

Abstract
Aberrant elevated Src activity is related to lung cancer growth and metastasis. Therefore, the development of potent small molecule inhibitors to target Src kinase is a potential therapeutic strategy for lung cancer. This study aimed to develop a computational model for the in silico screening of Src inhibitors and then assess the suppressive effect of candidate compounds on cellular functions. A 3D-quantitative structure-activity relationship (QSAR) pharmacophore model consisting of two hydrogen bond acceptors and two hydrophobic regions was constructed by using 28 structurally diverse compounds with IC50 values spanning four orders of magnitude. A National Cancer Institute (NCI) compound dataset was employed for virtual screening by applying the pharmacophore model and molecular docking. Candidate compounds were chosen from the top 20% of scored hits. Among these compounds, the suppressive effects of 30 compounds available in the NCI on Src phosphorylation were validated by using an enzyme-linked immunosorbent assay. Among these compounds, SJG-136, a pyrrolobenzodiazepine dimer, showed a significant inhibitory effect against Src activity in a dose-dependent manner. Further investigations showed that SJG-136 can inhibit lung cancer cell proliferation, clonogenicity, invasion and migration in vitro and tumour growth in vivo. Furthermore, SJG-136 also had an inhibitory effect on Src-related signaling pathways, including the FAK, paxillin, p130Cas, PI3K, AKT, and MEK pathways. In conclusion, we have established a pharmacophore-based virtual screening approach to identify novel Src inhibitors that can inhibit lung cancer cell growth and motility through suppressing Src-related pathways. These findings may contribute to the development of targeted drugs for lung cancer treatment, such as lead compounds.
Keywords: Lung cancer, Src inhibitor, pyrrolobenzodiazepine, pharmacophore model, virtual screening
Introduction
Non-small-cell lung cancer (NSCLC) is the predominant type of lung cancer and the leading cause of cancer-related mortality worldwide [1]. The low survival associated with NSCLC may result from treatment failure in patients with advanced, metastatic disease and recurrent disease [2]. Metastasis is a multi-step process that involves cell proliferation, migration, degradation of the endothelial basement membrane and invasion [3]. Currently, there is no effective therapy to prevent or control these metastatic processes. Hence, developing novel compounds by targeting both tumour growth and metastasis is a major mission for the improvement of lung cancer treatment.
Studies in lung cancer have shown that the dysregulated expression or genetic alteration of oncogenes and tumour suppressor genes, such as EGFR, KRAS, ALK, and TP53, is associated with tumour progression and metastasis [4-6]. In recent years, the development of therapeutic strategies to improve the prognoses of patients with NSCLC has focused on designing protein kinase inhibitors to suppress the oncogene addiction of cancer cells [7]. For example, crizotinib has been administered to NSCLC patients whose tumours harbour ALK fusions [8]. Moreover, the first-generation EGFR tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib have been associated with good responses in NSCLC patients with EGFR exon 19 deletions or L858R mutations [9]. However, approximately 10% of NSCLC patients display primary TKI resistance [10]. Furthermore, approximately 50% of EGFR-TKI-treated patients show EGFR T790M mutations at the time of acquired resistance to TKI therapy [11]. Drug resistance is a serious issue in NSCLC treatment; therefore, developing new targeted drugs or therapeutic strategies is an urgent goal.
Src is a proto-oncogene belonging to the non-receptor tyrosine kinase family [12]. The structure of Src includes an NH2-terminus, two conserved Src homology domains (including SH2 and SH3) and a tyrosine kinase domain (TKD) [13]. Regulation of Src is dependent upon phosphorylation of the C-terminal regulatory tail at Y530 by C-terminal Src kinase (CSK), resulting in transition to the inactive conformation and decreased activity [14]. In contrast, autophosphorylation in the ATP-binding pocket of TKD (Y418 and Y419) results in transition to the active conformation and increased Src activity [15,16]. The NH2-terminal region contains a myristoylation site that is important for localization to the cellular membrane. At this location, Src can interact with signaling proteins through its SH2 and SH3 domains to promote multiple cellular processes [17].
In recent decades, dozens of studies have demonstrated that Src plays a pivotal role in mediating signal transduction through its interactions with downstream proteins, such as FAK, PI3K and STAT3, to influence cell growth and motility, as well as in tumour progression, invasion, and metastasis [18-20]. Elevated Src expression was associated with various human malignancies, including NSCLC [18,21-23]. Previous studies have shown that Src overexpression is observed in approximately 60-80% of NSCLC patients [24,25] and related to poor prognosis [23]. Several clinically applicable small molecule Src inhibitors, such as dasatinib (BMS-354825), saracatinib (AZD0530), bosutinib (SKI-606), and ponatinib (AP24534), have been developed and evaluated for their clinical effectiveness against solid tumours [26,27]. However, there is no approved Src inhibitors in the treatment of lung cancer, yet. Given the crucial role of Src in tumour growth and metastasis, Src may be a potential therapeutic target in lung cancer treatment [28].
In this study, to identify novel Src inhibitors, we performed pharmacophore-based virtual screening and then assessed the capability of candidate compounds to inhibit the Src activity or expression in lung cancer cells. Finally, we determined that a pyrrolobenzodiazepine dimer (SJG-136; NCI/DTP NSC 694501) is a candidate Src-targeted inhibitor. In addition, we also investigated the functional roles underlying the ability of SJG-136 to suppress lung cancer progression. The overall workflow is shown in Figure 1. These results may benefit the development of new anticancer drugs and therapeutic strategies for the treatment of lung cancer in the future.
Figure 1.

Overall scheme of pharmacophore-based virtual screening implemented for the discovery of the novel Src inhibitor SJG-136.
Materials and methods
Data set preparation
A set of 51 compounds along with data on their experimental activity when tested with an Src kinase assay expressed as IC50 values were collected from the BindingDB database [29]. In addition, 5 in-house Src inhibitors were incorporated into the compound set. The 3D structures of the compounds were then energy minimized to the closest local minimum using the CHARMM force field [30] and the smart minimizer method, which performs 1000 steps of steepest descent followed by conjugate gradient minimization, available in Accelrys Discovery Studio 3.5 (DS) (Accelrys Inc., San Diego, USA). Of these 56 compounds, 28 comprised a training set, and the rest comprised a test set.
3D-QSAR pharmacophore model generation
To construct the pharmacophore model, we first explored the chemical features of compounds from the training set by utilizing the feature mapping module of DS. Six features (hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), ring aromatic (RA), hydrophobic region (HY), and positive and negative ionizable areas) were investigated to identify the vital features. A 3D-QSAR pharmacophore generation module (the HypoGen algorithm) [31] was used to build a series of pharmacophore hypotheses by considering the crucial chemical features and experimental activity values (IC50). The top 10 ranked pharmacophore hypotheses were generated with the following corresponding statistical parameters: cost values, root-mean-square (RMS), correlation coefficient, and fit values. The best hypothesis (i.e., top ranked model) was selected for large-scale virtual screening.
Pharmacophore model evaluation
To confirm the statistical significance and predictive validity of the pharmacophore hypotheses, Fischer’s randomization method and an independent test set were employed to assess the two independent objectives. In Fischer’s randomization method, to identify models at 95% and 99% confidence levels, the original training set was shuffled, and activity values were used to generate 19 and 99 different pharmacophore models, respectively. Then, the total cost values of randomized models were compared to those of the best model to judge the significance of the model. Compounds in the test set were used to estimate the correlation between the experimental and predicted activity values to understand the predictive power.
Pharmacophore-based virtual screening
Virtual screening of chemical databases can identify a limited number of candidate molecules that are likely to be active against a chosen biological receptor. The ligand pharmacophore mapping module of DS was utilized for this procedure. The best pharmacophore hypothesis was used as a structural query with which to search the NCI database (https://wiki.nci.nih.gov/display/NCIDTPdata/Chemical+Data) to retrieve molecules that fit all the features of the model well. Initially, Lipinski’s rules of five [32] and Veber’s rules [33] were used to evaluate the druglikeness of compounds from the NCI database. Furthermore, to screen the hit molecules, 255 conformers of each drug-like molecule were generated using the best conformer generation method and used to perform virtual screening with the pharmacophore model.
Structure-based molecular docking
In this experiment, molecular docking was conducted to simulate suitable binding modes of hit molecules in the Src protein active site using the LibDock algorithm [34] of DS software. Two crystal structures of the c-Src complex (PDB ID: 3G5D and 1Y57) [35,36] were retrieved from the Protein Data Bank to serve as the receptor protein. The water molecules and the bound inhibitor were removed from the protein structure, and then hydrogen atoms and potentials were added using the CHARMM force field. A radius of 10 Å around the inhibitor present in the crystal structure was defined as a binding site. The conformation of each hit compound was generated by applying the best conformer generation method and energy minimized by using the smart minimizer method. The best docked conformation for each compound based on the LibDock score was saved, and its binding affinity to Src was then evaluated by calculating the binding free energy [37] and various scoring functions. The following scoring functions were used: the potential of mean force (PMF) [38], potential of mean force 04 (PMF04) [39], piecewise linear potential 1 (PLP1) [40], piecewise linear potential 2 (PLP2), Jain [38], LigScore1 [41], LigScore2, Goldscore [42], and LUDI score [43]. The consensus scoring method [44] was employed taking all dock scores into account to rank each compound. Finally, candidate compounds were selected that showed good agreement with the following three scores: the Libdock, binding energy and consensus scores. The candidate compounds were subsequently subjected to further in vitro biological experiments.
Cell culture and drug treatment
The human lung adenocarcinoma cell lines A549 (ATCC CCL-185) and H358 (ATCC CRL-5807) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The two cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2. Cells were maintained in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco). The candidate compounds were obtained from the NCI drug repository and prepared as a stock solution at a concentration of 100 mM in dimethyl sulfoxide (DMSO). The vehicle control was treated with 0.1% DMSO.
Enzyme-linked immunosorbent assay
DuoSet Human Phospho-Src (Y419) ELISA reagent (R&D Systems, Minneapolis, MN, USA) was utilized according to the manufacturer’s protocol to investigate the effect of candidate compounds on the phosphorylation of Src. Briefly, cell lysates derived from compound- or vehicle-treated cells were added to a 96-well plate pre-coated with capture antibody to bind phosphorylated and unphosphorylated Src. A biotinylated detection antibody was subsequently added to detect Src phosphorylation at Y419. After washing away unbound antibodies, a streptavidin-HRP conjugate was added to detect the antibody. NeA-Blue tetramethylbenzidine substrate (Clinical Science Products, Bristol County, MA, USA) was employed to develop the signal according to the instruction manual. The absorbance was measured at 450 nm (with 570 nm as the reference) using a Victor3 spectrophotometer (Perkin-Elmer, Santa Clara, CA, USA). The vehicle control was used as a negative control. Dasatinib (1 µM)-treated cells served as a positive control.
Western blot analysis
Western blotting was performed to observe the phosphorylation and expression levels of Src and its associated proteins in lung cancer cells treated with candidate compounds, as described previously [45]. The primary antibody against Src was produced in our laboratory (ATCC CRL-2651). Antibodies against phospho-Src (Tyr418), phospho-FAK (Tyr576) and FAK were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies against STAT3, PI3K, paxillin, p130Cas, phospho-MEK1/2 (Ser218/Ser222), MEK, phospho-ERK (Tyr204) and ERK were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Antibodies against phospho-STAT3 (Tyr705), phospho-PI3K (Tyr458/Tyr199), phospho-paxillin (Tyr118), phospho-p130Cas (Tyr410), phospho-SAPK/Jun N-terminal kinase (JNK) (Thr183/Tyr185), SAPK/JNK, and AKT were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody against phospho-AKT (Ser473) was purchased from Millipore (Billerica, MA, USA). GAPDH (Upstate Biotechnology, Lake Placid, NY, USA) was used as a loading control.
Cell viability assay
PrestoBlue cell viability reagent (Invitrogen) was used to evaluate the effects of candidate compounds on cell cytotoxicity according to the manufacturer’s protocol. After the cells were treated with candidate compounds at different concentrations for different times, PrestoBlue reagent was added for reaction with the cells. The absorbance at 570 and 600 nm (the reference group) was measured using a Victor3 spectrophotometer (Perkin-Elmer).
Clonogenicity assay
An anchorage-dependent growth assay was performed to observe the effects of candidate compounds on cell colony formation. In this assay, 500 cells were resuspended in RPMI medium and then seeded in six-well plates containing a culture medium and drug solution. After 7-10 days, the cells were washed with 1 × PBS and fixed with methanol. The cells were subsequently stained with 0.05% crystal violet. Colonies with diameters greater than or equal to 0.2 mm were counted using an inverted microscope.
Migration and invasion assays
A Transwell device with a polycarbonate membrane (8-μm pore size; Corning Costar Corporation, Cambridge, MA, USA) that was or was not coated with Matrigel (R&D Systems) was used for Transwell invasion and migration assays, as described previously [46]. Cells were treated with various concentrations of candidate compound for 24 hours and seeded in Transwell chambers. The upper wells were filled with serum-free medium containing the drug of interest and lung cancer cells (5 × 103 or 2 × 104 cells per well). The lower wells of the Transwell device were filled with the same medium supplemented with 10% FBS. After incubation for 12 hours (migration) or 24 hours (invasion), the cells were swabbed from the upper wells and the upper surface of the membrane with a Q-tip. Then, methanol and a 10% Giemsa solution (Sigma Chemical, St. Louis, MO, USA) were used to fix and stain the cells, respectively. The number of cells attached to the lower surface of the polycarbonate filter was counted using a light microscope (magnification, 200 ×).
Tumourigenesis assay
Four-week-old nude mice (BALB/cAnN.Cg-Foxn1 nu/CrlNarl) were purchased from the National Laboratory Animal Center (NLAC, Taipei, Taiwan). Tumours were induced in nude mice according to a protocol described previously [47]. A total of 1 × 106 live A549 cells were subcutaneously injected into the nude mice. Subcutaneous tumour volumes were measured every 4 days until they reached an average size of 50 mm3. To evaluate the tumour suppressive effects of SJG-136, the mice were grouped into drug-treated or untreated groups. The animals in the untreated group were injected with 0.1% DMSO as vehicle control. In addition, by referring to a prior study [48], the animals in the drug-treated group were received separate intravenous injections with SJG-136 at two different drug administration schedules as follows: 122 μg/kg once daily for five treatments (schedule 1) and 400 μg/kg every seventh day for two treatments (schedule 2). The tumour volumes were estimated from calliper-measured lengths (a) and widths (b) using the following formula V = 0.4 × ab2 [49]. After 4-5 weeks, the mice were sacrificed and their tumour sizes were analysed. The mouse experiments were approved by the Institutional Animal Care and Use Committee of National Chung Hsing University.
Statistical analysis
All the experiments were performed at least in triplicate, and the significance of differences was analysed using a t-test (Excel, Microsoft). P values < 0.05 indicated statistical significance.
Results
3D-QSAR pharmacophore generation
The quality of a pharmacophore is directly proportional to the structural diversity and wide range of experimental activities of the compounds in the training set. The 28 structurally diverse training set compounds had IC50 values for Src inhibition spanning 4 nM-75000 nM (Figure 2) and were categorized into three groups: highly active (IC50 < 100 nM), moderately active (100 nM < IC50 < 1000 nM) and lowly active (IC50 > 1000 nM) compounds. In total, four crucial features (HBA, HBD, RA, and HY) were identified through the training set compounds and then used to produce the pharmacophore hypotheses by exploiting the 3D-QSAR Pharmacophore Generation module.
Figure 2.

Twenty-eight chemically diverse compounds used as a training set in 3D-QSAR pharmacophore modeling.
The top 10 ranked pharmacophore hypotheses generated confirmed the importance of the HBA, HY, and RA features, and the corresponding statistical parameters were calculated to reflect the model’s significance (Table 1). The top ranked hypothesis (i.e., Hypo1) was adopted as the best pharmacophore model with its corresponding parameters: the highest correlation coefficient (0.970), lowest RMS (0.678), lowest total cost (118.745), reliable configuration cost (16.649) and highest cost difference (83.461). The null cost and the fixed cost were 202.206 and 111.954, respectively.
Table 1.
Top 10 pharmacophore hypotheses generated by using a training set against Src inhibitors
| Hypothesis (Hypo) | Total cost | Cost difference* | RMS† | Correlation (r) | Features‡ |
|---|---|---|---|---|---|
| 1 | 118.745 | 83.461 | 0.678 | 0.970 | HBA, HBA, HY, HY |
| 2 | 121.852 | 80.354 | 0.838 | 0.953 | HBA, HBA, HY, HY, HY |
| 3 | 124.315 | 77.891 | 0.935 | 0.942 | HBA, HBA, HY, HY |
| 4 | 127.566 | 74.640 | 1.049 | 0.926 | HBA, HBA, HY, RA |
| 5 | 127.810 | 74.396 | 1.045 | 0.927 | HBA, HBA, HY, RA |
| 6 | 130.106 | 72.100 | 1.187 | 0.904 | HBA, HY, HY, RA |
| 7 | 131.824 | 70.382 | 1.187 | 0.904 | HBA, HY, HY, HY, RA |
| 8 | 134.117 | 68.089 | 1.245 | 0.894 | HBA, HBA, HY, HY, HY |
| 9 | 134.462 | 67.744 | 1.227 | 0.897 | HBA, HY, HY, HY, RA |
| 10 | 134.930 | 67.276 | 1.270 | 0.889 | HBA, HY, HY, HY, RA |
The difference between the null and total costs. The null cost, the fixed cost and the configuration cost were 202.206, 111.954 and 16.649, respectively. All costs are in units of bits.
RMS, root-mean-square deviation.
HBA, hydrogen bond acceptor; HY, hydrophobic; RA, ring aromatic.
The Hypo1 hypothesis was composed of two HBA and two HY chemical features; the distance between every two features is shown in Figure 3A. The most active compound, compound 1 (IC50: 4 nM), had a high fit value of 7.93 (Figure 3B) when mapped with Hypo1, while the least active compound, compound 28 (IC50: 75000 nM), showed a lower value of 4.51 (Figure 3C). The fit value represents how well the features in a pharmacophore overlap with the chemical groups present in the molecule, which is useful to understand the chemical meaning of a pharmacophore model. The correlation result indicated that the Hypo1 model showed good predictability between experimental and predicted activities, as shown in Figure 3D (r = 0.97). Furthermore, 9 of 11 highly active, 4 of 5 moderately active and 11 of 12 lowly active compounds in the same order of magnitude were predicted (Table 2).
Figure 3.
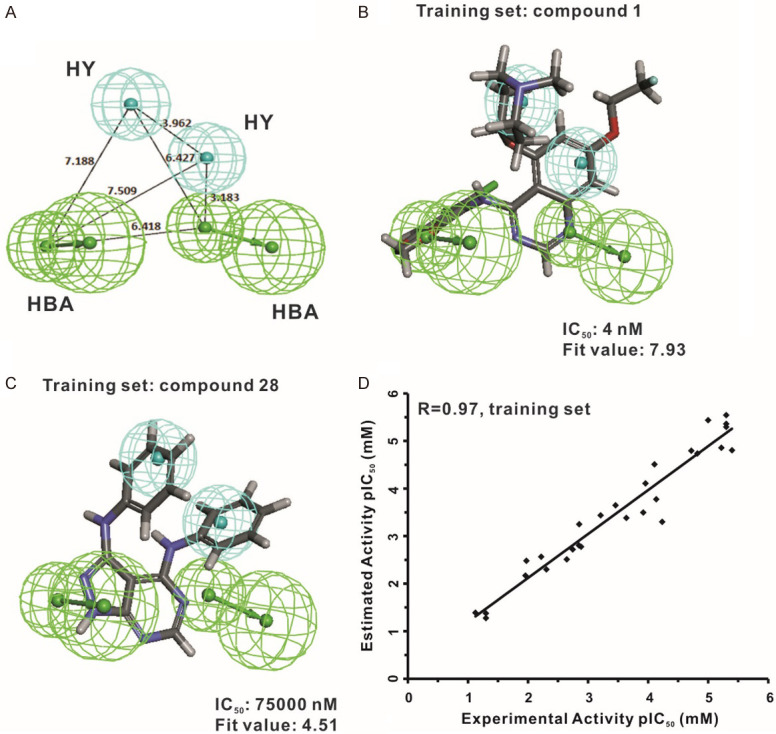
Hyop1 pharmacophore model of Src inhibitors. (A) Pharmacophore features of the Hypo1 model. HBA: hydrogen bond acceptor (green); HY: hydrophobic region (blue). The distance (Å) between every two features is shown. Hypo1 is mapped to the most active compound (IC50 = 4 nM; fit value = 7.93) (B) and the most inactive compound (IC50 = 75000 nM; fit value = 4.51) (C) by using the training set molecules. (D) Correlation analysis was used to evaluate the predictive validity of the Hypo1 model by calculating the correlation between the experimental activity and the estimated activity of the training set compounds (r = 0.97).
Table 2.
Actual and estimated activities of 28 training set compounds calculated on the basis of the Hypo1 pharmacophore model
| Compound no. | Experimental IC50 nM | Predicted IC50 nM | Fit value* | Error† | Experimental scale‡ | Predicted scale‡ |
|---|---|---|---|---|---|---|
| 1 | 4 | 15.70 | 7.93 | +3.93 | +++ | +++ |
| 2 | 5 | 2.87 | 8.67 | -1.74 | +++ | +++ |
| 3 | 5 | 4.37 | 8.48 | -1.14 | +++ | +++ |
| 4 | 5 | 5.07 | 8.42 | +1.01 | +++ | +++ |
| 5 | 6 | 13.92 | 7.98 | +2.32 | +++ | +++ |
| 6 | 10 | 3.66 | 8.56 | -2.73 | +++ | +++ |
| 7 | 15 | 18.29 | 7.86 | +1.22 | +++ | +++ |
| 8 | 19 | 16.04 | 7.92 | -1.18 | +++ | +++ |
| 9 | 58 | 503.67 | 6.42 | +8.68 | +++ | ++ |
| 10 | 73 | 168.16 | 6.90 | +2.30 | +++ | ++ |
| 11 | 78 | 31.02 | 7.63 | -2.51 | +++ | +++ |
| 12 | 110 | 78.19 | 7.23 | -1.41 | ++ | +++ |
| 13 | 120 | 319.36 | 6.62 | +2.66 | ++ | ++ |
| 14 | 230 | 415.89 | 6.50 | +1.81 | ++ | ++ |
| 15 | 347 | 225.04 | 6.77 | -1.54 | ++ | ++ |
| 16 | 616 | 367.88 | 6.56 | -1.67 | ++ | ++ |
| 17 | 1300 | 1678.35 | 5.90 | +1.29 | + | + |
| 18 | 1400 | 564.18 | 6.37 | -2.48 | + | ++ |
| 19 | 1410 | 1577.48 | 5.93 | +1.12 | + | + |
| 20 | 1800 | 1910.63 | 5.84 | +1.06 | + | + |
| 21 | 2250 | 3092.80 | 5.63 | +1.37 | + | + |
| 22 | 4920 | 5044.11 | 5.42 | +1.03 | + | + |
| 23 | 6000 | 2729.99 | 5.69 | -2.20 | + | + |
| 24 | 10600 | 3309.60 | 5.60 | -3.20 | + | + |
| 25 | 11000 | 6801.74 | 5.30 | -1.62 | + | + |
| 26 | 50000 | 42217.50 | 4.50 | -1.18 | + | + |
| 27 | 50000 | 52984.20 | 4.40 | +1.06 | + | + |
| 28 | 75000 | 41077.10 | 4.51 | -1.83 | + | + |
Fit value represents how well the features in the Hypo1 pharmacophore overlap with the chemical features in the molecule.
Difference between the experimental and predicted activities. “+”: the predicted IC50 is higher than the experimental IC50; “-”: the predicted IC50 is lower than the experimental IC50. A value of 1 indicates that the predicted IC50 is equal to the experimental IC50.
Activity scale: +++ (highly active, IC50 < 100 nM); ++ (moderately active, 100 nM < IC50 < 1000 nM); + (lowly active, IC50 > 1000 nM).
Pharmacophore validation
The Hypo1 pharmacophore model was validated by two different methods, test set validation and Fischer’s randomization method, to assess the predictive power and statistical significance. An independent test set was used to determine whether the Hypo1 hypothesis is capable of predicting the activity values of external test compounds. Twenty-eight test set compounds (Figure 4) with IC50 values spanning 3 nM-4740 nM were applied in this study. The Hypo1 hypothesis showed good predictive ability with a correlation coefficient value of 0.95 (Figure 5A). Overall, 9 of 11 highly active, 10 of 14 moderately active and all lowly active compounds were predicted in the same order of magnitude (Table 3). The two highly active and four moderately active compounds were separately underestimated as moderately active and lowly active compounds. Furthermore, the deviation between the experimental activity and predicted activity of each test compound was less than one order of magnitude. The above results reveal that the Hypo1 model contains the crucial chemical features required by inhibitors to block the activity of Src.
Figure 4.

Chemical structures of the test set compounds used to validate the Hypo1 pharmacophore.
Figure 5.
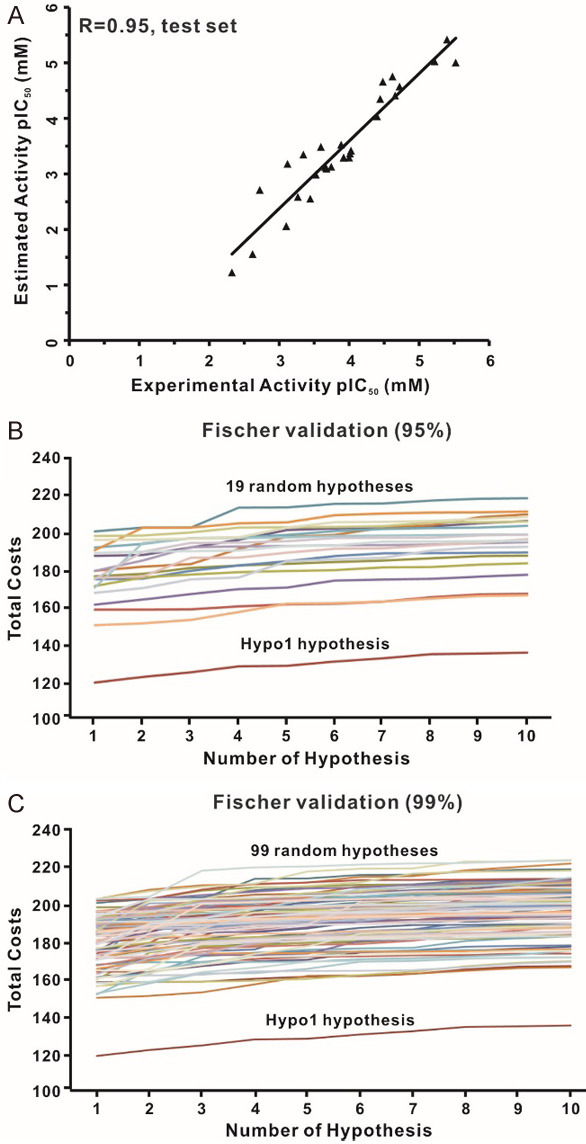
Evaluation of Hyop1 pharmacophore model. (A) Correlation analysis was used to evaluate the predictive power of the Hypo1 model by calculating the correlation between the experimental activity and the estimated activity of the test set compounds (r = 0.95). The differences in total costs between the Hypo1 hypothesis and the random pharmacophore hypotheses were examined by cross-validation. (B) The statistical significance at the 95% confidence level. (C) The statistical significance at the 99% confidence level.
Table 3.
Actual and estimated activities of 28 test set compounds calculated on the basis of the Hypo1 pharmacophore model
| Compound no. | Experimental IC50 nM | Predicted IC50 nM | Fit value* | Error† | Experimental scale‡ | Predicted scale‡ |
|---|---|---|---|---|---|---|
| 1 | 3 | 9.92 | 8.13 | +3.307 | +++ | +++ |
| 2 | 4 | 3.82 | 8.54 | -1.047 | +++ | +++ |
| 3 | 6 | 9.41 | 8.15 | +1.568 | +++ | +++ |
| 4 | 19 | 26.85 | 7.69 | +1.413 | +++ | +++ |
| 5 | 22 | 38.99 | 7.53 | +1.772 | +++ | +++ |
| 6 | 24 | 17.70 | 7.88 | -1.356 | +++ | +++ |
| 7 | 33 | 21.97 | 7.78 | -1.502 | +++ | +++ |
| 8 | 36 | 44.76 | 7.47 | +1.243 | +++ | +++ |
| 9 | 40 | 92.41 | 7.16 | +2.310 | +++ | +++ |
| 10 | 94 | 382.25 | 6.54 | +4.066 | +++ | ++ |
| 11 | 97 | 429.33 | 6.49 | +4.426 | +++ | ++ |
| 12 | 100 | 510.12 | 6.42 | +5.101 | ++ | ++ |
| 13 | 120 | 510.12 | 7.03 | +4.251 | ++ | ++ |
| 14 | 120 | 509.39 | 6.42 | +4.245 | ++ | ++ |
| 15 | 130 | 298.25 | 6.65 | +2.294 | ++ | ++ |
| 16 | 180 | 746.03 | 6.25 | +4.145 | ++ | ++ |
| 17 | 210 | 806.16 | 6.22 | +3.839 | ++ | ++ |
| 18 | 220 | 761.05 | 6.24 | +3.459 | ++ | ++ |
| 19 | 253 | 326.66 | 6.61 | +1.291 | ++ | ++ |
| 20 | 300 | 1034.43 | 6.11 | +3.448 | ++ | + |
| 21 | 360 | 2815.47 | 5.67 | +7.821 | ++ | + |
| 22 | 450 | 449.74 | 6.47 | -1.001 | ++ | ++ |
| 23 | 540 | 2607.24 | 5.71 | +4.828 | ++ | + |
| 24 | 760 | 662.23 | 6.30 | -1.148 | ++ | ++ |
| 25 | 790 | 8713.74 | 5.18 | +11.030 | ++ | + |
| 26 | 1890 | 1948.83 | 5.83 | +1.031 | + | + |
| 27 | 2400 | 27776.30 | 4.68 | +11.573 | + | + |
| 28 | 4740 | 59453.90 | 4.35 | +12.543 | + | + |
Fit value represents how well the features in the Hypo1 pharmacophore overlap with the chemical features in the molecule.
Difference between the experimental and predicted activities. “+”: the predicted IC50 is higher than the experimental IC50; “-”: the predicted IC50 is lower than the experimental IC50. A value of 1 indicates that the predicted IC50 is equal to the experimental IC50.
Activity scale: +++ (highly active, IC50 < 100 nM); ++ (moderately active, 100 nM < IC50 < 1000 nM); + (lowly active, IC50 > 1000 nM).
Moreover, the CatScramble method of DS was used to randomly scramble the activity values of training set molecules and construct new pharmacophore hypotheses using the same chemical features and parameters originated from the Hypo1 hypothesis. A total of 19 and 99 random spreadsheets with randomly shuffled activity values were generated and then used to model pharmacophores at 95% and 99% confidence levels, respectively. The total costs of the random pharmacophore hypotheses were much higher than that of the Hypo1 hypothesis at the two confidence levels (Figure 5B and 5C). The randomization test indicated that the Hypo1 hypothesis was not generated by chance and represents a true correlation in the training set activity data.
Identification of candidate compounds by virtual screening and molecular docking
A total of 46,872 NCI compounds were employed to identify potent Src inhibitors during the virtual screening progress. A total of 38,546 compounds with good druglikeness were selected by considering the drug-like properties and fit on the Hypo1 model for calculation of pharmacophore fit values. To identify more potential inhibitors against Src kinase, several clinically investigated or potential Src inhibitors [26,27,50] were utilized as positive controls to derive the fit values: dasatinib (fit value: 6.25), saracatinib (fit value: 8.16), bosutinib (fit value: 7.41), ponatinib (fit value: 5.96), and imatinib (fit value: 7.52). Only 186 molecules were better fitted to the Hypo1 model when the highest fit value derived from saracatinib was used as a criterion (i.e., fit value > 8.16 as the cut-off point). To avoid the loss of more candidates, we selected a fit value of > 8 as a cut-off point and then chose 339 hit molecules for further analysis (Figure 1).
The hit molecules were subjected to molecular docking studies with two crystallographic structures of the human c-Src kinase domain (PDB ID: 3G5D and 1Y57). Three different scores were calculated: the LibDock docking score, the binding free energy, and the consensus score. The docking scores of each hit compound from either of the best conformations docked on the 3G5D or 1Y57 structures were selected. Then, the 20% top scored candidates were separately chosen based on three individual rules: (1) only the LibDock score was considered in rule 1; (2) the LibDock score and consensus score were considered in rule 2; (3) the three different scores were simultaneously considered in rule 3. Among the candidates, 30 compounds were available from the NCI drug repository, and their inhibitory effects on the Src protein were assessed by further biological assays. The pharmacophore fit values and related docking scores of the available candidates are shown in Table 4.
Table 4.
Pharmacophoric fit values and docking-related scores of 30 available compounds
| Compounds | Fit value | Src Structure | LibDock score | Consensus score | Binding free Energy (kcal/mol) |
|---|---|---|---|---|---|
| Rule 1: top 20% of LibDock score | |||||
| N1 | 8.531 | 3G5D | 151.917 | - | - |
| N2 | 8.461 | 1Y57 | 162.011 | - | - |
| N3 | 8.417 | 3G5D | 135.924 | - | - |
| N4 | 8.372 | 3G5D | 136.926 | - | - |
| N5 | 8.370 | 1Y57 | 152.941 | - | - |
| N6 | 8.120 | 1Y57 | 149.732 | - | - |
| N7 | 8.027 | 1Y57 | 151.610 | - | - |
| N8 | 8.010 | 1Y57 | 150.854 | - | - |
| N9 | 8.003 | 1Y57 | 150.885 | - | - |
| Rule 2: top 20% of LibDock score and consensus score | |||||
| N10 | 8.352 | 1Y57 | 153.470 | 8 | - |
| N11 | 8.338 | 3G5D | 136.506 | 8 | - |
| N12 | 8.256 | 3G5D | 131.119 | 6 | - |
| N13 | 8.249 | 3G5D | 152.550 | 9 | - |
| N14 | 8.225 | 3G5D | 151.310 | 10 | - |
| N15 | 8.107 | 1Y57 | 137.128 | 7 | - |
| N16 | 8.107 | 3G5D | 140.889 | 9 | - |
| N17 | 8.093 | 3G5D | 136.202 | 8 | - |
| N18 | 8.052 | 3G5D | 132.178 | 8 | - |
| N19 | 8.027 | 1Y57 | 139.131 | 10 | - |
| N20 | 8.006 | 1Y57 | 149.570 | 9 | - |
| N21 | 8.001 | 1Y57 | 140.602 | 8 | - |
| Rule 3: top 20% of LibDock score, binding free energy and consensus score | |||||
| N22 | 8.262 | 1Y57 | 134.588 | 6 | -100.370 |
| N23 | 8.257 | 1Y57 | 129.853 | 9 | -93.870 |
| N24 | 8.218 | 1Y57 | 141.000 | 8 | -77.760 |
| N25 | 8.166 | 3G5D | 122.503 | 7 | -61.591 |
| N26 | 8.125 | 1Y57 | 149.207 | 7 | -110.110 |
| N27 | 8.084 | 1Y57 | 136.532 | 6 | -111.970 |
| N28 | 8.080 | 3G5D | 129.406 | 7 | -48.448 |
| N29 | 8.062 | 1Y57 | 155.506 | 9 | -95.640 |
| N30 | 8.012 | 1Y57 | 146.410 | 11 | -104.010 |
Evaluation of the inhibitory effect of candidate compounds on Src phosphorylation
To evaluate the suppressive effect of the 30 NCI compounds on Src-Y419 phosphorylation, an ELISA was performed in the A549 lung cancer cell line. In total, 11 of 30 compounds at a 1 µM concentration showed an inhibitory rate > 50% against Src phosphorylation (Figure S1). Among these compounds, compounds N1 and N25 could inhibit Src activity up to 60% and 63%, respectively, compared with Src activity in the vehicle control. Dasatinib was used as a positive control (inhibitory rate: 74%).
To further confirm the effect of the two compounds on Src activity, A549 and H358 cells were treated with various nonlethal concentrations of N1 and N25 for 24 hours and then subjected to Western blotting. The results indicated that N1 could significantly diminish the phosphorylation of Src-Y418 and Stat3-Y705 in a dose-dependent manner (Figure 6A and 6B). Compound N25 exhibited a similar but smaller effect on the inhibition of pSrc and pStat3 in A549 cells (Figure 6C) compared with N1, while it showed opposite or no effect on pSrc and pStat3 expressions in H358 cells (Figure 6D). However, both N1 and N25 had no significant or less effect on Src expression in A549 and H358 cells. Furthermore, compound N1 showed a better fit to the pharmacophoric features of the Hypo1 model than N25, as indicated by the pharmacophore fit values (fit values = 8.531 and 8.166, respectively; Figure 6E and 6F). Taken together, these results prompted us to choose compound N1, i.e., SJG-136, for further investigation of cellular functions and the involved signaling pathways, especially those related to tumour cell growth and motility.
Figure 6.
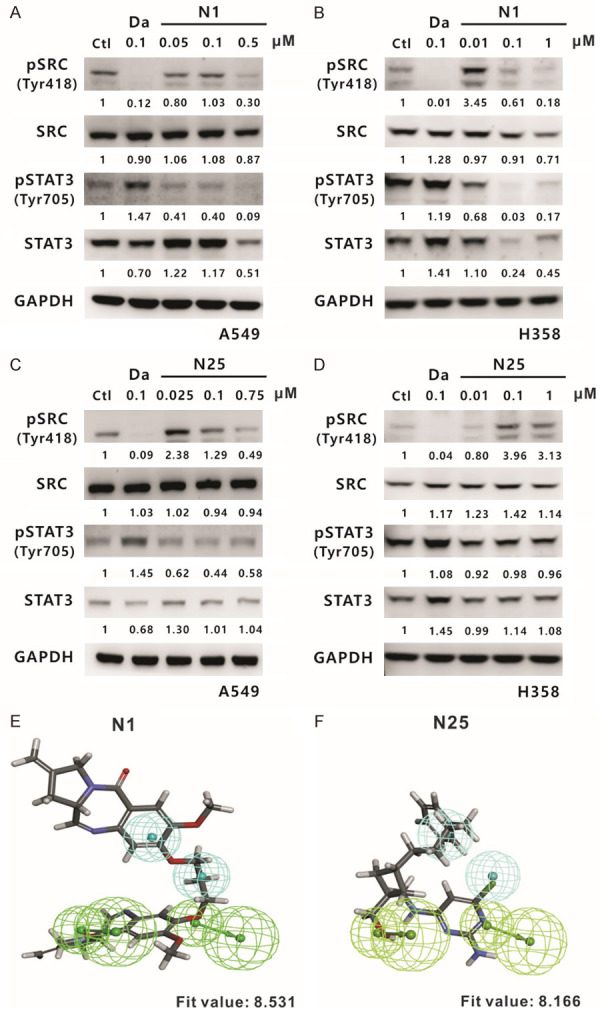
Evaluation of N1 and N25 compounds by investigating their suppressive effect on Src expression. Western blotting of Src and STAT3 was performed in A549 (A) and H358 (B) cells at 24 hours after treatment with compound N1, respectively. The phosphorylated and total protein levels of Src and STAT3 were also analysed in A549 (C) and H358 (D) cells after treatment with compound N25 for 24 hours. Ctl: 0.1% DMSO; Da: dasatinib, a positive control; and GAPDH: an internal control. Compound N1 (fit value = 8.531) (E) and compound N25 (fit value = 8.166) (F) were aligned to the Hypo1 pharmacophore model.
SJG-136 inhibits lung cancer cell growth and motility in vitro, as well as tumourigenesis in vivo
To determine the proper concentration of SJG-136 prior to performing further experiments, we performed a cytotoxicity assay with the A549 and H358 lung cancer cell lines at 24, 48, and 72 hours after exposure to various concentrations of SJG-136. The survival rates of both cell lines decreased in a dose-dependent manner. The IC50 values at three time points in A549 and H358 cells are shown in Figure 7A and 7B. Notably, at 72 hours, the IC50 values were as low as nanomolar scale, as in A549 cells.
Figure 7.
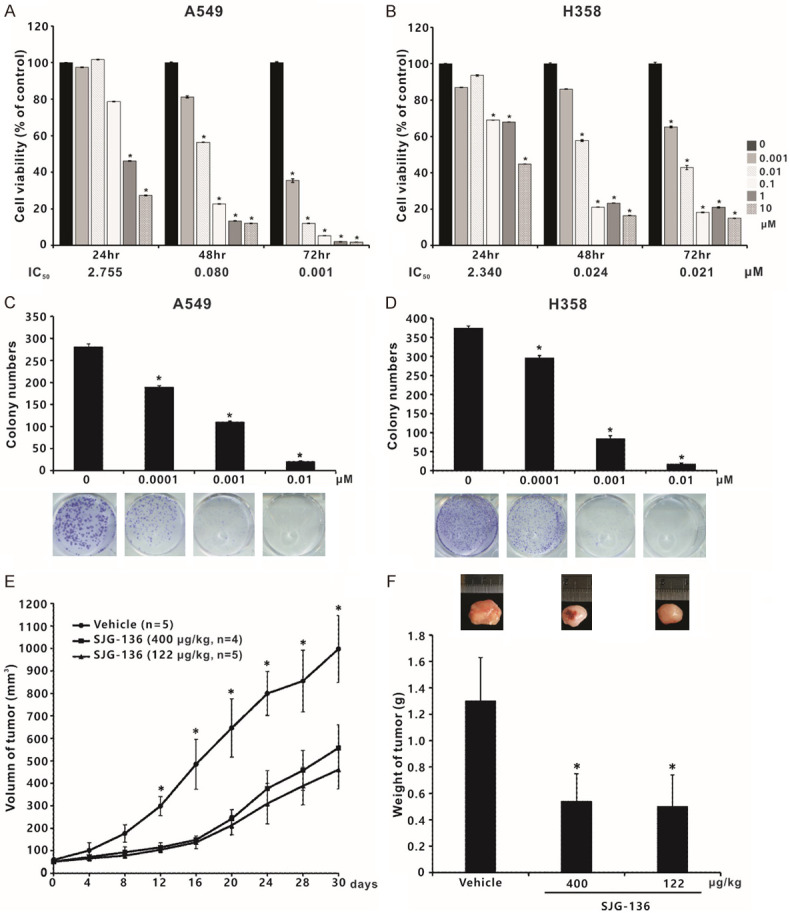
Suppressive effects of SJG-136 (compound N1) on tumour cell viability and growth. The cytotoxicity of SJG-136 was evaluated in A549 (A) and H358 (B) NSCLC cell lines by using a cell viability assay. The results are shown as the percentages of the control response (0 µM). The IC50 values at 72 hours in A549 and H358 cells were 1 and 21 nM, respectively. Colony formation assays in A549 (C) and H358 (D) cells. Cells grown in anchorage-dependent conditions were treated with SJG-136 and evaluated in clonogenic assays. Colonies with diameters ≥ 0.2 mm were counted. Each treatment was independently performed in triplicate; 0 µM indicates 0.1% DMSO. (E) Tumourigenesis assay. The indicated number of live A549 cells was subcutaneously injected into nude mice. The mice were then divided into three groups, including dosing schedule 1 (122 μg/kg once daily for five treatments; n = 5), dosing schedule 2 (400 μg/kg every seventh day for two treatments; n = 4) and vehicle-treated group (n = 5). Tumour volumes were measured every 4 days. (F) SJG-136 decreased tumour weights, which were presented as the mean ± standard deviation. *P < 0.05 compared with vehicle-treated control.
To investigate the anticancer effects of SJG-136, colony formation in vitro and tumour growth in vivo were carried out. The colony formation ability of both A549 and H358 cells was significantly inhibited by SJG-136 (0.0001, 0.001 and 0.01 µM) in a concentration-dependent manner (Figure 7C and 7D). In the in vivo experiments, the mean size of the tumours in the control group was 998 mm3 (95% CI 867-1128 mm3), however, the mean size of the tumours in the schedule 1 and schedule 2 groups were 461 mm3 (95% CI 386-536 mm3) and 558 mm3 (95% CI 457-658 mm3), respectively (Figure 7E). In addition, the mean weight of tumours in the three groups were 1300 mg (control group), 500 mg (schedule 1 group) and 540 mg (schedule 2 group), respectively (Figure 7F). Moreover, SJG-136 also significantly inhibited the ability of migration and invasion in A549 and H358 cancer cells, especially SJG-136 at a concentration of 0.5 µM (Figure 8A and 8B). Taken together, our data indicated that SJG-136 can inhibit not only the tumour growth but also tumour motility and invasion.
Figure 8.
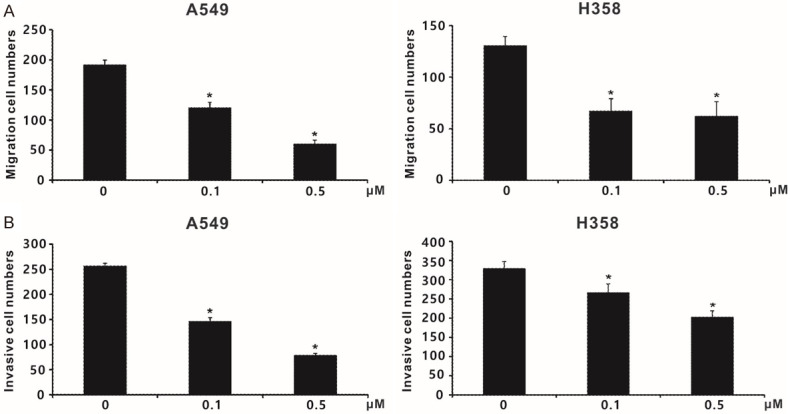
The impact of SJG-136 (compound N1) on lung cancer cell motility and invasion. (A) The effect of SJG-136 on A549 and H358 cell migration, as assessed by using non-coated transwell devices. (B) Inhibitory effect of SJG-136 on cancer cell invasion, as determined by the transwell assay with Matrigel. Each treatment was independently performed in triplicate; 0 µM indicates 0.1% DMSO. *P < 0.05 compared with vehicle-treated control (0 µM, 0.1% DMSO).
Effects of SJG-136 on Src downstream proteins
Src activity can affect the expression and activation of many downstream proteins, such as STAT3, FAK, JNK, paxillin, p130Cas, PI3K, AKT, MEK, and ERK [51]; therefore, we further investigated whether SJG-136 influences any of these proteins. The effects of SJG-136 on Src downstream targets in the H358 and A549 cell lines were detected by Western blotting and used as a demonstration. In H358 cells, the significant inhibitory effects of SJG-136 on the phosphorylation of FAK, paxillin, and p130Cas were increased as the concentration of SJG-136 increased from 0.1 µM to 0.5 µM (Figure 9). Moreover, the phosphorylation levels of PI3K, AKT, and MEK were slightly but still significantly reduced in a dose-dependent manner. In addition, JNK phosphorylation levels were slightly reduced following treatment with 0.5 µM SJG-136. Furthermore, the total protein expression levels of these Src downstream proteins, except paxillin and PI3K, showed no significant change. In A549 cells, the phosphorylation levels of FAK, AKT, PI3K, JNK, paxillin, and p130Cas were significantly reduced in an appropriate dose-dependent response, except phospho-MEK (Figure 10). In addition, the total protein levels of FAK, MEK, paxillin, and p130Cas were slightly or strongly reduced in A549 cells, especially for the treatment of SJG-136 at a concentration of 0.5 µM. However, no significant change in the phosphorylation status of ERK was detected in both cell lines. The inhibitory capacity of SJG-136 on the tested signaling pathways was equal to even better than that of dasatinib. These results suggested that SJG-136 inhibits the expression or activation of downstream proteins to regulate cell growth and motility, especially for the FAK, PI3K, paxillin, and p130Cas signaling pathways.
Figure 9.
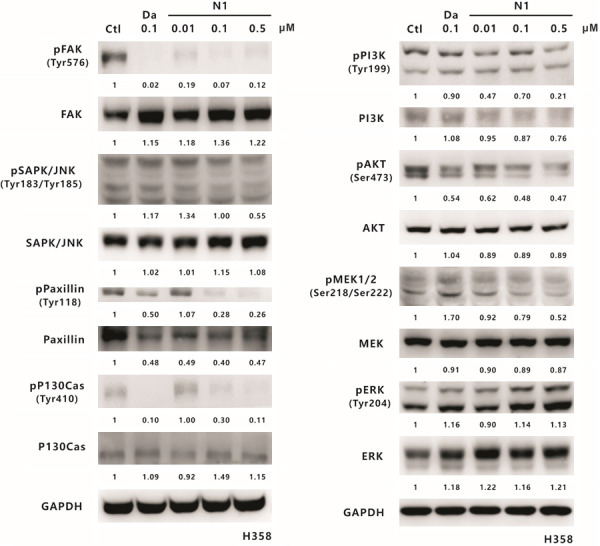
The effect of SJG-136 on Src-associated signaling pathways in H358 cells. Western blotting analyses of Src downstream proteins in the H358 cell line were performed after treatment with 0.01, 0.1 or 0.5 µM SJG-136 for 24 h. The phosphorylated and total protein levels of FAK, JNK, paxillin, p130Cas, PI3K, AKT, MEK1/2, and ERK were detected. GAPDH served as the loading control. Protein expression was quantified by ImageJ software (NIH), and the results are shown directly below the gel graph.
Figure 10.
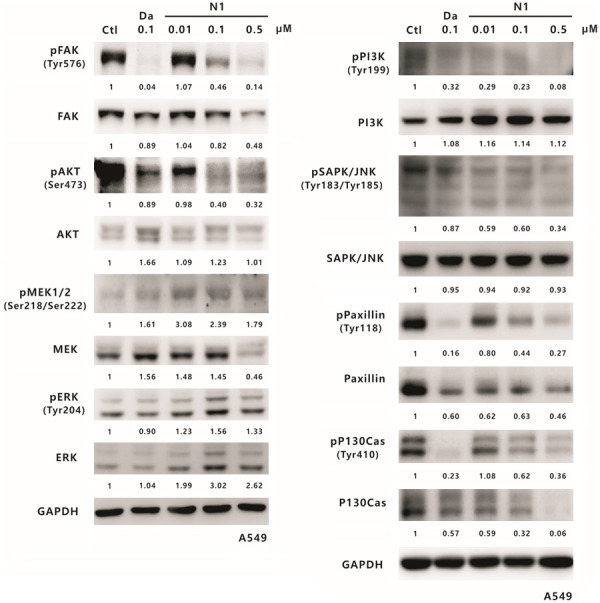
The effect of SJG-136 on Src downstream proteins in A549 cells. Lung cancer A549 cells were treated with SJG-136 at various concentrations such as 0.01, 0.1 or 0.5 µM for 24 h and then analysed on Western blots. The phosphorylation and total protein levels of FAK, AKT, MEK1/2, ERK, PI3K, JNK, paxillin, and p130Cas were detected. GAPDH was the loading control. Protein expression was calculated by using the ImageJ software (NIH), and the results are shown below the gel graph.
Discussion
NSCLC is the most common malignant neoplasm in lung carcinoma and exhibits a high mortality rate [1]. A treatment regimen of EGFR-TKIs, including gefitinib and erlotinib, can be used as the standard first-line treatment for advanced NSCLC with activating EGFR mutations (i.e., exon 19 deletion and the L858R point mutation) [9]. Despite recent successes in targeted therapy and kinase inhibitors, this therapeutic strategy inevitably promotes drug resistance to TKIs. Src, a well-known oncogene, has been implicated in multiple signaling pathways [21] and participates in the tumour progression of various cancers, including lung cancer [23]. Because of the crosstalk between Src and EGFR, the suppression of Src may induce apoptosis in EGFR-mutant cell lines by interrupting the downstream EGFR signaling pathway [52] and even improve the effectiveness of EGFR-TKIs through increasing the expression level of E-cadherin [53]. Therefore, Src can be a suitable target to treat lung cancer [28].
Several previous studies employed computer-aided drug design to develop Src inhibitors [54-57]. In this study, we constructed a 3D-QSAR pharmacophore model and used combined molecular docking to identify new potent scaffolds that could inhibit Src activity. A pharmacophore model is needed to explore the critical chemical features of structurally diverse ligands that can influence Src activity. In this study, 28 training set compounds with IC50 values determined by the Src kinase assay or cell viability assessment were collected from a public database and our unpublished data. Many previous studies indicated that training set compounds should fulfil the necessary criteria to construct a good hypothesis [31,54,58] by (i) covering a wide activity range of four orders of magnitude (4-75000 nM); (ii) binding to the same target in a similar mechanism, such as competitive inhibition; and (iii) adopting homogeneous procedures to measure IC50 values as far as possible. Under these criteria, the 10 top scored pharmacophore hypotheses were generated through pharmacophore modeling. A good pharmacophore hypothesis should follow the criteria of parameters and possess the following [31,54]: (a) the highest correlation coefficient, (b) the lowest RMS value, (c) the lowest total cost, (d) a fixed cost close to the total cost, (e) a configuration cost value less than 17, and (f) a high cost difference (the difference between the null and total cost values). Hence, Hypo1 was selected based on the above rules and evaluated by using standard methods, including Fischer’s randomization and test set methods. Typically, the ligand-receptor interaction depends on three kinds of functional groups: hydrophobic regions, polar positive regions or polar negative regions [59]; therefore, a well-defined pharmacophore model may include both hydrophobic volumes and hydrogen bond vectors. Two HBA and two HY chemical groups are included in the Hypo1 model, which is evidence to support the above contention.
The Hypo1 model was used to search the NCI drug database, and the hits were filtered by druglikeness rules and molecular docking to refine the retrieved hits. A total of 339 hits were screened to perform the docking studies. Structures of active (PDB ID: 3G5D; co-crystallized with dasatinib) and inactive (PDB ID: 1Y57; co-crystallized with imatinib) c-Src kinase domain were chosen, and compounds were docked against their ATP-binding sites. Dasatinib is an FDA-approved Src-Abl inhibitor for use in patients with chronic myelogenous leukaemia (CML) or Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukaemia (ALL) [27]. Imatinib is also an approved medication against CML [60] and Ph+ ALL [61]. Some efforts are currently focused on discovering the structural characteristics of imatinib that allow it to bind to human c-Src kinase [50,62]. In our study, superimposition of the two structures on the basis of their backbone atoms was performed, which yielded a root-mean-square deviation of 1.56 Å, suggesting a slight difference between the ATP-binding sites. To identify credible binding poses, the best docked conformations of each hit compound with either the 3G5D or 1Y57 structure during the docking progress were selected. Finally, 30 compounds were selected and obtained from NCI to investigate their suppressive effect on Src phosphorylation at Y418 and Y419 residues which associated with increased Src activity [15,16].
To demonstrate the feasibility and availability of the identified pharmacophore model, the two most effective compounds in ELISAs, N1 and N25, were selected. Subsequent Western blotting and fit value analysis indicated that SJG-136 is more effective than N25. Thus, we identified SJG-136 as a promising candidate compound for further investigation of functionality as an antitumour drug. SJG-136 (NSC 694501) is a pyrrolobenzodiazepine (PBD) dimer that was synthesized by joining two DC-81 monomer PBDs via their aromatic A-ring phenol C8-positions in the early 1990s [63]. PBDs are a class of sequence-selective DNA cross-linking agents based on the naturally occurring anthramycin family of antitumour antibiotics produced by Streptomyces species [64,65]. Previous studies indicated that the mechanism of SJG-136 biological activity might be through binding to six base pairs (5’-purine-GATC-pyrimidine-3’) in the minor groove of double-stranded DNA and producing covalently binding to induce the formation of interstrand cross-links [66,67]. As a result, SJG-136 exhibited potent in vitro activity and broad spectrum in vivo efficacy against various cancers [48,68], including ovarian carcinoma, colon carcinoma, melanoma, breast carcinoma, and NSCLC. However, the functionality underlying SJG-136 antitumour activity against lung cancer is largely unknown. In this study, we found that the IC50 value of SJG-136 against A549 lung cancer cells was 1 nM at 72 hours; however, a lower cytotoxic effect was observed in H358 cancer cells with an IC50 of 21 nM. A previous study of the in vitro antitumour activity of SJG-136 on NCI 60 cell lines indicated that the GI50 (i.e., 50% growth inhibitory concentration) value for SJG-136 in A549 lung cancer cells was approximately 14 nM after 48-hour exposure to SJG-136 [68]. Furthermore, another report showed that the IC50 values of SJG-136 following the exposure of five cell lines, including melanoma, prostate and ovarian cancer cells, for 96 hour ranged from 0.4 to 2.5 nM [69]. Our results indicated that the cytotoxic concentration of SJG-136 in both lung cancer lines tested is in the nanomolar range, which is also consistent with previous reports. A549 cells were also shown to be more sensitive to SJG-136 than H358 cells. This difference might be due to differences in the genetic backgrounds of the cell lines, such as the p53 status, as the former contains wild-type p53, while the latter contains null-type p53. Moreover, we also demonstrated that SJG-136 can suppress lung cancer cell clonogenicity, migration, and invasiveness in the in vitro experiments. A prior study of the NCI-H522 human lung cancer xenograft model showed that SJG-136 has a significant antitumour effect at two dosing schedules such as 122 μg/kg once daily for five treatments and 400 μg/kg every seventh day for three treatments [48]. In our study, the significant antitumour capacity of SJG-136 was also demonstrated in A549 human lung cancer xenografts of nude mice by adopting the similar dosing schedules.
In the past, research on the mechanism of SJG-136 or its analogues and conjugates has mainly focused on their ability to serve as a DNA-targeting agent that subsequently blocks the transcription of certain genes [70]. For example, the conjugated modification of SJG-136 with 4-(1-methyl-1H-pyrrol-3-yl) benzenamine (i.e., PBD-MPB hybrids) might inhibit the transcription factor NF-κB or its transcriptional activity [71]. Nevertheless, little is known about the signaling pathways involved in the action of SJG-136. It is well-known that Src can modulate the activities of STAT3, FAK, JNK, PI3K, AKT, paxillin, p130Cas, MEK, and ERK, which are widely considered to be essential for cancer cell growth, angiogenesis, migration, and invasion [22]. In this report, we found that SJG-136 inhibits the phosphorylation of Src and its downstream signaling pathways, except for ERK activity, in the tested lung cancer cell lines, which to the best of our knowledge has not been previously reported. Therefore, we speculated that SJG-136 or its associated derivatives have the potential to act as a single- or multi-target drug based on the above-mentioned reports and our data.
In conclusion, we constructed a pharmacophore-based virtual screening workflow that can accurately identify features of Src inhibitors and discovered that SJG-136 may be a potential Src-targeted inhibitor. The in vitro experiments provided strong evidence that SJG-136 can inhibit lung cancer cell viability, proliferation, invasion and migration by suppressing Src-related signaling pathways. Moreover, the tumourigenesis experiments in nude mice also revealed that SJG-136 can inhibit lung tumour growth in vivo. However, the issue of kinase inhibitor selectivity still cannot be excluded. For example, a previous study has revealed that two marketed drugs, sunitinib and dasatinib, have high affinities for multiple kinases [72]. Taken together, the findings of this study may be useful for the development of a novel compound for lung cancer therapeutics.
Acknowledgements
This study was supported by grants from the Ministry of Science and Technology, Taiwan, R.O.C. (MOST 103-2314-B-005-001-MY3 and MOST 107-2314-B-005-003-MY3) and Taichung Veterans General Hospital and National Chung-Hsing University (TCVGH-NCHU-1087609). The authors also acknowledge to the NCI Developmental Therapeutics Program as the source of the data (http://dtp.cancer.gov) and providing the tested compounds for this study.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Wood SL, Pernemalm M, Crosbie PA, Whetton AD. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat Rev. 2014;40:558–566. doi: 10.1016/j.ctrv.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 3.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728:23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsen JE, Minna JD. Molecular biology of lung cancer: clinical implications. Clin Chest Med. 2011;32:703–740. doi: 10.1016/j.ccm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen J, Fu J, Zhang W, Guo M. Genetic and epigenetic changes in lung carcinoma and their clinical implications. Mod Pathol. 2011;24:932–943. doi: 10.1038/modpathol.2011.46. [DOI] [PubMed] [Google Scholar]
- 6.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pao W. New approaches to targeted therapy in lung cancer. Proc Am Thorac Soc. 2012;9:72–73. doi: 10.1513/pats.201112-054MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 10.Giuliani J, Martelli S, Remo A, Bonetti A. Primary TKI resistance in advanced non-small cell lung cancer with EGFR mutation: an open question. Tumori. 2015;101:e115–117. doi: 10.5301/tj.5000317. [DOI] [PubMed] [Google Scholar]
- 11.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct. 2011;2011:865819. doi: 10.1155/2011/865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopetz S. Targeting SRC and epidermal growth factor receptor in colorectal cancer: rationale and progress into the clinic. Gastrointest Cancer Res. 2007;1(Suppl 2):S37–41. [PMC free article] [PubMed] [Google Scholar]
- 14.Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 15.Roskoski R Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Sato K. Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int J Mol Sci. 2013;14:10761–10790. doi: 10.3390/ijms140610761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–998. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 18.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 19.Niit M, Hoskin V, Carefoot E, Geletu M, Arulanandam R, Elliott B, Raptis L. Cell-cell and cell-matrix adhesion in survival and metastasis: Stat3 versus Akt. Biomol Concepts. 2015;6:383–399. doi: 10.1515/bmc-2015-0022. [DOI] [PubMed] [Google Scholar]
- 20.Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courtneidge SA. Role of Src in signal transduction pathways. The Jubilee Lecture. Biochem Soc Trans. 2002;30:11–17. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 22.Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–678. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, Ma L, Wistuba II, Johnson FM, Kurie JM. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170:366–376. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masaki T, Igarashi K, Tokuda M, Yukimasa S, Han F, Jin YJ, Li JQ, Yoneyama H, Uchida N, Fujita J, Yoshiji H, Watanabe S, Kurokohchi K, Kuriyama S. pp60c-src activation in lung adenocarcinoma. Eur J Cancer. 2003;39:1447–1455. doi: 10.1016/s0959-8049(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 25.Mazurenko NN, Kogan EA, Zborovskaya IB, Kisseljov FL. Expression of pp60c-src in human small cell and non-small cell lung carcinomas. Eur J Cancer. 1992;28:372–377. doi: 10.1016/s0959-8049(05)80056-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci. 2012;33:122–128. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist. 2011;16:566–578. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung EL, Tam IY, Tin VP, Chua DT, Sihoe AD, Cheng LC, Ho JC, Chung LP, Wong MP. SRC promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular-targeted therapy. Mol Cancer Res. 2009;7:923–932. doi: 10.1158/1541-7786.MCR-09-0003. [DOI] [PubMed] [Google Scholar]
- 29.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 31.Li H, Sutter J, Hoffmann R. HypoGen: an automated system for generating 3D predictive pharmacophore models. In: Güner F, editor. Pharmacophore perception, development and use in drug design. 2000. pp. 171–189. [Google Scholar]
- 32.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 33.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 34.Rao SN, Head MS, Kulkarni A, LaLonde JM. Validation studies of the site-directed docking program LibDock. J Chem Inf Model. 2007;47:2159–2171. doi: 10.1021/ci6004299. [DOI] [PubMed] [Google Scholar]
- 35.Getlik M, Grutter C, Simard JR, Kluter S, Rabiller M, Rode HB, Robubi A, Rauh D. Hybrid compound design to overcome the gatekeeper T338M mutation in cSrc. J Med Chem. 2009;52:3915–3926. doi: 10.1021/jm9002928. [DOI] [PubMed] [Google Scholar]
- 36.Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13:861–871. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 37.Tirado-Rives J, Jorgensen WL. Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J Med Chem. 2006;49:5880–5884. [Google Scholar]
- 38.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 39.Muegge I. PMF scoring revisited. J Med Chem. 2006;49:5895–5902. [Google Scholar]
- 40.Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, Freer ST. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol. 1995;2:317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 41.Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. LigScore: a novel scoring function for predicting binding affinities. J Mol Graph Model. 2005;23:395–407. doi: 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 43.Bohm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 44.Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 45.Wang CC, Lin SY, Lai YH, Liu YJ, Hsu YL, Chen JJ. Dimethyl sulfoxide promotes the multiple functions of the tumor suppressor HLJ1 through activator protein-1 activation in NSCLC cells. PLoS One. 2012;7:e33772. doi: 10.1371/journal.pone.0033772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai YH, Yu SL, Chen HY, Wang CC, Chen HW, Chen JJ. The HLJ1-targeting drug screening identified Chinese herb andrographolide that can suppress tumour growth and invasion in non-small-cell lung cancer. Carcinogenesis. 2013;34:1069–1080. doi: 10.1093/carcin/bgt005. [DOI] [PubMed] [Google Scholar]
- 47.Lai YH, Lin SY, Wu YS, Chen HW, Chen JJW. AC-93253 iodide, a novel Src inhibitor, suppresses NSCLC progression by modulating multiple Src-related signaling pathways. J Hematol Oncol. 2017;10:172. doi: 10.1186/s13045-017-0539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alley MC, Hollingshead MG, Pacula-Cox CM, Waud WR, Hartley JA, Howard PW, Gregson SJ, Thurston DE, Sausville EA. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: part 2: efficacy evaluations. Cancer Res. 2004;64:6700–6706. doi: 10.1158/0008-5472.CAN-03-2942. [DOI] [PubMed] [Google Scholar]
- 49.Conway TF Jr, Sabel MS, Sugano M, Frelinger JG, Egilmez NK, Chen F, Bankert RB. Growth of human tumor xenografts in SCID mice quantified using an immunoassay for tumor marker protein in serum. J Immunol Methods. 2000;233:57–65. doi: 10.1016/s0022-1759(99)00190-8. [DOI] [PubMed] [Google Scholar]
- 50.Tsutsui Y, Deredge D, Wintrode PL, Hays FA. Imatinib binding to human c-Src is coupled to inter-domain allostery and suggests a novel kinase inhibition strategy. Sci Rep. 2016;6:30832. doi: 10.1038/srep30832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 52.Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006;66:5542–5548. doi: 10.1158/0008-5472.CAN-05-4620. [DOI] [PubMed] [Google Scholar]
- 53.Coldren CD, Helfrich BA, Witta SE, Sugita M, Lapadat R, Zeng C, Baron A, Franklin WA, Hirsch FR, Geraci MW, Bunn PA Jr. Baseline gene expression predicts sensitivity to gefitinib in non-small cell lung cancer cell lines. Mol Cancer Res. 2006;4:521–528. doi: 10.1158/1541-7786.MCR-06-0095. [DOI] [PubMed] [Google Scholar]
- 54.Sakkiah S, Arullaperumal V, Hwang S, Lee KW. Ligand-based pharmacophore modeling and Bayesian approaches to identify c-Src inhibitors. J Enzyme Inhib Med Chem. 2014;29:69–80. doi: 10.3109/14756366.2012.753881. [DOI] [PubMed] [Google Scholar]
- 55.Raj K, Rekha R, Pathak L, Muttineni R, Ramachandran D. Using pharmacophore and docking models to gain insight into structural binding and virtual screening: an application study with c-Src kinase. International Journal of Environmental Science and Development. 2010;1:40–46. [Google Scholar]
- 56.Tintori C, Magnani M, Schenone S, Botta M. Docking, 3D-QSAR studies and in silico ADME prediction on c-Src tyrosine kinase inhibitors. Eur J Med Chem. 2009;44:990–1000. doi: 10.1016/j.ejmech.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 57.Manetti F, Locatelli GA, Maga G, Schenone S, Modugno M, Forli S, Corelli F, Botta M. A combination of docking/dynamics simulations and pharmacophoric modeling to discover new dual c-Src/Abl kinase inhibitors. J Med Chem. 2006;49:3278–3286. doi: 10.1021/jm060236z. [DOI] [PubMed] [Google Scholar]
- 58.Qing XY, Lee S, De Raeymaecker J, Tame J, Zhang K, De Maeyer M, Voet A. Pharmacophore modeling: advances, limitations, and current utility in drug discovery. 2014 [Google Scholar]
- 59.Scholz SW, Mhyre T, Ressom H, Shah S, Federoff HJ. Genomics and bioinformatics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009449. doi: 10.1101/cshperspect.a009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fischer T, O’Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 61.Hoelzer D, Gokbuget N, Ottmann OG. Targeted therapies in the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Semin Hematol. 2002;39:32–37. doi: 10.1053/shem.2002.36927. [DOI] [PubMed] [Google Scholar]
- 62.Dar AC, Lopez MS, Shokat KM. Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem Biol. 2008;15:1015–1022. doi: 10.1016/j.chembiol.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bose DS, Thompson AS, Ching J, Hartley JA, Berardini MD, Jenkins TC, Neidle S, Hurley LH, Thurston DE. Rational design of a highly efficient irreversible DNA interstrand cross-linking agent based on the pyrrolobenzodiazepine ring system. J Am Chem Soc. 1992;114:4939–4941. [Google Scholar]
- 64.Leimgruber W, Stefanović V, Schenker F, Karr A, Berger J. Isolation and characterization of anthramycin, a new antitumor antibiotic. J Am Chem Soc. 1965;87:5791–5793. doi: 10.1021/ja00952a050. [DOI] [PubMed] [Google Scholar]
- 65.Thurston DE. Advances in the Study of Pyrrolo[2,1-c] [1,4] benzodiazepine (PBD) Antitumour Antibiotics. In: Neidle S, Waring MJ, editors. Molecular Aspects of Anticancer Drug-DNA Interactions. London: Macmillan Education UK; 1993. pp. 54–88. [Google Scholar]
- 66.Jenkins TC, Hurley LH, Neidle S, Thurston DE. Structure of a covalent DNA minor groove adduct with a pyrrolobenzodiazepine dimer: evidence for sequence-specific interstrand cross-linking. J Med Chem. 1994;37:4529–4537. doi: 10.1021/jm00052a012. [DOI] [PubMed] [Google Scholar]
- 67.Smellie M, Bose DS, Thompson AS, Jenkins TC, Hartley JA, Thurston DE. Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers. Biochemistry. 2003;42:8232–8239. doi: 10.1021/bi034313t. [DOI] [PubMed] [Google Scholar]
- 68.Hartley JA, Spanswick VJ, Brooks N, Clingen PH, McHugh PJ, Hochhauser D, Pedley RB, Kelland LR, Alley MC, Schultz R, Hollingshead MG, Schweikart KM, Tomaszewski JE, Sausville EA, Gregson SJ, Howard PW, Thurston DE. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: part 1: cellular pharmacology, in vitro and initial in vivo antitumor activity. Cancer Res. 2004;64:6693–6699. doi: 10.1158/0008-5472.CAN-03-2941. [DOI] [PubMed] [Google Scholar]
- 69.Wilkinson GP, Taylor JP, Shnyder S, Cooper P, Howard PW, Thurston DE, Jenkins TC, Loadman PM. Preliminary pharmacokinetic and bioanalytical studies of SJG-136 (NSC 694501), a sequence-selective pyrrolobenzodiazepine dimer DNA-cross-linking agent. Invest New Drugs. 2004;22:231–240. doi: 10.1023/B:DRUG.0000026249.97007.60. [DOI] [PubMed] [Google Scholar]
- 70.Mantaj J, Jackson PJ, Rahman KM, Thurston DE. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody-drug conjugates (ADCs) Angew Chem Int Ed Engl. 2017;56:462–488. doi: 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rahman KM, Jackson PJ, James CH, Basu BP, Hartley JA, de la Fuente M, Schatzlein A, Robson M, Pedley RB, Pepper C, Fox KR, Howard PW, Thurston DE. GC-targeted C8-linked pyrrolobenzodiazepine-biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J Med Chem. 2013;56:2911–2935. doi: 10.1021/jm301882a. [DOI] [PubMed] [Google Scholar]
- 72.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.