

Abstract
Molecular events activating the phosphoinositide-3 kinase (PI3K) pathway are frequently detected in human tumors and the activation of PI3K signaling alters numerous cellular processes including tumor cell proliferation, survival, and motility. More recent studies have highlighted the impact of PI3K signaling on the cellular response to interferons and other immunologic processes relevant to anti-tumor immunity. Given the ability of interferon (IFN)-γ to regulate antigen processing and presentation and the pivotal role of major histocompatibility complex (MHC) class I (MHCI) and II (MHCII) expression in T-cell mediated anti-tumor immunity, we sought to determine the impact of PI3K signaling on MHCI and MHCII induction by IFN-γ. We found that the induction of cell surface MHCI and MHCII molecules by IFN-γ is enhanced by the clinical grade PI3K inhibitors dactolisib and pictilisib. We also found that PI3K inhibition increases STAT1 protein levels following IFN-γ treatment and increases accessibility at genomic STAT1 binding motifs. Conversely, we found that pharmacologic activation of PI3K signaling can repress the induction of MHCI and MHCII molecules by IFN-γ and likewise the loss of PTEN attenuates the induction of MHCI, MHCII, and STAT1 by IFN-γ. Consistent with these in vitro studies, we found that within human head and neck squamous cell carcinomas, intra-tumoral regions with high phospho-AKT immunohistochemical (IHC) staining had reduced MHCI IHC staining. Collectively, these findings demonstrate that MHC expression can be modulated by PI3K signaling and suggest that activation of PI3K signaling may promote immune escape via effects on antigen presentation.
INTRODUCTION
The development of small molecule kinase inhibitors to counter aberrant growth factor signaling within human tumors has been a major advancement in the treatment of cancer (1). In addition to targeting the intrinsic enzymatic activity of growth factor receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR) and other members of the human EGFR (HER2–4) family, two RTK-downstream pathways that have received significant attention in this regard are the mitogen-activated protein kinase (MAPK) pathway and the phosphoinositide 3-kinase (PI3K) pathway (2, 3). The major paradigm driving the development of these inhibitors has centered on the role of these pathways in tumor cell autonomous processes such as the regulation of cellular proliferation, apoptosis, and metastasis. However, a growing number of studies have highlighted the immunologic impact of oncogenic signaling and underscored the need to understand how these pathways and the medications designed to inhibit them influence processes governing cell-mediated anti-tumor immunity (4).
A key process regulating anti-tumor immunity and the response to immune-based therapy is the presentation of peptide antigens by major histocompatibility class I (MHCI) and class II (MHCII) molecules (5, 6). This notion is supported by the fact that tumors frequently down regulate the expression of MHCI and/or MHCII molecules and that generally speaking, a loss of MHC expression is associated with a poor prognosis and/or a worse response to immunotherapy (7). Conversely, high MHCI and/or MHCII expression is typically associated with an improved clinical response to immunotherapy (7, 8). Given the above paradigms, it is important to fully understand how oncogenic signaling and the small molecule kinase inhibitors used to inhibit this signaling alters the expression of MHCI and MHCII molecules. In contrast to prior studies linking EGFR/HER and MAPK signaling to MHC expression, the impact of PI3K signaling on MHC expression is largely undefined (9, 10). This is intriguing given the fact that the PI3K pathway has been implicated in mediating resistance to some of the effects of interferon (IFN)-γ a potent inducer of both MHCI and MHCII expression (11). As such, we sought here to determine if PI3K activation and/or inhibition could influence the ability of IFN-γ to modulate MHC expression. We provide evidence that PI3K inhibition augments whereas PI3K activation represses the induction of MHC molecules by IFN-γ in some cellular contexts.
MATERIALS AND METHODS
Cell lines, chemical reagents and cytokines
SqCCY/Y1 cells were a gift from Dong Shin (Winship Cancer Institute, Atlanta Georgia) and HaCaT cells were purchased from AddexBio (San Diego, CA). FaDu and Detroit 562 cells were purchased from the ATCC (Manassas, VA) and used for experiments immediately after thawing at less than 20 passages. The above cells were grown in Dulbecco’s Modified Eagle Medium with 1 g/L D-glucose and L-Glutamine (ThermoFisher Scientific, Waltham, MA) with 10% fetal bovine serum at 5% CO2 without antibiotics. SqCC/Y1 and HaCaT cells were validated by short tandem repeat analysis at the Emory Integrated Genomics Core and mycoplasma testing was performed multiple times using a commercially available PCR-based assay per manufacture instruction over the time of these experiments (Millipore Sigma, St. Louis, MO). HCT116 colon carcinoma cells and HCT116 cells lacking PTEN were purchased from Horizon Discovery (Cambridge, United Kingdom) and were grown in RPMI 1640 Medium with L-glutamine (ThermoFisher Scientific, Waltham, MA) with 10% fetal bovine serum at 5% CO2 without antibiotics. Dactolisib (NVP-Bez235) was purchased from LC Laboratories (Woburn, MA). Pictilisib (GDC-0941) was purchased from Selleck Chemical (Houston, TX). SC79 was purchased from (Millipore Sigma, St. Louis, MO). VO-OHPIC was purchased from Tocris (Minneapolis, MN). All small molecules were dissolved in dimethylsulfoxide and stored at – 80 degrees C until use. Interferon-γ purchased from PeproTech (Rocky Hill, NJ).
Antibodies and Flow cytometry
MHC class I antibody clone G46–2.6 (Figure 1A) was purchased from Becton Dickinson (Franklin Lakes, NJ) and clone W6/32 (used in all other flow cytometry figures) was purchased from Biolegend (San Diego, CA). MHC class II antibody clone L203 (Figures 1A) was purchased from R & D Systems (Minneapolis, MN), and L243 (used in all other figures) purchased from Biolegend (San Diego, CA). Antibodies recognizing total STAT1, phospho-STAT1-Y701, phospho-STAT1-S727, total AKT, phospho-AKT-S473, phospho-AKT-T308, and PTEN were all purchased from Cell Signaling (Danvers, MA). Antibodies against MHC class I (clone EMR8–5) used for immunohistochemistry were purchased from MBL (Woburn, MA) and that for phospho-AKT-S473 from Abcam (Cambridge, United Kingdom).
Figure 1. PI3K inhibitors augment the induction of MHCI and MHCII molecules by IFN-γ.
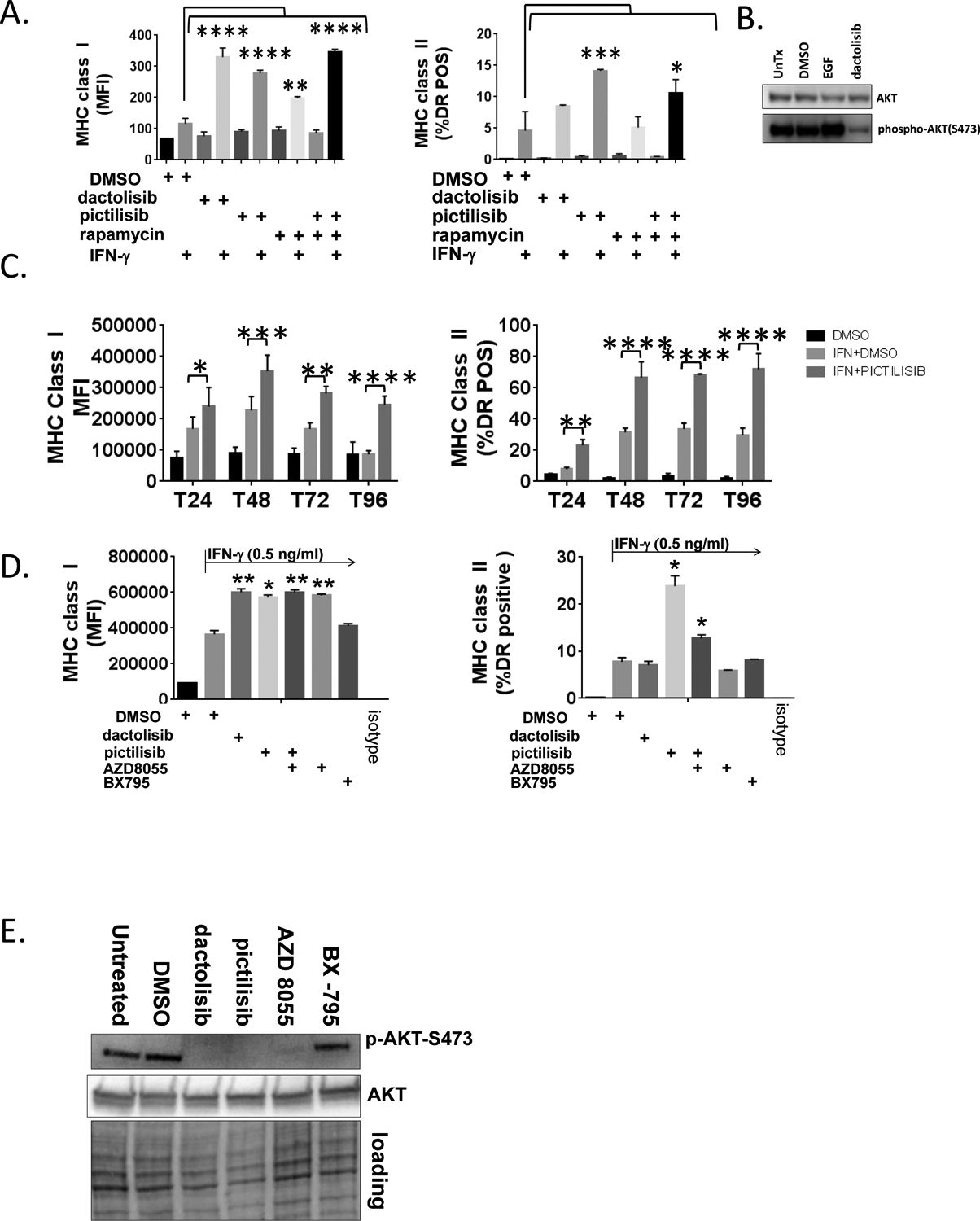
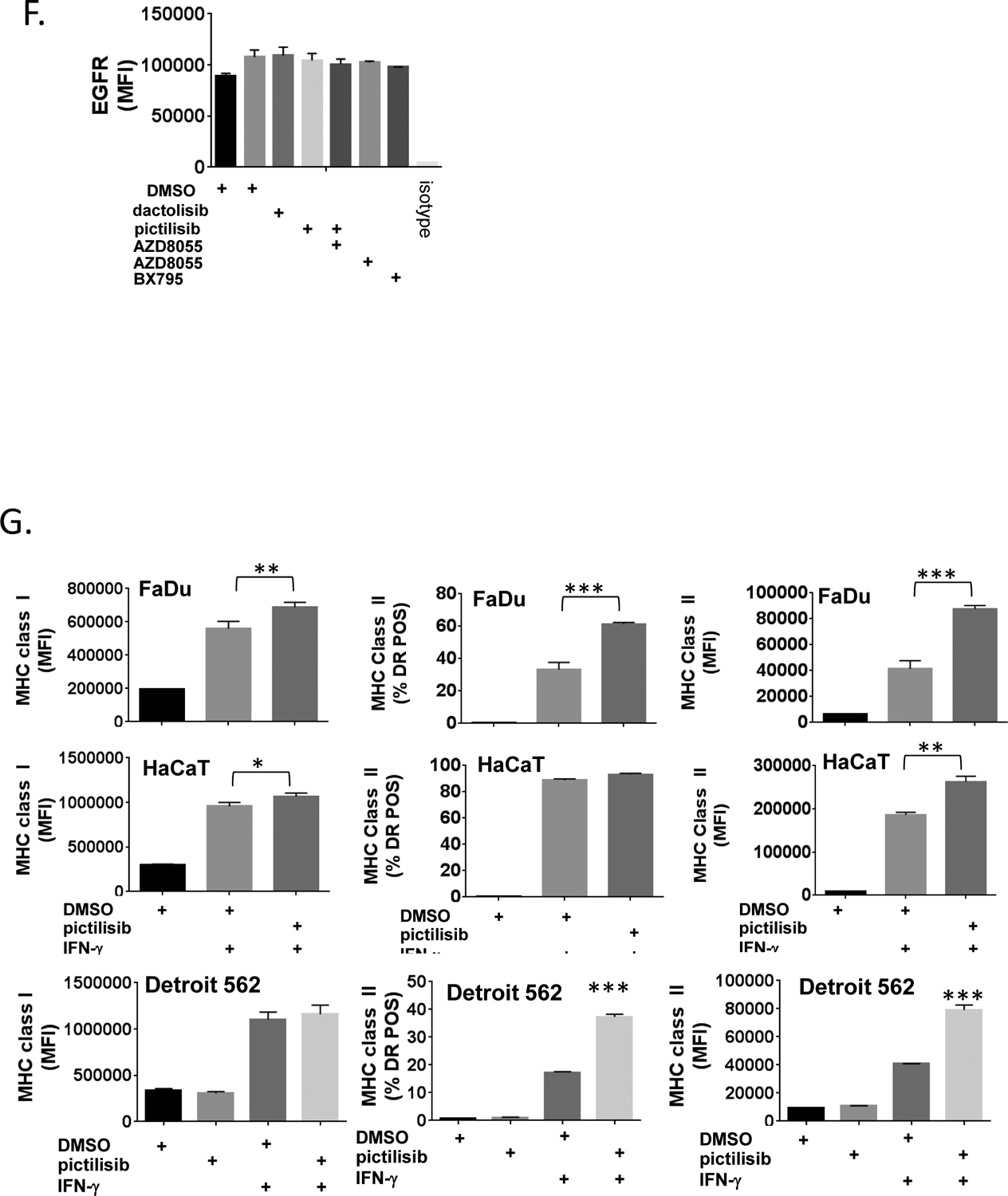
A., SqCC/Y1 cells were treated with vehicle (DMSO), dactolisib (0.5 mM), pictilisib (0.5 mM), rapamycin (1 mM), or both pictilisib and rapamycin alone or with IFN-γ (0.5 ng/ml) as indicated. Cell surface MHC class I and class II were analyzed by flow cytometry 48 hours later. The y-axes represent average mean fluorescence intensity (MFI) or averaged percent MHC class II positive (HLA-DR) cells from two independent experiments. B., Western blots of whole cell lysates from SqCC/Y1 cells treated with EGF (10 ng/ml for 15 minutes) or for 24 hours with vehicle (DMSO) or dactolisib (0.5 mM). C., Experiments were performed as indicated in panel A using 100 ng/ml of IFN-γ at the time points indicated. D., SqCC/Y1 cells were treated with vehicle (DMSO) alone or IFN-γ (0.5 ng/ml) plus vehicle, dactolisib (0.5 mM), pictilisib (0.5 mM), AZD8055 (0.5 mM), or Bx-795 and cell surface MHC class I and II analyzed 48 hours later. E., Western blots from SqCC/Y1 cell lysates treated for 60 minutes with the indicated inhibitors (all 0.5 mM). F., Cell surface expression of the EGFR from cells shown in D. G., FaDu, HaCaT, and Detroit 562 cells were treated as above using IFN-γ (0.1 ng/ml for HaCaT and FaDu and 0.5 ng/ml for Detroit 562) and MHC class I and II analyzed 48 hours later. The y-axes represent averages for two (Detroit 562) or three (HaCaT and FaDu) experiments. (*, p<0.05, **, p<0.01, ***, p<0.005, **** p<0.0001, 2way ANOVA)
Real-time RT-PCR
RNA isolation was carried out as previously described and reverse transcription was carried out using AzuraQuant cDNA synthesis kit from Azura Genomics (Raynham, MA) according to manufacturer instructions. Real-time PCR was performed on the Bio-Rad CFX96 platform using SYBR green and analyzed using Bio-Rad CFX Maestro software (Hercules, CA). All primers were purchased from RealTimePrimers.com (Elkins Park, PA).
Western blotting and western blot quantification
Whole cell protein lysates were prepared as previously described or using denaturing lysis buffer followed by column homogenization per manufacturers’ instructions (101BIO, Mountainview, CA) (12). SDS-PAGE was performed using Bio-Rad Criterion Stain Free Gels (Hercules, CA) and after electrophoresis stain-free gels underwent activation using ultraviolet radiation on the ChemiDOC XRS+ platform to allow for visualization and quantification of protein loading. Total protein was quantified from the gel and/or the blot following transfer per manufacturer instructions. Chemiluminescence signals from bands of the expected sizes were detected and quantified on the ChemiDOC XRS+ platform and normalized to the corresponding total protein loaded from the corresponding sample.
ATAC-seq
The ATAC-seq assay was performed as detailed previously (13). Briefly, 50,000 cells were resuspended in 25 μl Transposition Reaction (1x TD Buffer, 2.5 μl Tn5, 0.02% Digitonin, 0.1% Tween-20) and incubated for 60 min at 37°C. DNA was purified and PCR amplified using Nextera Indexing Primers (Illumina) and HiFi Polymerase (Kapa Biosystems). Libraries were sequenced at the Yerkes Genomics Core at the Emory Vaccine Center. Raw sequencing reads were mapped to the hg19 version of the human genome using Bowtie with the default parameters (14). Enriched accessible loci were identified by MACS2 and differentially accessible regions (DAR) determined between sample groups with edgeR (15, 16). Reads mapping to peaks were annotated and reads per million (rpm) normalized using custom R/Bioconductor scripts. For PCA analysis the vegan package in R/Bioconductor was used with Z-score normalized DAR as input. STAT1 motifs in DAR were identified using HOMER (17). All data processing scripts are available upon request. ATAC-seq data has been deposited in the NCBI GEO data base accession # GSE137719.
Mining of TCGA databases
TCGA genomic and RNA-seq expression data was accessed using cBioportal (www.cbioportal.org) (18, 19). Datasets used included the Head and Neck Squamous Cell Carcinoma (TCGA-HNSC, Provisional), Lung Squamous Cell Carcinoma (TCGA-LUSC, PanCancer), Pancreatic Adenocarcinoma (TCGA-PAAD, PanCancer), and Colon Adenocarcinoma (TCGA-COAD, PanCancer). Spearman and Pearson correlations between PTEN, PIK3CA and HLA genes were assessed using co-expression queries at cBioportal.org. Mutation, copy-number, and Z-score normalized RNAseq data for TCGA LUSC and PAAD were downloaded from cBioPortal and analyzed in R to generate violin plot distributions and compute p-values of differential HLA gene expression between cases with PTEN or PIK3CA alteration, and cases with no alterations in either gene.
Immunohistochemistry
Immunohistochemistry (IHC) was performed as previously reported and slide scanning was performed at the Emory Winship Core Pathology Laboratory (12). Formalin fixed paraffin embedded de-identified archived tumor samples were stained using antibodies against MHC class I (clone EMR8–5) and phospho-AKT-S473 (Abcam, catalogue number ab81283).
RESULTS
PI3K inhibitors enhance the induction of MHC class I and II molecules by IFN-γ in epithelial cells.
To understand the impact of PI3K inhibition on the induction of MHCI and MHCII by IFN-γ, we used SqCC/Y1 oral carcinoma cells as a model for squamous cell carcinoma of the head and neck (SCCHN) (20). To inhibit PI3K signaling, we used dactolisib, a dual PI3K / mammalian (also referred to as mechanistic) target of rapamycin (mTOR) inhibitor (also referred to as NVP-BEZ235) (21). We found that 0.5 μM dactolisib increased the average mean fluorescence intensity (MFI) for MHCI following IFN-γ (Figure 1A). In contrast, the impact of dactolisib on MHCII expression was not significantly different in these experiments and varied in other experiments depending on the cell line used and the conditions of the experiments (data not shown) (Figure 1A) (22). Since dactolisib inhibits mTOR in addition to being a pan-PI3K inhibitor, we also examined the impact of another PI3K inhibitor, pictilisib (also referred to GDC-0941), on the induction of MHCI and MHCII by IFN-γ (23). We included treatments with rapamycin, a canonical mTOR inhibitor to further define the roles of PI3K and mTOR on the induction of MHCI and MHCII by IFN-γ. We found that like dactolisib, pictilisib could enhance the induction of MHCI induction by IFN-γ (Figure 1A). Consistent with a prior study examining the effect of rapamycin on a murine model of head and neck squamous cell carcinoma (HNSCC), in our model, rapamycin augmented the induction of MHCI by IFN-γ though to a lesser extent than either of the PI3K inhibitors (Figure 1A) (24). In addition to MHCI, pictilisib repeatedly augmented MHCII induction (Figure 1B). In contrast to MHCI where rapamycin had a slight enhancing effect on pictilisb, rapamycin attenuated the ability of pictilisib to enhance MHCII induction to levels that approximated those of dactolisib. As expected for a PI3K inhibitor, dactolisib decreased levels of phosphorylated AKT-S473 as determined by western blotting (Figure 1B) as did pictilisib (Figure 1E). These results suggest that PI3K activity and/or mTOR activity can modulate the induction of MHCI and MHCII by IFN-γ in some cellular contexts.
To further define the impact of pictilisib on MHC induction, a time course was conducted over 96 hours. As shown in Figure 1C, in the presence of pictilisib there were sustained increases in cell surface levels of MHCI and MHCII over the entire course of the experiment. To better define the roles of mTOR in MHCI and MHCII induction, we repeated experiments using a pan mTOR inhibitor AZD8055 and a PDK1 inhibitor BX795. As shown in Figure 1D and as we observed with rapamycin, AZD8055 by itself augmented MHCI levels following IFN-γ and did not alter the effect of pictilisib on MHCI induction. In contrast and like rapamycin, AZD8055 attenuated the impact of pictilisib on MHCII levels. The PDK1 inhibitor BX795 had no effect on MHCI or II induction. As shown in Figure 1E, dactolisib, pictilisib and AZD8055 reduced levels of phospho-AKT-S473. In contrast to the changes seen in MHCI and/or MHCII we observed no significant changes in EGFR expression with these treatments (Figure 1F).
To determine the impact of pictilisib on MHCI/MHCII induction in other cellular models, we performed similar studies on Fadu cells which are derived from a tumor of the hypopharynx, HaCaT cells, an immortalized epidermal keratinocyte cell line, and Detroit 562 cells which are derived from a pharyngeal metastatic tumor and harbor an activating mutation in PIK3CA (25–27). Pictilisib augmented the induction of MHCII in all cell lines as measured by average MFI and/or the percentage of cells expressing the MHCII gene HLA-DR (Figure 1G). In contrast, the impact of pictilisib on MHCI induction was less robust though still significant in FaDu and HaCaT cells. These results support the notion that inhibition of PI3K using pictilisib can alter the ability of IFN-γ to induce genes involved in antigen presentation.
Signal transducer and activator of transcription 1 (STAT1) is a key regulator of both MHCI and MHCII expression and both STAT1 transcriptional activity and expression are increased by IFN-γ (28–31). Interestingly, STAT1 expression has been reported to be repressed by signaling mediated by oncogenic RAS which is upstream of PI3K signaling (32, 33). Therefore, we tested the impact of PI3K inhibition using pictilisib on STAT1 induction by IFN-γ and found that pictilisib increased the induction of total STAT1 by IFN-γ with concomitant increases in phospho-STAT1-Y701 and S727 (Figure 2A and 2B). These results suggest that in some contexts, PI3K signaling can act as a negative regulator of STAT1 expression. This underscores the complex interplay between the PI3K pathway and given that prior results have shown that the PI3K pathway phosphorylates STAT1 at serine 727; a modification that has been reported to be required for STAT1 to be fully active as a transcriptional regulator (34).
Figure 2. The PI3 kinase inhibitor pictilisib augments STAT1 protein induction by IFN-γ.
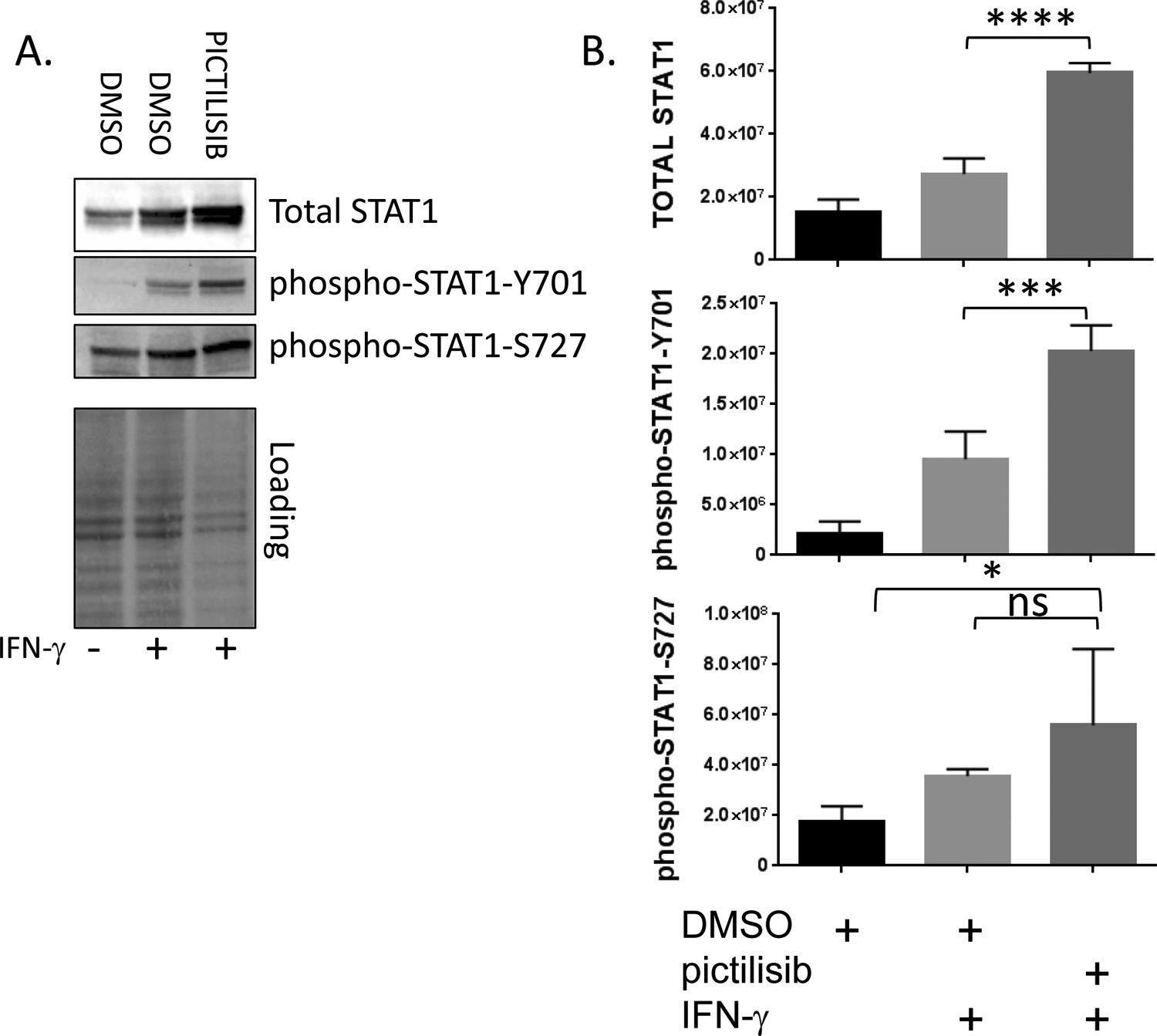
A., SqCC/Y1 cells were treated with vehicle (DMSO) alone, vehicle plus IFN-γ (1 ng/ml) or pictilisib (0.5 mM) plus IFN-γ. Whole cell lysates were prepared 24 hours after the addition of IFN-γ and total-STAT1, phospho-STAT1-Y701 and phospho-STAT1-S727 protein expression analyzed by western blot. Total protein loading on a representative blot is shown in the bottom panel. B., Averaged total-STAT1 (top panel), phospho-STAT1-Y701 (middle panel) and phospho-STAT1-S727 signal intensities (bottom panel) from four experiments are shown normalized to total protein. (*, p<0.05, ***, p<0.001, **** p<0.0001, adjusted P values using 1way ANOVA).
Pictilisib increases accessibility at genomic STAT1 consensus sequences.
The cellular response to IFN-γ involves changes in transcription that are associated with changes in chromatin structure (35, 36). To better define the impact of PI3K inhibition on this aspect of the cellular response to IFN-γ, we performed ATAC-seq on SqCC/Y1 cells treated with vehicle (DMSO) and pictilisib alone or in the presence of IFN-γ (37). ATAC-seq provides information about chromatin structure because the transposase used in this assay can access genomic DNA and place DNA linkers preferentially in regions of open chromatin (which are typically transcriptionally active) whereas this is less probable in regions with more compact chromatin. Principal component analysis of differentially accessible regions (DAR) highlights the impact of pictilisib on the response to IFN-γ (Figure 3A). Although there is small variation between cells treated with DMSO and pictilisib alone, in the presence of IFN-γ the variation is more pronounced. Importantly, and in line with our data showing that pictilisib increases STAT1 induction by IFN-γ, pictilisib treatment leads to significantly increased chromatin accessibility at regions surrounding STAT1 binding consensus sequences as revealed by ATAC-seq footprinting (Figure 3B and 3C). Thus, PI3K inhibition not only augments the induction of STAT1 by IFN-γ but in addition alters the changes to chromatin structure induced by IFN-γ including those at STAT1 binding sites.
Figure 3. The PI3 kinase inhibitor pictilisib augments chromatin accessibility at STAT1 binding sites.
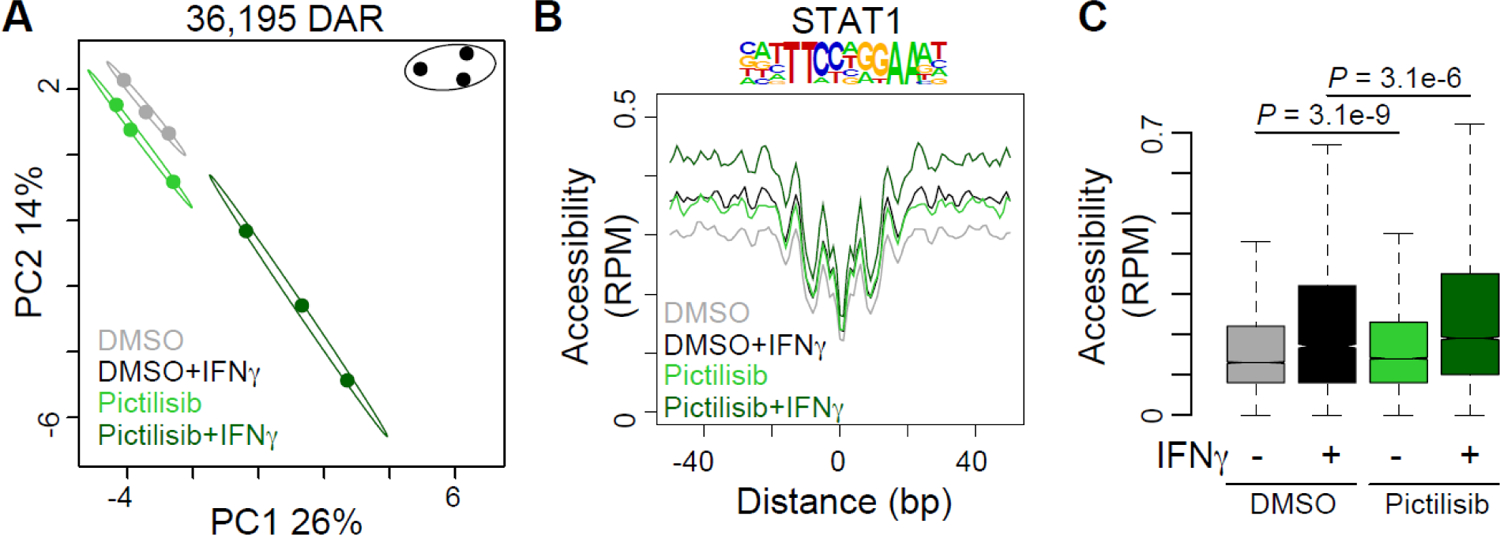
SqCC/Y1 cells were treated with vehicle (DMSO) alone, vehicle plus IFN-γ (100 ng/ml), pictilisib alone (0.5 mM) or pictilisib plus IFN-γ. Forty-eight hours after IFN-γ treatment, chromatin accessibility was assessed using ATAC-seq. A., Principle component analysis of differentially accessible regions (DAR) between the four treatment groups. Circles represent 95% confidence intervals for each group. B. Histogram of chromatin accessibility in each group at STAT1 consensus motifs located in DAR. 100bp surrounding each motif is shown. RPM, reads per million. C., Box plots summarizing accessibility at STAT1 motifs from B. Significance determined by two-tailed Student’s T-test.
Pharmacologic activation of PI3K signaling represses the induction of MHCI and MHCII by IFN-γ.
The above results whereby PI3K inhibitors increase MHC levels suggest that PI3K signaling has a repressive effect on the induction of MHCI and/or MHCII by IFN-γ. To further explore this notion, we examined how the activation of PI3K signaling impacts the induction of MHC molecules by IFN-γ. Because AKT is a key downstream effector of PI3K signaling, we employed a pan-AKT activator (named SC79) in our assay. SC79 binds to the pleckstrin homology domain of AKT and promotes its phosphorylation and activation by upstream kinases (38). We found that SC79 had the opposite effect of the PI3K inhibitors and repressed both MHCI and MHCII induction by IFN-γ in SqCC/Y1 cells (Figure 4A). As expected, SC79 promoted AKT phosphorylation at serine 473 and threonine 408 (Figure 4B). To complement these studies and activate PI3K signaling via a different mechanism, we targeted the activity of phosphatase and tensin homolog (PTEN), a pivotal negative regulator of PI3K signaling and a well-defined tumor suppressor (39). For these studies we used a PTEN inhibitor, VO-OHpic, which is a 3-hydroxypicolinate vanadium complex that inhibits the phosphatase activity of PTEN (40). As we observed with the AKT activator SC79, the PTEN inhibitor VO-OHpic repressed the induction of MHCI and MHCII by IFN-γ (Figure 4C). As expected, the PTEN inhibitor VO-OHpic activated PI3K signaling and promoted the phosphorylation of AKT at serine 473 and threonine 308 (Figure 4D).
Figure 4. Activation of PI3K signaling represses the induction of MHC molecules by IFN-γ.
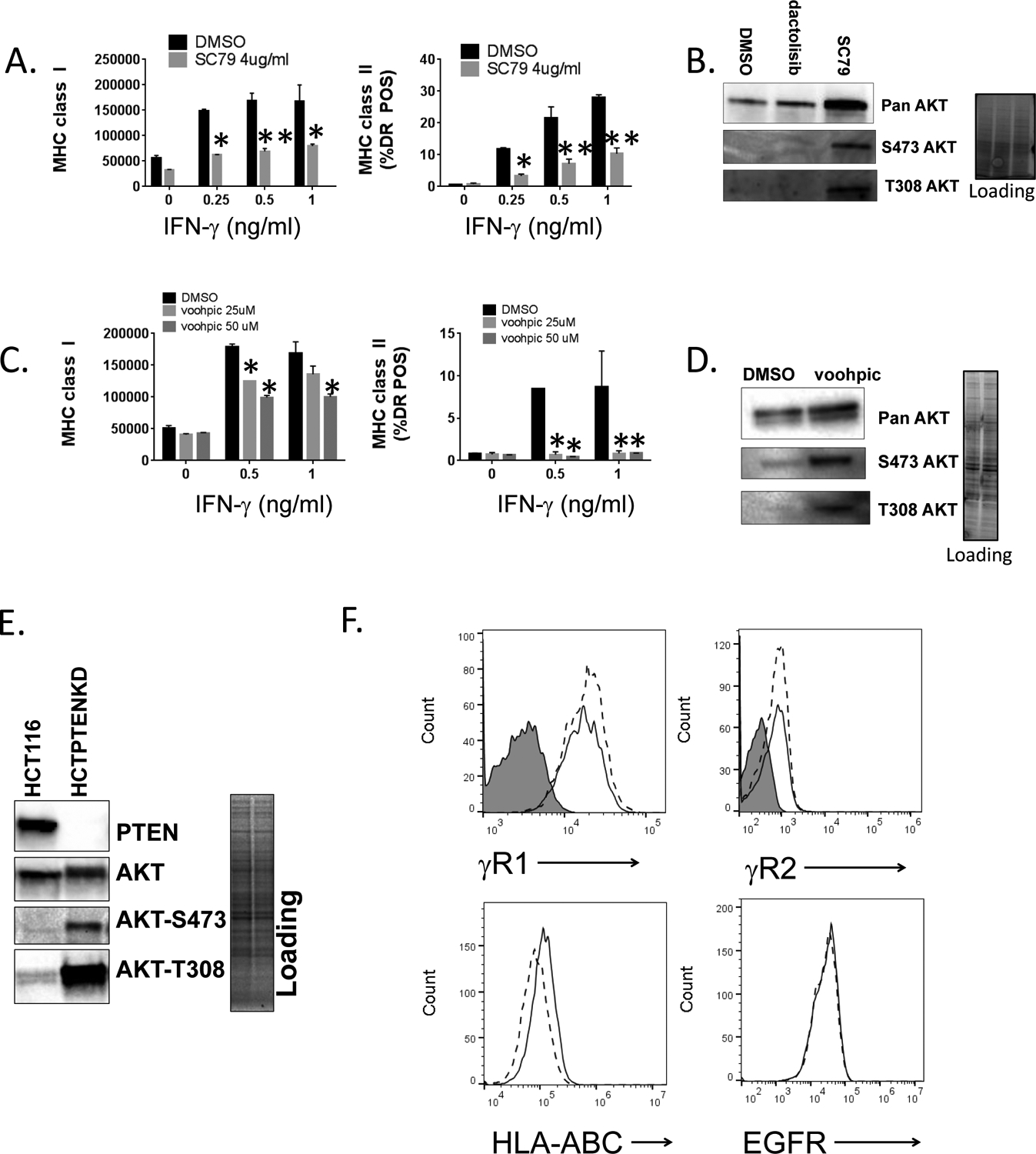
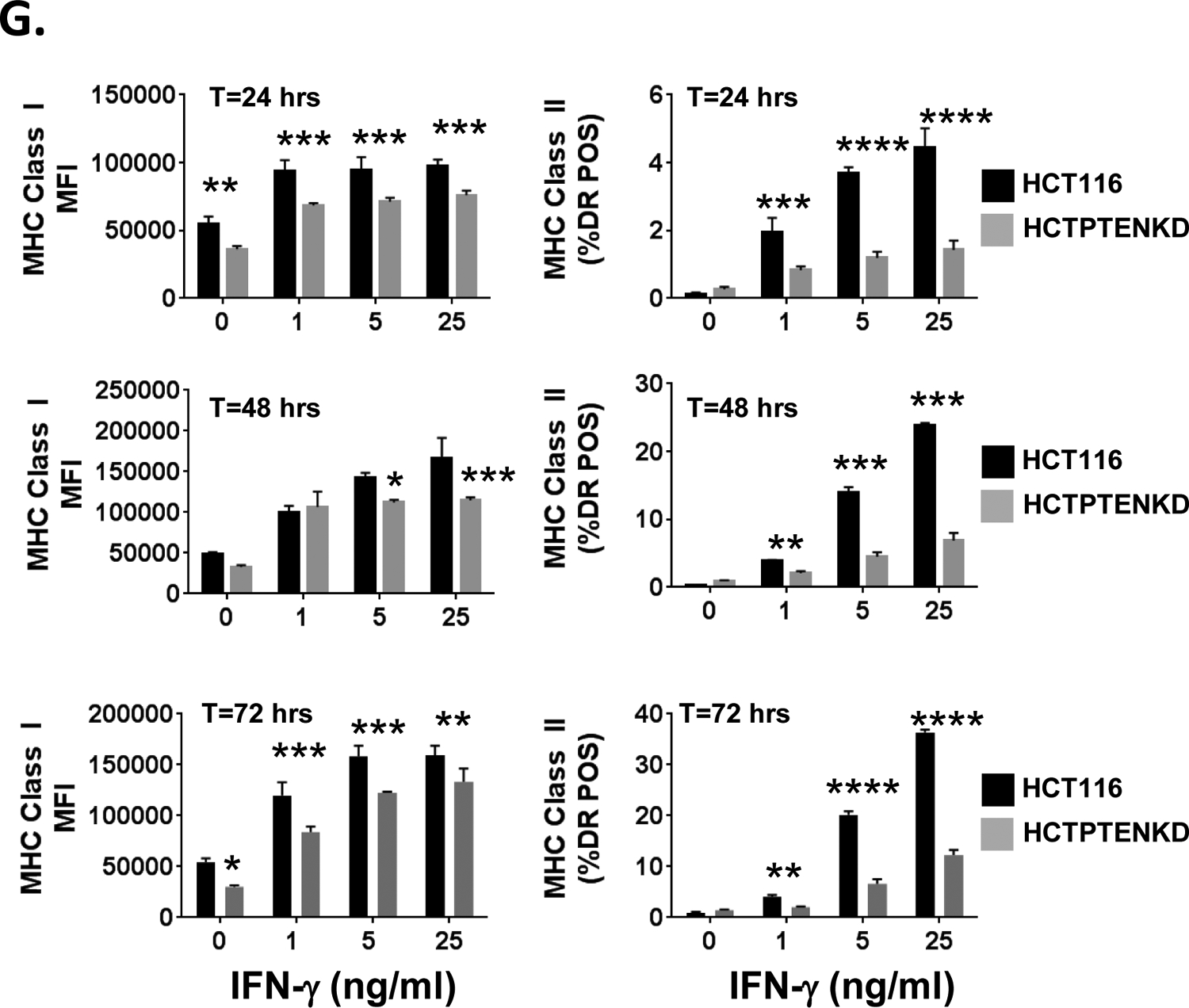
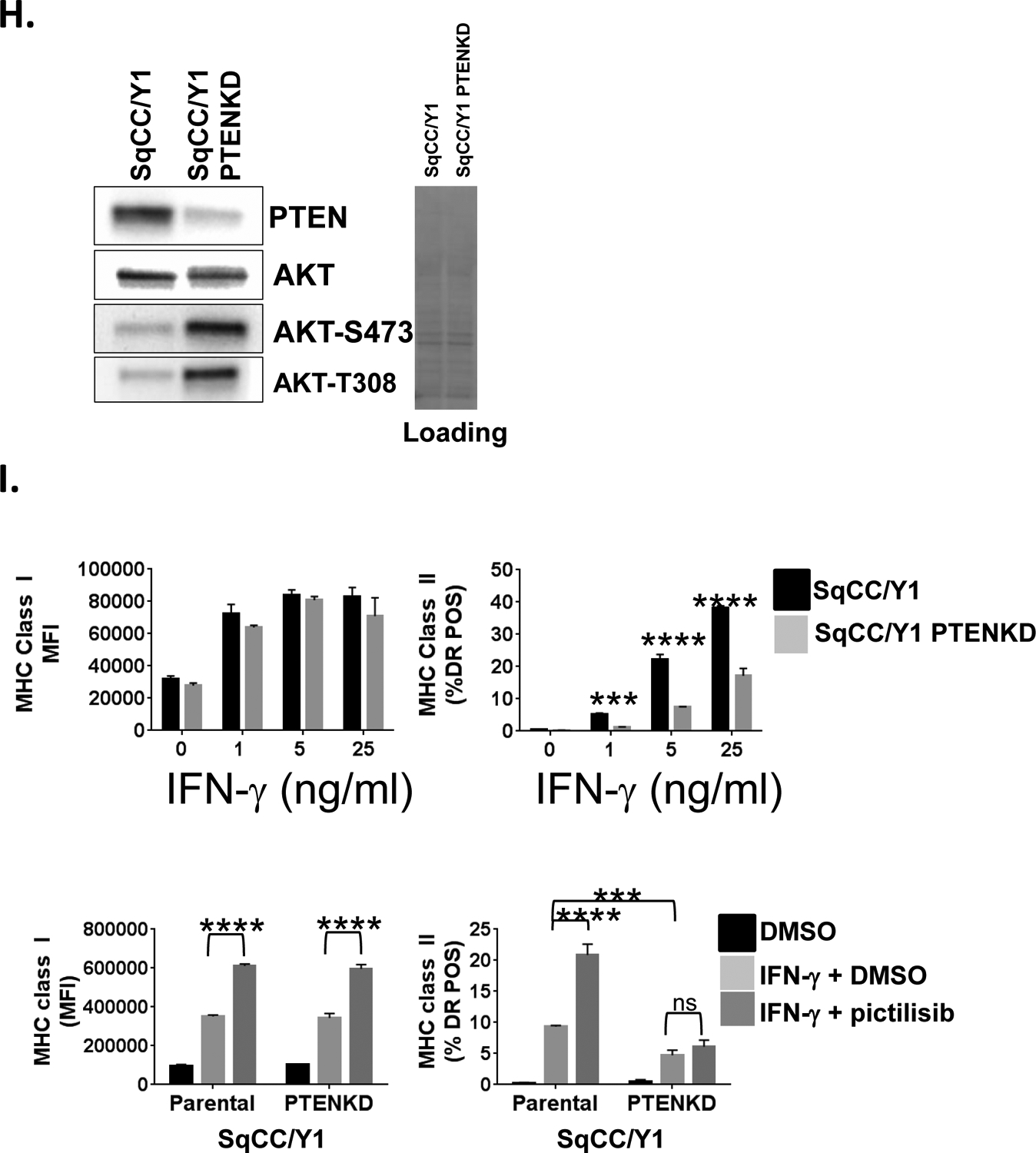
A., SqCC/Y1 cells were treated with IFN-γ plus vehicle (DMSO) or plus SC79 (4 mg/ml) and cell surface MHC class I (left panel) and MHC class II (right panel) measured by flow cytometry 48 hours later. The y-axes represent the averaged mean fluorescence intensity (MHCI) or the averaged % of MHC class II-positive cells from two experiments. B., Representative western blots of whole cell lysates from SqCC/Y1 cells treated for 30 minutes with DMSO, dactolisib, or SC79 (4 mg/ml). Total protein loading is shown in panel on right. C., SqCC/Y1 oral carcinoma cells were treated with IFN-γ plus vehicle (DMSO) or plus VO-OHpic (voohpic, 25 or 50 mM) and cell surface MHC class I (left panel) and MHC class II (right panel) measured by flow cytometry 48 hours later. D., Representative western blots of whole cell lysates from SqCC/Y1 cells treated for 24 hours with DMSO or VO-OHpic. Total protein loading is shown in panel on right. E., Western blots showing expression of PTEN, total AKT, and phospho-AKT in HCT116 cells and HCT116 cells lacking PTEN (HCTPTENKD). Total protein loading is shown in panel on right. F., Top panels, representative histograms of HCT116 (solid line) and HCTPTENKD cells (dashed line) stained with antibodies against IFNγR1, IFNγR2 or left unstained (gray filled). Bottom panels, HCT116 (solid line) and HCTPTENKD (dashed line) cells stained with antibodies against MHC class I (HLA-ABC) or the EGFR. G., Flow cytometric data from HCT116 cells (black bars) or those lacking PTEN (gray bars) which were treated with IFN-γ at the concentrations indicated along the x-axis. Cell surface MHC class I (left side) and II (right side) were measured 24 (top panels), 48 (middle panels) and 72 (bottom panels) hours later. The y-axis for MHC class I represents averaged mean fluorescence intensity from three experiments and that for MHC class II represents the averaged % HLA-DR positive cells from three independent experiments. H., Western blots of parental SqCC/Y1 cells and those with reduced PTEN expression (SqCC/Y1 PTENKD). I., Top panels, SqCC/Y1 and SqCC/Y1 PTENKD cells were treated with IFN-γ at the concentrations indicated and MHC class I and II expression measured 48 hours later. Bottom panels, SqCC/Y1 and SqCC/Y1 PTENKD cells were treated with vehicle (DMSO), IFN-γ (0.5 ng/ml) plus vehicle, or IFN-γ plus pictilisib (0.5 mM) and surface MHC class I and II levels measured 48 hours later. The y-axes represent averaged values for three experiments. (*, p<0.05, **, p<0.01, *** p<0.005 and **** < 0.001 adjusted P values using 2way ANOVA).
Loss of PTEN represses the induction of MHCI and MHCII by IFN-γ.
While the above pharmacologic studies targeting PI3K, AKT, and PTEN suggest that PI3K signaling can modulate MHCI and MHCII induction, we next sought to further assess these interactions using a genetic model. To this end, we assessed how the loss of PTEN expression would influence MHCI and MHCII induction by IFN-γ using commercially available parental HCT116 colon carcinoma cells and HCT116 cells that were rendered PTEN null by CRISPR/Cas9 gene editing (41). As expected, the HCT116 cells lacking PTEN showed evidence of increased PI3K signaling as measured by AKT phosphorylation at serine 473 and threonine 308 (Figure 4E). We confirmed that parental HCT116 cells and those lacking PTEN expressed both chains of the IFN-γ receptor complex (Figure 4F). In addition, while both cell lines expressed the same level of the EGFR, we repeatedly observed lower surface MHCI levels in untreated HCT116 cells lacking PTEN (Figure 4F). Consistent with our prior findings, the induction of MHCI and MHCII by IFN-γ was attenuated in HCT116 cells lacking PTEN at three different IFN-γ concentrations and at 24, 48, and 72 hour time points after IFN-γ treatment (Figure 4G). We also examined the impact of PTEN loss on SqCC/Y1 cells. While some PTEN expression remains, the decrease in PTEN lead to an increase in AKT phosphorylation (Figure 4H). In addition, the ability of IFN-γ to induce MHC class II expression was significantly decreased in SqCC/Y1 cells with decreased levels of PTEN though there were only small differences in MHCI induction (Figure 4I). In addition, the ability of pictilisib to enhance MHCII induction was lost in SqCC/Y1 cells with reduced PTEN expression. Taken together, these results demonstrate that activation of PI3K signaling can repress the induction of MHCI and/or MHCII by IFN-γ and alter the ability of PI3K inhibition to augment MHC induction.
Loss of PTEN predominantly impacts the expression of HLA-B.
While the loss of PTEN in HCT116 cells impacted basal MHCI expression and the induction of MHCI and MHCII by IFN-γ, the effect was most pronounced on MHCII induction and more modest on the induction of MHCI. While all three MHCI genes (HLA-A, B and C) are upregulated by IFN-γ, there are gene-specific differences in their transcriptional regulation and the expression status a single MHCI gene may have important clinical implications in the setting of immunotherapy or resistance to immunotherapy (7, 42, 43). To determine if the loss of PTEN has MHCI gene-specific effects, we assessed the expression of HLA-A, B, and C protein separately by western blot in untreated and IFN-γ treated HCT116 cells and those lacking PTEN. As shown in Figure 5A and 5B, the loss of PTEN had the most marked impact on HLA-B protein expression and its induction by IFN-γ.
Figure 5. Impact of PTEN loss of the expression of MHC class I genes and STAT1 in HCT116 cells.
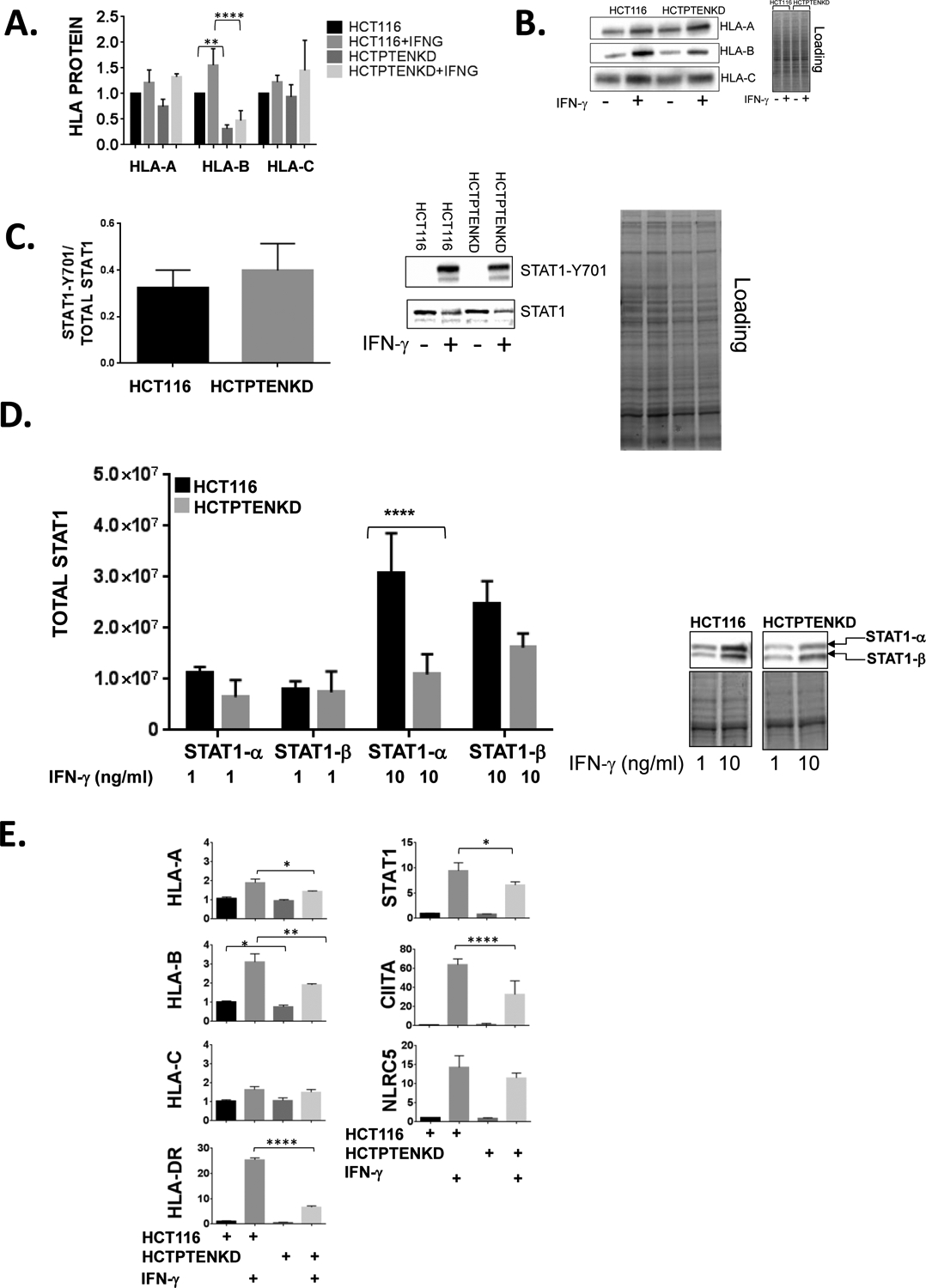
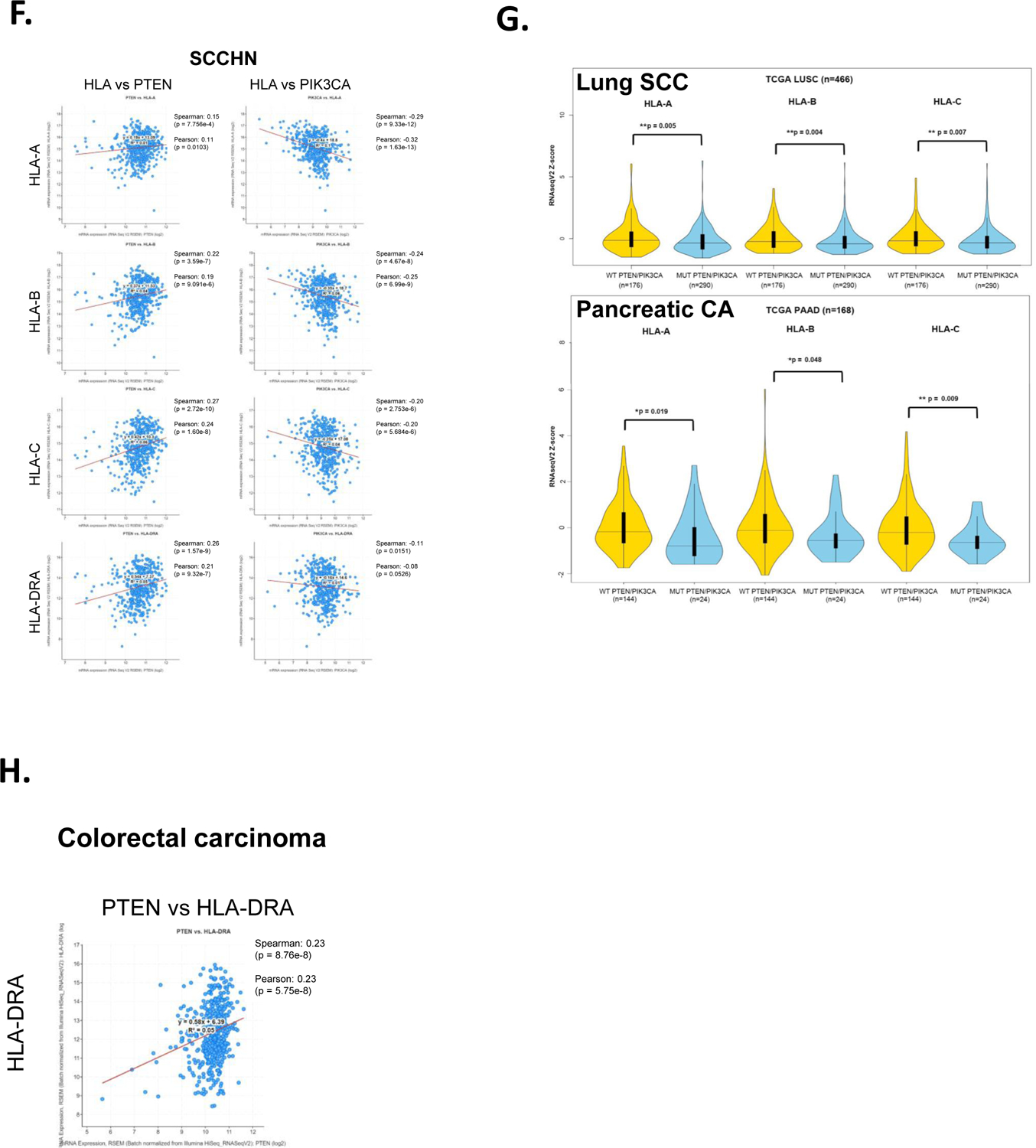
A., HCT116 cells and HCT116 cells lacking PTEN (HCTPTENKD) were left untreated or treated with IFN-γ (IFNG, 1ng/mL) and whole cell lysates prepared 24 hours later. The y-axis represents averaged, HLA-A, HLA-B and HLA-C values from 3 experiments and are normalized to total protein and are expressed relative to untreated parental HCT116 cells. (**, p<0.01, ****, p<0.0001, adjusted p values from 2way ANOVA). B., Representative western blots are shown for HLA-A, HLA-B and HLA-C protein expression and total protein loading for HLA-B western blot. C., STAT1 phosphorylation was assessed 90 minutes after IFN-γ treatment (5 ng/ml) of HCT116 and HCTPTENKD cells and was quantified by western blot. Values are normalized to total STAT1-a and represent the average of three replicate experiments. Representative western blots of STAT1-Y701 and total STAT1 are shown. D., Quantification of STAT1-a and STAT1-b protein levels in HCT116 cells (black bars) or HCTPTENKD cells (gray bars) treated with IFN-γ at the concentrations indicated along the x-axis for 48 hours. STAT1-a and STAT1-b expression are normalized to total protein. The y-axis represents averages from three experiments. Right panel, representative STAT1 western blots and protein loading. Error bars represent the standard deviation. (***, <0.001 adjusted P values using 2way ANOVA). E., Analysis of steady state mRNA levels in HCT116 and HCTPTENKD cells treated with IFN-γ (4 ng/ml). Expression of the genes indicated was measured using quantitative real-time RT-PCR. Values are normalized to beta glucuronidase and are expressed relative to untreated parental HCT116 cells. Values shown represent averages from three experiments. Error bars represent the standard deviation. (*, p<0.05, **, p < 0.01, ****, p<0.0001 using students t-test). F., TCGA RNA-seq data from squamous cell carcinoma of the head and neck showing correlation of expression between HLA-A, HLA-B and HLA-C with PTEN or PIK3CA. G., TCGA RNA-seq data from lung squamous cell carcinoma and pancreatic adenocarcinoma showing violin plots of HLA-A, HLA-B and HLA-C mRNA in patients with mutations or copy number changes in PTEN or PIK3CA (blue) compared to patients without those genetic alterations (yellow). H., TCGA colorectal carcinoma RNA-seq data showing correlation between PTEN mRNA expression and HLA-DRA expression.
Loss of PTEN represses the induction of STAT1 by IFN-γ.
Given the increase in STAT1 protein induction that we observed in the presence of the PI3K inhibitor pictilisib, we examined how the loss of PTEN would influence STAT1phosphorylation and its induction by IFN-γ in parental HCT116 cells and those lacking PTEN. As shown in Figure 5C, there were no differences between parental HCT116 and HCT116 PTEN knockout cells with regards to STAT-1 tyrosine 701 phosphorylation 90 minutes following IFN-γ treatment. In contrast, when we assessed STAT1 levels 48 hours after IFN-γ treatment, STAT1 levels were reduced in HCT116 cells lacking PTEN (Figure 5D). Interestingly, the lack of PTEN had a more prominent impact on the induction of the full length STAT1 protein (STAT1-α) as compared to the induction of STAT1-β which lacks the carboxy-terminal transactivation domain of 38 amino acids.
Effects of PTEN loss on MHC steady state mRNA levels.
To assess if the loss of PTEN impacts MHC and STAT1 expression at the mRNA level, real-time reverse transcriptase (RT)-PCR was performed on untreated and IFN-γ-treated parental HCT116 cells and those lacking PTEN. We also examined the expression of the key transcriptional co-activators of MHCI and MHCII known as NLRC5 and CIITA respectively (44, 45). With respect to MHCI genes, we found that the lack of PTEN had the largest impact on steady state mRNA levels of HLA-B, a smaller impact on HLA-A and no significant impacts on HLA-C or NLRC5 (Figure 5E). This pattern is similar to what we observed at the protein level where the loss of PTEN had the greatest impact on HLA-B expression and less of an effect on HLA-A and HLA-C. The lack of PTEN also had a significant impact on the induction of STAT1 and the MHCII gene HLA-DR as well as its co-activator CIITA, which drives MHCII expression (46). These findings suggest that some of the effects of PTEN loss on the response to IFN-γ are due to changes in steady state mRNA levels of IFN-γ-regulated genes.
To further define the interplay between PI3K/PTEN and MHC expression, we examined public databases containing genomic and RNA-seq data in SCCHN. As expected from the data outlined above, there were positive correlations between MHC class I and II expression with PTEN expression and negative correlations between MHC and PIK3CA expression (Figure 5F). However, we found no differences in MHC levels between SCCHN tumors with genetic alterations in PTEN or PIK3CA and tumors lacking alterations in these genes. In contrast, when we extended our analysis to include other cancers, we found that in both pancreatic cancer and squamous cell carcinoma (SCC) of the lung tumors with alteration in PTEN or PIK3CA had statistically significant lower expression levels of HLA-A, B and C (Figure 5G). With regards to colon carcinoma and consistent with our data using HCT116 cells, we found a positive correlation between PTEN expression and the expression of the MHCII gene HLA-DRA (Figure 5H).
Intra-tumoral regions with high levels of phospho-AKT have decreased MHCI staining.
The above studies provide evidence that there’s an inverse relationship between PI3K signaling and MHC expression. To further explore these interactions in vivo, we used immunohistochemistry (IHC) to asses MHCI protein expression and the expression of phosphorylated AKT-S473 (p-AKT) as a marker of activated PI3K signaling in squamous cell carcinomas of the head and neck. As shown in the panels within Figure 6, while MHCI expression is readily seen, there is regional variation of MHCI staining intensity within tumor nodules with regions of high MHCI staining intensity as well as areas with less MHCI staining intensity. Importantly, in tumors from different patients, we repeatedly observed that the staining pattern of phospho-AKT appeared as an inverse image of that for MHC class I. Namely, regions with the highest MHCI staining had low to no detectable phospho-AKT staining and conversely, regions with high phospho-AKT staining had conspicuously lower levels of MHCI staining (compared to other regions within the tumor). Thus, intra-tumoral regions with increased PI3K signaling are associated with a decrease in MHCI protein expression.
Figure 6. Inverse immunohistochemical staining of MHC class I and phospho-AKT-S473 in human squamous cell carcinoma.

Squamous cell carcinomas of the head and neck were stained for MHC class I (top panels of each example) and phospho-AKT-S473 (bottom panels of each example). Regions of lower staining (black outline) of MHCI correspond to regions of highest phospho-AKT staining (yellow outlines) and vice versa. In some images, such regions are indicated using double headed arrows. Areas of stroma are indicated with brackets in some of the tumor images.
DISCUSSION
In this study, we examined the impact of inhibition and activation of the PI3K pathway on the expression of MHC class I and II molecules and their induction by IFN-γ, a cytokine that plays a pivotal role in immune surveillance, anti-tumor immunity, and the response to immunotherapy (47, 48). Using human cell lines and archived squamous cell carcinomas of the head and neck, we demonstrate that activation of PI3K signaling is associated with a repression of MHC expression/induction. Importantly, during the time our manuscript was under review, two groups (Marijt et al., and Sivaram et al.) published studies that support our findings. In these studies, murine model systems were used demonstrating that: (1) PI3K activation (through low oxygen and/or glucose) can repress STAT1 signaling and MHCI induction/expression and (2) PI3K-mediated changes in MHCI levels can functionally impact T cell infiltration into tumors and anti-tumor immunity (49, 50). Collectively, our work and the work of these authors support a broader model of the PI3K pathway that links PI3K signaling to MHC expression (Figure 7). Given the frequency of PI3K signaling alterations in human cancer and the pivotal role of MHC expression in anti-tumor immunity, moving forward it will be critical to define PI3K-MHC interactions and develop approaches to reverse the repressive effects of PI3K signaling on MHC expression (2, 51).
Figure 7. Model of PI3K regulation of MHC expression.
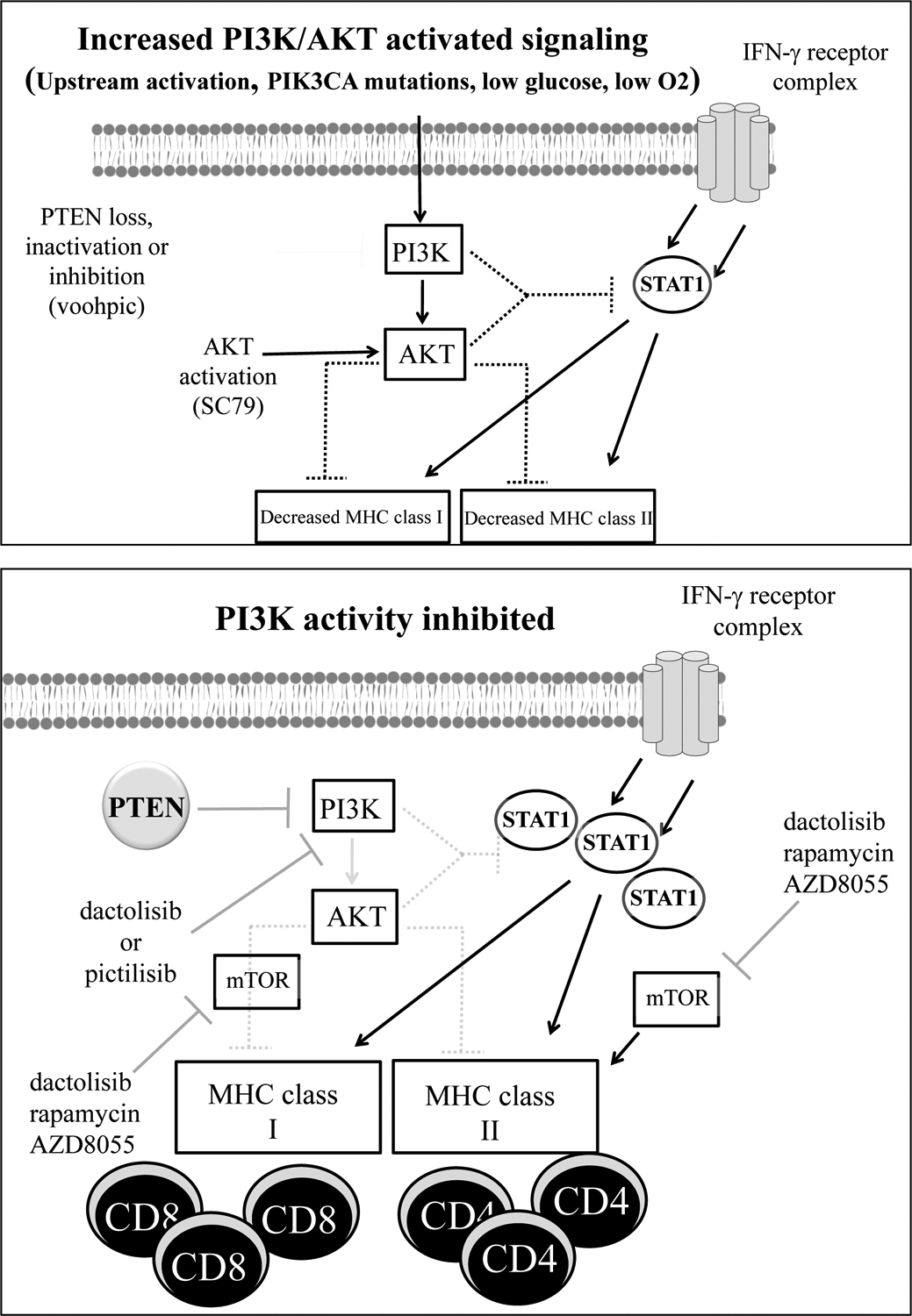
Top panel, PI3K-activated signaling can occur via upstream activating events, activating mutations in PIK3CA, or via low glucose/oxygen levels as reported by Marijt et al. In addition, PTEN loss or inhibition as well as activation of AKT can activate PI3K signaling. In some settings, these events can have a repressive effect on the expression of MHC molecules and/or their induction by IFN-γ possibly by attenuating the ability of IFN-γ to increase STAT1 protein levels and/or phosphorylation. Decreases in MHC levels may hinder CD8+ and/or CD4+ T-cell activation, tumor recognition, and anti-tumor immunity. Bottom panel, in the setting of PI3K inhibition using PI3K inhibitors such as dactolisib or pictilisib, or the loss of PIK3CA as recently reported by Sivaram et al., the repressive effects of PI3K signaling on MHC expression are diminished. This allows for increased MHC expression and/or induction by IFN-γ possibly mediated by increases in STAT1 protein levels and/or phosphorylation. The increases in MHC expression may promote CD4+ and/or CD8+ activation and anti-tumor immunity. mTOR inhibition by dactolisib, rapamycin or AZD8055 does not alter the impact of PI3K inhibition on MHCI levels and by itself can enhance MHCI levels induced by IFN-γ though mTOR inhibition attenuates the ability of PI3K inhibitors to enhance MHCII induction.
In contrast to the PI3K pathway, several prior studies have demonstrated that EGFR/HER activity and the activity of enzymes in the MAPK pathway (such as BRAFV600E and MEK1/2) can influence MHC expression. For example, expression of HER2 and BRAFV600E have been reported to downregulate MHCI levels in breast cancer and melanoma respectively and with regards to gastric cancer, an inverse correlation between MAPK activation (as measured by levels of phosphorylated extracellular signal-related kinase (ERK)) and MHCI has been reported (10, 22, 52–54). These studies are supported by a recent RNAi screen for HLA-regulating kinases where MAP2K1 (MEK), the EGFR and RET were validated as negative regulators of MHC expression (55). Conversely, EGFR, HER2, and MAPK inhibitors have been shown to increase levels of MHCI and/or MHCII expression (56). Our data combined with that of Marijt el al. and Sivaram et al. add to these studies and now demonstrate that in some contexts, PI3K signaling can also influence MHCI and/or MHCII induction.
Tumor cell MHC expression is pivotal to host anti-tumor immunity and the response to immune-based therapies such as immune checkpoint blockade (57). At present it is unclear if changes in MHCI and/or MHCII expression/induction mediated by PI3K signaling are functionally relevant to anti-tumor immunity in humans. However, our data supports other observations whereby activating events in PI3K signaling are associated with mechanisms of immune escape and/or resistance to the effects of IFN-γ (48, 58, 59). For example, prior studies have shown that the loss of PTEN can alter the anti-proliferative and cytotoxic effects of IFN-γ in lung and gastric carcinoma models (11, 59). Our work supports these observations and suggests that the loss of PTEN also represses the ability of IFN-γ to induce MHCI and/or MHCII expression. Moreover, our study suggests that in some contexts there are MHCI gene-specific effects of PTEN loss since in HCT116 cells we observed that the induction of HLA-B was the most sensitive (vs. HLA-A and HLA-C) to PTEN loss. In addition, the impact of PTEN loss may not be apparent in the absence of IFN-γ. Indeed, the two MHC genes impacted the most by PTEN loss (HLA-B and HLA-DR) in our study were the ones whose expression was most induced by IFN-γ.
STAT1 is downstream of IFN-γ and plays a complex yet critical role in anti-tumor immunity through its ability to regulate the expression of genes with immunologic function including those within the MHC (60). STAT1 control of MHCII was previously shown to be mediated by direct binding to the CIITA promoter and CIITA levels correlate with the levels of all MHCII genes (61). In a similar manner, STAT1 also regulates NLRC5 expression which is a key regulator of MHCI genes (62). IFN-γ regulates STAT1 function through the action of Jak kinases which phosphorylate STAT1 thereby promoting dimerization and nuclear translocation. In addition, IFN-γ induces the expression of STAT1 (and other members of the STAT family such as STAT2) via regulatory regions within the STAT1 promoter and more distal elements (31, 63). Our studies implicate the PI3K pathway as a negative regulator of STAT1 expression since inhibition of PI3K increased whereas activation of PI3K signaling repressed STAT1 levels following IFN-γ treatment. This is consistent with prior studies demonstrating that RAS, which is upstream of PI3K, can repress STAT1 expression (32). Moreover, other studies have shown that oncogenic signaling can influence the phosphorylation and functions of STAT1 via effects on phosphatases such as SHP2 (64). Another intriguing aspect of STAT1 biology centers on the role of unphosphorylated STAT1 which has been shown to have effects on gene expression that are distinct from those of phosphorylated STAT1 (65). Thus, through their ability to alter STAT1 expression and/or phosphorylation, PI3K signaling may disrupt the cellular response to cytokines and in doing so cell-mediated anti-tumor immunity.
It is worth noting that multiple groups, including our own, have shown that the effects of kinase inhibitors are variable and not universal with some cells responding to kinase inhibitors with increases in MHC expression whereas in other cells, the same inhibitors have no effect on MHC expression/induction (10, 22). Indeed, in a previous study, none of the ten melanoma cell lines tested responded to dactolisib with increases in MHC expression (22). Similar findings were found by Sivaram et al. where an AKT inhibitor increased MHCI levels in only three of eight human pancreatic carcinoma cell lines (49). These differences may reflect differences in cell lineage, signaling and/or the status of driver mutations. Likewise, the study by Peng et al., failed to detect any impact of PTEN loss on MHC expression in human melanoma. Interestingly, the recent study by Marijt demonstrates that in B16 melanoma cells PI3K inhibitors can reverse MHCI repression by mediated in response to low oxygen and glucose. Thus, metabolic state may influence the impact of PI3K signaling on MHCI expression.
In contrast to the variable effects on MHCI induction observed using different cell lines, the impact of PI3K inhibition and activation on MHCII induction was more consistent in our study. Indeed, in all the malignant cell lines used, PI3K inhibition doubled the number of MHCII positive cells and/or the levels of MHCII on the cell surface. Conversely, using several pharmacologic and genetic approaches/models, PI3K activation decreased the induction of MHCII by 50% or more. To our knowledge these effects of PI3K signaling on MHCII induction in human cancer cells have not been previously reported despite the fact that tumor cell MHCII expression is being increasingly recognized for its relevance to anti-tumor immunity (57, 66, 67). Along these lines, in the study Sivaram et al., the authors found that anti-tumor immunity against pancreatic tumors lacking PIK3CA required CD4+ T cells (in addition to CD8+ T cells) further implicating potential roles for tumor cell MHCII expression (49). These findings with MHCII may be due to the fact that in contrast to MHCI expression which is regulated by multiple signaling pathways, in most epithelial cells MHCII expression is regulated/induced almost exclusively by IFN-γ signaling (68). Moving forward it will be important to determine what governs PI3K-MHC interactions and the response to PI3K inhibition within human cell lines, tumors and tissues.
In summary, we provide additional evidence that the PI3K pathway can regulate MHC expression and/or induction by IFN-γ within human tumor cells and that within human SCCHN there’s an inverse relationship between phospho-AKT and MHCI expression. These findings extend the mechanisms through which the activation of PI3K signaling within tumor cells may influence T-cell mediated anti-tumor immunity and adds to the list of ways that the PI3K pathway can alter how cells respond to IFN-γ (39). Given their key roles in mediating responses to immunotherapy, it will be important to more precisely define how alterations within the PI3K pathway impact cellular responses to IFNs and the induction and/or expression of MHC molecules.
ACKNOWLEDGEMENTS
This work was supported by Merit Review Award (#1I01BX001922-01A1), by NIH/NCI grant U54 CA119338-04, P50 CA128613 (Head and Neck Cancer SPORE), the Melanoma and Skin Cancer Fund from the Winship Cancer Institute and the Melanoma Innovation Fund from the Winship Cancer Institiute (to B.P.P.). Additional support was provided by NIH NRSA T32 CA160040 (to S.C.) and NIH Award 5R01CA207619-03 (to S.N.T.). This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454 and P30CA138292. “The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.” Research reported in this publication was supported in part by the Cancer Tissue and Pathology shared resource of Winship Cancer Institute of Emory University and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Hynes NE, Lane HA. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5: 341–54 [DOI] [PubMed] [Google Scholar]
- 2.Thorpe LM, Yuzugullu H, Zhao JJ. 2015. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15: 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santarpia L, Lippman SM, El-Naggar AK. 2012. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets 16: 103–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. 2015. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 28: 690–714 [DOI] [PubMed] [Google Scholar]
- 5.Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. 2013. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 105: 1172–87 [DOI] [PubMed] [Google Scholar]
- 6.Accolla RS, Lombardo L, Abdallah R, Raval G, Forlani G, Tosi G. 2014. Boosting the MHC Class II-Restricted Tumor Antigen Presentation to CD4+ T Helper Cells: A Critical Issue for Triggering Protective Immunity and Re-Orienting the Tumor Microenvironment Toward an Anti-Tumor State. Front Oncol 4: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, Greenbaum B, Carroll J, Garon E, Hyman DM, Zehir A, Solit D, Berger M, Zhou R, Rizvi NA, Chan TA. 2018. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359: 582–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, Sanders ME, Lovly CM, Frederick DT, Kelley MC, Richmond A, Irish JM, Shyr Y, Sullivan RJ, Puzanov I, Sosman JA, Balko JM. 2016. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 7: 10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vertuani S, Triulzi C, Roos AK, Charo J, Norell H, Lemonnier F, Pisa P, Seliger B, Kiessling R. 2009. HER-2/neu mediated down-regulation of MHC class I antigen processing prevents CTL-mediated tumor recognition upon DNA vaccination in HLA-A2 transgenic mice. Cancer Immunol Immunother 58: 653–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimura K, Shiraishi K, Mueller A, Izawa S, Kua LF, So J, Yong WP, Fujii H, Seliger B, Kiessling R, Kono K. 2013. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol 191: 6261–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tseng PC, Huang WC, Chen CL, Sheu BS, Shan YS, Tsai CC, Wang CY, Chen SO, Hsieh CY, Lin CF. 2012. Regulation of SHP2 by PTEN/AKT/GSK-3beta signaling facilitates IFN-gamma resistance in hyperproliferating gastric cancer. Immunobiology 217: 926–34 [DOI] [PubMed] [Google Scholar]
- 12.Pollack BP, Sapkota B, Cartee TV. 2011. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res 17: 4400–13 [DOI] [PubMed] [Google Scholar]
- 13.Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD. 2018. EZH2 Represses the B Cell Transcriptional Program and Regulates Antibody-Secreting Cell Metabolism and Antibody Production. J Immunol 200: 1039–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiss M, Pitman SW, Sartorelli AC. 1985. Modulation of the terminal differentiation of human squamous carcinoma cells in vitro by all-trans-retinoic acid. J Natl Cancer Inst 74: 1015–23 [PubMed] [Google Scholar]
- 21.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. 2008. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7: 1851–63 [DOI] [PubMed] [Google Scholar]
- 22.Sapkota B, Hill CE, Pollack BP. 2013. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology 2: e22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. 2008. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 51: 5522–32 [DOI] [PubMed] [Google Scholar]
- 24.Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. 2016. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol Res 4: 611–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rangan SR. 1972. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer 29: 117–21 [DOI] [PubMed] [Google Scholar]
- 26.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106: 761–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu W, Schonleben F, Li X, Ho DJ, Close LG, Manolidis S, Bennett BP, Su GH. 2006. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res 12: 1441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotenko SV, Izotova LS, Pollack BP, Mariano TM, Donnelly RJ, Muthukumaran G, Cook JR, Garotta G, Silvennoinen O, Ihle JN, et al. 1995. Interaction between the components of the interferon gamma receptor complex. J Biol Chem 270: 20915–21 [DOI] [PubMed] [Google Scholar]
- 29.Lee YJ, Benveniste EN. 1996. Stat1 alpha expression is involved in IFN-gamma induction of the class II transactivator and class II MHC genes. J Immunol 157: 1559–68 [PubMed] [Google Scholar]
- 30.Shuai K, Schindler C, Prezioso VR, Darnell JE Jr., 1992. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 258: 1808–12 [DOI] [PubMed] [Google Scholar]
- 31.Yuasa K, Hijikata T. 2016. Distal regulatory element of the STAT1 gene potentially mediates positive feedback control of STAT1 expression. Genes Cells 21: 25–40 [DOI] [PubMed] [Google Scholar]
- 32.Klampfer L, Huang J, Corner G, Mariadason J, Arango D, Sasazuki T, Shirasawa S, Augenlicht L. 2003. Oncogenic Ki-ras inhibits the expression of interferon-responsive genes through inhibition of STAT1 and STAT2 expression. J Biol Chem 278: 46278–87 [DOI] [PubMed] [Google Scholar]
- 33.Castellano E, Downward J. 2011. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2: 261–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen H, Ramana CV, Bayes J, Stark GR. 2001. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem 276: 33361–8 [DOI] [PubMed] [Google Scholar]
- 35.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–7 [DOI] [PubMed] [Google Scholar]
- 36.Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, Dey A, Wakabayashi Y, Sakaguchi K, Fujita T, Tamura T, Zhu J, Ozato K. 2018. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci U S A 115: E9162–e71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, Tejeda AO, Man H, Rigby AC, Luo HR. 2012. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci U S A 109: 10581–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eissing M, Ripken L, Schreibelt G, Westdorp H, Ligtenberg M, Netea-Maier R, Netea MG, de Vries IJM, Hoogerbrugge N. 2019. PTEN Hamartoma Tumor Syndrome and Immune Dysregulation. Transl Oncol 12: 361–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosivatz E, Matthews JG, McDonald NQ, Mulet X, Ho KK, Lossi N, Schmid AC, Mirabelli M, Pomeranz KM, Erneux C, Lam EW, Vilar R, Woscholski R. 2006. A small molecule inhibitor for phosphatase and tensin homologue deleted on chromosome 10 (PTEN). ACS Chem Biol 1: 780–90 [DOI] [PubMed] [Google Scholar]
- 41.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gobin SJ, van Zutphen M, Woltman AM, van den Elsen PJ. 1999. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. J Immunol 163: 1428–34 [PubMed] [Google Scholar]
- 43.Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, Valente WJ, Koelle SJ, Church CD, Vandeven N, Thomas H, Colunga AG, Iyer JG, Yee C, Kulikauskas R, Koelle DM, Pierce RH, Bielas JH, Greenberg PD, Bhatia S, Gottardo R, Nghiem P, Chapuis AG. 2018. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun 9: 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreno CS, Beresford GW, Louis-Plence P, Morris AC, Boss JM. 1999. CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity 10: 143–51 [DOI] [PubMed] [Google Scholar]
- 45.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A 107: 13794–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beresford GW, Boss JM. 2001. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat Immunol 2: 652–7 [DOI] [PubMed] [Google Scholar]
- 47.Ivashkiv LB. 2018. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin CF, Lin CM, Lee KY, Wu SY, Feng PH, Chen KY, Chuang HC, Chen CL, Wang YC, Tseng PC, Tsai TT. 2017. Escape from IFN-gamma-dependent immunosurveillance in tumorigenesis. J Biomed Sci 24: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sivaram N, McLaughlin PA, Han HV, Petrenko O, Jiang YP, Ballou LM, Pham K, Liu C, van der Velden AW, Lin RZ. 2019. Tumor-intrinsic PIK3CA represses tumor immunogenecity in a model of pancreatic cancer. J Clin Invest 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marijt KA, Sluijter M, Blijleven L, Tolmeijer SH, Scheeren FA, van der Burg SH, van Hall T. 2019. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNgamma receptor signaling. J Immunother Cancer 7: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. 2016. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 39: 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrmann F, Lehr HA, Drexler I, Sutter G, Hengstler J, Wollscheid U, Seliger B. 2004. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res 64: 215–20 [DOI] [PubMed] [Google Scholar]
- 53.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, Liu S, Whittington M, Deng W, Li F, Bernatchez C, Radvanyi LG, Davies MA, Hwu P, Lizee G. 2015. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol Res 3: 602–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, Watanabe M, Maruyama T, Kawaguchi Y, Inoue S, Kawasaki T, Choudhury A, Katoh R, Fujii H, Kiessling R, Kono K. 2012. Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology 1: 1104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, Mondello P, Han JE, Jarvis CA, Ulmert D, Xiang Q, Chang AY, Garippa RJ, Merghoub T, Wolchok JD, Rosen N, Lowe SW, Scheinberg DA. 2016. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res 4: 936–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kersh AE, Sasaki M, Cooper LA, Kissick HT, Pollack BP. 2016. Understanding the Impact of ErbB Activating Events and Signal Transduction on Antigen Processing and Presentation: MHC Expression as a Model. Front Pharmacol 7: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, Chang H, Lovitch SB, Horak C, Weber JS, Weirather JL, Wolchok JD, Postow MA, Pavlick AC, Chesney J, Hodi FS. 2018. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 10 [DOI] [PubMed] [Google Scholar]
- 58.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL Jr., Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P. 2016. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 6: 202–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen CL, Chiang TH, Tseng PC, Wang YC, Lin CF. 2015. Loss of PTEN causes SHP2 activation, making lung cancer cells unresponsive to IFN-gamma. Biochem Biophys Res Commun 466: 578–84 [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Liu Z. 2017. STAT1 in cancer: friend or foe? Discov Med 24: 19–29 [PubMed] [Google Scholar]
- 61.Morris AC, Beresford GW, Mooney MR, Boss JM. 2002. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol Cell Biol 22: 4781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. 2010. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A 107: 13794–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong LH, Hatzinisiriou I, Devenish RJ, Ralph SJ. 1998. IFN-gamma priming up-regulates IFN-stimulated gene factor 3 (ISGF3) components, augmenting responsiveness of IFN-resistant melanoma cells to type I IFNs. J Immunol 160: 5475–84 [PubMed] [Google Scholar]
- 64.Leibowitz MS, Srivastava RM, Andrade Filho PA, Egloff AM, Wang L, Seethala RR, Ferrone S, Ferris RL. 2013. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin Cancer Res 19: 798–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang J, Stark GR. 2008. Roles of unphosphorylated STATs in signaling. Cell Res 18: 443–51 [DOI] [PubMed] [Google Scholar]
- 66.Accolla RS, Tosi G. 2012. Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: the key issue for development of anti-tumor vaccines. J Transl Med 10: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bou Nasser Eddine F, Ramia E, Tosi G, Forlani G, Accolla RS. 2017. Tumor Immunology meets…Immunology: Modified cancer cells as professional APC for priming naive tumor-specific CD4+ T cells. Oncoimmunology 6: e1356149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van den Elsen PJ, Holling TM, Kuipers HF, van der Stoep N. 2004. Transcriptional regulation of antigen presentation. Curr Opin Immunol 16: 67–75 [DOI] [PubMed] [Google Scholar]