

Abstract
Background
Cerebrospinal fluid (CSF) has been demonstrated as a better source of circulating tumor DNA (ctDNA) than plasma for brain tumors. However, it is unclear whether whole exome sequencing (WES) is qualified for detection of ctDNA in CSF. The aim of this study was to determine if assessment of ctDNA in CSF by WES is a feasible approach to detect genomic alterations of glioblastoma.
Methods
CSFs of ten glioblastoma patients were collected pre-operatively at the Department of Neurosurgery, Sun Yat-sen University Cancer Center. ctDNA in CSF and genome DNA in the resected tumor were extracted and subjected to WES. The identified glioblastoma-associated mutations from ctDNA in CSF and genome DNA in the resected tumor were compared.
Results
Due to the ctDNA in CSF was unqualified for exome sequencing for one patient, nine patients were included into the final analysis. More glioblastoma-associated mutations tended to be detected in CSF compared with the corresponding tumor tissue samples (3.56 ± 0.75 vs. 2.22 ± 0.32, P = 0.097), while the statistical significance was limited by the small sample size. The average mutation frequencies were similar in CSF and tumor tissue samples (74.1% ± 6.0% vs. 73.8% ± 6.0%, P = 0.924). The R132H mutation of isocitrate dehydrogenase 1 and the G34V mutation of H3 histone, family 3A (H3F3A) which had been reported in the pathological diagnoses were also detected from ctDNA in CSF by WES. Patients who received temozolomide chemotherapy previously or those whose tumor involved subventricular zone tended to harbor more mutations in their CSF.
Conclusion
Assessment of ctDNA in CSF by WES is a feasible approach to detect genomic alterations of glioblastoma, which may provide useful information for the decision of treatment strategy.
Keywords: Circulating tumor DNA, Cerebrospinal fluid, Glioblastoma, Mutation, Whole exome sequencing
Introduction
Glioblastoma (GBM) is the most prevalent primary malignant brain tumor with bleak prognosis in adults.[1] Despite the standard of care therapy, the median survival duration is only 14.6 months.[2] The diagnosis of GBM has stepped into a molecular era since the 2016 World Health Organization (WHO) Classification of Tumors of the Central Nervous System (CNS) incorporates molecular parameters into the classification of CNS tumor entities.[3] Some of these molecular parameters have a prognostic role for GBM patients.[4,5] Meanwhile, these molecular parameters may also serve as predictive biomarkers of therapeutic effect, including extent of surgical resection and sensitivity of chemotherapy.[6–8] Therefore, the early diagnosis of molecular parameters may be helpful for the decision of treatment strategy.
Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA circulating in body fluid and may reflect the entire tumor genome. Cerebrospinal fluid (CSF) directly contacts the brain tumor and has been demonstrated as a better source of ctDNA than plasma for brain tumors.[9,10] Recent studies have shown that ctDNA detected using targeted deep sequencing in CSF could represent the genomic alterations of brain tumors[10] and monitor the evolution of the glioma genome.[9] However, most of these previous results were based on Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets, a custom sequencing-based tumor sequencing assay that captures all protein-coding exons of 341, 410, or 468 cancer-associated genes with deep coverage.[11] However, this assay is not so accessible and affordable as whole exome sequencing (WES) overall the world. However, it is unclear whether WES is qualified for detection of ctDNA in CSF.
Here, we aimed to determine if assessment of ctDNA in CSF by WES is a feasible approach to detect genomic alterations of GBM.
Methods
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the Medical Ethics Committees of Sun Yat-sen University Cancer Center (No. GZR2018-244), followed the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consents to review medical data and use biological samples were obtained from all patients before the study.
Study cohort and sample collection
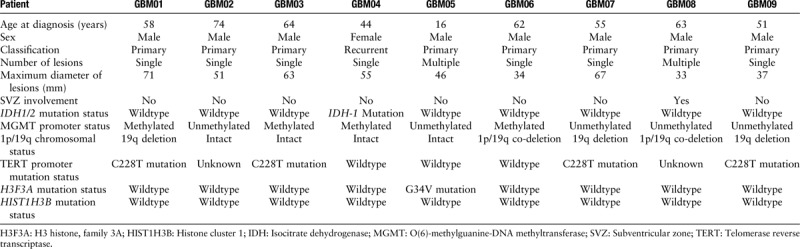
Ten patients who were diagnosed with GBM and underwent lumbar puncture pre-operatively as part of their clinical evaluation for neurological signs or symptoms and surgical resection were included in this study. The study subjects were enrolled at the Department of Neurosurgery, Sun Yat-sen University Cancer Center. Collection of CSF was performed through lumbar puncture. To avoid the contamination of blood, only the clear CSF was collected. After spinning down, cell-free supernatants were collected and stored at −80°C. Then, all patients underwent surgical resection and fresh tumor tissue samples were stored in liquid nitrogen. The diagnosis of each patient was confirmed by pathologist according to the 2016 WHO Classification of Tumors of the CNS. Clinicopathological data including patient's age at diagnosis, sex, number of lesions, size of lesions, subventricular zone (SVZ) involvement described by Lim et al,[12] isocitrate dehydrogenase 1 (IDH1) mutation status, O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation status, 1p/19q chromosomal status, telomerase reverse transcriptase (TERT) promoter mutation status, H3 histone, family 3A (H3F3A) mutation status, and Histone cluster 1, H3b (HIST1H3B) mutation status were collected from medical records.
Extraction of ctDNA and genome DNA
For each patient, 5 to 10 mL of CSF was subjected to ctDNA extraction using QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA, USA). Genome DNA from the resected tumor was extracted using a DNeasy Blood and Tissue Kit (Qiagen). DNA concentration was quantified by the Qubit 4.0 Fluorometer (Invitrogen, Singapore). Bioanalyzer agilent 2100 (Agilent Technologies, Palo Alto, CA, USA) defined the sample DNA qualities with high sensitivity DNA chip.
Exome-sequencing analysis
Exome sequencing libraries were prepared with the SureSelect Human All Exon 50 Mb Targeted exome enrichment kit v4, targeting all protein-coding exons, exon-intron boundaries, and untranslated regions of all protein-coding genes. The library quality and quantity were controlled by KAPA SYBR FAST LightCycler 480 qPCR kit (KAPA Biosystems, Wilmington, MA, USA), on BioAnalyzer agilent 2100 High Sensitivity DNA chip (Agilent Technologies). Libraries containing captured DNA fragments were then sequenced on the Illumina HiSeq 2500 system as paired-end 150 bp. The mean sequencing depth was 93X for all samples.
Single nucleotide variants (SNVs) and small insertions and deletions (INDELs) calling
First, raw Fastq reads were trimmed with Trimmomatic as previously described,[13] reads were then aligned in paired-end mode to the human genome build 19 (hg19) version using Burrow-Wheeler Aligner-maximal exact match (BWA-MEM) with default parameters to generate a binary sequence alignment map file.[14] SNVs were called with MuTect, a Bayesian framework for the detection of somatic mutations, and small INDELs (<30 bp in length) were detected by SomaticINDELDetector, a tool in GATK version 2.3.9, according to the pipeline previously described.[11,15,16] Variants identified within CSF and tumor tissue were filtered out if found in the 1000 Genomes East Asian cohort (1000g2014oct_eas), avsnp150, cosmic or esp6500sic2_all (NHLBI-ESP project) databases. Finally, we only kept functional variants predicted as damaging by Polyphen2, LRT, MutationTaster, FATHMM and Sift for further analysis.[17–20]
Statistical analyses
SPSS 22.0 (IBM Corp, Armonk, NY, USA). USA) was used for statistical analyses. The independent Student's t test was used to assess the statistical significance between any two preselected groups. The results were presented as mean ± standard deviation (SD). A two-sided P <0.05 was considered as statistically significant.
Results
Patients’ clinicopathological characteristics
Because the ctDNA was unqualified for exome sequencing for one patient, nine patients were included into the final analysis. The clinicopathological characteristics of patients were summarized in Table 1. Median age at diagnosis was 58 years (range, 16–74 years). Only one patient was female who was diagnosed with recurrent GBM and had received surgery followed by radiotherapy plus concomitant and adjuvant temozolomide (TMZ) before CSF collection. All of the eight male patients were diagnosed with primary therapy-naïve GBM. Two patients had multiple lesions. The median maximum diameter of lesions was 5.1 cm. One patient had the tumor involving SVZ. In pathological diagnosis, R132H mutation of IDH1 and G34V of H3F3A were identified in one patient, respectively. C228T mutation of TERT promoter and MGMT promoter methylation were identified in four patients, respectively. Deletion of chromosome arm 19q was detected in three patients and 1p/19q co-deletion occurred in two patients. Nobody had HIST1H3B-mutant tumor.
Table 1.
Clinicopathological characteristics of nine glioblastoma patients.
Landscape of genetic alterations in CSF and tumor tissues
Deep sequencing that achieved a coverage of approximately 74× in CSF and over 100× in tumor identified and validated averagely 1211 non-silent variants per patient (range 62–7482). Across these nine pairs of CSF and corresponding tumor tissue samples, a total of 10,690 SNVs and 212 INDELs were called. The exons and surrounding noncoding genomic regions of 27 protein-coding genes that were most frequently mutated in GBM defined by the Catalogue of Somatic Mutations In Cancer (COSMIC) and the Cancer Genome Atlas (TCGA)-GBM database were captured [Table 2]. After excluding those identified in matched germline DNA, the non-silent coding variants (missense, nonsense, short INDELs, splicing variants) of 15 GBM-associated genes were enriched in CSF [Figure 1A] and the corresponding tumor tissue samples [Figure 1B]. We have identified novel SNVs and INDELs in epidermal growth factor receptor (EGFR), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1), and fibroblast growth factor receptor 3 (FGFR3), which are the major genes of the receptor tyrosine kinase (RTK)/Ras GTPase/phosphatidylinositol 3-kinase (PI3K) pathway, and IDH1, ATRX chromatin remodeler (ATRX), and tumor protein P53 (TP53), genes well known to be altered in GBM. A detailed table including the identified non-silent coding variants in both CSF and tumor tissues was listed [Table 3].
Table 2.
GBM-associated genes defined by the Catalogue of Somatic Mutations In Cancer (COSMIC) and the Cancer Genome Atlas (TCGA)-GBM database.

Figure 1.
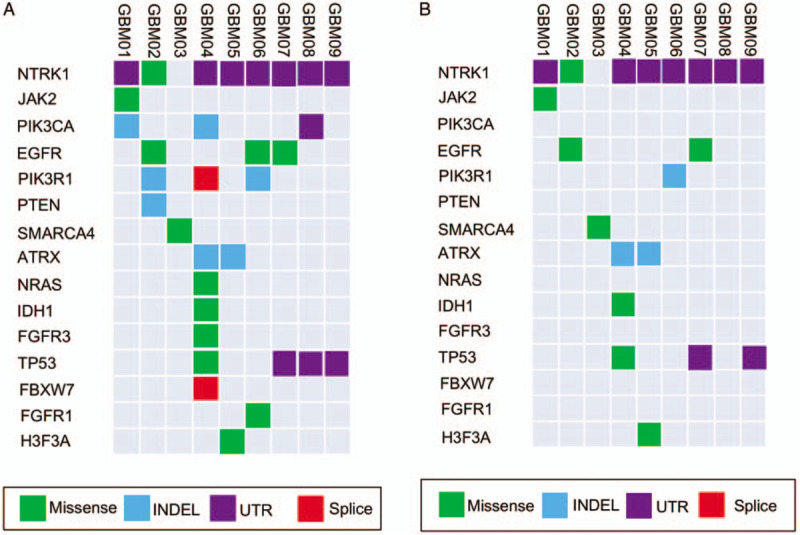
Landscape of GBM-associated mutations detected in CSF (A) and tumor tissue (B). ATRX: ATRX chromatin remodeler; CSF: Cerebrospinal fluid; EGFR: Epidermal growth factor receptor; FBXW7: F-box and WD repeat domain containing 7; FGFR1: Fibroblast growth factor receptor 1; FGFR3: Fibroblast growth factor receptor 3; GBM: Glioblastoma; H3F3A: H3 histone, family 3A; INDEL: Insertion and deletion; IDH1: Isocitrate dehydrogenase 1; JAK2: Janus kinase 2; NRAS: NRAS proto-oncogene, GTPase; NTRK1: Neurotrophic receptor tyrosine kinase 1; PIK3CA: Phosphatidylinositol-4,5-bisphosphate 3-Kinase Catalytic Subunit Alpha; PIK3R1: Phosphoinositide-3-Kinase Regulatory Subunit 1; PTEN: Phosphatase And Tensin Homolog; SMARCA4: SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 4; TP53: Tumor Protein P53; UTR: Untranslated region.
Table 3.
Non-silent mutational profiles in the CSF and tumor tissues.
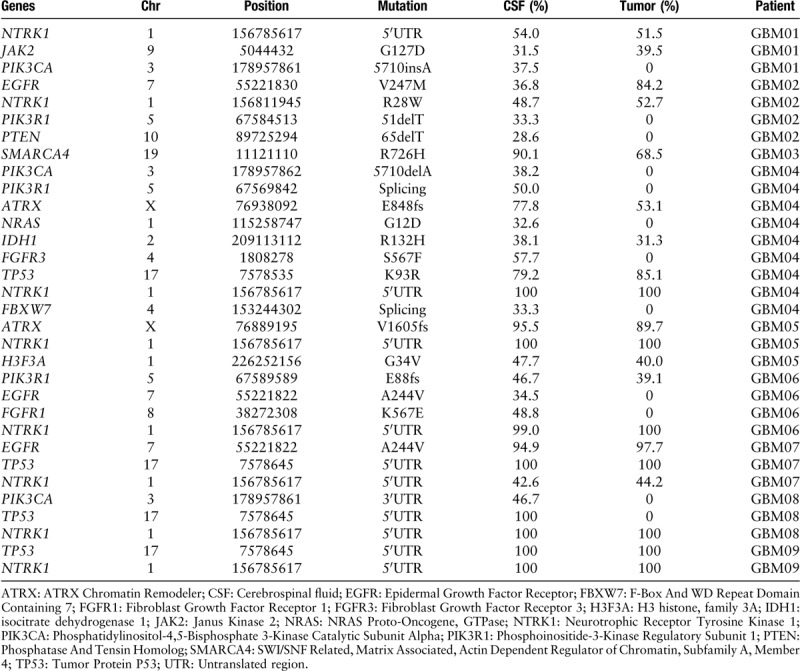
In general, more glioblastoma-associated mutations tended to be detected in CSF comparing with the corresponding tumor tissue samples (3.56 ± 0.75 vs. 2.22 ± 0.32, P = 0.097), while the statistical significance was limited by the small sample size. The average mutation frequencies were similar in CSF and tumor tissue samples (74.1% ± 6.0% vs. 73.8% ± 6.0%, P = 0.924). In patient GBM01, PIK3CA mutation was only detected in CSF but not in the corresponding tumor tissue sample. In patient GBM02, PIK3R1 mutation and phosphatase and tensin homolog (PTEN) mutation were uniquely detectable in the CSF. In patient GBM04, PIK3CA mutation, neuroblastoma RAS viral oncogene homolog (NRAS) mutation, FGFR3 mutation, PIK3R1 mutation, and F-Box and WD repeat domain containing 7 (FBXW7) mutation were all exclusively found in the CSF. In the CSF of patient GBM06, we found the EGFR mutation and FGFR1 mutation which were undetectable in the corresponding tumor tissue sample. Similarly for patient GBM08, PIK3CA mutation and TP53 mutation were only detectable in the CSF. Moreover, patient GBM04 who received TMZ chemotherapy previously and patient GBM08 whose tumor involved SVZ harbored the biggest number of SNVs and INDELs in their CSF [Figure 2]. Most of mutations identified in patient GBM04 had higher mutation frequencies in CSF than tumor (except K93R of EGFR). Meanwhile, the R132H mutation of IDH1 which had been reported in the pathological diagnosis of patient GBM04 was also detected in both CSF and tumor by WES. Likewise, the G34V mutation of H3F3A which had been reported in the pathological diagnosis of patient GBM05 was detected a higher mutation frequency in CSF than tumor.
Figure 2.
Landscape of total SNVs and INDELs detected in cerebrospinal fluid of all patients. GBM: Glioblastoma; INDELs: Insertions and deletions; SNVs: Single nucleotide variants.
Discussion
In this study, we found that gene mutations in GBM could be effectively detected in CSF ctDNA by WES pre-operatively. IDH1 R132H mutation and H3F3A G34V mutation which had been respectively reported in the pathological diagnoses of patient GBM04 and patient GBM05 were detected in their CSF and confirmed in the corresponding tumor tissue samples by WES. It has reported that IDH1 mutant GBM is more amenable to surgical resection and has a survival benefit associated with maximal surgical resection.[6] The determination of gene mutations pre-operatively may facilitate individualized surgical strategies for GBM in the future.
The standard tissue biopsy is the traditional way to determine gene mutations in GBM. However, this method is limited by the difficult access to the tumor and especially the tumor heterogeneity. The molecular heterogeneity within tumors has been increasingly recognized.[21–23] Intriguingly, we found that more gene mutations were detected in CSF ctDNA than in tumor tissue samples, which indicated that CSF ctDNA was better to represent the tumor genome than tumor tissue samples.
Compared with other patients, the numbers of gene mutations detected in CSF ctDNA were extremely higher for Patient 4 and Patient 8. Since Patient 4 suffered recurrent GBM, we speculated the hypermutation of Patient 4 was induced by the treatment of TMZ before CSF collection.[24,25] As the tumor of Patient 8 had involved ventricle, the hypermutation in CSF ctDNA may be due to the direct exposure of tumor into CSF.
The main limitation of this study was the small sample size. Although we found more GBM-associated mutations were detected in CSF comparing with the corresponding tumor tissue samples, the difference was not statistically significant. Future studies with large sample size are warranted to confirm these preliminary results.
In summary, assessment of ctDNA in CSF by WES is a feasible approach to detect genomic alterations of GBM, which may provide useful information for the decision of treatment strategy.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81872324), Science and Technology Planning Project of Guangdong Province, China (No. 2018A030313754), Science and Technology Program of Guangzhou, China (Nos. 201704020133, 201707010169), and Science and Technology Planning Project of Jiangmen, China (No. 2018630100110019805).
Conflicts of interest
None.
Footnotes
How to cite this article: Duan H, Hu JL, Chen ZH, Li JH, He ZQ, Wang ZN, Zhang GH, Guo XY, Liang L, Mou YG. Assessment of circulating tumor DNA in cerebrospinal fluid by whole exome sequencing to detect genomic alterations of glioblastoma. Chin Med J 2020;133:1415–1421. doi: 10.1097/CM9.0000000000000843
Hao Duan and Ji-Long Hu contributed equally to this work.
References
- 1.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 2013; 15: Suppl 2: ii1–56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016; 131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 4.Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 2015; 372:2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang JL, Zhong XS, Yang SB, Kang X, Li Y, Chen JX, et al. Features and therapeutic potential of T-cell receptors in high-grade glioma. Chin Med J 2019; 132:1435–1440. doi: 10.1097/CM9.0000000000000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M, et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol 2014; 16:81–91. doi: 10.1093/neuonc/not159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arita H, Yamasaki K, Matsushita Y, Nakamura T, Shimokawa A, Takami H, et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol Commun 2016; 4:79.doi: 10.1186/s40478-016-0351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen HN, Lie A, Li T, Chowdhury R, Liu F, Ozer B, et al. Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy. Neuro Oncol 2017; 19:394–404. doi: 10.1093/neuonc/now189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019; 565:654–658. doi: 10.1038/s41586-019-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martinez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 2015; 6:8839.doi: 10.1038/ncomms9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015; 17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S, et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol 2007; 9:424–429. doi: 10.1215/15228517-2007-023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997 2013. [Google Scholar]
- 15.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31:213.doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009; 19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 20.Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018; 34:511–513. doi: 10.1093/bioinformatics/btx536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019; 178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-Cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep 2017; 21:1399–1410. doi: 10.1016/j.celrep.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014; 344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, et al. Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol 2019; 9:41.doi: 10.3389/fonc.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi S, Yu Y, Grimmer MR, Wahl M, Chang SM, Costello JF. Temozolomide-associated hypermutation in gliomas. Neuro Oncol 2018; 20:1300–1309. doi: 10.1093/neuonc/noy016. [DOI] [PMC free article] [PubMed] [Google Scholar]