliminated. 32 The rat was euthanized under CO2. Two testes were taken out and placed in cold M199 medium and decapsulated. STs were mechanically separated using a fine forceps under a microscope. 27 STs were cut to about 3‐cm‐long fragments and distributed randomly into 12‐well plates, with each well containing equal amount of ST fragments. The STs were cultured at 34°C and 5% CO2 for up to 14 days in LDM for induction of SLCs into ALCs to produce T as previously described. 27 , 30 Twelve isolations were performed. To study the effects of EGF on the proliferation of SLCs on the surface of STs, EGF (0, 1, and 10 ng/mL) was added to M199 medium and cultured at 34°C and 5% CO2 for 7 days, during which SLCs have the highest capacity of proliferation. 30
2.5. EdU incorporation into SLCs
The proliferative capacity of SLCs after EGF treatment was measured by EdU kit as previously described. 33 Media containing EGF and its inhibitor were removed for both CD90+ SLCs and STs above, and 2 µL of 1:1000 diluted EdU was added in the well and incubated at 34°C and 5% CO2 for 24 hours. STs were washed twice with 500 µL phosphate‐buffered saline (PBS) containing 3% BSA. Fragments of STs were also brought down in 2% argarose gel after centrifugation at 750 g, and the cross sections were cut. CD90+ SLCs and ST cross sections were then fixed in 4% paraformaldehyde at room temperature for 30 minutes. The CD90+ SLCs and STs were washed and incubated with reaction solution in the dark for 45 minutes. CD90+ antibody was used for staining CD90+ SLCs in purified SLCs. DAPI served the counterstaining. Then, CD90+ SLCs and STs were washed again and mounted on slide for visualization under fluorescence microscope (Olympus) and images were captured. EdU‐positive cells (green fluorescence in cell nucleus) were counted and calculated by the total surface area of STs using the ImageProPlus 7.0 software (Media Cybernetics).
2.6. SLC differentiation
Our previous study has demonstrated that STs are cultured in LDM for 14 days, and SLCs on the surface of STs are capable of differentiating into ALCs, which robustly produce T. 30 To study the effects of EGF on the differentiation of SLCs, EGF (0, 1, and 10 ng/mL) with or without EGF antagonist (erlotinib, E, 100 nmol/L) was added to LDM and cultured at 34°C and 5% CO2 for 14 days. Then, media containing EGF were removed for measurement of T levels. Fragments of STs were also brought down in 2% argarose gel after centrifugation at 750 g, and the cross sections were cut. ST cross sections were then fixed in 4% paraformaldehyde at room temperature for 30 minutes. The STs were stained immunohistochemically after incubating HSD11B1 antibody (for ALC biomarker) and smooth actin muscle (SMA for peritubular myoid cells) and then Alexa Fluor 488 (green colour for HSD11B1) or Fluor 594 (red colour for α‐SMA) for 1 hour. The sections were stained with DAPI for the nucleus of the cells. The slides were covered with 50% glycerol. The fluorescence was visualized under a fluorescent microscope (Olympus).
2.7. SLC proliferation
Our previous study has demonstrated that the STs are cultured during the first week of culture, and the number of SLCs is greatly amplified. 30 Then, STs were switched to LDM for additional 7 days, the amplified SLCs could be differentiated into ALCs, and thus, the increased number of ALCs could contribute into the robust increase of T level in the medium. 30 Using this approach, EGF (0, 1 and 10 ng/mL) with or without EGF antagonist (E, 100 nmol/L) was added to M199 and cultured at 34°C and 5% CO2 for 7 days, and then, STs were switched into LDM for additional 7 days. Media were collected for the measurement of T level.
2.8. Isolation of PLCs
The procedure for PLC isolation and cultured was performed as previously described. 9 Briefly, forty 21‐day‐old rats were euthanized under CO2 for isolation of PLCs. All testes were taken out and decapsulated and put in sterile 50‐mL tube (10 mL medium). Testes were digested in medium 199 containing 0.25 mg/mL collagenase‐D at 34°C in the shaking bath (75 rpm) for 10 minutes. The testis fragments were gently shaken and filtered through two layers of nylon mesh (200 μm) and washed with medium 199. The filtered cells were centrifuged at 250 g for 10 minutes. Crude cell preparations were resuspended in the 55% isotonic Percoll. Following density gradient centrifugation at 25 000 g at 4°C for 45 minutes, the PLC fraction was gently collected between densities of 1.064 and 1.070 g/mL. The cells were washed with HBSS and centrifuged at 250 g for 10 minutes. PLCs were resuspended in phenol red‐free 1:1 DMEM: F12 supplemented with 1 mg/mL BSA. Purity of PLCs was judged after histochemical staining of HSD3B1 activity with 0.4 mmol/L etiocholanolone and 0.4 mmol/L NAD+ as previously described. 34 The purity of PLCs was typically more than 95%. The purifications of PLCs were repeated for four times.
2.9. [3H]‐Thymidine incorporation into PLCs
[3H]‐Thymidine incorporation into PLCs was used to assess cell proliferation as previously described. 17 1 × 106 PLCs were cultured with DMEM: F12 (1:1) alone or in combination with 1 and 10 ng/mL EGF at 34°C 5% CO2 for 24 hours. Cells were incorporated with [3H]‐thymidine at 1 µCi/mL during the last 24 hours of incubation at 34°C. After the incorporation, PLCs were washed twice with PBS and harvested. PLCs were lysed in 0.5 mL hyamine hydroxide, and radioactivity was measured in a liquid scintillation counter (PE, USA). Cpm per 106 PLCs was calculated for thymidine incorporation into PLCs.
2.10. Measurement of cellular H2O2‐induced reactive oxygen species in PLCs
Reactive oxygen species (ROS) production was measured with the fluorescence dye 2′7′‐dichlorofluorescin diacetate (DCFH‐DA) assay kit (Qcbio Science and Technologies Co., Ltd.). Briefly, 1.5 × 105 cells/mL isolated PLCs were plated into the 6‐well plates and incubated for 24 hours. Then, cells were divided into four groups: control, 10 ng/mL EGF, 200 μmol/L H2O2, and 10 ng/mL EGF + 200 μmol/L H2O2. 200 μmol/L H2O2 was used as a positive inducer of ROS. Cells were cultured for 48 hours. Thereafter, cells were harvested and suspended with 200 μL DCFH‐DA for 20 minutes at 37°C in the dark. Cells were washed twice with PBS, and fluorescence intensity determined by flow cytometer was used to measure ROS.
2.11. Annexin V and PI assay for apoptosis of PLCs
Isolated PLCs were planted into the 6‐well plates with the density of 2.5 × 106 cells/mL and incubated for 24 hours. Cells were divided into four groups: control, 10 ng/mL EGF, 200 μmol/L H2O2, and 10 ng/mL EGF + 200 μmol/L H2O2. 200 μmol/L H2O2 was used as a positive inducer of cell apoptosis. Cells were cultured for 48 hours. To evaluate early and lately apoptotic activity, an Annexin V‐FITC/PI Apoptosis Detection Kit (Nanjing KeyGEN Biotech) was used as previously described. 35 Cells were harvested and washed with cold PBS and then were resuspended in 200 μL the annexin V‐binding buffer. After cells were stained with FITC‐labelled annexin V and PI, they were instantly measured using flow cytometer.
2.12. PLC steroidogenesis after EGF treatment
Progenitor Leydig cells with a density of 0.5 × 106 cells per cell were cultured with DMEM: F12 (1:1) alone or in combination with 1 and 10 ng/mL EGF at 34°C 5% CO2 for 24 hours. Media were collected for measurement of AO and T. PLCs were washed twice with PBS and harvested for isolation of RNAs and proteins.
2.13. Medium T and androsterone analysis
Medium concentrations of T and AO were measured by the tritium‐based radioimmunoassay validated for the use of rat antiserum as using either anti‐T antibody (Fitzgerald, MA) or anti‐AO antibody. 9 Standards ranging between 10 and 2000 pg/mL T or AO were prepared in triplicate. Standards and samples were incubated with respective tracer and antibody at 4°C overnight, and charcoal‐dextran suspension was used to separate the bound and free steroids. The bound steroids were mixed with a scintillation buffer and counted in a β‐scintillation counter (PE, USA). The minimum detectable concentration of the assay for either T or AO was 5 pg/mL. The quality control had either 100 pg/mL T or 100 pg/mL AO dissolved in the same culture media. Interassay and intra‐assay coefficients of variation for T and AO were within 10%.
2.14. Microarray hybridization and scanning
Progenitor Leydig cells were treated with 0, 1 and 10 ng/mL EGF as well as 1 ng/mL LH. LH serves the positive control for induction of PLC proliferation and differentiation. Total RNAs were harvested from PLCs after EGF treatment using a Trizol kit (Invitrogen) for microarray analysis. The RatRef‐12 Expression BeadChip containing 21 910 rat genes was used as previously described.36 Genes are selected from the NCBI RefSeq database to cover the whole rat transcriptome. Four groups of samples were used: 0, 1 and 10 ng/mL EGF‐treated as well as 1 ng/mL LH‐treated PLCs. Four replicates per group were performed. Probe labelling, hybridization, washing and scanning were performed using the Illumina Total Prep Kit (Applied Biosystems) as previously described. 37 First strand of cDNA was synthesized in a total volume of 20 μL with the supplied reagents. The first‐strand product was used for the second‐strand synthesis, followed by column purification. The purified product was then used for in vitro transcription using T7 polymerase. Biotin‐16‐dUTP was incorporated and the biotinylated complementary RNA (cRNA) probe as prepared. The probe integrity was verified using the Agilent 2100 Bioanalyzer. Labelled cRNA (750 ng) was hybridized to the array chip overnight at 58°C in a total volume of 30 μL of hybridization buffer, followed by post‐hybridization stringency washing. The chip was scanned in the NextSeq 550 System (Illumina).
2.15. Microarray data analysis
Microarray data analysis was performed as previously described. 36 Briefly, after scanning, the microarray data were imported into the BeadStudio software (Illumina) for normalization, preliminary analysis and filtering. The background subtraction was performed, and the Illumina custom error model was used to generate present/absent calls for each probe (“present” defined as P < .01 for signal detection) and to call differentially expressed genes (defined as P < .05 after false discovery rate correction). For each array, all probe sets were normalized to a mean signal intensity value of 100. Normalized data from BeadStudio were filtered to exclude genes not expressed in PLCs (ie data from probes that were classed as “absent” in all samples). All of the 21 910 genes were present in the data based on which further analyses were carried out. The data were further imported into Microsoft Access 2010, and queries to find the increased and decreased genes after EGF treatment when compared to the control were generated to find the expression levels.
2.16. Biological pathway analysis
Biological pathway analysis was performed as previously described. 36 The Gene MicroArray Pathway Profiler 2.1 (GenMAPP2.1) software was used to find the biological pathway, and GO pathway was generated according to the software developer's instruction. The GenMAPP2.1 was used to create a map of signal pathways for the potential pathways. We imported our statistical results into the program and illustrated biological pathways containing differentially expressed genes. The results of the differential gene expression profile were validated by RT‐qPCR.
2.17. Quantitative real‐time PCR (RT‐qPCR)
Briefly, first‐strand synthesis of DNA and RT‐qPCR were performed as previously described. 38 qPCR was carried out in a 20 µl volume in a 96‐well plate using the SYBR Green PCR Kit from Applied Biosystems. Primer titration was performed with the concentration of 300 nmol/L. Fluorescence was detected using the ABI 7700 system (PE Applied Biosystems). Each sample was run in duplicate and in parallel with no template controls. The relative mRNA levels of targeted genes were adjusted to housekeeping gene, ribosomal protein S16 (Rps16), as the internal control. Rsp16 in LCs has been used as the internal control in many studies because it showed consistent expression. 32 , 39 The Ct value was read, and the levels of the target mRNAs were calculated using the standard curve method as previously described. 36 All primers in the present study were designed by Primer 3 software (Whitehead Institute for Biomedical Research). Forward and reverse primers were placed in different exons to minimize the effects of possible DNA contamination. These genes are as follows: luteinizing hormone receptor (Lhcgr), scavenger receptor class B member 1 (Scarb1), Star, Cyp11a1, Hsd3b1, Cyp17a1, Hsd17b3, Srd5a1, Akr1c9, insulin‐like 3 (Insl3), sulfotransferase 1A1 (Sult1a1) and cyclin D1 (Ccnd1). The primers were listed in Table S1.
2.18. Western blotting
Progenitor Leydig cells were homogenized and lysed. Protein concentrations in samples were measured using the Bio‐Rad Protein Assay Kit (Bio‐Rad) as previously described. 33 BSA was used as the protein standard. Samples (50 µg protein) were boiled in equal volumes of sample loading buffer, containing 20% glycerol, 5% sodium dodecyl sulfate, 3.1% dithiothreitol and 0.001% bromophenol blue. Samples were electrophoresed on 10% polyacrylamide gels containing sodium dodecyl sulfate. Proteins were electrophoretically transferred onto nitrocellulose membrane. After one‐hour exposure to 5% non‐fat milk to block nonspecific binding, the membranes were incubated with the primary antibodies (1:1000 dilution) against LHCGR (Santa Cruz), CYP11A1 (Santa Cruz, Santa Cruz, CA), CYP17A1 (Santa Cruz), StAR (Pterosaur Biotech) and actin β (ACTB, Cell Signaling Technology). The membranes were then washed and incubated with a 1:1000 dilution of anti‐rabbit or anti‐goat antiserum (R&D Systems, Inc) that was conjugated to horseradish peroxidase. The washing step was repeated, and immunoreactive bands were visualized by chemiluminescence using an ECL kit (Amersham). The density was scanned by ImageJ software.
2.19. Statistics
Data were subjected to analysis by Student's t test to identify significant differences whenever two groups (a single concentration of EGF vs. control) were compared. Data were subjected to analysis by one‐way ANOVA test followed by ad hoc Tukey multiple comparisons to identify significant differences between the tested group and the controls whenever there were three or more groups (multiple concentrations of etomidate vs control) were compared. All data are expressed as means ± SEM. Differences were regarded as significant at P < .05.
3. RESULTS
3.1. EGF increases EdU incorporation into SLCs
To investigate the effects of EGF on SLC proliferation, we treated STs for 7 days with EGF and/or EGF inhibitor in vitro. SLCs reside on the surface of the ST (Figure 1A‐C). 30 EGF concentration‐dependently increased EdU incorporation into SLCs (Figure 1D). This indicates that EGF stimulates SLC proliferation. Our previous study has shown that during the first week SLCs had the highest proliferative capacity and LDM was capable of inducing the differentiation of SLCs into ALCs, which robustly produced T after additional 7 days in LDM. 30 Using this indirect approach, we treated STs with 0‐10 ng/mL EGF for 7 days and then switched STs in LDM for additional 7 days to induce the formation of ALCs, which produced T. As shown in Figure 1E, EGF concentration‐dependently increased medium T levels after 7‐day culture, indicating that EGF is capable of increasing the number of SLCs that are differentiated into ALCs, which produce more T. We also tested EGF action using EGF antagonist (Erlotinib, E). Indeed, E (100 nmol/L) alone did not affect T level but it can reverse EGF (10 ng/mL)‐induced action (Figure 1F). This indicates that EGF acts via EGF receptor. In order to test the effects of EGF on SLC proliferation, purified CD90+ SLCs were treated with EGF and/or its antagonist (Cetuximab, Cet) for 24 hours. EGF (10 ng/mL) significantly increased EdU incorporation into CD90+ SLCs (Figure 1H,K) when compared to the control (Figure 1G). Cet (5 µg/mL, Figure 1I) alone did not affect EdU incorporation but it can reverse EGF (10 ng/mL)‐induced action (Figure 1J,K). Using the indirect approach, we treated CD90+ SLCs with 0‐10 ng/mL EGF for 7 days and then switched CD90+ SLCs in LDM for additional 7 days to induce the formation of ALCs. As shown in Figure 1L, EGF increased medium T levels after 7‐d culture, indicating that EGF is capable of increasing the number of SLCs that are differentiated into ALCs. This further confirms that EGF acts via EGF receptor to stimulate SLC proliferation.
FIGURE 1.
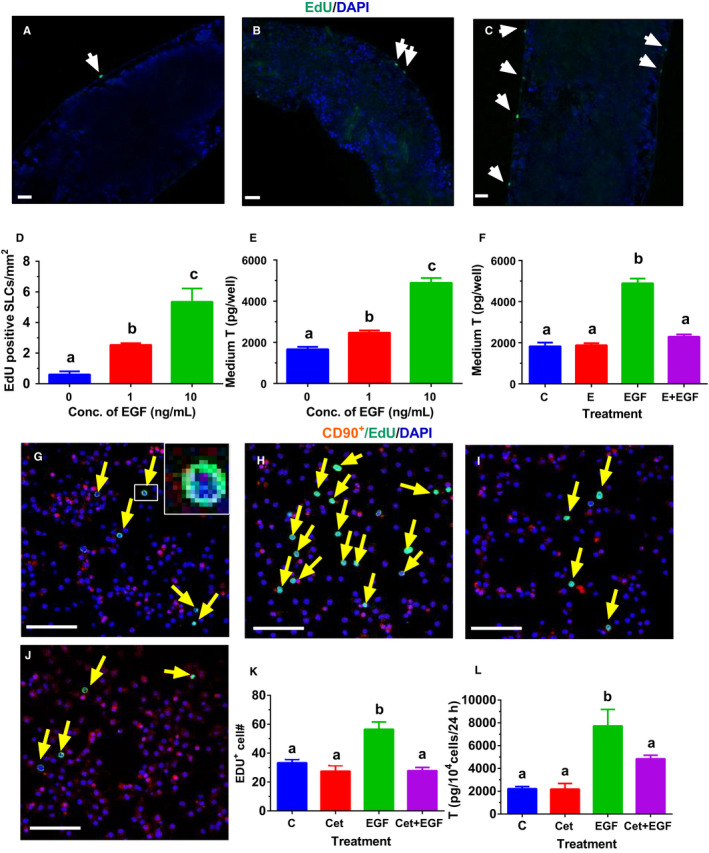
Effects of EGF on proliferation of stem Leydig cells (SLCs). Leydig cell (LC)‐depleted seminiferous tubules (STs) were cultured without (Panel A) or with 1 ng/mL EGF (Panel B), or 10 ng/mL EGF (Panel C) for 7 d. CD90+ SLCs were cultured without (Panel G) or with 10 ng/mL EGF (Panel H), or 5 μg/mL Cet (cetuximab, Panel I), or 10 ng/mL EGF + 5 μg/mL Cet (Panel J) for 24 h. EdU incorporation into the nucleus of SLC on the STs (white arrow) or CD90+ SLC (yellow arrow) was observed. DAPI serves as the counterstaining. EdU‐positive cells were located outside of STs. Bar = 50 μm. The EdU‐positive SLCs per mm2 STs were presented in Panel D (mean ± SEM, n = 5). In Panel E, STs were cultured without or with 1 or 10 ng/mL EGF for 7 d, and then, STs were switched to Leydig cell differentiation medium (LDM) for additional 7 d to produce testosterone (T), and the effects of EGF on SLC proliferation were indirectly analysed (mean ± SEM, n = 4). In Panel F, STs were treated with 0 ng/kg EGF (control, C) or 100 nmol/L EGF antagonist (erlotinib, E), or 10 ng/mL EGF (EGF) or 100 nmol/L E plus 10 ng/mL EGF (E + EGF) for 7 d, and then, STs were switched to LDM for 14 d to produce T (mean ± SEM, n = 4). The EdU‐positive CD90+ SLCs were presented in Panel K (mean ± SEM, n = 6). In Panel L, CD90+ SLCs were treated with 0 ng/kg EGF (control, C) or 5 μg/mL EGF antagonist (Cet), or 10 ng/mL EGF (EGF) or 5 μg/mL Cet plus 10 ng/mL EGF (Cet + EGF) for 7 d, and then, cells were switched to LDM for 7 d to produce T (mean ± SEM, n = 4). Identical letters designate no significant difference between two groups at P < .05
3.2. EGF inhibits differentiation of SLCs into the LC lineage
To examine whether EGF is capable of affecting SLC differentiation, STs were cultured in M199 for 7 days and then were switched to EGF (0, 1, and 10 ng/mL)‐containing LDM for additional 7 days. We used HSD11B1 as the biomarker of ALCs. After 7‐day culture in LDM, many HSD11B1 positive ALCs were formed on the surface of STs, designating ALCs (Figure 2A). EGF antagonist (E, 100 nmol/L) did not affect the number of LCs (Figure 2B). However, EGF (10 ng/mL) significantly reduced the number of HSD11B1 positive LCs, while EGF antagonist E reversed EGF‐mediated inhibition (Figure 2D). EGF concentration‐dependently lowered LC number per ST (Figure 2E) and medium T levels (Figure 2F). EGF antagonist E did not affect medium T level but reversed EGF (10 ng/mL)‐induced suppression of T synthesis (Figure 2G). We further cultured CD90+ SLCs in the presence of EGF (10 ng/mL) and/or Cet (5 μg/mL) for 14 days. EGF antagonist Cet did not affect medium T output but reversed EGF (10 ng/mL)‐induced suppression of T synthesis (Figure 2H). These data indicate that EGF inhibits SLC differentiation into the LC lineage via EGF receptor.
FIGURE 2.
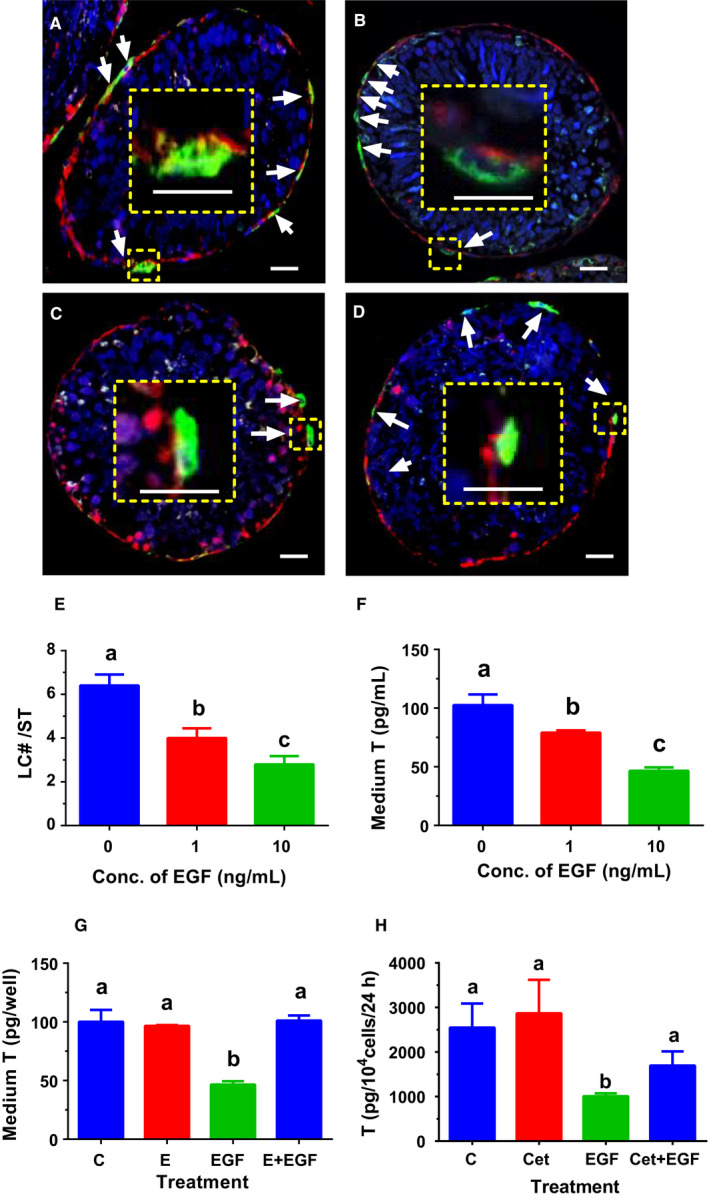
Effects of EGF on differentiation of stem Leydig cells (SLCs) in vitro. Leydig cell (LC)‐depleted seminiferous tubules (STs) were cultured LC differentiation medium (LDM) containing EGF (0 ng/mL, Panel A), EGF antagonist (erlotinib, E, 1 µmol/L, Panel B), EGF (10 ng/mL, Panel C) and EGF (10 ng/mL) + E (1 µmol/L, Panel D) for 14 d. LCs were identified by biomarker 11β‐hydroxysteroid dehydrogenase 1 (HSD11B1, green colour in the cytoplasm, white arrow). Peritubular myoid cells were stained by smooth muscle actin (red colour in the cytoplasm), drawing a boundary for the ST. DAPI serves as counterstaining. Inset is the magnified picture, showing that HSD11B1 positive LCs existed outside of STs. Bar = 10 μm. The HSD11B1‐positive LCs per ST in the cross section were presented in Panel E (mean ± SEM, n = 4). In Panel F, STs were cultured in LDM without or with 1 or 10 ng/mL EGF for 14 d, and medium T levels were measured (mean ± SEM, n = 4). In Panel G, STs were treated in LDM with 0 ng/kg EGF (control, C) or 100 nmol/L EGF antagonist (erlotinib, E), or 10 ng/mL EGF (EGF) or 100 nmol/L E plus 10 ng/mL EGF (E + EGF) for 14 d, and medium T levels were measured (mean ± SEM, n = 4). In Panel H, CD90+ SLCs were treated with 0 ng/kg EGF (control, C) or 5 μg/mL EGF antagonist (Cet), or 10 ng/mL EGF (EGF) or 5 μg/mL Cet plus 10 ng/mL EGF (Cet + EGF) for 14 d to produce T (mean ± SEM, n = 4). Identical letters designate no significant difference between two groups at P < .05
3.3. EGF inhibits differentiation of PLCs
To examine whether EGF can affect PLC differentiation, PLCs were cultured with EGF (0, 1, and 10 ng/mL) in DMEM: F12 medium for 48 hours. EGF (10 ng/mL) significantly lowered AO and total androgen (T plus AO) production by PLCs (Figure 3A). The data indicate that EGF inhibits PLC differentiation.
FIGURE 3.
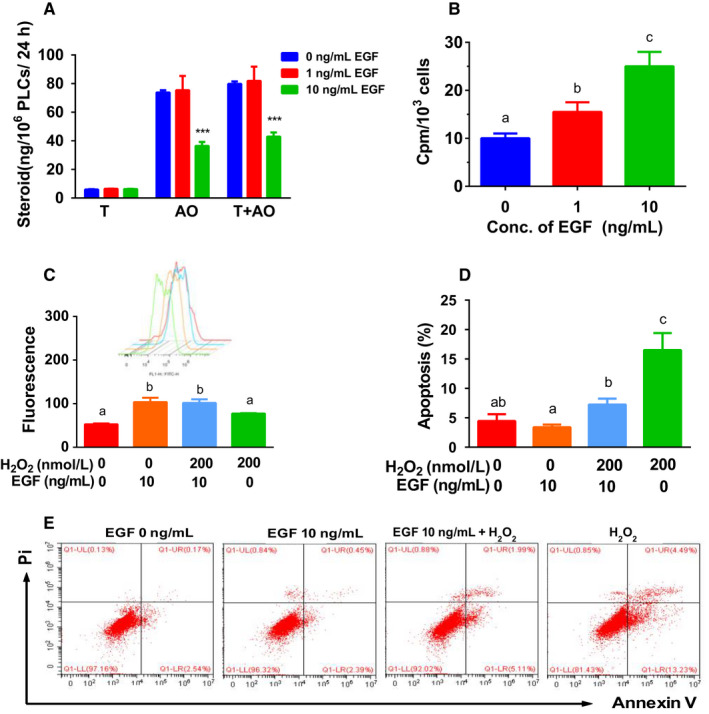
Effects of EGF on androgen production, proliferation, ROS generation and apoptosis by progenitor Leydig cells (PLCs) in vitro. Panel A, PLCs were cultured with 0, 1 and 10 ng/mL EGF for 24 h, and medium levels of testosterone (T) and androsterone (AO) were measured; mean ± SEM, n = 8. Panel B, PLCs were cultured with 0, 1 and 10 ng/mL EGF for 24 h, and PLCs were incorporated with [3H]‐thymidine for additional 24 h, and CPM was counted; mean ± SEM, n = 4. Panel C, PLCs were treated with control, 10 ng/mL EGF, 200 μmol/L H2O2 and 10 ng/mL EGF + 200 μmol/L H2O2 for 24 h, and ROS was counted, mean ± SEM, n = 3, inset is ROS fluorescence image; Panel D, PLCs were treated with control, 10 ng/mL EGF, 200 μmol/L H2O2 and 10 ng/mL EGF + 200 μmol/L H2O2 for 24 h, and apoptosis was counted; mean ± SEM, n = 4. Panel E, apoptosis image. Identical letters designate no significant difference between two groups at P < .05
3.4. EGF increases thymidine incorporation into PLCs
To investigate the effects of EGF on PLC proliferation, we treated PLCs for 48 hours in vitro. Both concentrations of EGF significantly increased thymidine incorporation into PLCs (Figure 3B). This indicates that EGF stimulates PLC proliferation.
3.5. EGF and H2O2 induce ROS generation in PLCs
To investigate the effects of EGF on ROS generation by PLCs, we treated PLCs with EGF for 48 hours. H2O2 was used as a positive control (Figure 3C). Indeed, H2O2 (200 μmol/L) induced ROS generation in PLCs. Interestingly, EGF (10 ng/mL) also induced ROS generation in PLCs (Figure 3C). EGF plus H2O2 did not further increased ROS production in PLCs (Figure 3C). This indicates that EGF and H2O2 induce ROS production via different mechanisms.
3.6. EGF antagonizes H2O2‐induced PLC apoptosis
To investigate the effects of EGF on PLC apoptosis, we treated PLCs with EGF for 48 hours. H2O2 was used as the positive control to induce apoptosis (Figure 3D,E). Indeed, H2O2 induced PLC apoptosis at 200 μmol/L. EGF (10 ng/mL) did not affect PLC apoptosis. However, EGF antagonized H2O2‐induced PLC apoptosis (Figure 3D,E). This indicates that EGF protects PLCs from H2O2‐mediated apoptosis.
3.7. EGF‐mediated gene expression of PLCs
Luteinizing hormone is an important hormone for PLC development. 2 We used LH (1 ng/mL) as the positive control. PLCs were cultured with EGF (0, 1 and 10 ng/mL) in DMEM: F12 medium for 24 hours. Microarray of gene profiles of PLCs after EGF treatment were compared to LH (1 ng/mL). A genome‐wide expression containing 21 910 probes was analysed. Of these probes, 8667 probes were detected in the control group. Of them, 33 genes were up‐regulated and 53 genes down‐regulated more than twofold in 1 ng/mL LH control group. Eighteen genes were up‐regulated (Table 1) and 81 genes down‐regulated (Table 2) more than twofold in 1 ng/mL EGF group, while 66 genes were up‐regulated (Table 1) and 198 genes down‐regulated (Table 2) more than twofold in 10 ng/mL EGF group. GO analysis showed that up‐regulation of genes by EGF (10 ng/mL) includes the following 10 major categories: lipid metabolism, response to stimulus, steroid metabolism, hormone metabolic process, complement activation, activation of plasma proteins, humoral immune response, lipid transport, positive regulation of Wnt, axon guidance, response to nutrient, and immune response (Figure 4A). The down‐regulation of genes by EGF (10 ng/mL) includes the following 10 major categories: motor axon guidance, collagen catabolism, peptidoglycan metabolism, neuron migration, regulation of apoptosis, cell adhesion, brain development, G‐protein, amino acid dephosphorylation and organismal development (Figure 4C). When compared to LH (1 ng/mL), most of categories up‐regulated by EGF (10 ng/mL) were overlapped with those of LH (Figure 4B), except one (humoral immune response) for EGF and one (steroid catabolism) for LH. Of 33 up‐regulated genes by LH, 12 genes were also up‐regulated by EGF (Figure 4E). When compared to LH, the categories down‐regulated by EGF were more diversified (Figure 4D).
TABLE 1.
Genes were up‐regulated after EGF treatment
| Gene | Gene name | Expression levels | ||||
|---|---|---|---|---|---|---|
| EGF (ng/mL) | LH (ng/mL) | |||||
| 0 | 1 | 10 | 1 | |||
| Cell cycle regulation/ DNA repair | ||||||
| Ercc1 | Endonuclease non‐catalytic subunit | 641 ± 35 | 1138 ± 30 (1.8) | 1750 ± 115 (+2.7) | 951 ± 41 (+1.5) | |
| Ccnd1 | Cyclin D1 | 85 ± 3 | 120 ± 2 (+1.4) | 219 ± 1 (+2.6) | 93 ± 3 (+1.1) | |
| Phlda11 | Pleckstrin homology like domain A | 246 ± 11 | 438 ± 17 (+1.8) | 621 ± 3 (+2.5) | 528 ± 2 (+2.2) | |
| Reln | Reelin | 415 ± 6 | 643 ± 26 (+1.5) | 1038 ± 43 (+2.5) | 819 ± 29 (+2.0) | |
| Schip1 | Schwannomin interacting protein 1 | 140 ± 2 | 245 ± 8 (+1.8) | 319 ± 6 (+2.3) | 211 ± 5 (+0.6) | |
| Cd82 | Metastasis suppressor kangai‐1 | 478 ± 6 | 785 ± 14 (+1.6) | 1070 ± 28 (+2.2) | 754 ± 16 (+1.6) | |
| Cell metabolism | ||||||
| Upp1 | Uridine phosphorylase 1 | 1006 ± 54 | 3816 ± 41 (+3.8) | 5182 ± 99 (+5.1) | 2897 ± 38 (+2.8) | |
| Car2 | Carbonic anhydrase II | 250 ± 3 | 721 ± 29 (+2.9) | 1093 ± 72 (+4.4) | 1033 ± 20 (+4.2) | |
| Hs3st1 | heparan sulfate‐glucosamine 3‐sulfotransferase 1 | 146 ± 14 | 393 ± 5 (+2.7) | 577 ± 14 (+4.0) | 275 ± 8 (+2.0) | |
| Dusp6 | Dual specificity phosphatase 6 | 1150 ± 25 | 3183 ± 195 (+2.8) | 4408 ± 81 (+3.8) | 202 848± (+1.9) | |
| Aadat | Aminoadipate aminotransferase | 304 ± 4 | 763 ± 31 (+2.5) | 971 ± 15 (+3.2) | 1963 ± 36 (+6.2) | |
| Smpd1 | Sphingomyelin phosphodiesterase 1 | 6626 ± 242 | 14 384± (+2.2) | 18 775± (+2.8) | 12 677 ± 807 (+2.0) | |
| Plscr1 | Phospholipid scramblase 1 | 166 ± 6 | 280 ± 25 (+1.7) | 423 ± 28 (+2.6) | 459 ± 4 (2.7) | |
| Lipg | Endothelial lipase | 145 ± 1 | 254 ± 13 (+1.8) | 337 ± 24 (+2.3) | 201 ± 3 (+0.6) | |
| Ligand/receptors | ||||||
| Adm | Adrenomedullin | 1662 ± 46 | 3557 ± 71 (+2.1) | 4828 ± 68 (+2.9) | 2131 ± 51 (+1.2) | |
| Gal | Galanin | 542 ± 23 | 1229 ± 48 (+2.3) | 1524 ± 100 (+2.8) | 558 ± 28 (+1.1) | |
| Gchfr | GTP cyclohydrolase I | 175 ± 8 | 347 ± 3 (+2.0) | 465 ± 12 (+2.7) | 150 ± 10 (+1.0) | |
| Serpine1 | Plasminogen activator inhibitor 1 | 805 ± 41 | 1819 ± 66 (+2.3) | 2124 ± 29 (+2.6) | 1482 ± 29 (+1.6) | |
| Plcd4 | Phospholipase C delta 4 | 422 ± 13 | 1014 ± 23 (+2.4) | 1081 ± 24 (+2.6) | 447 ± 24 (+1.0) | |
| Tnfrsf12a | TNF receptor superfamily 12A | 1230 ± 17 | 2237 ± 88 (+1.8) | 3146 ± 120 (+2.6) | 2189 ± 69 (+1.6) | |
| Cxcr4 | C‐X‐C motif chemokine receptor 4 | 253 ± 9 | 491 ± 19 (+1.9) | 626 ± 11 (+2.5) | 444 ± 23 (+1.6) | |
| Mmp10 | Matrix metalloproteinases 10 | 3312 ± 113 | 5671 ± 135 (+1.7) | 12 386 ± 255 (+3.7) | 4322 ± 103 (+1.3) | |
| Alcam | CD166 antigen | 560 ± 7 | 971 ± 27 (+1.7) | 1367 ± 51 (+2.4) | 641 ± 25 (+1.3) | |
| Procr | Protein C receptor | 466 ± 17 | 712 ± 10 (+1.5) | 1097 ± 96 (+2.4) | 1088 ± 38 (+2.0) | |
| Ednrb | Endothelin receptor type B | 2304 ± 73 | 4360 ± 61 (+1.9) | 5238 ± 232 (+2.3) | 7117 ± 278 (+3.0) | |
| Areg | Amphiregulin | 238 ± 9 | 385 ± 18 (+1.6) | 488 ± 4 (+2.2) | 406 ± 22 (+1.8) | |
| Pdgfa | Platelet‐derived growth factor A | 1734 ± 37 | 2475 ± 84 (+1.4) | 3736 ± 43 (+2.2) | 2530 ± 81 (+1.4) | |
| Stc1 | Stanniocalcin 1 | 1818 ± 86 | 2899 ± 92 (+1.6) | 3809 ± 110 (+2.1) | 3773 ± 265 (+2.0) | |
| Transcription factor activity | ||||||
| Maf | Proto‐oncogene c‐Maf | 697 ± 17 | 2198 ± 68 (+3.2) | 2914 ± 55 (+4.2) | 1420 ± 15 (+2.2) | |
| Hmga1 | High mobility group AT‐hook 1 | 370 ± 11 | 812 ± 1 (+2.2) | 1011 ± 57 (+2.7) | 662 ± 38 (+1.8) | |
| Ehd4 | EH domain containing 4 | 459 ± 17 | 820 ± 50 (+1.8) | 1123 ± 62 (+2.4) | 710 ± 29 (+1.6) | |
| Runx1 | Runt related transcription factor 1 | 607 ± 25 | 1276 ± 40 (+2.1) | 1476 ± 59 (+2.4) | 1542 ± 65 (+2.5) | |
| Arhgap22 | Rho GTPase activating protein 22 | 148 ± 4 | 259 ± 1 (+1.7) | 349 ± 10 (+2.4) | 194 ± 16 (+1.3) | |
| Abhd3 | Abhydrolase domain containing 3 | 980 ± 34 | 1876 ± 60 (+1.9) | 2092 ± 164 (+2.1) | 1608 ± 56 (+1.6) | |
| Twist1 | Twist BHLH transcription factor | 275 ± 13 | 464 ± 8 (+1.7) | 587 ± 46 (+2.1) | 434 ± 3 (+1.5) | |
| Transporter/Protein binding | ||||||
| Slc17a1 | solute carrier family 1 member 17a1 | 510 ± 3 | 1423 ± 27 (+2.8) | 1845 ± 70 (+3.6) | 569 ± 48 (+1.0) | |
| Slc1a5 | solute carrier family 1 member 5 | 935 ± 38 | 2153 ± 41 (+2.3) | 3114 ± 35 (+3.3) | 1663 ± 26 (+1.7) | |
| Slc16a3 | Solute carrier family 16 member 3 | 1858 ± 23 | 3451 ± 207 (+1.9) | 4645 ± 250 (+2.5) | 5015 ± 284 (+2.8) | |
| Igfbp3 | IGF‐1 binding protein 3 | 589 ± 33 | 905 ± 33 (+1.5) | 1400 ± 30 (+2.4) | 917 ± 48 (+1.2) | |
| Sh2b3 | SH2B adaptor protein 3 | 382 ± 16 | 727 ± 9 (+1.9) | 785 ± 18 (+2.2) | 520 ± 11 (+1.3) | |
| Ak3l1 | Adenylate kinase isoenzyme 4 | 462 ± 28 | 885 ± 24 (+1.9) | 1004 ± 31 (+2.2) | 1089 ± 16 (+2.1) | |
| Ube2q2 | Ubiquitin conjugating enzyme E2Q2 | 518 ± 10 | 875 ± 23 (+1.7) | 1125 ± 45 (+2.2) | 720 ± 16 (+1.4) | |
() fold up‐regulated when compared to control.
TABLE 2.
Genes were down‐regulated after EGF treatment
| Gene symbol | Expression level | ||||
|---|---|---|---|---|---|
| EGF (ng/mL) | LH (ng/mL) | ||||
| 0 | 1 | 10 | 1 | ||
| Cell metabolism | |||||
| Hmgcs2 | 3hydroxy3methylglutarylCoA synthase 2 | 5387 ± 119 | 867 ± 52 (−6.2) | 454 ± 15 (−11.9) | 1019 ± 33 (−5.3) |
| Aldh1a1 | Aldehyde dehydrogenase 1A1 | 18 447 ± 563 | 4873 ± 165 (−3.8) | 1872 ± 103 (−9.9) | 2162 ± 185 (−8.6) |
| Adh1 | Alcohol dehydrogenase 1 | 1927 ± 52 | 616 ± 11 (−3.1) | 397 ± 6 (−4.9) | 558 ± 34 (−3.4) |
| Srpx | Sushi repeat‐containing protein | 1356 ± 44 | 458 ± 22 (−3.0) | 284 ± 6 (−4.8) | 920 ± 40 (−1.5) |
| Es1 | ES1 protein | 2088 ± 23 | 804 ± 22 (−2.6) | 442 ± 31 (−4.7) | 908 ± 51 (−2.3) |
| Ddah2 | Dimethylarginine dimethylaminohydrolase2 | 4845 ± 117 | 1906 ± 63 (−2.5) | 1205 ± 16 (−4.0) | 2512 ± 61 (−1.9) |
| Aox1 | Aldehyde oxidase 1 | 6698 ± 140 | 2892 ± 137 (−2.3) | 1724 ± 77 (−3.9) | 2316 ± 57 (−2.9) |
| Ces3 | Carboxylesterase 3 | 515 ± 19 | 174 ± 6 (−3.0) | 134 ± 1 (−3.9) | 173 ± 5 (−3.0) |
| Ssg1 | Steroid‐sensitive gene 1 | 1226 ± 37 | 389 ± 15 (−3.2) | 321 ± 13 (−3.8) | 517 ± 22 (−2.3) |
| Ldhb | Lactate dehydrogenase‐B | 2753 ± 18 | 1153 ± 51 (−2.4) | 795 ± 19 (−3.5) | 1663 ± 98 (−1.7) |
| Cyp27a1 | Cytochrome P450 family 27 A1 | 2436 ± 67 | 1046 ± 30 (−2.3) | 716 ± 17 (−3.4) | 1473 ± 55 (−1.6) |
| Dpep1 | Dipeptidase 1 | 949 ± 9 | 395 ± 7 (−2.4) | 296 ± 21 (−3.2) | 459 ± 22 (−2.0) |
| Pah | Phenylalanine hydroxylase | 335 ± 3 | 138 ± 3 (−2.4) | 109 ± 1 (−3.1) | 202 ± 1 (−1.7) |
| Gstm2 | Glutathione S‐transferase mu 2 | 1611 ± 95 | 704 ± 30 (−2.3) | 542 ± 16 (−3.0) | 744 ± 67 (−2.1) |
| Ligand | |||||
| Sectm1a | Secreted and transmembrane protein 1A | 1904± | 470± (−4.0) | 237± (−8.1) | 832± (−2.3) |
| Sectm1 | Secreted and transmembrane 1 | 820± | 287± (−2.9) | 143 (−5.9) | 734± (−1.1) |
| Cx3cl1 | C‐X3‐C motif chemokine ligand 1 | 4969± | 1865± (−2.7) | 892± (−5.6) | 3050± (−1.6) |
| CD302 | C‐type lectin domain family 13 A | 3907± | 1253± (−3.1) | 745± (−5.2) | 2476± (−1.6) |
| Akr1c9 | 3α‐Hydroxysteroid dehydrogenase | 960± | 201± (−4.8) | 128± (−7.5) | 344± (−2.8) |
| Wnt6 | Wnt6 | 1805± | 501± (−3.6) | 358± (−5.1) | 414± (−4.4) |
| Wnt4 | Wnt4 | 456± | 148± (−3.1) | 108± (−4.2) | 217± (−2.1) |
| Cxcl12 | C‐X‐C motif chemokine ligand 12 | 472± | 147 ± (−3.2) | 117± (−4.0) | 215± (−2.0) |
| Ogn | Osteoglycin | 3438± | 1482 ± (−2.3) | 1064 ± (−3.2) | 1313± (−2.6) |
| Mdk | Neurite growth‐promoting factor 2 | 1107± | 492 ± (−2.3) | 355 ± (−3.1) | 683± (−1.6) |
| Plxdc2 | Plexin domain containing 2 | 5029± | 2348± (−2.3) | 1659± (−3.1) | 3075 ± (−1.6) |
| Ly6e | Lymphocyte antigen 6 family member E | 2887± | 1371± (−2.1) | 957± (−3.1) | 1463± (−2.0) |
| Protein binding | |||||
| Selenbp1 | Selenium binding protein 1 | 7799 ± 275 | 2319 ± 144 (−3) | 1323 ± 75 (−5.9) | 1757 ± 102 (−4.4) |
| Itm2a | Integral membrane protein 2A | 4833 ± 235 | 1571 ± 40 (−3.1) | 945 ± 79 (−5.1) | 1469 ± 31 (−3.3) |
| Mx2 | Interferon‐induced GTP‐binding protein | 940 ± 30 | 442 ± 15 (−2.1) | 262 ± 16 (−3.6) | 519 ± 6 (−1.8) |
| Serping1 | Serpin family G member 1 | 2185 ± 63 | 910 ± 12 (−2.4) | 612 ± 9 (−3.5) | 1384 ± 11 (−1.6) |
| C4‐2 | MHC‐linked complement C4 | 487 ± 23 | 190 ± 5 (−2.6) | 139 ± 1 (−3.5) | 244 ± 6 (−2.0) |
| Rcan2 | Regulator of calcineurin 2 | 385 ± 17 | 140 ± 1 (−2.8) | 115 ± 5 (−3.4) | 168 ± 8 (−2.2) |
| Lrrc17 | Leucine rich repeat containing 17 | 1498 ± 32 | 630 ± 20 (−2.3) | 448 ± 22 (−3.3) | 571 ± 22 (−2.6) |
| Wbp5 | Transcription elongation factor A9 | 3374 ± 102 | 1633 ± 28 (−2.1) | 1105 ± 35 (−3.1) | 1787 ± 145 (−1.8) |
| Serpina3n | Serpin family A member 3 | 1036 ± 25 | 514 ± 19 (−2.1) | 343 ± 16 (−3.1) | 844 ± 14 (−1.2) |
| Signal transduction | |||||
| Rhoj | Ras homolog family member J | 1261 ± 51 | 511 ± 18 (−2.5) | 378 ± 13 (−3.3) | 546 ± 15 (−2.3) |
| Ngfrap11 | Protein BEX3 | 1501 ± 54 | 672 ± 17 (−2.2) | 458 ± 24 (−3.3) | 789 ± 46 (−1.9) |
| Gas7 | Growth arrest specific 7 | 1307 ± 12 | 598 ± 13 (−2.3) | 429 ± 11 (−3.1) | 638 ± 37 (−2.1) |
| C1qtnf7 | C1q And TNF Related 7 | 1535 ± 45 | 640 ± 10 (−2.4) | 347 ± 8 (−4.4) | 807 ± 14 (−1.9) |
| Structure | |||||
| Col3a1 | Collagen type III alpha 1 | 1469 ± 24 | 462 ± 22 (−3.2) | 302 ± 19 (−4.9) | 542 ± 33 (−2.7) |
| Apoe | Apolipoprotein E | 8793 ± 489 | 3588 ± 44 (−2.5) | 2711 ± 162 (−3.2) | 4733 ± 183 (−2.9) |
| Epb4.1l3 | Erythrocyte membrane protein band 4.1 | 602 ± 15 | 271 ± 7 (−2.1) | 203 ± 6 (−3.0) | 358 ± 11 (−1.8) |
() fold changes when compared to control.
FIGURE 4.
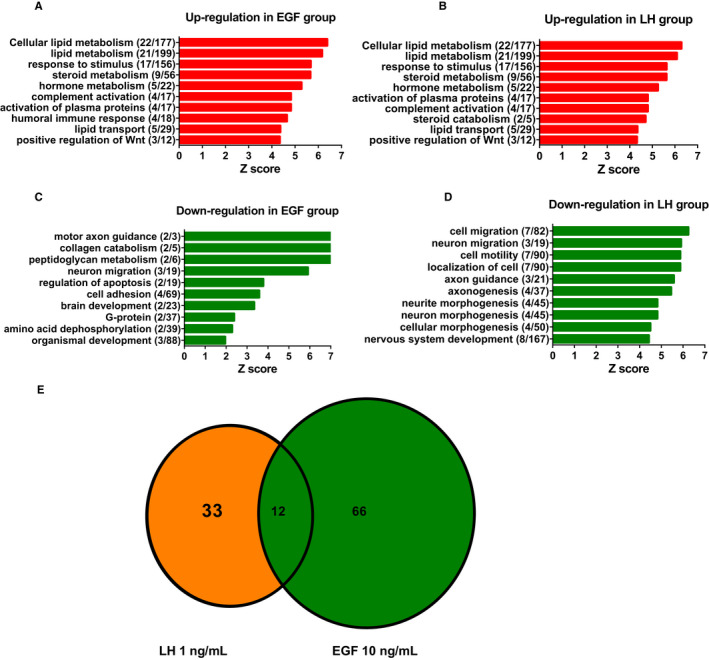
Effects of EGF and LH on gene expression in progenitor Leydig cells: GO analysis. After rat progenitor Leydig cells (PLCs) were cultured with 0, 1 and 10 ng/mL EGF as well as 1 ng/mL LH for 24 h, cells were subjected to gene microarray analysis. Panels A and B: up‐regulation by EGF and LH, respectively; Panels C and D: down‐regulation by EGF and LH, respectively; and Panel E: overlapping genes in EGF or LH up‐regulated genes
3.8. PCR analysis of gene expression after EGF treatment
We used PCR to analyse gene expression in EGF‐treated PLCs. We compared EGF with LH (Figure 5). The following patterns were shown: (a) no change in EGF but up‐regulation in LH (Scarb1, Cyp11a1, and Cyp17a1); (b) down‐regulation in EGF but up‐regulation in LH (Star, Hsd3b1, and Tsp2); (c) down‐regulation in both EGF and LH groups (Pdgfra and Gstm2); (d) up‐regulation in EGF but no change in LH (Ccnd1, Mmp10, and Lef1); and (e) no change in both EGF and LH groups (Pcna). PCR results were similar to that from microarray data.
FIGURE 5.
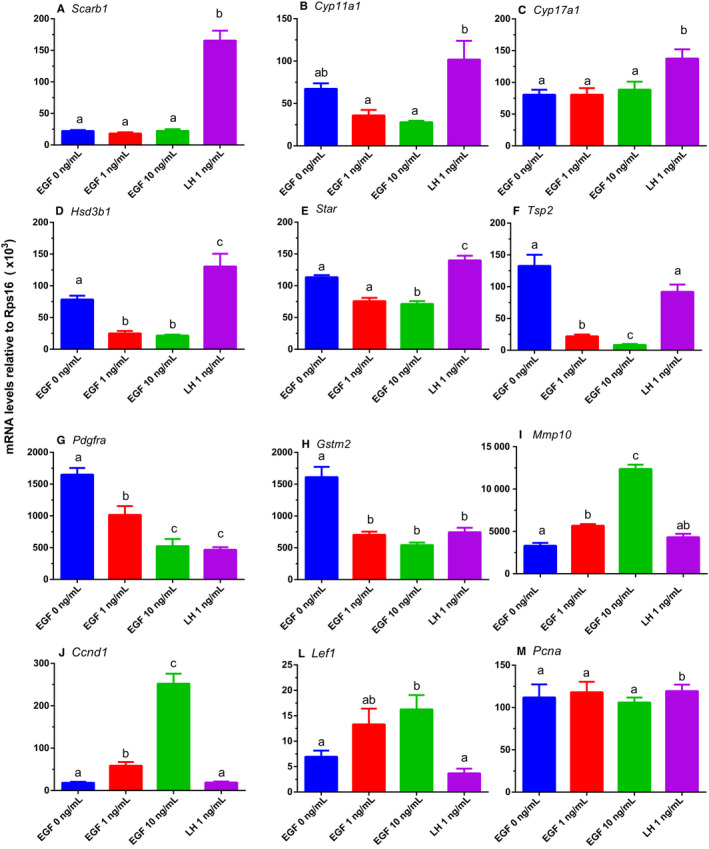
Effects of EGF on the expression levels of steroidogenesis‐related genes in rat immature Leydig cells. After rat progenitor Leydig cells (PLCs) were cultured with 0, 1 and 10 ng/mL EGF as well as 1 ng/mL LH for 24 h, cells were subjected to qPCR analysis. The expression levels of steroidogenesis‐related genes were measured and calculated adjusted to Rps16, the internal control. Mean ± SEM, n = 4; identical letters indicate that there is no significant difference between two groups at P < .05
3.9. Major pathways after EGF treatment
Using GenMAPP2, we discovered several pathways that were specific to the regulation of EGF. The expression of matrix metalloproteinases (Mmp3 and Mmp10) was significantly up‐regulated by more than twofold by EGF (Figure 6). Interestingly, the antioxidant genes (Ugt1a6, Idh1, Gsst1, Gstm2 and Sod3) were down‐regulated by EGF (Figure 6), indicating that EGF might lead to ROS accumulation. Of these 5 genes, two (Gsst1 and Gstm2) were also down‐regulated by LH (Figure 6).
FIGURE 6.
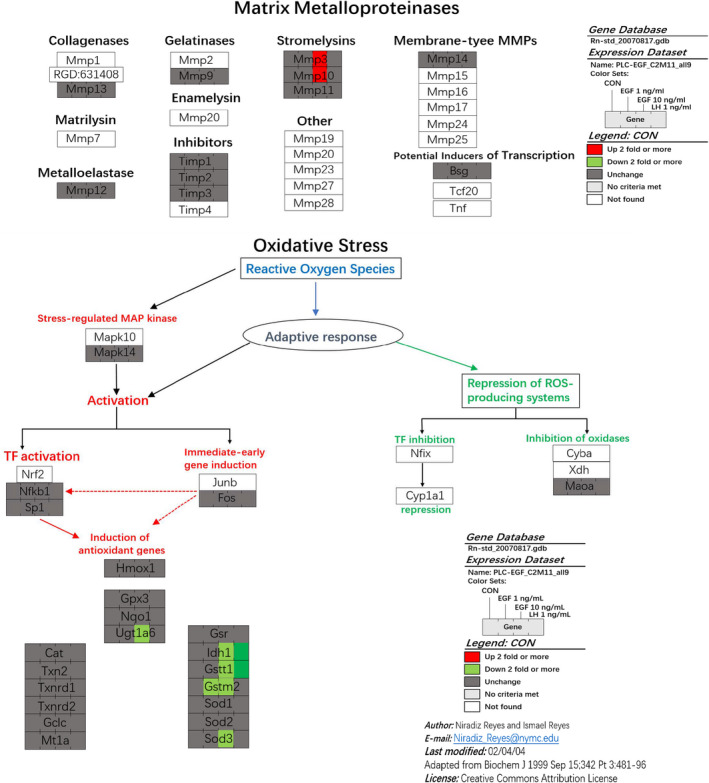
EGF up‐regulates some matrix metalloproteinase expression and down‐regulates some antioxidative protein expression in progenitor Leydig cells. After rat progenitor Leydig cells (PLCs) were cultured with 0, 1 and 10 ng/mL EGF as well as 1 ng/mL LH for 24 h, cells were subjected to gene microarray analysis. Expression of matrix metalloproteinases was analysed. Expression of antioxidative proteins was analysed
3.10. EGF lowers steroidogenesis‐related protein levels via EGFR signalling
We performed Western blot to identify changes of steroidogenesis‐related proteins (including StAR, HSD3B1 and CYP11A1) in PLCs after treatment with EGF and/or EGFR antagonist (E) for 24 hours. As shown in Figure 7A, EGF concentration‐dependently lowered StAR and HSD3B1 protein levels without affecting CYP11A1 and E can reverse the action of EGF (10 ng/mL). This confirms the EGF‐induced mRNA changes.
FIGURE 7.
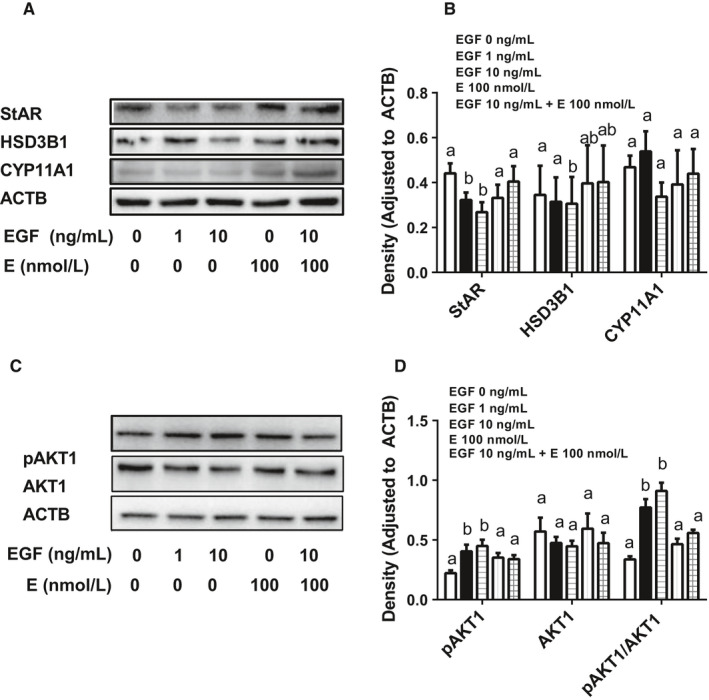
Effects of EGF on steroidogenesis‐related proteins and AKT1 phosphorylation in rat progenitor Leydig cells. After rat progenitor Leydig cells (PLCs) were cultured with 0, 1 and 10 ng/mL EGF as well as 1 ng/mL LH for 24 h, cells were subjected to Western blot. The levels of steroidogenesis‐related proteins (StAR, HSD3B1 and CYP11A1, Panels A and B) as well as AKT1 and its phosphorylated protein (Panel C and D) were analysed. Proteins were measured and calculated adjusted to β‐actin (ACTB), the internal control. Mean ± SEM, n = 4‐7; identical letters indicate that there is no significant difference between two groups at P < .05
3.11. EGF lowers the phosphorylation of AKT1 signalling
We performed Western blot to identify the changes of AKT1 as well as its phosphorylated protein in PLCs after treatment with EGF and/or E for 24 hours. As shown in Figure 7B, EGF concentration‐dependently increased phosphorylation of AKT1 without affecting total AKT1 level (Figure 7B). This suggests that the phosphorylation of AKT1 is involved in EGFR pathway.
4. DISCUSSION
In the present study, we found that EGF stimulated SLC/PLC proliferation but blocked their differentiation.
There is a growing evidence to indicate that EGF is a critical proliferative growth factor for SLCs or PLCs during pubertal development. 13 , 40 , 41 , 42 Apparently, EGF stimulated SLC and PLC proliferation in two‐type LC precursor cells: SLCs on STs and CD90+ SLCs (Figure 1) and isolated PLCs (Figure 3). Previous studies have demonstrated that CD90+ cells on the surface of STs were SLCs and can be induced into ALCs in LDM in vitro. 30 CD90+ SLCs also contain other stem cell biomarkers including nestin, Cd51, Coup‐tf2, Arx, Pdgfra and Tcf21. 43 This indicated that EGF is required for the amplification of SLCs/PLCs for LC pool. EGF exerts its action via binding to EGF receptor, because EGF antagonists (E and Cet) can completely reverse EGF‐mediated effects (Figure 1). Previous studies have demonstrated SLCs and PLCs as well as mouse LC cell lines (MA‐10) possessed both EGF receptors, and the LC was responsive to EGF stimulation. 13 , 24 , 26 , 40 , 44 , 45 , 46 EGF stimulated the proliferation of SLCs/PLCs, possibly via the activation of Cyclin D1 (encoded by Ccnd1) and lymphoid enhancer factor (encoded by Lef1), as Ccnd1 and Lef1 were significantly up‐regulated in PLCs after EGF treatment. Cyclin D1 has been reported to be activated by EGF in various cells. 47 , 48 In the nucleus, LEF1 drives the expression of genes involved in cancer stem cell proliferation. 49 LEF protein also mediates WNT signalling in adult tissues for stem cell proliferation. 49 Interestingly, besides Ccnd1, other cell cycle regulatory genes (Ercc1, Phlda11, Reln, Schip1 and Cd82) were also up‐regulated by EGF (Table 1). Apparently, SLCs did not possess LHCGR, 13 and LH was not able to increase EdU incorporation into SLCs. 30 Thus, at SLC stage, growth factors like EGF are critical for maintaining SLC pool via stimulating their proliferation.
Previous studies indicated that EGF activated phosphatidylinositol 3‐kinase (PI3K). 50 PI3K is able to phosphorylate AKT1, thus in turn activating cyclin D1 pathway. 50 , 51 The data presented here show that EGF indeed increased AKT1 phosphorylation without affecting total AKT1 protein levels (Figure 7). This regulation of phosphorylation of AKT1 in PLCs is dependent on EGFR, because the EGF antagonist (E) can reverse EGF action (Figure 7).
The new data presented here also showed that activation of EGF inhibited H2O2‐induced apoptosis in PLCs (Figure 3). Indeed, EGF inhibited apoptosis in ILCs. 46 The present data confirmed what was found for EGF action in ILCs. As AKT1 signalling mediates both proliferative and anti‐apoptotic effects, EGF‐mediated anti‐apoptotic action might be also mediated by AKT1.
Effects of EGF on T production in LCs are still controversial. Several in vitro studies found that EGF inhibited LH or cAMP‐induced steroid production in LCs. 52 , 53 Other studies indicated that EGF stimulated T production in LCs. 45 , 46 , 54 , 55 , 56 The effects of EGF on androgen production depend on cell maturity, cell types of LCs, and EGF treatment duration.
In the current study, we clearly demonstrated that EGF blocked SLC/PLC differentiation into ALCs: (a) EGF concentration‐dependently lowered ALC number (Figure 2E) and medium T levels after induction of CD90+ SLC differentiating into ALCs in LDM (Figure 2) via EGFR signalling (Figure 2G and H); (b) EGF concentration‐dependently lowered AO and T production in PLCs (Figure 3A); (c) EGF down‐regulated Hsd3b1 and Star expression in PLCs (Figure 5) and their protein levels (Figure 7A); and (d) EGF significantly down‐regulated Pdgfra (Figure 5), which was increased during Leydig cell development. 27 EGF lowered the transcription of Star, which encodes StAR. StAR protein mediates the rate limiting in androgen synthesis, in which it serves the critical carrier of cholesterol to transport it for steroidogenesis from LC mitochondrial outer to the inner membrane. 57 , 58 After cholesterol is transported to inner membrane, CYP11A1 cleaves the cholesterol side‐chain to form pregnenolone. The exact mechanism of EGF‐induced down‐regulation of Star expression is unclear. Many signalling pathways, including protein kinase A, protein kinase C and nuclear receptor, are involved in the positive regulation of transcription of Star gene. 59 EGF might interfere with one or more of these pathways. EGF also down‐regulated Hsd3b1 (Figure 5), which encodes HSD3B1, catalysing pregnenolone into progesterone. Interestingly, EGF‐mediated regulation of steroidogenesis in PLCs is quite different from LH, which actually up‐regulated all steroidogenesis‐related genes, including Scarb1, Star, Cyp11a1, Cyp17a1 and Hsd3b1. LH primarily acts on LHCGR, thus activating cAMP/PKA signalling.
Interestingly, the expression levels of many genes in antioxidative proteins (Ugt1a6, Idh1, Gsst1, Gstm2 and Sod3) were significantly down‐regulated more than twofold after EGF treatment. This could lead to ROS accumulation in PLCs (Figure 6). Indeed, EGF significantly led to ROS accumulation (Figure 3C). Several studies demonstrated that the accumulation of ROS is capable of damaging testis function 60 and inhibiting T production by LCs. 61 Mitochondrion is an important organelle for regulation of T synthesis and is sensitive to ROS attack. 62 , 63 , 64 ROS is capable of disturbing StAR level. 65 Although ROS accumulation might lead to cell apoptosis, 66 the concentration of EGF (10 ng/mL) tested in the current study was not enough to induce PLC apoptosis (Figure 3). Actually, EGF can antagonize H2O2‐induced apoptosis (Figure 3).
TSP2 (encoded by Tsp2) was significantly down‐regulated by EGF (Figure 5). TSP2 functions the differentiation of mesenchymal stem cells into bones. 67 The down‐regulation of Tsp2 by EGF could lead to stem/progenitor Leydig cells stay in the Leydig cell lineage not into other cell lineage as SLCs were multipotent and SLCs can also differentiate into bone cells. 8
In conclusion, the present study demonstrated that EGF induced the proliferation of SLCs/PLCs but blocks their differentiation into LCs.
CONFLICT OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
AUTHOR CONTRIBUTIONS
Ren‐Shan Ge designed research; Xiaoheng Li, Yiyan Wang and Fei Ge performed research; Qiqi Zhu and Kaiming Yuan contributed new reagents; Ren‐Shan Ge and Zhijian Su analysed data, wrote the paper and revised the manuscript; and Yadong Huang supervised the experiments, revised and approved the manuscript.
Supporting information
Table S1‐S2
ACKNOWLEDGEMENT
The present study was supported by NSFC (81730042, 81601266, 81871150, 81871155) and Department of Science and Technology of Zhejiang Province (2019C03035).
Li X, Wang Y, Zhu Q, et al. Epidermal growth factor regulates the development of stem and progenitor Leydig cells in rats. J Cell Mol Med. 2020;24:7313–7330. 10.1111/jcmm.15302
Contributor Information
Ren‐Shan Ge, Email: tydhuang@jnu.edu.cn, Email: r_ge@yahoo.com.
Yadong Huang, Email: tydhuang@jnu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Chen P, Zirkin BR, Chen H. Stem Leydig cells in the adult testis: characterization, regulation and potential applications. Endocr Rev. 2020;41(1):22‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ye L, Li X, Li L, Chen H, Ge RS. Insights into the development of the adult Leydig cell lineage from stem Leydig cells. Front Physiol. 2017;8:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ge R, Dong Q, Sottas C, Papadopoulos V, Zirkin BR, Hardy MP. In search of rat stem Leydig cells: identification, isolation, and lineage‐specific development. Proc Natl Acad Sci USA. 2006;103(8):2719‐2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stanley E, Lin CY, Jin S, et al. Identification, proliferation, and differentiation of adult Leydig stem cells. Endocrinology. 2012;153:5002‐5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang MH, Cai B, Tuo Y, et al. Characterization of Nestin‐positive stem Leydig cells as a potential source for the treatment of testicular Leydig cell dysfunction. Cell Res. 2014;24:1466‐1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zang ZJ, Wang J, Chen Z, et al. Transplantation of CD51+ stem Leydig cells: a new strategy for the treatment of testosterone deficiency. Stem Cells. 2017;35:1222‐1232. [DOI] [PubMed] [Google Scholar]
- 7. Chen P, Guan X, Zhao X, et al. Characterization and differentiation of CD51(+) Stem Leydig cells in adult mouse testes. Mol Cell Endocrinol. 2019;493:110449. [DOI] [PubMed] [Google Scholar]
- 8. Zhang M, Wang J, Deng C, et al. Transplanted human p75‐positive stem Leydig cells replace disrupted Leydig cells for testosterone production. Cell Death Dis. 2017;8:e3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ge RS, Hardy MP. Variation in the end products of androgen biosynthesis and metabolism during postnatal differentiation of rat Leydig cells. Endocrinology. 1998;139:3787‐3795. [DOI] [PubMed] [Google Scholar]
- 10. Mendis‐Handagama SM, Ariyaratne HB. Differentiation of the adult Leydig cell population in the postnatal testis. Biol Reprod. 2001;65:660‐671. [DOI] [PubMed] [Google Scholar]
- 11. Phillips DM, Lakshmi V, Monder C. Corticosteroid 11β‐dehydrogenase in rat testis. Endocrinology. 1989;125:209‐216. [DOI] [PubMed] [Google Scholar]
- 12. Guo J, Zhou H, Su Z, et al. Comparison of cell types in the rat Leydig cell lineage after ethane dimethanesulfonate treatment. Reproduction. 2013;145:371‐380. [DOI] [PubMed] [Google Scholar]
- 13. Ge RS, Dong Q, Sottas CM, Papadopoulos V, Zirkin BR, Hardy MP. In search of rat stem Leydig cells: identification, isolation, and lineage‐specific development. Proc Natl Acad Sci USA. 2006;103:2719‐2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lei ZM, Mishra S, Zou W, et al. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol. 2001;15:184‐200. [DOI] [PubMed] [Google Scholar]
- 15. Zhang F‐P, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LHRKO) mice. Mol Endocrinol. 2001;15:172‐183. [DOI] [PubMed] [Google Scholar]
- 16. Shan LX, Hardy MP. Developmental changes in levels of luteinizing hormone receptor and androgen receptor in rat Leydig cells. Endocrinology. 1992;131:1107‐1114. [DOI] [PubMed] [Google Scholar]
- 17. Ge RS, Hardy MP. Decreased cyclin A2 and increased cyclin G1 levels coincide with loss of proliferative capacity in rat Leydig cells during pubertal development. Endocrinology. 1997;138:3719‐3726. [DOI] [PubMed] [Google Scholar]
- 18. Tan K, Song HW, Wilkinson MF. Single‐cell RNAseq analysis of testicular germ and somatic cell development during the perinatal period. Development. 2020;147(3):dev183251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo J, Nie X, Giebler M, et al. The dynamic transcriptional cell atlas of testis development during human puberty. Cell Stem Cell. 2020;26(2):262–276.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coulter CL, Read LC, Barry SJ, Tarantal AF, Styne DM. Role of hypothalamic‐pituitary axis in EGF action on maturation of adrenal gland in fetal rhesus monkey in vivo. Pediatr Res. 2001;50:210‐216. [DOI] [PubMed] [Google Scholar]
- 21. Levine E, Cupp AS, Miyashiro L, Skinner MK. Role of transforming growth factor‐alpha and the epidermal growth factor receptor in embryonic rat testis development. Biol Reprod. 2000;62:477‐490. [DOI] [PubMed] [Google Scholar]
- 22. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59: S21‐S26. [DOI] [PubMed] [Google Scholar]
- 23. Foresta C, Caretto A, Varotto A, Rossato M, Scandellari C. Epidermal growth factor receptors (EGFR) localization in human testis. Arch Androl. 1991;27:17‐24. [DOI] [PubMed] [Google Scholar]
- 24. Saez JM, Benahmed M, Reventos J, Bommelaer MC, Monbrial C, Haour F. Hormonal regulation of pig Leydig cells in culture. J Steroid Biochem. 1983;19:375‐384. [PubMed] [Google Scholar]
- 25. Suarez‐Quian CA, Niklinski W. Immunocytochemical localization of the epidermal growth factor receptor in mouse testis. Biol Reprod. 1990;43:1087‐1097. [DOI] [PubMed] [Google Scholar]
- 26. Melner MH, Lutin WA, Puett D. Epidermal growth factor and cyclic AMP stimulation of distinct protein kinase activities in Leydig cell tumor membranes. Life Sci. 1982;30:1981‐1986. [DOI] [PubMed] [Google Scholar]
- 27. Odeh HM, Kleinguetl C, Ge R, Zirkin BR, Chen H. Regulation of the proliferation and differentiation of leydig stem cells in the adult testis. Biol Reprod. 2014;90:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rommerts FF, Teerds KJ, Hoogerbrugge JW. In vitro effects of ethylene‐dimethane sulfonate (EDS) on Leydig cells: inhibition of steroid production and cytotoxic effects are dependent on species and age of rat. Mol Cell Endocrinol. 1988;55:87‐94. [DOI] [PubMed] [Google Scholar]
- 29. Teerds KJ, De Rooij DG, Rommerts FF, Wensing CJ. The regulation of the proliferation and differentiation of rat Leydig cell precursor cells after EDS administration or daily HCG treatment. J Androl. 1988;9:343‐351. [DOI] [PubMed] [Google Scholar]
- 30. Li X, Wang Z, Jiang Z, et al. Regulation of seminiferous tubule‐associated stem Leydig cells in adult rat testes. Proc Natl Acad Sci USA. 2016;113:2666‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug‐resistance mechanisms can emerge from drug‐tolerant cancer persister cells. Nat Commun. 2016;7:10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang YF, Yuan KM, Liang Y, et al. Alterations of gene profiles in Leydig‐cell‐regenerating adult rat testis after ethane dimethane sulfonate‐treatment. Asian J Androl. 2015;17:253‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li L, Wang Y, Li X, et al. Regulation of development of rat stem and progenitor Leydig cells by activin. Andrology. 2016;5:125‐132. [DOI] [PubMed] [Google Scholar]
- 34. Payne AH, Downing JR, Wong KL. Luteinizing hormone receptor and testosterone synthesis in two distinct populations of Leydig cells. Endocrinology. 1980;106:1424‐1429. [DOI] [PubMed] [Google Scholar]
- 35. Guo X, Wang H, Wu X, et al. Nicotine affects rat Leydig cell function in vivo and vitro via down‐regulating some key steroidogenic enzyme expressions. Food Chem Toxicol. 2017;110:13‐24. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y, Yuan K, Li X, et al. Leukemia inhibitory factor stimulates steroidogenesis of rat immature Leydig cells via increasing the expression of steroidogenic acute regulatory protein. Growth Factors. 2016;34:166‐176. [DOI] [PubMed] [Google Scholar]
- 37. Griffin DK, Ellis PJ, Dunmore B, Bauer J, Abel MH, Affara NA. Transcriptional profiling of luteinizing hormone receptor‐deficient mice before and after testosterone treatment provides insight into the hormonal control of postnatal testicular development and Leydig cell differentiation. Biol Reprod. 2010;82:1139‐1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin H, Lian QQ, Hu GX, et al. In utero and lactational exposures to diethylhexyl‐phthalate affect two populations of leydig cells in male long‐evans rats. Biol Reprod. 2009;80:882‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ge RS, Dong Q, Sottas CM, Chen H, Zirkin BR, Hardy MP. Gene expression in rat leydig cells during development from the progenitor to adult stage: a cluster analysis. Biol Reprod. 2005;72:1405‐1415. [DOI] [PubMed] [Google Scholar]
- 40. Geiger TL, Khan M, Whisnant CS, Prien SD, Khan SA. Regulation of DNA synthesis in Leydig cells obtained from neonatal pig testes. Domest Anim Endocrinol. 1999;17:65‐75. [DOI] [PubMed] [Google Scholar]
- 41. Khan S, Teerds K, Dorrington J. Growth factor requirements for DNA synthesis by Leydig cells from the immature rat. Biol Reprod. 1992;46:335‐341. [DOI] [PubMed] [Google Scholar]
- 42. Shiraishi K, Ascoli M. Lutropin/choriogonadotropin stimulate the proliferation of primary cultures of rat Leydig cells through a pathway that involves activation of the extracellularly regulated kinase 1/2 cascade. Endocrinology. 2007;148:3214‐3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guan X, Chen P, Zhao X, et al. Characterization of stem cells associated with seminiferous tubule of adult rat testis for their potential to form Leydig cells. Stem Cell Res. 2019;41:101593. [DOI] [PubMed] [Google Scholar]
- 44. Shiraishi K, Ascoli M. A co‐culture system reveals the involvement of intercellular pathways as mediators of the lutropin receptor (LHR)‐stimulated ERK1/2 phosphorylation in Leydig cells. Exp Cell Res. 2008;314:25‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evaul K, Hammes SR. Cross‐talk between G protein‐coupled and epidermal growth factor receptors regulates gonadotropin‐mediated steroidogenesis in Leydig cells. J Biol Chem. 2008;283:27525‐27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tai P, Shiraishi K, Ascoli M. Activation of the lutropin/choriogonadotropin receptor inhibits apoptosis of immature Leydig cells in primary culture. Endocrinology. 2009;150:3766‐3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen DG, Zhu B, Lv SQ, et al. Inhibition of EGR1 inhibits glioma proliferation by targeting CCND1 promoter. J Exp Clin Cancer Res. 2017;36:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ding D, Huang H, Jiang W, et al. Reticulocalbin‐2 enhances hepatocellular carcinoma proliferation via modulating the EGFR‐ERK pathway. Oncogene. 2017;36:6691‐6700. [DOI] [PubMed] [Google Scholar]
- 49. Basu S, Haase G, Ben‐Ze'ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Res. 2016;5:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606‐619. [DOI] [PubMed] [Google Scholar]
- 51. Liu W, Ren H, Ren J, et al. The role of EGFR/PI3K/Akt/cyclinD1 signaling pathway in acquired middle ear cholesteatoma. Mediators Inflamm. 2013;2013:651207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsueh AJ, Welsh TH, Jones PB. Inhibition of ovarian and testicular steroidogenesis by epidermal growth factor. Endocrinology. 1981;108:2002‐2004. [DOI] [PubMed] [Google Scholar]
- 53. Ascoli M, Segaloff DL. Regulation of the differentiated functions of Leydig tumor cells by epidermal growth factor. Ann N Y Acad Sci. 1989;564:99‐115. [DOI] [PubMed] [Google Scholar]
- 54. Sordoillet C, Chauvin MA, de Peretti E, Morera AM, Benahmed M. Epidermal growth factor directly stimulates steroidogenesis in primary cultures of porcine Leydig cells: actions and sites of action. Endocrinology. 1991;128:2160‐2168. [DOI] [PubMed] [Google Scholar]
- 55. Syed V, Khan SA, Nieschlag E. Epidermal growth factor stimulates testosterone production of human Leydig cells in vitro. J Endocrinol Invest. 1991;14:93‐97. [DOI] [PubMed] [Google Scholar]
- 56. Verhoeven G, Cailleau J. Stimulatory effects of epidermal growth factor on steroidogenesis in Leydig cells. Mol Cell Endocrinol. 1986;47:99‐106. [DOI] [PubMed] [Google Scholar]
- 57. Lemaire G, Mnif W, Mauvais P, Balaguer P, Rahmani R. Activation of alpha‐ and beta‐estrogen receptors by persistent pesticides in reporter cell lines. Life Sci. 2006;79:1160‐1169. [DOI] [PubMed] [Google Scholar]
- 58. Zhou DJ, Pompon D, Chen SA. Structure‐function studies of human aromatase by site‐directed mutagenesis: kinetic properties of mutants Pro‐308––Phe, Tyr‐361––Phe, Tyr‐361––Leu, and Phe‐406––Arg. Proc Natl Acad Sci USA. 1991;88:410‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De Jong WH, Tentij M, Spiekstra SW, Vandebriel RJ, Van Loveren H. Determination of the sensitising activity of the rubber contact sensitisers TMTD, ZDMC, MBT and DEA in a modified local lymph node assay and the effect of sodium dodecyl sulfate pretreatment on local lymph node responses. Toxicology. 2002;176:123‐134. [DOI] [PubMed] [Google Scholar]
- 60. Tseng LH, Lee CW, Pan MH, et al. Postnatal exposure of the male mouse to 2,2′,3,3′,4,4′,5,5′,6,6′‐decabrominated diphenyl ether: decreased epididymal sperm functions without alterations in DNA content and histology in testis. Toxicology. 2006;224:33‐43. [DOI] [PubMed] [Google Scholar]
- 61. Weissman BA, Niu E, Ge R, et al. Paracrine modulation of androgen synthesis in rat leydig cells by nitric oxide. J Androl. 2005;26:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hales DB, Allen JA, Shankara T, et al. Mitochondrial function in Leydig cell steroidogenesis. Ann N Y Acad Sci. 2005;1061:120‐134. [DOI] [PubMed] [Google Scholar]
- 63. Allen JA, Diemer T, Janus P, Hales KH, Hales DB. Bacterial endotoxin lipopolysaccharide and reactive oxygen species inhibit Leydig cell steroidogenesis via perturbation of mitochondria. Endocrine. 2004;25:265‐275. [DOI] [PubMed] [Google Scholar]
- 64. Li M, Sun J, Zou F, et al. Glycidamide inhibits progesterone production through reactive oxygen species‐induced apoptosis in R2C Rat Leydig Cells. Food Chem Toxicol. 2017;108:563‐570. [DOI] [PubMed] [Google Scholar]
- 65. Diemer T, Allen JA, Hales KH, Hales DB. Reactive oxygen disrupts mitochondria in MA‐10 tumor Leydig cells and inhibits steroidogenic acute regulatory (StAR) protein and steroidogenesis. Endocrinology. 2003;144:2882‐2891. [DOI] [PubMed] [Google Scholar]
- 66. Zhang Z, Zhang X, Sun Z, et al. Cytochrome P450 3A1 mediates 2,2',4,4'‐tetrabromodiphenyl ether‐induced reduction of spermatogenesis in adult rats. PLoS ONE. 2013;8:e66301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jeong SY, Ha J, Lee M, et al. Autocrine action of thrombospondin‐2 determines the chondrogenic differentiation potential and suppresses hypertrophic maturation of human umbilical cord blood‐derived mesenchymal stem cells. Stem Cells. 2015;33:3291‐3303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.