

Abstract
Amyloid formation of full-length TTR involves dissociation of the native tetramers into misfolded monomers that self-assemble into amyloid. In addition to the full-length TTR, C-terminal fragments including residue 49 – 127 were also observed in vivo, implying the presence of additional misfolding pathways. It was previously proposed that a proteolytic cleavage might lead to the formation of the C-terminal fragment TTR amyloid. Here, we report mechanistic studies of misfolding and aggregation of a TTR variant (G53A) in the absence and presence of a serine protease. A proteolytic cleavage of G53A in the CD loop (K48-T49) with agitation promoted TTR misfolding and aggregation, suggesting that the proteolytic cleavage may lead to the aggregation of the C-terminal fragment (residue 49 – 127). In order to gain more detailed insights into TTR misfolding promoted by proteolytic cleavage, we investigated structural changes of G53A TTR in the presence and absence of trypsin. Our combined biophysical analyses revealed that the proteolytic cleavage accelerated the formation of spherical small oligomers, which exhibited cytotoxic activities. However, the truncated TTR appeared to maintain native-like structures, rather than the C-terminal fragment (49 – 127) being released and unfolded from the native state. In addition, our solid-state NMR and FT-IR structural studies showed that the two aggregates derived from the full-length and cleaved TTR exhibited nearly identical molecular structural features, suggesting that the proteolytic cleavage in the CD loop destabilizes the native tetrameric structure and accelerates oligomer formation through a common TTR misfolding and aggregation mechanism rather than through a distinct molecular mechanism.
Keywords: TTR, Amyloid, Solid-state NMR, oligomer, misfolding, G53A
Graphical Abstract
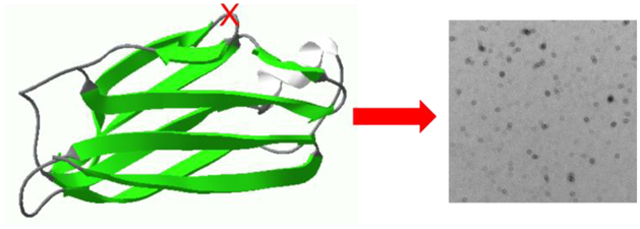
Transthyretin (TTR) is a 55 kDa homo-tetrameric transporter protein that carries the retinol binding protein and thyroxine (T4) in the plasma and cerebrospinal fluid.1, 2, 3, 4 Each monomer consisting of 127 residues adopts a β-sandwich structure formed by two four-β-stranded anti-parallel sheets (CBEF and DAGH) (Fig. 1).2, 5, 6, 7 The β-sheet rich monomers form a dimer through anti-parallel H-H’ and F-F’ interactions, and the dimers are then assembled into a tetramer through interactions between AB and GH loops
Figure 1.

Crystal structure of native tetrameric TTR with the monomeric form of TTR. The arrow indicates the K48 – T49 peptide bond in the CD loop. A pdb code of 1F416 was used to draw the crystal structure.
Misfolding and aggregation of transthyretin (TTR) is associated with several human amyloid diseases (ATTR amyloidosis) including cardiomyopathy and polyneuropathy.8, 9, 10, 11 ATTR amyloidosis is characterized by a wide range of phenotypes. For example, deposition of wild-type TTR is implicated in a late onset senile systemic amyloidosis (cardiac disorder), which affects nearly 25% of the population over age 80.12, 13, 14, 15 More than 100 single-point mutations have been identified in the 127-residue protein, and most of the TTR variants were shown to accelerate misfolding and aggregation, leading to earlier onset of various amyloidosis such as familial amyloidotic polyneuropathy, familial amyloid cardiomyopathy, and rarely central nervous system selective amyloidosis.15, 16, 17, 18
Previous biochemical analyses of TTR misfolding and aggregation process revealed that the full-length native tetrameric TTR is dissociated into monomers, which undergo conformational transition to aggregation-prone monomers.19, 20, 21, 22, 23, 24, 25 The amyloidogenic monomers form oligomeric intermediate states that self-assemble into bigger aggregates.26 In addition to the full-length TTR amyloid, C-terminal fragments were detected in TTR amyloids in vivo,14, 27, 28 implying the presence of additional misfolding and aggregation pathway. A major component of the TTR fragments is the residue 49 – 127 C-terminal fragment,29, 30 indicating that a proteolytic cleavage of the peptide bond K48 – T49 in the CD loop might lead to the aggregation of the C-terminal fragment. Indeed, trypsin digestion of the K48 – T49 peptide bond in TTR variants (S52P and E51_S52dup) was demonstrated to accelerate the formation of TTR amyloids containing the residue 49 – 127 C-terminal fragment.31, 32 It was also shown that increased sheer stress produced by excessive stirring (900 rpm) facilitated the proteolytic cleavages of other TTR variants (V30M and L55P), triggering misfolding and aggregation of the TTR variants.33 The sheer force generated by stirring is equivalent to that in the heart in vivo. In addition, a ubiquitous enzyme, plasmin, was able to cleave the K48 – T49 peptide bond,34 suggesting the proteolytic cleavage and subsequent aggregation may take place in vivo. However, detailed molecular mechanism of the aggregation induced by the cleavage remains elusive. The cleaved C-terminal fragment (49 – 127) that might be released from the folded native TTR may undergo misfolding transition. The misfolded fragments may then form distinct TTR aggregates with different molecular conformations from those of the full-length TTR. Indeed, the recombinant C-terminal fragment (49 – 127) was shown to readily form non-fibrillar aggregates at the physiological pH.35 On the other hand, the proteolytic cleavage may just destabilize native tetrameric structures, accelerating dissociation of the tetramers. The dissociated monomers with the cleaved C-terminal fragment (49 – 127) may self-assemble into amyloid, like full-length TTR. In this study, we investigated misfolding and aggregation of a TTR variant (G53A) in the presence and absence of trypsin to elucidate aggregation pathways of the full-length and C-terminal fragment (49 – 127) TTR with a combined use of various biophysical techniques. Circular dichroism (CD), dynamic light scattering (DLS), and transmission electron microscopy (TEM) were used to examine an early stage of aggregation. Solid-state NMR and FT-IR spectroscopy were employed to compare structural features of G53A TTR aggregates formed in the presence and absence of trypsin. Our comprehensive biophysical studies suggest that the proteolytic cleavage of the K48 – T49 peptide bond in the CD loop accelerates the formation of cytotoxic oligomers. The G53A TTR aggregates formed in the presence and absence of trypsin, however, exhibited similar molecular structures, indicating that the cleaved G53A TTR variant forms aggregates through a common TTR misfolding and aggregation pathway.
Materials and Methods
Protein expression and purification:
The recombinant human G53A TTR was produced using BL21(DE3) expression system using the pMMHa plasmid as described previously.20 Briefly, an isolated colony from the LB agar plate was inoculated into five milliliters of LB media and the small culture was added to the larger culture (0.5 L) after 4 hours of incubation. Protein expression was induced at an OD600 of 0.6-0.7 with 1 mM IPTG and the bacteria cells were grown overnight (12-14 hrs) at 25 °C. The cells were harvested by centrifugation at 8000 rpm at 4 °C and the bacterial pellet was stored at −80 °C for at least 1 hr. The pellet was then resuspended in lysis buffer (10 mM Tris buffer, 150 mM NaCl, pH 8) and sonicated to disrupt the cells. After the sonication, supernatant was collected by centrifugation and ammonium sulphate was added (up to 50 %) to precipitate out other cellular proteins. The precipitates were removed by centrifugation, and the supernatant was dialyzed against 20 mM Tris buffer (1 mM EDTA and 1mM PMSF, pH 8) overnight at 4 °C. The G53A TTR was further purified with HiTrap Q HP column (GE Healthcare) followed by size exclusion gel chromatography using HiLoad 16/60 Superdex 200 column (GE Healthcare). The protein concentration was calculated using an extinction coefficient of 7.76 × 104 M−1 cm−1 at 280 nm.
ThT fluorescence:
Wild-type and mutant forms of TTR (0.3 and 0.7 mg/ml) were incubated in duplicates in 10 mM phosphate buffer with 10 μM thioflavin T (ThT). A 200 μL of each sample was added to a 96 well black clear bottom microplate in the absence and presence of trypsin. The microplate was incubated at 37 °C under constant agitation at 250 rpm in an orbital shaker. Using a SpectraMax® microplate reader, ThT fluorescence emission was monitored every 30 min at 482 nm with excitation of 440 nm.
Aggregation Kinetics:
In addition to the ThT fluorescence, aggregation kinetics of the TTR variants (0.25 – 0.7 mg/ml, pH 7.4) in the presence and absence of trypsin were monitored by measuring optical density at 400 nm using 1 mm quartz cuvette. Dynamic light scattering (DLS, DynaPro Nano Star) was also used to probe oligomerization of the TTR variant (G53A) at an early stage of aggregation. An average of 10 scans was recorded with an acquisition time of 3 sec for each scan. The kinetic experiments were carried out in duplicate using two different protein concentrations.
SDS-PAGE:
G53A TTR incubated at 0.7 mg/mL in the presence and absence of trypsin was analyzed by SDS-PAGE. Twenty microliters of the reaction mixture was removed from the TTR samples at different incubation times. For the G53A TTR samples incubated in the presence of trypsin, the digestion reaction was quenched by adding phenylmethylsulfonyl fluoride (PMSF) to a final concentration of 1.5 mM. For the SDS-PAGE analyses, the TTR samples collected at different incubation times were denatured by heating at 70 °C in the presence of SDS sample buffer for 10 min. The denatured TTR samples were saved at −80 °C until they were run on 4-12 % gradient bis tris SDS-PAGE gel using MES running buffer and the gel was stained with Coomassie blue.
MTT assay:
Human neuroblastoma SH-SY5Y cells were used to measure cytotoxic activities of G53A TTR incubated in the presence and absence of trypsin at 37 °C. The mammalian cells were grown in 1:1 mixture of DMEM/F12 medium with 10 % FBS and 1 % Pen-Strep at 37 °C in 5 % CO2. Every 4-5 days the growth medium was renewed.
To measure the cell viability, about 10,000 cell/well were plated in 96 well black clear bottom plate. After 24 hrs of incubation, the cell medium was replaced by fresh medium and the cells were treated with G53A TTR samples (0.25 and 0.5 mg/mL) incubated in the presence and absence of trypsin. After 48 hrs of the treatment, cell medium was replaced by the fresh medium. The cells were then treated with MTT and incubated for 4 hrs. SDS was added to the wells to dissolve formazan and optical density at 570 nm was measured using a SpectraMax® microplate reader. The cell viability (%) was calculated as (ODsample - ODblank) * 100/ (ODcell medium alone - ODblank). The experiments were carried out in duplicate using two different protein concentrations.
Mass spectrometry:
G53A TTR was incubated in the presence of trypsin at 0.7 mg/mL at 37°C under constant agitation at 250 rpm. After 8 hrs of incubation, the reaction mixture was desalted using Millipore C18 Zip-Tips. One microliter of desalted sample was mixed with one microliter of matrix (sinapinic acid) and the mixture was loaded onto the MALDI plate. The MALDI mass spectrum was obtained using Bruker Autoflex Speed MALDI-TOF mass spectrometry.
Transmission Electron Microscopy:
G53A TTR (0.5 mg/mL) was incubated in the presence and absence of trypsin at 37 °C under constant agitation at 250 rpm. To examine the oligomer morphologies, G53A TTR samples were diluted by 20 times with 10 mM phosphate buffer and five microliters of the sample was placed on the formvar/carbon coated copper 400 mesh grid. After 30 sec, excess solution was blotted off with filter paper and the grid was stained with 10 μL of 0.2 % uranyl acetate buffer for 30 sec. Excess stain was blotted off by the filter paper and the grids were air-dried. TEM images of the samples were acquired using Philips CM12 Transmission Electron Microscope at 80 kV.
CD Spectroscopy:
CD spectra were recorded using Jasco J-815 CD spectrometer at room temperature using 1 mm quartz cuvette. Protein samples were incubated (in the presence and absence of trypsin) at the concentration of 0.5 mg/mL at 37 °C under constant agitation at 250 rpm. At every 10 min interval, 10 μL of the incubated sample was collected and diluted to 0.25 mg/mL with 10 mM phosphate buffer (pH 7.4) before the data collection. Each CD spectrum was recorded with an average of 10 scans. Protein secondary structure analysis was carried out using the CDSSTR secondary structure analysis program on DICHROWEB36, 37.
FT-IR spectroscopy:
G53A TTR was incubated at 0.7 mg/mL in the presence and absence of trypsin for one week and two weeks, respectively. Amyloid precipitates from the TTR samples were collected by centrifugation. The precipitates were then washed with water to remove any soluble proteins. The protein samples were placed on the ATR crystal and air-dried for 30 min. A background scan was performed before the data collection and the IR spectra for the protein precipitates were recorded using Nicolet iS50 ATR.
Solid-state NMR:
Solid-state NMR spectra were recorded using Bruker 800 MHz spectrometers equipped with a 3.2 mm MAS probe. Two-dimensional 13C-13C correlation NMR spectra were recorded using a dipolar-assisted rotational resonance (DARR)38 mixing scheme at a spinning frequency of 12.5 kHz. The 90° pulse-lengths for 1H and 13C were 3.0 and 3.5 μs, respectively, and the two-pulse phase-modulated (TPPM)39 decoupling scheme was employed with a radio-frequency field strength of 90 kHz. For the 2D DARR spectra, complex data points of 2048 × 512 were collected for the amyloid states with an acquisition delay of 2 s, and 64 FIDs were accumulated for each t1 data point.
Preparation of TTR aggregates for solid-state NMR:
The TTR variant (0.7 mg/ml) was incubated at 37 °C in 10 mM phosphate buffer (pH 7.4) under constant agitation at 250 rpm in the presence and absence of trypsin to prepare proteolyzed and full-length TTR amyloids, respectively. To obtain full-length G53A TTR amyloid, the protein sample was incubated at the above condition in the absence of trypsin for two weeks and the amyloid was collected via centrifugation. For the proteolyzed TTR amyloid, G53A TTR was treated with trypsin at a ratio of 1:200 enzyme to protein ratio by weight and incubated for one week. The TTR aggregates were collected by centrifugation and their structural features were initially examined with FT-IR. The structural analyses revealed that the secondary structure of the TTR aggregates obtained at different incubation times were identical (Fig. S1). Thus, the longer incubation time was used for structural characterization of the full-length G53A TTR amyloid due to the slower aggregation kinetics in the absence of trypsin. TTR aggregates were prepared from different batches of the TTR solution and solid-state NMR spectra were collected and averaged for the TTR aggregates.
Results
Proteolytic cleavage triggers aggregation of G53A TTR
Previous biochemical studies showed that mutations in the CD loop (S52P and E51_S52dup) render the TTR variants susceptible to the proteolytic cleavage, which triggers amyloid formation of the TTR variants.31, 32 In this study, another mutant form, G53A TTR associated with leptomeningeal amyloidosis,40 was used to investigate misfolding and aggregation pathway of the TTR variant in the presence and absence of proteolytic enzyme, trypsin. Figure S2 shows aggregation kinetics of G53A TTR at the physiological pH of 7.4 in the presence and absence of trypsin. The aggregation kinetics was initially monitored by measuring optical density (OD) at 400 nm as a function of incubation times (Fig. S2). In the absence of trypsin, the TTR variant remains soluble without notable precipitation for 10 hours at a protein concentration of 0.7 mg/ml. Upon addition of trypsin, aggregation of the TTR variant was greatly accelerated, as demonstrated for the other mutants (S52P and E51_S52dup).
The aggregation behavior of G53A TTR was compared to those of wild-type (WT) and another mutant form of TTR (L55P) in the presence and absence of trypsin at the physiological pH of 7.4 (Fig. 2). Aggregation of the WT and two mutant forms of TTR was promoted in the presence of trypsin (Fig. 2). It is also notable that G53A TTR exhibited a stronger aggregation propensity than L55P and WT TTR. Aggregation of WT TTR was also observed after 4 hrs of incubation in the presence of trypsin. The accelerated aggregation kinetics in the presence of trypsin was reproduced at a lower protein concentration of 0.3 mg/ml (Fig. S3).
Figure 2.

Aggregation kinetics of WT and mutant forms of TTR (0.7 mg/ml) in the presence and absence of trypsin. Thioflavin T (ThT) fluorescence emission spectra were recorded at 482 nm with an excitation of 440 nm. The uncertainty was calculated from two independent experiments.
G53A TTR variant was chosen for more detailed analyses because of its stronger aggregation behavior in the presence of trypsin. In order to investigate whether the mutant TTR (G53A) is cleaved by trypsin, the TTR variant incubated in the presence of trypsin was analyzed by polyacrylamide gel electrophoresis (SDS-PAGE). The SDS-PAGE analysis of the TTR variant in the presence and absence of trypsin clearly shows that G53A TTR is cleaved upon addition of trypsin (Fig. S4). Mass spectrometry was used to identify TTR fragments produced by the trypsin digestion, and the residue 49 – 127 C-terminal fragment was the major component (Fig. S5a). Protein precipitates formed in the presence of trypsin were collected by centrifugation and analyzed with SDS-PAGE (Fig. S5b). The G53A precipitates contained small fragments as well as full-length TTR. These results indicate that the proteolytic cleavage of the K48 – T49 peptide bond in the CD loop promotes amyloid formation of G53A TTR. It also appears that the C-terminal fragments co-aggregate with full-length TTR on the basis of SDS-PAGE shown in Fig. S5b. These results are consistent with recent cryo-EM structural studies of ex vivo TTR fibrils that revealed the TTR fibrils consist of N-terminally truncated and full-length TTR41.
G53A TTR remains largely folded after the proteolytic cleavage
In order to examine an early stage of misfolding and aggregation for the full-length and truncated G53A TTR, CD spectroscopy was used to probe initial conformational transition of the TTR variant during aggregation process at the physiological pH of 7.4. The initial CD spectrum of G53A TTR exhibits a minimum at 213 nm and maximum at 195 nm, corresponding to those of β-structured proteins. In the absence of trypsin, the CD spectra remain nearly identical for 3 hours of incubation at 37 °C. Upon addition of trypsin, the CD signal at 195 nm gradually decreases, while other regions at 200 – 220 nm are not affected, suggesting that the TTR variant becomes slightly more disordered by the proteolytic cleavage.
The CD spectra were analyzed to obtain secondary structure information using the software DICHROWEB36, 37 (Table 1). The secondary structural analyses showed that the TTR variant has slightly less β-sheet content (38 %) than WT TTR (42 %)26. The less structured or more dynamic nature of the TTR variant might be related to its stronger aggregation propensity than WT TTR.26 In the presence of trypsin, relative content of the β-sheet structure slightly decreases by 2 %, while the disordered regions increase by 2 % after 3 hours of incubation. These small changes in the secondary structure indicate that the N- and C-terminal fragments produced by trypsin cleavage remain bound together and the cleaved TTR is still largely folded.
Table 1.
Relative content of the secondary structure for G53A TTR in the presence and absence of trypsin obtained from the analyses of the CD spectra in Figure 3 using the software DICHROWEB.
| α-helix (%) | β-strand (%) | disordered (%) | |
|---|---|---|---|
| G53A (0 min) | 5 | 38 | 56 |
| G53A + Trypsin (0 min) | 5 | 38 | 57 |
| G53A + Trypsin (10 min) | 6 | 36 | 58 |
| G53A + Trypsin (30 min) | 5 | 36 | 59 |
| G53A + Trypsin (90 min) | 4 | 36 | 58 |
| G53A + Trypsin (180 min) | 4 | 36 | 58 |
Proteolytic cleavage promotes the formation of small cytotoxic oligomers
The changes in the CD spectra upon addition of trypsin may originate from the formation of small oligomers. Dynamic light scattering (DLS) was used to probe oligomerization in the presence and absence of trypsin (Fig. 4). In the absence of trypsin, the autocorrelation function decays slowly at longer time intervals (> 500 μs) after 2 hrs of incubation, suggesting the formation of small oligomers (Fig. 4a). Upon addition of trypsin, the correlation of the signal decays more slowly even after shorter incubation times (Fig. 4b), clearly suggesting that the oligomerization process is greatly accelerated in the presence of trypsin.
Figure 4.

Autocorrelation function measured by using dynamic light scattering for G53A TTR in the absence (a) and presence (b) of trypsin at different incubation times. The TTR variant (0.25 mg/ml) was incubated in the phosphate buffer (pH 7.4) at 37 °C.
Transmission electron microscopy (TEM) was used to examine morphology of the oligomers formed in the presence and absence of trypsin (Fig. S6). The TEM images reveal that the TTR variant forms small spherical oligomers at the physiological pH (Fig. S6a). Similar types of oligomers were also observed in the presence of trypsin, suggesting that the full-length as well as cleaved TTR variant form similar oligomeric species (Fig. S6b). It appears that the spherical oligomers are clustered to form bigger less-ordered aggregates rather than fibrillar aggregates in the presence and absence of trypsin.
It is widely believed that small oligomeric species formed at an early state of aggregation are real cytotoxic agents.42, 43, 44, 45, 46, 47, 48 Cytotoxic activities were investigated for the TTR variant incubated for 2 hours using (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 5). The TTR variant incubated in the presence of trypsin exhibits a stronger cytotoxic activity (blue in Fig. 5). Taken together with the DLS and TEM results that indicated addition of trypsin accelerated the formation of small oligomeric species (Fig. S6 and Fig. 4), the proteolytic cleavage by trypsin appears to promote the formation of cytotoxic G53A oligomers at the physiological pH.
Figure 5.

Cell viability measured with MTT assay for the G53A (0.25 and 0.5 mg/ml) TTR incubated in the presence and absence of trypsin in phosphate buffer (pH 7.4) at 37 °C. The uncertainty was calculated from two independent measurements for each protein concentration.
G53A TTR forms nearly identical aggregates in the presence and absence of trypsin
The TEM images of the TTR variant aggregates formed in the presence and absence of trypsin show that the two oligomeric species have similar structural features, implying that the full-length G53A and truncated TTR may have similar misfolding and aggregation pathways. In order to compare molecular structures of the G53A aggregates formed in the presence and absence of trypsin, FT-IR and solid-state NMR were employed (Fig. 6). Amyloid fibrils have absorption spectra with broad amide-I bands ranging from 1600 to 1700 cm−1, which are sensitive to the secondary structures and dihedral torsional angles φ/ψ of the amide backbone.49, 50 The amide-I bands from both G53A aggregates produced in the presence and absence of trypsin exhibit nearly identical maxima at 1630 and 1675 cm−1. The absorption peaks at the amide-II (1,500 – 1600cm−1) and -III (1,300 – 1,450 cm−1) regions (Fig. 6a and Fig. S7a) also appear at the same wavenumbers, suggesting that the two aggregates have similar β-structures.
Figure 6.

(a) Overlaid FT-IR spectra for the full-length (red) and truncated (black) G53A TTR aggregates obtained with an incubation time of 3 days. (b) Aliphatic regions of the 2D 13C-13C correlation solid-state NMR spectra of the two aggregates obtained with a DARR mixing time of 25 ms. The cross-peaks from the valine and threonine sidechains (Cβ-Cγ) are indicated by V and T, respectively.
Morphology of the two TTR aggregates incubated in the presence and absence of trypsin was examined by using TEM (Fig. S7b and S7c). The TEM images reveal that G53A TTR forms non-fibrillar aggregates in the presence and absence of trypsin. It appears that the small spherical oligomers observed at an early stage of aggregation (Fig. S6) self-assemble into the less ordered aggregates, suggesting the oligomers are on the aggregation pathway. Molecular structural features of the two TTR aggregates were also compared by solid-state NMR spectroscopy. Fig. 6b shows overlaid 13C-13C correlation solid-state NMR spectra for G53A TTR aggregates formed in the presence (black) and absence (red) of trypsin. The 2D DARR spectra for the aliphatic regions are overlapped well, suggesting that the 13C NMR chemical shifts of aliphatic carbons including Cα and Cβ carbons are very similar for the two oligomeric states. The 13C NMR chemical shifts of the aliphatic carbons are highly sensitive to the secondary structures and φ/ψ dihedral angles,51 and thus the overlaid NMR spectra suggests that the two oligomeric species adopt similar molecular conformations consistent with the FT-IR spectra shown in Fig. 6a. The cross-peaks with different chemical shifts may come from the residues close to the cleavage site in the CD loop.
It is, also, notable that the cross-peaks from the truncated G53A oligomers (black) are better resolved than those of the full-length G53A oligomers (red). The higher resolution is more evident in the one-dimensional slice along 55 ppm in the 2D DARR spectra (Fig. S8). Relatively sharp resonances that are absent in the full-length G53A oligomers are also observed in the truncated G53A oligomers. These results suggest that the two oligomeric states have overall similar secondary structures and morphology, but the truncated oligomers contain more ordered local structures.
Discussion
There is an increasing body of evidence that suggests aggregation-prone proteins behave like a prion that can form diverse misfolded protein aggregates with distinct molecular conformations and different biological activities, which might be associated with diverse disease phenotypes of protein misfolding disorders. TTR amyloidosis is also characterized by various disease phenotypes, suggesting the presence of distinct TTR amyloids. Indeed, at least two different types of TTR amyloids have been detected in vivo.27 One of the two amyloids consists only of full-length TTR, while the other amyloid was formed by a mixture of the full-length and truncated TTR. A major component of the fragmented TTR is the residue 49 – 127 C-terminal fragment. The presence of the two types of TTR amyloids suggests that the full-length and C-terminal fragment TTR may have distinct misfolding pathways, leading to the different types of TTR amyloids.
Previous studies showed that a proteolytic cleavage of the K48 – T49 peptide bond in the CD loop promotes TTR misfolding and aggregation of the TTR variants (S52P and Glu51_Ser52dup).31, 32 The cleaved C-terminal fragment (49 – 127) may be released from the folded TTR and subsequently unfolded, leading to the formation of amyloid. The TTR aggregates formed by the C-terminal fragment may have distinct molecular conformations from those of full-length TTR. On the other hand, the N- and C-terminal fragments may still be bound together due to the hydrogen-bonding interactions between the β-strands in the natively folded TTR. The proteolytic cleavage of the CD loop may just destabilize the tetrameric structure of TTR, accelerating dissociation of the tetramers to monomers that is the rate determining step in the TTR aggregation process24, 52. In this mechanism, TTR amyloid formed in the presence of trypsin would have similar molecular conformations to those of intact full-length TTR.
In this study, we used various biophysical techniques to investigate an early stage of TTR aggregation in the presence and absence of trypsin. Structural features of the final product (oligomeric aggregates) formed in the presence and absence of trypsin were also examined using FT-IR and solid-state NMR in order to compare aggregation pathway of the full-length and truncated TTR. In the presence of trypsin, the peptide bond K48-T49 was cleaved, and subsequently TTR oligomerization was accelerated (Fig. 4). The small spherical oligomeric species detected in TEM (Fig. S6) exhibited cytotoxic activities (Fig. 5). Structural changes during the oligomerization process in the presence of trypsin were probed by using CD spectroscopy (Fig. 3b). Notable changes in the CD signals at low wavelength region (< 200 nm) were observed during TTR oligomerization promoted by the proteolytic cleavage, suggesting a local unfolding of the protein at the early stage of oligomerization. However, FT-IR and solid-state NMR spectra for the G53A TTR oligomers formed in the presence and absence of trypsin revealed that the two oligomeric states adopt nearly identical molecular conformations. The combined structural analyses of G53A TTR at an early stage of oligomerization process and the oligomeric states indicate that the TTR variant in the presence and absence of trypsin forms oligomeric aggregates via a similar misfolding and aggregation pathway. The proteolytic cleavage in the CD loop appears to induce a local unfolding transition, accelerating oligomerization of the TTR variant without inducing major conformational changes. These results are consistent with our previous solid-state NMR structural studies of TTR oligomers that revealed the oligomeric species maintain extensive native-like β-sheet structures.26, 53
Figure 3.

Experimental CD spectra of G53A TTR in the absence (a) and presence (b) of trypsin at different incubation times. Calculated spectra derived from the simulated secondary structure in Table 1 are plotted with solid squares. The TTR variant (0.5 mg/ml) was incubated in 10 mM sodium phosphate buffer (pH 7.4) at 37 °C and the CD spectra were recorded at 20 °C.
The crystal structures of WT and mutant forms of TTR revealed that the CD loop adopts a tight turn on the residue G53 (Fig. S9). Glycine is commonly found in the turn position because of its high steric flexibility. Substitution of the glycine with the smallest sidechain by alanine may increase flexibility of the CD loop in the native TTR and destabilize the native tetrameric structure of G53A TTR. Perturbations of the CD loop in the other TTR variants (S52P and Glu51_Ser52dup) were also shown to reduce the thermodynamic stability of tetrameric native TTR presumably due to the enhanced flexibility of the CD loop. In addition, a single-point mutation in the strand D (L55P) disrupts β-strand D, affecting the stability of the CD loop.54, 55 The structural, dynamic perturbation of the CD loop by the mutations renders the three TTR variants (S52P, Glu51_Ser52dup, and L55P) more susceptible to the proteolytic cleavage. Thus, the single-point mutation (G53A) in the CD loop may also affect the stability of the CD loop, destabilizing the native tetrameric TTR. Indeed, the TTR variant is more easily dissociated from tetramers to dimers and monomers in SDS-PAGE compared to WT TTR (Fig. S10). In addition, our previous biochemical studies of misfolding and aggregation of G53A TTR variant revealed that tetrameric forms of the native TTR variant are readily dissociated into monomers and dimers even at the physiological pH of 7.4.26
In summary, a proteolytic cleavage of the K48-T49 bond in the CD loop promoted the formation of cytotoxic spherical oligomers. Structural analyses of the TTR variant (G53A) in the presence and absence of trypsin at an early stage oligomerization revealed that the full-length and cleaved TTR did not undergo major structural changes during the oligomerization process. In addition, our FT-IR and solid-state NMR structural studies of the TTR oligomers formed in the presence and absence of trypsin suggested that the two oligomeric species adopt similar molecular conformations. These results indicate that the full-length and cleaved TTR variants form the aggregates through a similar misfolding and aggregation mechanism. The proteolytic cleavage appears to destabilize the CD loop, accelerating dissociation of the native tetramers into monomers that is the rate determining step in the TTR aggregation process.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge Dr. Fink (ECU) for assistance in the TEM and Dr. Pender (ECU) for assistance in FT-IR.
Funding Sources
This work was supported by NIH grant (NS097490, KHL). The solid-state NMR spectra were acquired at Francis Bitter Magnet Laboratory, Massachusetts Institute of Technology, Cambridge, MA.
ABBREVIATIONS
- NMR
nuclear magnetic resonance
- DARR
dipolar assisted rotational resonance
- TTR
transthyretin
- CP
cross-polarization
- WT
wild-type
- CD
circular dichroism
- TEM
transmission electron microscopy
- FT-IR
Fourier transform infrared
- DLS
dynamic light scattering
- MAS
magic-angle spinning
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium bromide
- ATTR
TTR amyloidosis
- PAGE
polyacrylamide gel electrophoresis
- ThT
thioflavin T
- DARR
dipolar-assist rotational resonance
Footnotes
Supporting Information. The following files are available free of charge. SDS-PAGE of the G53A TTR in the presence and absence of trypsin. MALDI spectrum of G53A TTR in the presence of trypsin. TEM images of G53A TTR incubated in the presence and absence of trypsin. FTIR spectra of G53A TTR amyloids obtained in the presence and absence of trypsin. Crystal structure of monomeric subunit in WT TTR. SDS-PAGE of WT TTR and G53A TTR.
The authors declare no competing financial interests.
Accession Codes
Transthyretin: UniProtKB entry P02766
REFERENCES
- 1.Buxbaum JN, and Reixach N (2009) Transthyretin: The servant of many masters. Cell Mol. Life Sci 66, 3095–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blake CC, Geisow MJ, Oatley SJ, Rerat B, and Rerat C (1978) Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by fourier refinement at 1.8 A. J. Mol. Biol 121, 339–356. [DOI] [PubMed] [Google Scholar]
- 3.Herbert J, Wilcox JN, Pham KT, Fremeau RT, Zeviani M, Dwork A, Soprano DR, Makover A, Goodman DS, and Zimmerman EA (1986) Transthyretin: A choroid plexus-specific transport protein in human brain. the 1986 S. weir mitchell award. Neurology. 36, 900–911. [DOI] [PubMed] [Google Scholar]
- 4.Hagen GA, and Elliott WJ (1973) Transport of thyroid hormones in serum and cerebrospinal fluid. J. Clin. Endocrinol. Metab 37, 415–422. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton JA, Steinrauf LK, Braden BC, Liepnieks J, Benson MD, Holmgren G, Sandgren O, and Steen L (1993) The x-ray crystal structure refinements of normal human transthyretin and the amyloidogenic val-30-->met variant to 1.7-A resolution. J. Biol. Chem 268, 2416–2424. [PubMed] [Google Scholar]
- 6.Hornberg A, Eneqvist T, Olofsson A, Lundgren E, and Sauer-Eriksson AE (2000) A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol 302, 649–669. [DOI] [PubMed] [Google Scholar]
- 7.Connelly S, Choi S, Johnson SM, Kelly JW, and Wilson IA (2010) Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr. Opin. Struct. Biol 20, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekijima Y (2015) Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 86, 1036–1043. [DOI] [PubMed] [Google Scholar]
- 9.Galant NJ, Westermark P, Higaki JN, and Chakrabartty A (2017) Transthyretin amyloidosis: An under-recognized neuropathy and cardiomyopathy. Clin. Sci. (Lond) 131, 395–409. [DOI] [PubMed] [Google Scholar]
- 10.Kelly JW (1998) The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol 8, 101–106. [DOI] [PubMed] [Google Scholar]
- 11.Adams D, Koike H, Slama M, and Coelho T (2019) Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol 15, 387–404. [DOI] [PubMed] [Google Scholar]
- 12.Westermark P, Sletten K, Johansson B, and Cornwell GG (1990) Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 87, 2843–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coelho T, Maurer MS, and Suhr OB (2013) THAOS - the transthyretin amyloidosis outcomes survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin 29, 63–76. [DOI] [PubMed] [Google Scholar]
- 14.Westermark P, Bergstrom J, Solomon A, Murphy C, and Sletten K (2003) Transthyretin-derived senile systemic amyloidosis: Clinicopathologic and structural considerations. Amyloid. 10 Suppl 1, 48–54. [PubMed] [Google Scholar]
- 15.Herrick MK, DeBruyne K, Horoupian DS, Skare J, Vanefsky MA, and Ong T (1996) Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology. 47, 988–992. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, and Buxbaum JN (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black americans. N. Engl. J. Med 336, 466–473. [DOI] [PubMed] [Google Scholar]
- 17.Connors LH, Lim A, Prokaeva T, Roskens VA, and Costello CE (2003) Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 10, 160–184. [DOI] [PubMed] [Google Scholar]
- 18.Saraiva MJ (1995) Transthyretin mutations in health and disease. Hum. Mutat 5, 191–196. [DOI] [PubMed] [Google Scholar]
- 19.Sun X, Dyson HJ, and Wright PE (2018) Kinetic analysis of the multi-step aggregation pathway of human transthyretin. Proc. Natl. Acad. Sci. U. S. A 115, E6201–E6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colon W, and Kelly JW (1992) Partial denaturation of transthyretin is sufficient for amyloid fibril formation invitro. Biochemistry. 31, 8654–8660. [DOI] [PubMed] [Google Scholar]
- 21.Lai ZH, Colon W, and Kelly JW (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 35, 6470–6482. [DOI] [PubMed] [Google Scholar]
- 22.Hammarstrom P, Schneider F, and Kelly JW (2001) Trans-suppression of misfolding in an amyloid disease. Science. 293, 2459–2462. [DOI] [PubMed] [Google Scholar]
- 23.Hammarstrom P, Wiseman RL, Powers ET, and Kelly JW (2003) Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 299, 713–6. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, and Kelly JW (2001) An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 40, 11442–11452. [DOI] [PubMed] [Google Scholar]
- 25.Quintas A, Saraiva MJ, and Brito RM (1999) The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J. Biol. Chem 274, 32943–32949. [DOI] [PubMed] [Google Scholar]
- 26.Dasari AKR, Hughes RM, Wi S, Hung I, Gan Z, Kelly JW, and Lim KH (2019) Transthyretin aggregation pathway toward the formation of distinct cytotoxic oligomers. Sci. Rep 9, 33–018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gustavsson A, Jahr H, Tobiassen R, Jacobson DR, Sletten K, and Westermark P (1995) Amyloid fibril composition and transthyretin gene structure in senile systemic amyloidosis. Lab. Invest 73, 703–708. [PubMed] [Google Scholar]
- 28.Kingsbury JS, Theberge R, Karbassi JA, Lim A, Costello CE, and Connors LH (2007) Detailed structural analysis of amyloidogenic wild-type transthyretin using a novel purification strategy and mass spectrometry. Anal. Chem 79, 1990–1998. [DOI] [PubMed] [Google Scholar]
- 29.Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, Ikeda S, Lavatelli F, Obici L, Quarta CC, Leone O, Jono H, Ueda M, Lorenzini M, Liepnieks J, Ohshima T, Tasaki M, Yamashita T, and Westermark P (2013) Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 20, 142–150. [DOI] [PubMed] [Google Scholar]
- 30.Bergstrom J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson BO, and Westermark P (2005) Amyloid deposits in transthyretin-derived amyloidosis: Cleaved transthyretin is associated with distinct amyloid morphology. J. Pathol 206, 224–232. [DOI] [PubMed] [Google Scholar]
- 31.Mangione PP, Porcari R, Gillmore JD, Pucci P, Monti M, Porcari M, Giorgetti S, Marchese L, Raimondi S, Serpell LC, Chen W, Relini A, Marcoux J, Clatworthy IR, Taylor GW, Tennent GA, Robinson CV, Hawkins PN, Stoppini M, Wood SP, Pepys MB, and Bellotti V (2014) Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. U. S. A 111, 1539–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klimtchuk ES, Prokaeva T, Frame NM, Abdullahi HA, Spencer B, Dasari S, Cui H, Berk JL, Kurtin PJ, Connors LH, and Gursky O (2018) Unusual duplication mutation in a surface loop of human transthyretin leads to an aggressive drug-resistant amyloid disease. Proc. Natl. Acad. Sci. U. S. A 115, E6428–E6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcoux J, Mangione PP, Porcari R, Degiacomi MT, Verona G, Taylor GW, Giorgetti S, Raimondi S, Sanglier-Cianferani S, Benesch JL, Cecconi C, Naqvi MM, Gillmore JD, Hawkins PN, Stoppini M, Robinson CV, Pepys MB, and Bellotti V (2015) A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med 7, 1337–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangione PP, Verona G, Corazza A, Marcoux J, Canetti D, Giorgetti S, Raimondi S, Stoppini M, Esposito M, Relini A, Canale C, Valli M, Marchese L, Faravelli G, Obici L, Hawkins PN, Taylor GW, Gillmore JD, Pepys MB, and Bellotti V (2018) Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem 293, 14192–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuguchi M, Hayashi A, Takeuchi M, Dobashi M, Mori Y, Shinoda H, Aizawa T, Demura M, and Kawano K (2008) Unfolding and aggregation of transthyretin by the truncation of 50 N-terminal amino acids. Proteins. 72, 261–269. [DOI] [PubMed] [Google Scholar]
- 36.Whitmore L, and Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitmore L, and Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 89, 392–400. [DOI] [PubMed] [Google Scholar]
- 38.Takegoshi K, Nakamura S, and Terao T (2003) 13C–1H dipolar-driven 13C–13C recoupling without 13C rf irradiation in nuclear magnetic resonance of rotating solids. The Journal of Chemical Physics. 118, 2325–2341. [Google Scholar]
- 39.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, and Griffin RG (Invalid date) Heteronuclear decoupling in rotating solids. J. Chem. Phys 103, 6951–6958. [Google Scholar]
- 40.Douglass C, Suvarna K, Reilly MM, Hawkins PN, and Hadjivassiliou M (2007) A novel amyloidogenic transthyretin variant, Gly53Ala, associated with intermittent headaches and ataxia. J. Neurol. Neurosurg. Psychiatry 78, 193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt M, Wiese S, Adak V, Engler J, Agarwal S, Fritz G, Westermark P, Zacharias M, and Fandrich M (2019) Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun 10, 5008–019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TP, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM, and Cremades N (2015) Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. U. S. A 112, E1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breydo L, and Uversky VN (2015) Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 589, 2640–2648. [DOI] [PubMed] [Google Scholar]
- 44.Fandrich M (2012) Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol 421, 427–440. [DOI] [PubMed] [Google Scholar]
- 45.Tipping KW, van Oosten-Hawle P, Hewitt EW, and Radford SE (2015) Amyloid fibres: Inert end-stage aggregates or key players in disease? Trends Biochem. Sci. 40, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reixach N, Deechongkit S, Jiang X, Kelly JW, and Buxbaum JN (2004) Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. U. S. A 101, 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, and Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 300, 486–489. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez Camargo DC, Garg D, Buday K, Franko A, Rodriguez Camargo A, Schmidt F, Cox SJ, Suladze S, Haslbeck M, Mideksa YG, Gemmecker G, Aichler M, Mettenleiter G, Schulz M, Walch AK, Hrabe de Angelis M, Feige MJ, Sierra CA, Conrad M, Tripsianes K, Ramamoorthy A, and Reif B (2018) hIAPP forms toxic oligomers in plasma. Chem. Commun. (Camb) 54, 5426–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moran SD, and Zanni MT (2014) How to get insight into amyloid structure and formation from infrared spectroscopy. J. Phys. Chem. Lett 5, 1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zandomeneghi G, Krebs MR, McCammon MG, and Fandrich M (2004) FTIR reveals structural differences between native beta-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, and Jardetzky O (2002) Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang X, Buxbaum JN, and Kelly JW (2001) The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. U. S. A 98, 14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dasari AKR, Hung I, Michael B, Gan Z, Kelly JW, Connors LH, Griffin RG, and Lim KH (2020) Structural characterization of cardiac ex vivo transthyretin amyloid: Insight into the transthyretin misfolding pathway in vivo. Biochemistry. 59, 1800–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cendron L, Trovato A, Seno F, Folli C, Alfieri B, Zanotti G, and Berni R (2009) Amyloidogenic potential of transthyretin variants: Insights from structural and computational analyses. J. Biol. Chem. 284, 25832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sebastiao MP, Saraiva MJ, and Damas AM (1998) The crystal structure of amyloidogenic leu(55) -> pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 273, 24715–24722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.