

Abstract
Essential tremor (ET) is one of the most common movement disorders and the prototypical disorder for abnormal rhythmic movements. However, the pathophysiology of tremor generation in ET remains unclear. Here, we used autoptic cerebral tissue from patients with ET, clinical data, and mouse models to report that synaptic pruning deficits of climbing fiber (CF)-to-Purkinje cell (PC) synapses, which are related to glutamate receptor delta 2 (GluRδ2) protein insufficiency, cause excessive cerebellar oscillations and might be responsible for tremor. The CF-PC synaptic pruning deficits were correlated with the reduction in GluRδ2 expression in the postmortem ET cerebellum. Mice with GluRδ2 insufficiency and CF-PC synaptic pruning deficits develop ET-like tremor that can be suppressed with viral rescue of GluRδ2 protein. Step-by-step optogenetic or pharmacological inhibition of neuronal firing, axonal activity, or synaptic vesicle release confirmed that the activity of the excessive CF-to-PC synapses is required for tremor generation. In vivo electrophysiology in mice showed that excessive cerebellar oscillatory activity is CF dependent and necessary for tremor and optogenetic-driven PC synchronization was sufficient to generate tremor in wild-type animals. Human validation by cerebellar electroencephalography confirmed that excessive cerebellar oscillations also exist in patients with ET. Our findings identify a pathophysiologic contribution to tremor at molecular (GluRδ2), structural (CF-to-PC synapses), physiological (cerebellar oscillations), and behavioral levels (kinetic tremor) that might have clinical applications for treating ET.
INTRODUCTION
Essential tremor (ET) is the most common movement disorder (1) and a prototypical disease model for human motor rhythm control (2). However, the pathophysiology of ET remains poorly understood, probably due to its complex etiology. Genome-wide association studies revealed several candidate genes but lacked consistent results across sites (3–6). Environmental toxins, such as β-carboline alkaloids, also play a role in ET (7–10). The complicated genetic-environmental interactions in ET pose a major obstacle to generate animal models to probe ET pathophysiology (11). There is an unmet need for an animal model that can capture the adult-onset, chronic, and progressive action tremor that is observed in patients with ET (12).
Despite the complex etiology, the consistent core clinical feature of ET is a kinetic tremor that is likely to reflect common underlying brain circuitry alterations. In Parkinson’s disease (PD), the discovery of dopamine neuronal loss and α-synuclein aggregation greatly advanced the therapeutic paradigm and provided the conceptual framework for the subsequent genetic and microbiome studies (13, 14). Therefore, studying structural and molecular substrates by detailed postmortem pathological examination may offer invaluable information to understand the pathophysiology of ET.
Recently, we observed the pruning deficits of climbing fiber (CF)-to-Purkinje cell (PC) synapses in the postmortem ET cerebellum (15, 16). Specifically, cerebellum from patients with ET showed an increased number of CF synapses on the PC dendrites within the parallel fiber synaptic territory, which is normally more distally situated than the CF synaptic territory (17). The excessive CF-PC synapses are a prominent pathological feature observed in ET and not in other cerebellar degenerative disorders (15). Moreover, the CF-PC synaptic pathology is consistently observed in ET with varying clinical features (such as independent of family history or age of onset) (18), suggesting that this synaptic pathology, among others, might be a core feature of ET. The PC synaptic changes in patients with ET provide a starting point to investigate the molecular candidates for ET pathophysiology.
The synaptic distribution on PC dendrites is strictly regulated by a set of molecules expressed on PCs (19). Metabotropic glutamate receptor 1 (mGluR1) and glutamate receptor delta 2 (GluRδ2) are two key proteins controlling the territorial distribution of CF and parallel fiber synapses on the PC dendrites (19, 20). This regulatory mechanism provides an opportunity to understand the synaptic changes observed in ET. It provides molecular targets for animal studies to bridge pure human observation into mechanistic investigation.
In this study, we identified the association between GluRδ2 protein expression and CF-PC synaptic pruning deficits in the postmortem cerebellum from patients with ET and further used a mouse model with ET-like cerebellar GluRδ2 deficiency, synaptic pruning deficits, and tremor. We applied optogenetic and pharmacological approaches with simultaneous electrophysiology in vivo to identify the circuitry mechanism of tremor in freely moving mice. Moreover, cerebellar electroencephalography (EEG) technique was developed to validate mouse discoveries in humans. Understanding the pathophysiology of tremor could help the development of effective therapeutic approaches for treating ET.
RESULTS
GluRδ2 protein reduction in the human cerebellum is correlated with PC synaptic pathology
We first investigated the PC synaptic pathology in a cohort of patients with ET (table S1, patient demographics). Our cohort constituted diverse clinical features of ET, including cases with and without head tremor, voice tremor, and a family history of tremor. Consistent with our previous observation (18), we found that patients with ET, as compared to age-matched controls, had more CF synapses in the parallel fiber synaptic territory on PC dendrites (Fig. 1, A to D).
Fig. 1. PC synaptic pathology in ET correlates with reduced GluRδ2 expression.
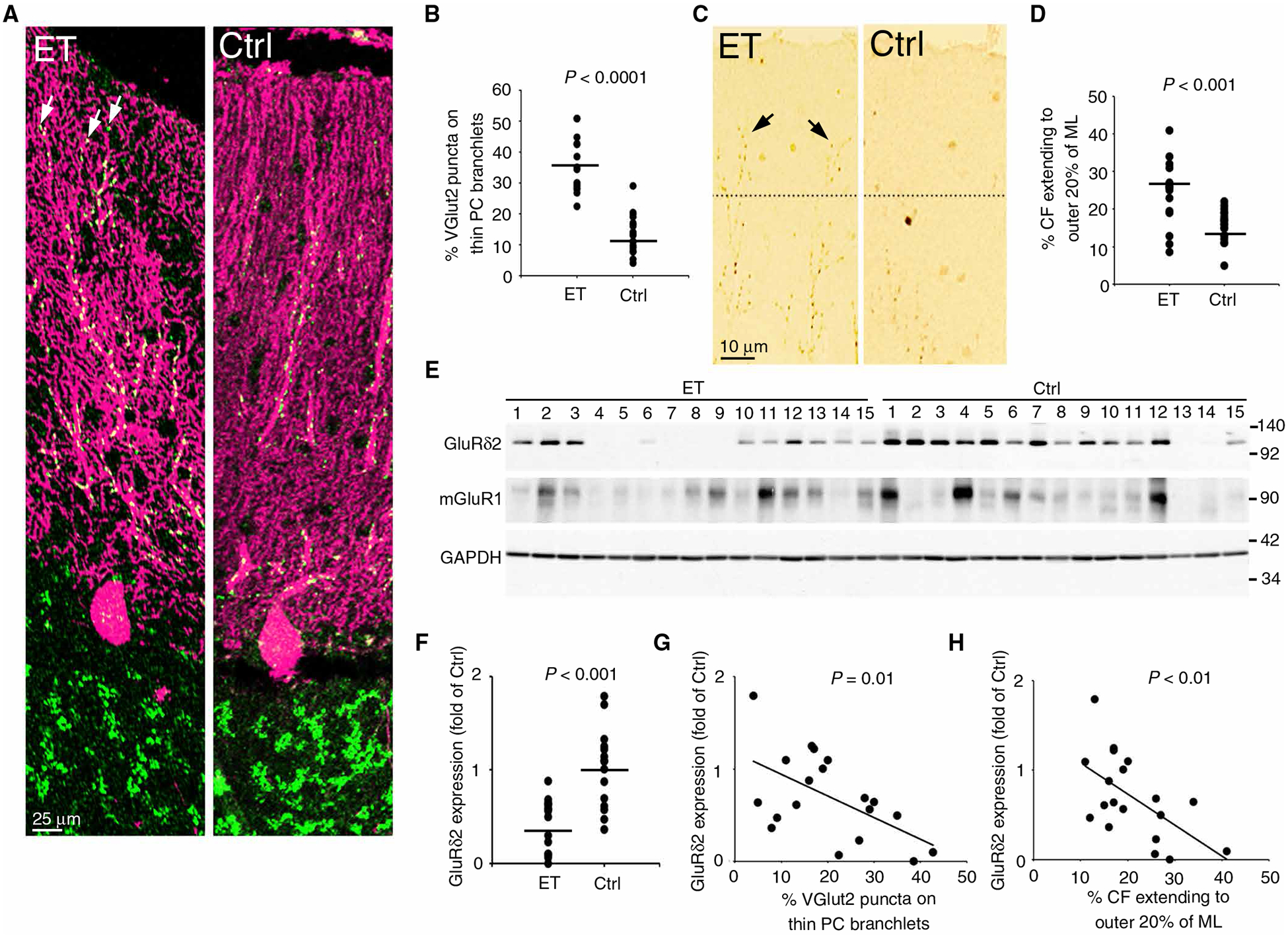
(A) Representative images of CF synapses in the postmortem human cerebellar cortex of a patient with ET and a control (Ctrl). Patients had more CF synapses, as visualized by VGlut2 puncta (green), on the thin, spiny branchlet of Purkinje cell (PC) dendrites (arrows). (B) Quantified VGlut2 puncta counts (15 ET and 19 controls, Student’s t test). (C and D) Representative cerebellar cortical sections (C) and quantification (D) of VGlut2 immunohistochemistry for CF-PC synapses. The dotted lines indicated the border between the outer 20% and the inner 80% of the molecular layer. (E to H) Images and quantification of Western blot analysis of GluRδ2, mGluR1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in frozen cerebellar cortex [(F) 15 ET and 15 controls, Student’s t test; (G and H) available in 11 ET and 8 controls, Spearman’s correlation]. CF, climbing fiber; ET, essential tremor; VGlut2, vesicular glutamate transporter type 2; PC, Purkinje cell; ML, molecular layer.
We next studied the expression of GluRδ2 and mGluR1, two key molecules that regulate PC synaptic organization, in the postmortem cerebellum of patients with ET. As compared to controls, patients with ET had approximately 75% reduction in the mean GluRδ2 expression (Fig. 1, E and F). A subset of patients and controls had both formalin-fixed tissues for the quantification of PC synaptic pathologies and frozen tissues for the Western blot determination of GluRδ2 expression, allowing us to perform correlation analysis. Consistent with the fact that GluRδ2 deficiency in mice will lead to abnormal PC synaptic pathology (specifically, CF synapses in the parallel fiber synaptic territory) (19, 20), we found that the amount of GluRδ2 inversely correlated with the percentage of CFs extending to parallel fiber synaptic territory (Fig. 1, G and H). In contrast, expression of mGluR1 protein did not differ between patients and controls (Fig. 1E and fig. S1, A to C). Our results suggest that the PC synaptic pathology in ET might be related to the reduced GluRδ2 expression.
A mouse model has GluRδ2 insufficiency
To further study the interaction between GluRδ2 insufficiency and tremor, we identified a natural mutant mouse line (hotfoot17J) with partially preserved GluRδ2 expression mimicking ET. Reverse transcription polymerase chain reaction (RT-PCR) of different segments of Grid2 complementary DNA (cDNA), the gene that encodes GluRδ2, of the cerebellar cortex in hotfoot17J mice revealed that the PCR products including Grid2 exon 1–4, but not exon 1–2, had a band shift to a higher molecular weight, although there was still a small amount of normal-length PCR products of Grid2 exon 1–4, suggesting an alternative splicing mechanism (Fig. 2A). We performed sequence analysis of the RT-PCR product and found that hotfoot17J mice carry a complete duplication of the exon 3 of Grid2 (Fig. 2B and fig. S2). We next performed quantitative PCR of the genomic DNA obtained from hotfoot17J mouse tails and determined the copy number of individual Grid2 exons. As compared with the wild-type (WT) littermates, homozygous hotfoot17J mice had a twofold increase in Grid2 exon 3, whereas heterozygous hotfoot17J mice had 1.5-fold increase. On the other hand, the copy number of Grid2 exon 16 remained unchanged (Fig. 2C). Hotfoot17J mice thus have duplication of exon 3 of Grid2. Although hotfoot strains usually imply mice with deleterious Grid2 mutation that produces no functional GluRδ2 protein, the alternative splicing mechanism in hotfoot17J mice leads to the production of approximately 10% of full-length GluRδ2 protein (see below). We therefore used the term Grid2dupE3/dupE3 or Grid2dupE3 mice hereafter to indicate the homozygous status of hotfoot17J mice to avoid the impression of total loss of GluRδ2 protein in hotfoot strains.
Fig. 2. GluRδ2 protein insufficiency in mice recapitulates ET-like CF synaptic pathology.
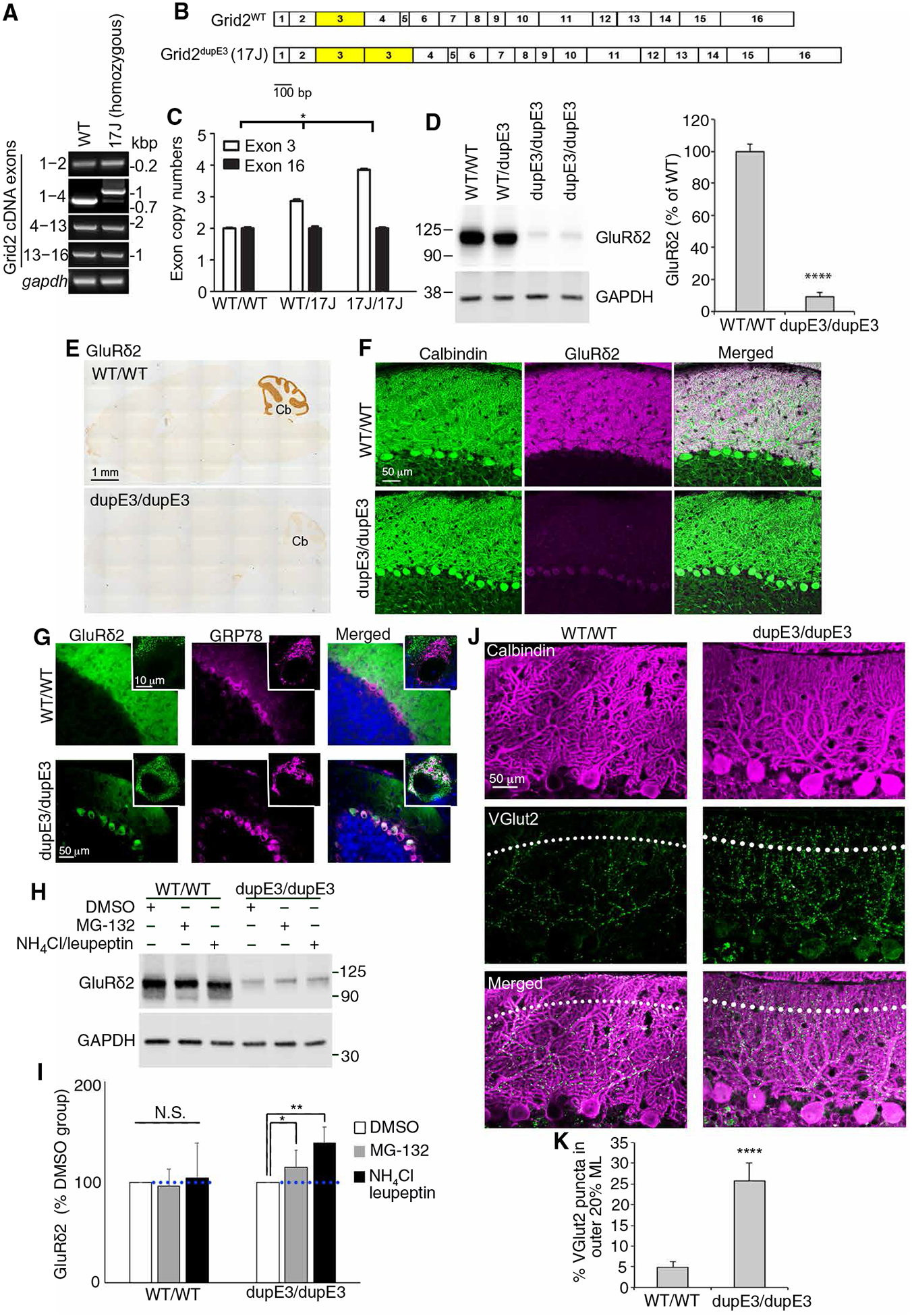
(A) RT-PCR of the cerebellar cortex of Grid2 cDNA from hotfoot17J mice. PCR fragments that include exon 3 showed an increase-sized fragment. kbp, kilo base pair. (B) Diagrams of WT and hotfoot17J Grid2 cDNA allele. bp, base pair. (C) Quantitative PCR of genomic DNA of exon 3 and exon 6 of Grid2 gene from WT or homozygous hotfoot17J (Grid2dupE3) mice (n = 3 in each group, Kruskal-Wallis, one-way ANOVA). (D) Western blots of the cerebellar cortex of WT and homozygous hotfoot17J (Grid2dupE3) mice [(D) n = 5 in each group, Student’s t test). (E) Representative images of GluRδ2 immunohistochemistry. (F) Representative images of dual immunofluorescence of calbindin to visualize PCs (green) and GluRδ2 (magenta). (G) Representative images of dual immunofluorescence of GRP78 to visualize endoplasmic reticulum (magenta) and of GluRδ2 (green). (H) Western blots of cerebellar slices incubated with either proteasomal (MG-132) or lysosomal (NH4Cl and leupeptin) inhibition in a WT mouse or a hotfoot17J (Grid2dupE3) mouse and (I) the quantification of GluRδ2 expression [(I) n = 8 in each group, Kruskal-Wallis one-way ANOVA). DMSO, dimethyl sulfoxide. (J) Representative images of dual immunofluorescence of calbindin and VGlut2 in a WT mouse or a Grid2dupE3 mouse. The dotted lines indicate the border between the outer 20% and the inner 80% of the molecular layer. (K) Quantification of the percentage CF synapses in the outer 20% of the molecular layer in WT and Grid2dupE3 mice (n = 8 in each group, Student’s t test). Error bars denote SEM. *P < 0.05, **P < 0.01, and ****P < 0.001. dupE3, Grid2dupE3; WT, wild type.
We found that Grid2dupE3 mice had a marked reduction in GluRδ2 expression in the cerebellum, but there was approximately 10% of full-length GluRδ2 protein being produced (Fig. 2D), suggesting that the alternatively spliced mRNA transcript of the full-length GluRδ2 could still make a certain amount of GluRδ2 protein. The result suggests that the long isoform of mutant GluRδ2 protein might be unstable and degraded in the cell body and the endoplasmic reticulum at the protein level, despite the presence of the long isoform GluRδ2 mRNA (Fig. 2A). GluRδ2 immunohistochemistry revealed a marked reduction in GluRδ2 in the cerebellar cortex of Grid2dupE3 mice (Fig. 2E and fig. S3A). Consistently, the dual immunofluorescence of calbindin and GluRδ2 also demonstrated a marked reduction in GluRδ2 in the PC dendrites of Grid2dupE3 mice (Fig. 2F and fig. S3B). We found weak immunofluorescence of GluRδ2 in the PC soma that partially colocalized with a marker of endoplasmic reticulum, GRP78 (Fig. 2G and fig. S3C). This supports the hypothesis that the long isoform of GluRδ2 containing two exon 3 regions might induce endoplasmic reticulum-associated protein degradation. We further compared the speed of protein degradation between WT mice and Grid2dupE3 mice by incubating cerebellar slices with either proteasomal (MG-132) or lysosomal inhibitors (NH4Cl and leupeptin). In WT mice, GluRδ2 expression did not change with either proteasomal or lyosomal inhibition of protein degradation during a 6-hour incubation period (Fig. 2, H and I), consistent with the long half-life of GluRδ2 protein (17). In contrast, Grid2dupE3 mice had 15 and 39% increase in GluRδ2 protein expression during a 6-hour proteasomal and lysosomal inhibition, respectively, indicating accelerated protein degradation both by proteasomes and lysosomes (Fig. 2, H and I). GluRδ2 is the key molecule restricting CFs from extending into parallel fiber synaptic territory on PC dendrites (19, 20). Consistently, Grid2dupE3 mice also developed CF synapses innervating distal, thin PC dendrites (Fig. 2, J and K).
Together, Grid2dupE3 mice have a genetic mutation of Grid2, leading to mislocalization of GluRδ2 protein, accelerated protein degradation, and, consequently, a GluRδ2-insufficient state. Because of this unique genetic mutation, Grid2dupE3 mice are capable of producing some full-length GluRδ2 protein, which mimics the reduced expression of GluRδ2 protein and CF-PC synaptic pruning deficits in the cerebellum of patients with ET and might play a role in tremor.
Grid2dupE3 mice develop ET-like tremor
We next determined the tremor behaviors in our mouse model with GluRδ2 insufficiency. Using frequency spectrum analysis by fast Fourier transformation, we studied the frequency of tremor in freely moving mice on a sensitive force plate (Fig. 3A). We found that Grid2dupE3 mice developed a robust 20-Hz tremor (Fig. 3, B and C, and movie S1). We coregistered the tremor measurement with a real-time video capturing system to detect mouse movements (Fig. 3A) and found that the tremor occurred predominantly during action and minimally at rest (Fig. 3, D to F, and movie S1). The tremor developed approximately at 12 weeks of age (3 months) and progressively worsened over time (Fig. 3, G and H). Robust mouse tremor was reliably observed at the age of 18 weeks (P < 0.01; Fig. 3I). Since ET is characterized by age-related kinetic tremor (21, 22), the mouse model with GluRδ2 insufficiency can recapitulate key tremor characteristics of ET.
Fig. 3. Reduced expression of GluRδ2 protein in mice causes progressive kinetic tremor.
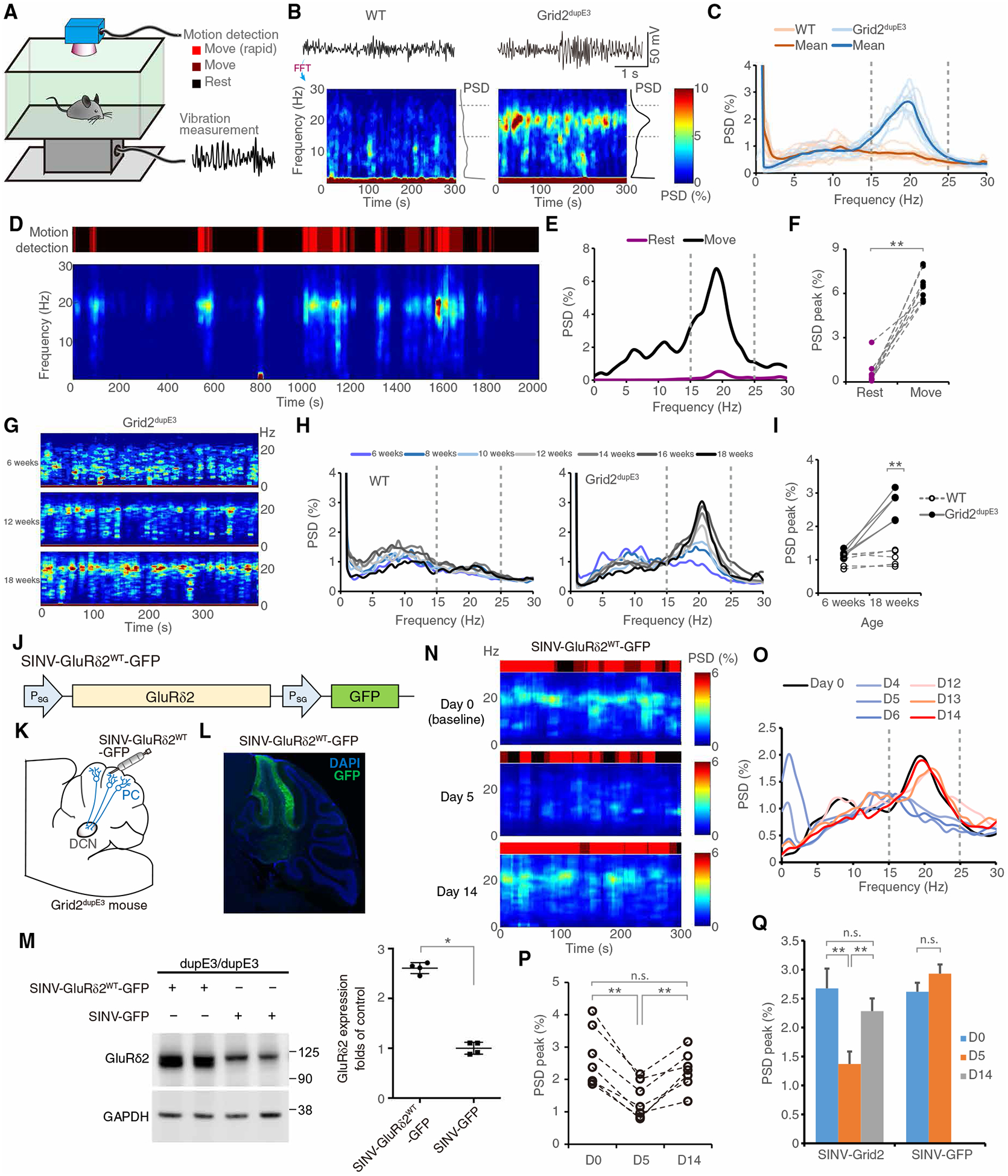
(A) Scheme showing tremor recording with simultaneous motion monitoring in a freely moving mouse. (B and C) Representative time-frequency plots (B) and normalized power spectral density (PSD) diagram [(C) n = 10 mice in each group] of tremor in 3-month-old mice (see Materials and Methods). FFT, fast Fourier transformation. (D to F) Kinetic tremor in Grid2dupE3 mice. A time-frequency plot and the corresponding PSD diagram, coregistered by video-based motion detection, showed kinetic predominant tremor [(F) n = 9 mice]. (G to I) Tremor progression with age. Representative time-frequency plots (G) and corresponding PSD diagrams (H) in Grid2dupE3 mice and group analysis of peak PSDs (I) from WT or Grid2dupE3 mice in different ages (n = 5 mice in each group). (J) Construct design for Sindbis virus (SINV) expressing GluRδ2 for the rescue experiment. (K) Schematic demonstrating SINV-mediated GluRδ2 expression in the cerebellar cortex. DAPI, 4′,6-diamidino-2-phenylindole. (L) GFP expression in a Grid2dupE3 mouse motor cerebellum injected with SINV-GluRδ2WT-GFP. (M) GluRδ2 protein expression in Grid2dupE3 mouse cerebellum transfected with either SINV-GluRδ2WT-GFP or control SINV-GFP at postinjection day 5. (N) Representative time-frequency plots of tremor in a Grid2dupE3 mouse before SINV-GluRδ2WT-GFP injection in the cerebellum (day 0) and at postinjection day 5 and day 14. (O) Representative PSD diagram of tremor in a Grid2dupE3 mouse injected with SINV-GluRδ2WT-GFP at different time points. (P) Group analysis of tremor PSD peaks by SINV-GluRδ2WT-GFP intervention (n = 7 mice). (Q) Quantification of peak tremor PSD of Grid2dupE3 mice transfected with SINV-GluRδ2WT-GFP (n = 7 mice) when compared with control SINV-GFP at different time points (n = 6 mice). n.s., not significant. *P < 0.05, **P < 0.01 by Wilcoxon signed-rank tests. See also fig. S4.
To further validate our mouse model, we next tested the pharmacological responses of the mouse tremor to primidone and propranolol, two first-line therapies for ET (22, 23). In addition, we also determined the tremor responsiveness to ethanol, for which many patients with ET report that alcohol can suppress their tremor (21). We found that systemic administration of primidone, propranolol, or ethanol, but not saline, suppressed tremor (fig. S4). Together, the GluRδ2-insufficient mouse model recapitulates core clinical features of ET, including prominent kinetic tremor with minimal rest tremor, chronic tremor that is adult onset and progressive, and similar pharmacological responses to ET.
GluRδ2 rescue suppresses tremor in the mouse model
To further establish the causative contribution of GluRδ2 insufficiency in tremor generation, we used a viral approach to rescue GluRδ2 protein in Grid2dupE3 mice. We took advantage of rapid and peak protein expression around days 3 to 5 and the disappearance of protein expression at days 12 to 14 with Sindbis virus (SINV) infection (24) to assess rescue and reversibility of GluRδ2 in Grid2dupE3 mice. SINV carrying Grid2 mRNA (SINV-GluRδ2WT-GFP) was injected into lobules IV to VI (motor cerebellum) of Grid2dupE3 mice (Fig. 3, J to L). We found that WT GluRδ2 protein could be reliably expressed in the motor cerebellum by postinjection day 5 (Fig. 3M). Consistent with the time frame of GluRδ2 expression, tremor was reduced in Grid2dupE3 mice by postinjection days 4 to 6 (Fig. 3, N to P) and returned to baseline by postinjection days 12 to 14. The timing of tremor suppression coincided with the corresponding changes of CF-PC synaptic pathology (fig. S5). To exclude a nonspecific effect of SINV in tremor modulation, we also injected SINV that only carries green fluorescent protein (GFP) (SINV-GFP). This control virus did not have effects on mouse tremor (Fig. 3Q and fig. S6), confirming that GluRδ2 insufficiency plays an essential role in Grid2dupE3 mouse tremor.
In addition to Grid2dupE3 mice, we also studied another strain of mice with spontaneous mutation of Grid2 gene, hotfoot4J, which also has GluRδ2 insufficiency (25). Homozygous hotfoot4J mice also developed 20-Hz tremor (fig. S7), supporting the role of GluRδ2 deficiency in the pathophysiology of tremor.
CF-PC-deep cerebellar nuclei circuit contributes to tremor generation
To understand the circuitry mechanism from CF synaptic pruning deficits toward tremor generation, we investigated the tremor modulatory roles in the CF-PC-DCN (deep cerebellar nuclei) axis (Fig. 4A). We first removed the cerebellar cortex by applying cryoinjury to the brain surface in Grid2dupE3 mice (Fig. 4, B and C, and fig. S8). Dry ice exposure for 30 s immediately abolished mouse tremor (Fig. 4, D to E, and movie S2), suggesting a modulatory role of the cerebellum in tremor.
Fig. 4. CF-to-PC pathway contributes to tremor generation.
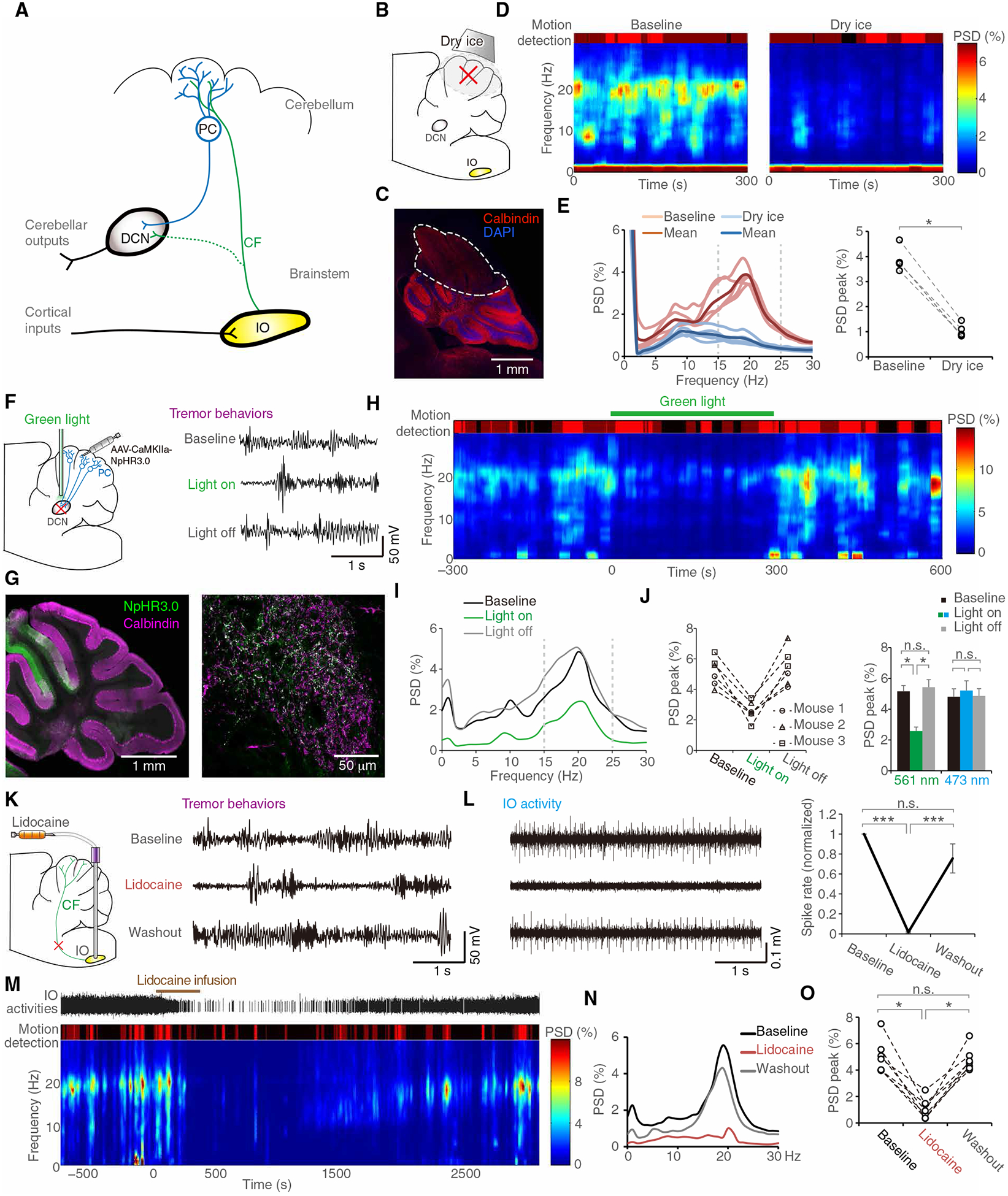
(A) Scheme showing the IO-CF-PC-DCN axis of the cerebellar pathway. (B to E) Cryoinjury of cerebellar cortex. Thirty seconds of dry ice exposure (B) created loss of anatomical architecture of lobules IV to VI of the cerebellar cortex (C). A representative time-frequency plot of a Grid2dupE3 mouse before and after cryoinjury to the cerebellum (D). Group data of PSD diagrams (left) and quantification of corresponding peak PSD (right) in Grid2dupE3 mice before and after cryoinjury to the cerebellum [(E) n = 5 mice]. (F to J) Optogenetic inhibition of PC outputs. We virally expressed NpHR 3.0 in PCs and suppressed PC outputs by optic inhibition of PC axonal terminals in DCN (F). NpHR expression was colocalized with calbindin, a specific marker of PCs, in both cerebellar cortex and DCN [(G) right and left panels, respectively]. Green light suppressed tremor instantaneously, and the light removal caused immediate reappearance of tremor (H and I). Quantitative spectrum analysis demonstrated significant and reversible effects of PC outputs in tremor generation [(J) n = 3 mice, each had two runs]. (K to O) IO silencing by in situ lidocaine microinfusion. Lidocaine suppressed IO firings in representative multiunit traces (L, right) and quantitative single-unit analysis (L, left, n = 15 units in two mice). Behaviorally, lidocaine effects on tremor followed the time course of IO activity changes shown in representative plots (M to N) and group analysis [(O) n = 7 mice]. *P < 0.05 and ***P < 0.001 by Wilcoxon signed-rank tests. Error bars denote SEM. DCN, deep cerebellar nuclei; IO, inferior olive.
By expressing the inhibitory opsin halorhodopsin in PCs of the mouse motor cerebellum, we next optogenetically inhibited PC outputs by illuminating PC axonal terminals at the DCN (Fig. 4F). Green light (561 nm) illumination caused immediate suppression of tremor, and removal of the inhibition induced instantaneous rebounds (Fig. 4, G to J, and movie S3). In contrast, blue light (473 nm) did not activate halorhodopsin and had no effects on tremor (Fig. 4J, right). To further probe the function of CFs, we subsequently targeted the inferior olive (IO), the origin of CFs. IO neurons have gap junction-mediated electrotonic coupling, making optogenetic interventions targeting ion currents unreliable in IO (26). We therefore inhibited IO activity by lidocaine microinfusion in situ. Tremor was not only suppressed by IO inhibition (fig. S9) but also followed the time courses of IO activity changes identified by single-unit recording (Fig. 4, K to O). We also analyzed the spike-phase coupling between single-unit activity of IO and the corresponding phase profiles of the oscillatory signals from the force plate (fig. S10). As compared with the WT mice, IO simple spikes developed correlation with tremor phases. IO bursts, which have shown greater contribution to CF signaling to PC (27), revealed much stronger spike-phase coupling with tremor (fig. S10, B and C). Together, the data show that neuronal activity from IO to PCs and PCs to DCN contributes to tremor generation.
CF-PC synaptic activity generates tremor
Besides CFs, IO neurons also have sparse collaterals to DCN (28). These olivonuclear fibers, in theory, may directly modulate cerebellar outputs at DCN and bypass the CF-PC effects. To dissect the potential contributions between olivocerebellar and olivonuclear projections in tremor mice, we chose synaptophysin (SYP)-anchored mini singlet oxygen generator (miniSOG), an optogenetic tool for synapse-specific inhibition (29). Blue light triggered miniSOG-dependent generation of free radicals that could destroy nearby vesicle docking proteins and disturb synaptic vesicle release for hours (Fig. 5A) (29). Moreover, this is a method free from rebound firings or back firings by optogenetic ion current modulators that may cause remote effects on axonal collaterals (30). We injected bilateral IOs with adeno-associated virus (AAV)-carried SYP-miniSOG, which resulted in broad distribution of miniSOG at CF presynaptic terminals (Fig. 5, B and C), allowing for optogenetic manipulation of neurotransmission. Taking advantage of the long-lasting effects of SYP-miniSOG, we achieved diffuse CF inhibition in vivo by scanning the cerebellar surface with blue light via a transparent cranial window (Fig. 5D), which preferentially inhibited the CF synapses extending to the outer surface of the molecular layer, where we observed the excessive CF synapses in the parallel fiber territory. Inhibition of the CF-PC synaptic transmission sufficiently suppressed tremor up to 1.5 hours (Fig. 5, E to H, and movie S4). Scanning the cerebellar surface with nonactivating green light did not result in tremor suppression (Fig. 5H). In addition, synaptic silencing of IO-to-DCN olivonuclear fibers did not modulate tremor (Fig. 5, I to N). These results clarified the specificity of tremor mechanism and confirmed that the activity of overgrown CF-to-PC synapses contributes to tremor generation.
Fig. 5. Optogenetic silencing of CF synapses suppresses tremor.
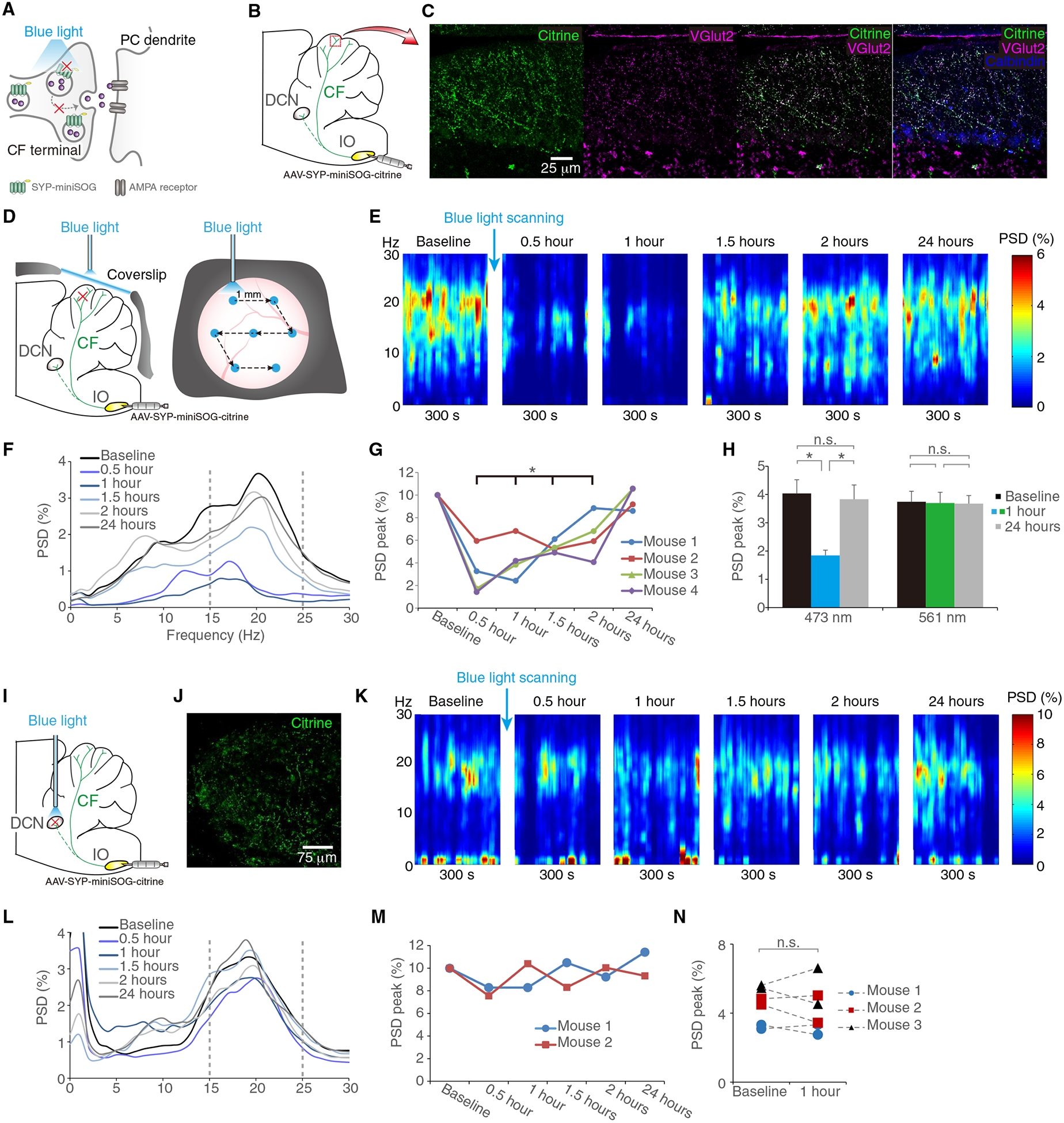
(A) Schematic illustration of SYP-miniSOG functions. Illumination of miniSOG with blue light causes synaptic protein damage and, thus, inhibits synaptic vesicle release. (B and C) Expression of SYP-miniSOG in the CF synaptic terminals. Viral injection of SYP-miniSOG-citrine in the IO (B) and the expression of citrine in the CF synaptic terminals across the cerebellar cortex, confirmed by the colocalization with VGlut2 on PCs, visualized by calbindin (C). (D to H) Blue light scanning of lobules V and VI of the cerebellar surface and tremor in Grid2dupE3 mice. We scanned the cerebellar surface with blue light through a cranial window in the sequence indicated by arrows [(D) see Supplementary Materials and Methods for details] and measured mouse tremor in different time points (E to G). Effective blue light and controlled green light illumination showed different effects on tremor [(H) n = 4 mice in two runs]. (I to N) Effect of slicing synaptic terminals from IO-to-DCN collaterals. Virus was injected into IO and optic fiber was placed in DCN (I), confirmed by fluorescence imaging showing citrine (+) synaptic terminals in DCN (J). Representative time-frequency plot (K) and corresponding PSD (L and M) of mouse tremor are shown in different time points. PSD peaks were compared in between baseline and postillumination hour 1 in a group of Grid2dupE3 mice [(N) n = 3 mice in two runs]. *P < 0.05 by Wilcoxon signed-rank tests. Error bars denote SEM. SYP, synaptophysin.
CF-PC synaptic pruning deficits create excessive cerebellar oscillations
Recently, synchronization of PC neuronal activity has been observed in multijoint movement control (31), and CFs can generate PC synchronization in microbands during action (32–35). If CFs regulate behavioral rhythm via PC synchronization, the CF hyper-innervation on PCs may augment the synchronization beyond the microbands, leading to large-scale cerebellar oscillations that are electrophysiologically detectable by local field potentials (LFPs) and coherent with tremor. To test this hypothesis, we next examined the LFPs in the mouse cerebellum. We observed robust cerebellar oscillations at 20 Hz, and the oscillations were coherent with tremor in Grid2dupE3 mice (Fig. 6, A to D). In contrast, WT mice with normal CF innervations did not generate excessive cerebellar oscillations (Fig. 6, B to D).
Fig. 6. Cerebellar oscillations correlate to tremor generation.
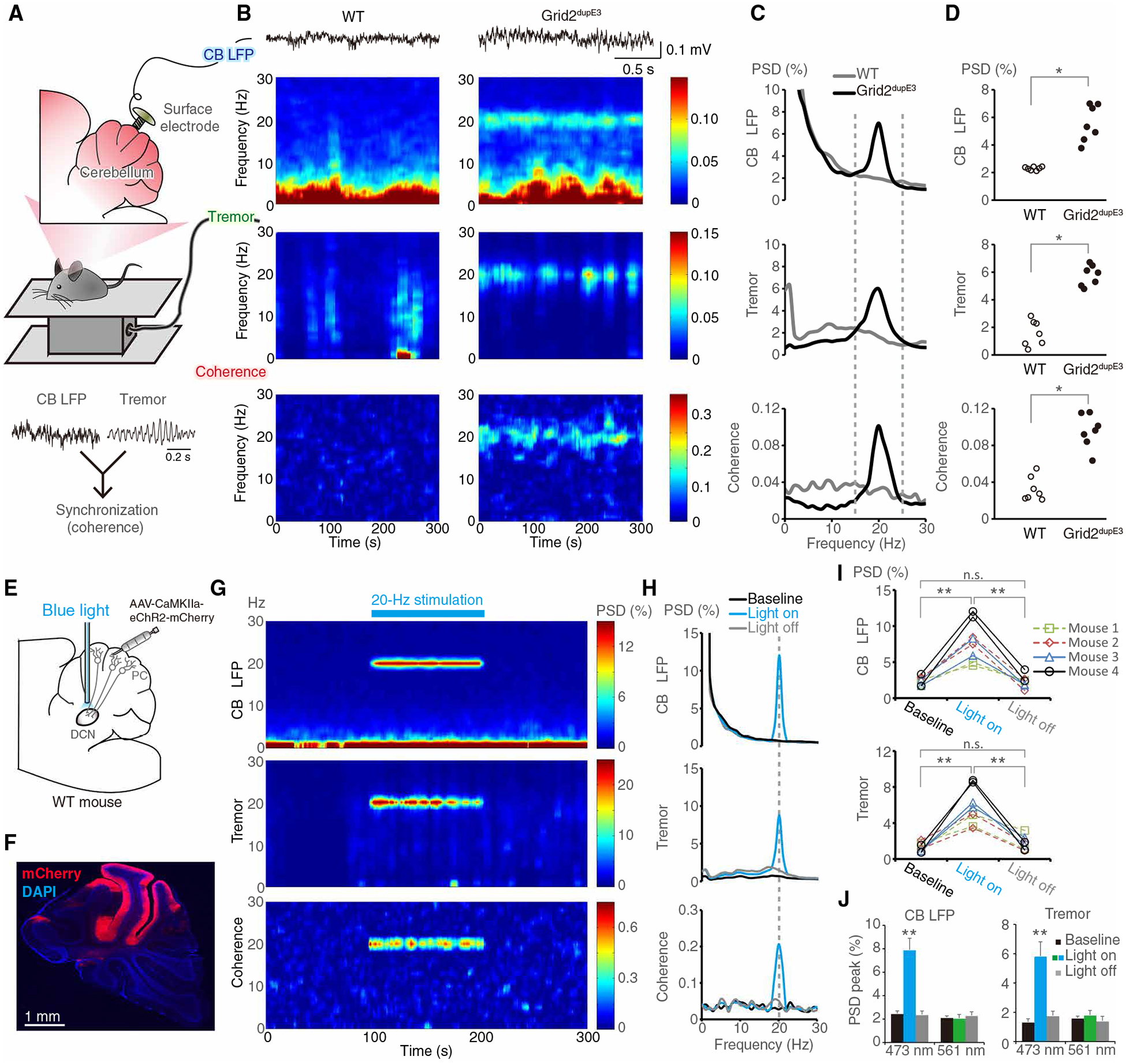
(A) Scheme showing simultaneous recordings of cerebellar LFPs of lobules V and VI and tremor in a freely moving mouse. (B and C) Representative time-frequency plots and corresponding PSD diagrams in a WT mouse and a Grid2dupE3 mouse. (D) Group analysis of PSD peaks (n = 7 mice in each group, *P < 0.05 by Mann-Whitney tests). (E) Scheme demonstrating AAV-mediated ChR2 expression in the PCs, and an optic fiber was implanted in the DCN to activate PC axonal terminals. (F) AAV-CaMKIIa-eChR2-mCherry expression in lobules IV to VI. In the cerebellar cortex, CaMKIIa is a promoter specific for PCs. (G and H) Representative time-frequency plots and PSD diagrams of cerebellar oscillations, tremor, and coherence for optogenetic PC stimulation in a WT mouse. (I and J) Quantitative data of cerebellar LFP and tremor before, during, and after blue light stimulation (I) and also nonactivating green light control (J) (n = 4 mice in each group; **P < 0.01 by Wilcoxon signed-rank tests). Error bars denote SEM. CB, cerebellum. LFP, local field potential.
Excessive cerebellar oscillations are linked to mouse tremor
To establish the relationship between excessive CF innervations, cerebellar oscillations, and tremor, we first measured the spike-phase coupling between IO single-unit activity and cerebellar oscillations around tremor frequency (15 to 25 Hz). Similar to the results observed between IO spikes and tremor (fig. S10), Grid2dupE3 mice developed spike-phase coupling between cerebellar oscillations and IO spikes, especially for the bursting spikes (fig. S11). We next measured cerebellar LFPs in Grid2dupE3 mice for which IO activity was suppressed by lidocaine microinfusion in situ (fig. S12A). IO silencing abolished cerebellar oscillations that tightly followed the chronological changes of tremor (fig. S12, B to D), demonstrating that cerebellar rhythm and oscillatory activity are CF dependent.
To further investigate the causal relationship between excessive cerebellar oscillations and tremor, we used optogenetic approaches to force synchronous and rhythmic PC firings in WT mice by transfecting PCs in the motor cerebellum with AAV-CaMKIIa-ChR2 (Fig. 6, E and F), an excitatory opsin. The rhythmic blue light illumination at the PC axonal terminals generated synchronous and rhythmic PC outputs that lead to mouse tremor, as well as cerebellar oscillations by PC synchronization via back-propagating axonal activity (Fig. 6, G to J, figs. S13 and S14, and movie S5). Cerebellar oscillations and tremor were coherent with each other and reversibly regulated by synchronous PC activation (Fig. 6, G and H). In contrast, control green light at the same frequency and intensity did not generate any cerebellar oscillatory or behavioral effects (Fig. 6J and fig. S14). The results suggest that excessive cerebellar oscillations by synchronous and rhythmic PC activity are sufficient to generate tremor. Putting the evidence together, GluRδ2 insufficiency causes CF synaptic pruning deficits, and the surplus CF-PC synaptic activity generates excessive cerebellar oscillations, which drive tremor.
Patients with ET can have excessive cerebellar oscillations
The mouse model reveals how CFs regulate cerebellar oscillations and tremor. Patients with ET have prominent CF synaptic pathology and predominant action tremor and may also have excessive cerebellar oscillations. To test this prediction, we first performed cerebellar EEG in 10 patients with ET and 10 age-matched controls (patient demographics in table S2). We observed that patients with ET had robust cerebellar oscillations at the human tremor frequencies (4 to 12 Hz) (Fig. 7, A to D). Source localization analysis confirmed that oscillations originated from the cerebellum but not the adjacent occipital cortex (Fig. 7, E to H).
Fig. 7. Patients with ET develop excessive cerebellar oscillations.
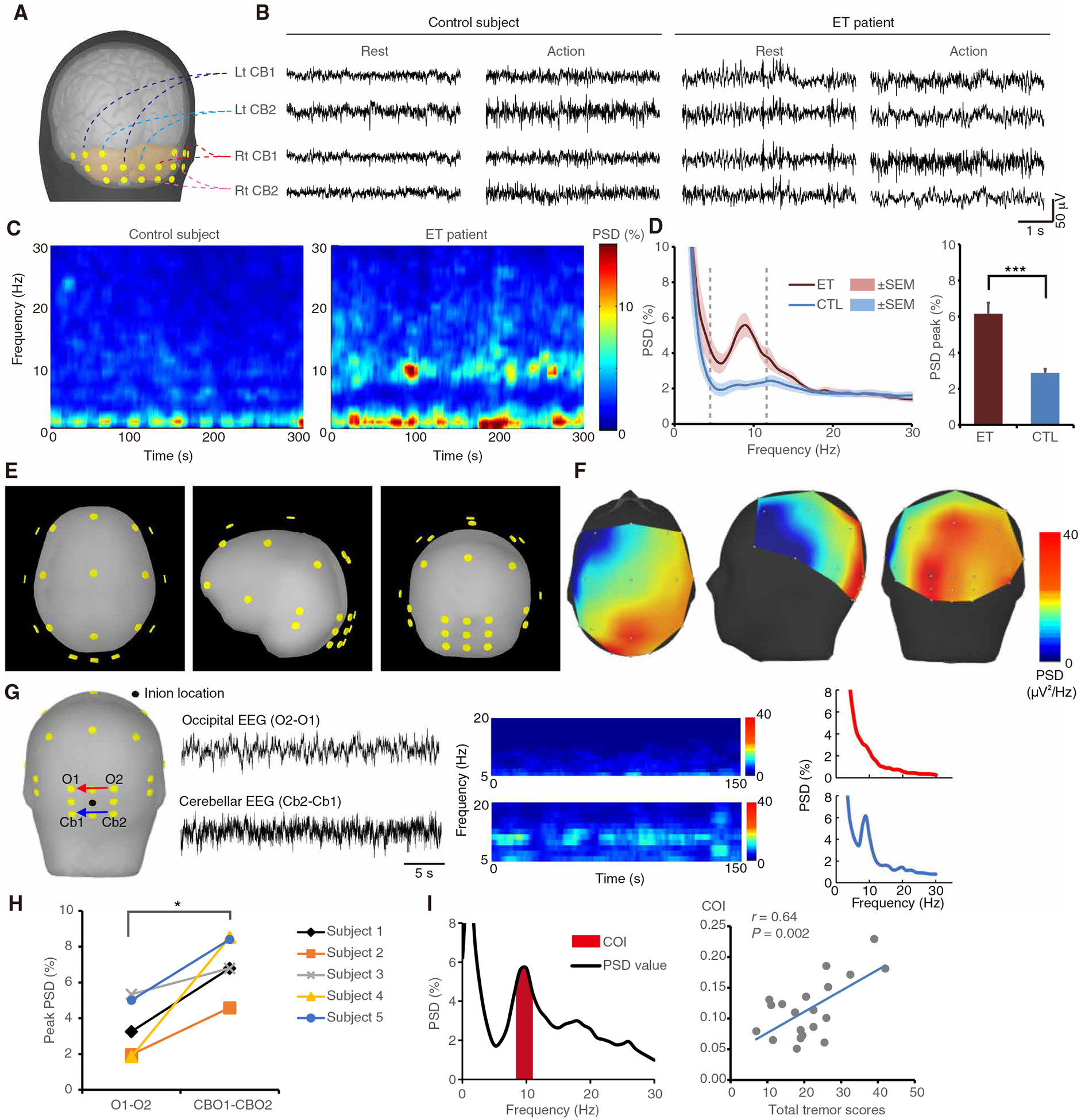
(A and B) Human cerebellar electroencephalogram (EEG) recorded in the different cerebellar regions in both patients and controls. (C and D) Representative time-frequency plots (C), and corresponding PSD diagrams and group analysis [(D) n = 10 patients and 10 age-matched controls; ***P < 0.001 by Mann-Whitney test]. (E to H) Source localization and bipolar comparison of cerebellar oscillations in patients with ET. EEG was recorded in awake patients under eyes-open condition to suppress occipital alpha rhythm. Location of EEG leads (E) and representative PSD diagrams (F) with color-coded EEG intensity at human tremor frequency (4 to 12 Hz). The highest intensity (red) is located in the cerebellar region. Direct comparison between cerebellar leads (Cb2-Cb1) and occipital leads (O2-O1) in a representative patient (G) and group analysis [(H) n = 5 patients; *P < 0.05 by Wilcoxon signed-rank test]. (I) Cerebellar oscillatory index (COI) and its correlation with tremor severity in patients in an extended cohort (n = 20 patients, r = 0.64, P = 0.002 by Pearson’s correlation coefficient). Error bars denote SEM.
EEG recording at the cerebellar region is a new technique. It is therefore crucial to exclude other potential artifacts and signal sources. Muscle artifacts, motion artifacts, and alpha rhythm from occipital lobes are major candidates that may lead to false-positive signals. For muscle artifacts, we recorded wide-band (0.3 to 250 Hz) signals from nearby capitis muscles simultaneously to identify frequency-dependent contamination from muscles to EEG leads. Wires from cerebellar EEG leads and muscle leads were bundled together to ensure that motion artifacts affect these leads equally. It is clear that cerebellar EEG and muscle signals have different spectral distribution (fig. S15, A and B) and characteristics (fig. S15, C and D). Moreover, the power of cerebellar EEG signals at the range of tremor frequencies (4 to 12 Hz) are five times larger than those recorded in the nearby muscle leads (fig. S16), suggesting that the smaller muscle and motion artifacts are likely not major contributors to the much larger cerebellar EEG oscillatory signals at the tremor frequencies.
Cerebellar EEG oscillations in our patients with ET fall into 4 to 12 Hz, which covers alpha (8–12) frequencies. We next determined whether cerebellar EEG oscillations are volume conduction from occipital alpha rhythm. Several lines of evidence showed that occipital alpha and cerebellar rhythms are different. First, all EEGs were recorded during eyes-open condition, which suppresses occipital alpha activity. Bipolar montage comparison (Fig. 7G) in recorded EEG suggested that oscillations in patients come from the cerebellar but not the occipital region. Second, occipital alpha activity should exist in both patients and normal subjects, but cerebellar oscillations were only observed in patients with ET (Fig. 7, C to E). Third, direct evidence showed that frequencies of cerebellar oscillations and occipital alpha rhythms in the same patient are distinct (fig. S17).
Cerebellar oscillations in patients with ET correlate with tremor severity
To further validate the EEG findings and evaluate the correlation between oscillatory power and ET severity in human, we expanded the cohort to 20 patients with ET and 20 age-matched controls (table S3 for demographics). The oscillatory power can be better described by the area under curve near the oscillatory peak value rather than the single peak value. We therefore defined the cerebellar oscillatory index (COI), as the area under the curve of the oscillatory peak ±1 Hz (Fig. 7I, left). As expected, COIs were higher in patients with ET than in controls (table S3). Moreover, COIs were correlated with tremor scores in patients, showing that COI could be an index reflecting tremor severity (Fig. 7I, right).
Taking the evidence together, excessive cerebellar oscillations in patients with ET, as predicted by the mouse model, can be captured by cerebellar EEG and are correlated with tremor severity. Currently, diagnosis of ET is based on pure clinical tremor phenomenology and direct tremor measurement (36), without a physiological marker indicating the underlying brain circuitry abnormalities. Cerebellar oscillations can be a physiological signature and a therapeutic target for ET.
DISCUSSION
In summary, this study identified a pathophysiology of tremor with evidence spanning molecular, structural, physiological, and behavioral levels in mouse models of tremor and patients with ET (graphical summary in fig. S18). Reduced GluRδ2 protein in PCs can lead to pruning deficits of CF-to-PC synapses in mice, and the activity of these surplus CF-PC synapses contributes to excessive cerebellar oscillations that generate tremor. With the cerebellar EEG technique, we validated that excessive cerebellar oscillations also exist in patients with ET and correlate with tremor severity. The translational aspect of the current study provides the first physiological signature of ET and a new tool, cerebellar EEG, applicable to living patients for clinical diagnosis and future research.
One major unanswered question in this study is the neuronal determinant of tremor frequency. As compared with patients with ET, the tremor frequency in Grid2dupE3 mice is almost doubled. The frequency may depend on the intrinsic firing properties of each oscillatory node, the interactions between nodes, and the size of the oscillatory circuit. Our data showed that optical synchronization of PC outputs can also generate 10-Hz tremor in addition to the 20-Hz tremor in Grid2dupE3 mice, suggesting the capability of PC and tremor in other frequencies. The mechanism of frequency selection requires future study.
This study provides an approach to ET based on cerebellar EEG. However, EEG is not the only tool to probe cerebellar electric activity. Magnetoencephalography (MEG), including recently developed optic-pumped MEG, has been validated in cerebellar research (37–40). As compared with EEG, MEG has better source-localizing ability and higher signal-to-noise ratio in high frequencies but preferentially loses electric signals from perpendicular dipoles (specifically, electric signals projecting from cerebellar cortex to the deep nuclei). Therefore, combination of the two technologies may provide a better picture of human cerebellar activity.
One important argument is that Grid2 gene mutation has not been identified in previous genetic studies for ET (3–6). A complete loss of GluRδ2 protein due to deleterious Grid2 mutations in human can lead to a specific disease entity, autosomal recessive spinocerebellar ataxia type 18 (41), with clinical manifestations of ataxia, in addition to tremor, whereas patients with ET often have prominent tremor and subtle ataxia signs, such as difficulty in performing tandem gait (42). Our data suggest that a partial loss of GluRδ2, rather than a complete loss, can create prominent action tremor, suggesting dose-dependent regulation of the cerebellar phenotypes. Protein homeostasis is critically important for cerebellar disorders. For instance, disturbance of ataxin-1 protein expression has a marked impact on the disease phenotypes in the mouse model of spinocerebellar ataxia type 1 (43, 44). GluRδ2 may be an important contributing molecule for ET, and studying the regulatory mechanism of GluRδ2 expression in the adult cerebellum, by either genetic or environmental factors, can potentially yield additional molecular targets for ET.
ET is considered a group of diseases rather than a single disease entity. On the basis of the mouse model and corresponding cerebellar EEG findings in human, excessive cerebellar oscillations in patients with ET may be originated from the CF synaptic pathology and can be a noninvasive biomarker to identify a subgroup of patients potentially beneficial from GluRδ2- or CF-based therapy. However, there are possibilities that other mechanisms could also generate excessive cerebellar oscillations. Future studies are required to identify the detailed parameters of cerebellar oscillations across different behavioral scenarios in Grid2dupE3 mice and patients with ET. These parameters may help for better identification of a subset of patients that are amendable to therapy targeting the CF synaptic pruning mechanism.
Oscillatory activity is common in the brain for movement control and cognitive processing. However, oscillations in different brain regions can lead to diverse behaviors. Cerebellar oscillations can drive tremor, whereas oscillatory activity in the basal ganglia may correlate with bradykinesia in parkinsonian state (45, 46). The detailed mechanism will shed light on common principles about how the brain controls movements with therapeutic implications.
The current study identified the link between neuronal oscillations and synaptic pruning in the cerebellum in Grid2dupE3 mice with potential clinical implications for tremor. Excessive cerebral oscillations have been found in other neurological disorders, such as autistic spectrum disorder (ASD) (47–51), and there are mounting successful experiences in patients with ASD receiving deep brain stimulations in various brain regions (52–54). Currently, repairing the structural abnormality of synaptic pruning deficits is a major research focus for therapies in ASD (55–57). Our findings provide a different therapeutic prospect that pruning disorders may also be treated via rhythm correction, which might be exploited for treating tremor.
There are several limitations of this study. Although our study indicates that CF-to-PC synapses are essential for cerebellar oscillations in tremor, the contribution of parallel fiber-to-PC synapses still needs to be considered in future studies. Granule cell and PC activities are known to influence cerebellar LFPs (58–62) and could interact with CF-to-PC synapses at the circuit level to determine tremor frequency or amplitude. In addition, whether tremor is generated in relation to the zone-specific organization of the cerebellum deserves further study. The neuronal determinant of tremor frequency remains elusive and requires future exploration. Although pathological and electro-physiological findings were observed in both tremor mice and patients in this study, most of the mechanisms were only investigated in mice. To identify the tremor mechanism in humans, further research is required to examine both cerebellar oscillations and CF pathology in the same patient. Our works mainly focus on the circuitry mechanism; the molecular mechanism responsible for GluRδ2 down-regulation remains to be explored in patients with ET. Autoantibodies targeted on GluRδ2 have been reported to cause acute cerebellar ataxia and tremor (63), but there is still lack of direct evidence that tremor is linked to the decreased expression or functional deficit of GluRδ2 protein. All these future research directions will advance our deeper understanding of tremor pathophysiology.
MATERIALS AND METHODS
Study design
Our study uses multidisciplinary approaches to explore the mechanism of tremor in both mouse models and patients with ET. We used postmortem cerebellar pathology in patients with ET and controls to identify the candidate microstructural changes and the corresponding molecular targets. We applied the mouse models with similar molecular changes and examined the corresponding structural, electrophysiological, and behavioral correlates to patients. With animal models, we probed the circuitry mechanism and the cerebellar EEG signatures by tremor measurement, cryoinjury, optogenetics, synaptic silencing, and simultaneous electrophysiology in vivo. The EEG signatures identified in the mouse model were used to guide the development of cerebellar EEG technology in humans and validate the abnormal cerebellar oscillations in patients with ET. All the mice and human experiments are single-blind design without randomization. The animals or subjects were preassigned to their groups, power precalculated for sample size, and analyzed by team members blind to the animal or subject status.
Statistical analysis
For categorical variables, we used chi-square analysis. For continuous variables, we first determined the normality using Kolmogorov-Smirnov test. For normally distributed variables, we tested the statistical significance of the differences between experimental groups in instances of single comparisons by the two-tailed Student’s t test. In instances of multiple comparisons, we used one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests to determine statistical significance. For non-normally distributed variables, we used Mann-Whitney analysis to compare between groups, and Wilcoxon signed-rank test to compare the tremor before and after drug administration and also tremor before and after SINV-mediated rescue. We performed Spearman’s correlation for non-normally distributed variables. We used GraphPad Prism 5 and SPSS for the statistical analysis. All tests of statistical significance were conducted at the two-tailed α level of 0.05.
Supplementary Material
Fig. S1. mGluR1 expression in the postmortem ET cerebellum.
Fig. S2. The amino acid alignment of the protein products of WT and hotfoot17J GluRδ2 cDNA.
Fig. S3. Quantification of cellular and subcellular expression of GluR?2 in WT and Grid2dupE3 cerebellum.
Fig. S4. Suppression of mouse tremor by ET medications.
Fig. S5. Regression of CF synapses after rescue of GluRδ2 expression.
Fig. S6. Tremor in Grid2dupE3 mice receiving intracerebellar injection of control SINV.
Fig. S7. Tremor in hotfoot4J mice.
Fig. S8. Cryoinjury of mouse cerebellum by dry ice.
Fig. S9. Frequency profiles of tremor in Grid2dupE3 mice by IO silencing.
Fig. S10. Spike-phase coupling between IO spikes and tremor phases.
Fig. S11. Spike-phase coupling between IO spikes and cerebellar LFPs.
Fig. S12. Suppression of cerebellar oscillations and tremor by IO silencing.
Fig. S13. Optogenetic stimulation of PC outputs at 10 Hz.
Fig. S14. Optogenetic stimulation of PC outputs by nonactivating green light.
Fig. S15. Signals and characteristics of cerebellar EEG leads and nearby muscle leads.
Fig. S16. Signal comparison between cerebellar EEG leads and nearby muscle leads in patients with ET.
Fig. S17. Occipital alpha rhythm and cerebellar oscillations in a patient with ET.
Fig. S18. CF synaptic pruning deficits, cerebellar oscillations, and tremor.
Table S1. Clinical and pathological features of postmortem cerebellum in patients with ET and control subjects.
Table S2. Demographic data of patients with ET and control subjects for the EEG study.
Table S3. Demographic data of patients with ET and control subjects for cerebellar oscillation index.
Movie S1. Tremor characteristics in WT versus Grid2dupE3 mice.
Movie S2. Tremor modulation by dry ice-mediated cerebellar lesioning.
Movie S3. Tremor modulation by optogenetic inhibition of cerebellar outputs.
Movie S4. Tremor modulation by synaptic inhibition of CFs.
Movie S5. Tremor induction by PC stimulation in a WT mouse.
Data file S1. Raw data (provided as separate excel file).
Acknowledgments:
We thank M. Yuzaki for the Sindbis viral constructs. We thank A. H. Koeppen for providing the rabbit polyclonal anti-VGlut2 antibody. We thank S. M. Pulst and K. P. Figueroa for providing advice to identify genetic mutations of hotfoot17 mice. We also thank the patients and families for the donation of brains, the New York Brain Bank for processing of autopsy tissues, and the people who participated in the cerebellar EEG studies.
Funding:
This research is supported by the NIH [grants K08NS083738 and R01NS104423 (to S.-H.K.); R01NS086736, R01NS073872, R01NS085136, and R01NS088257 (to E.D.L.); and R01NS04289 and R21NS077094 (to P.L.F.)], Louis V. Gerstner Jr. Scholar Award, Parkinson’s Foundation (to S.-H.K.), International Essential Tremor Foundation (to S.-H.K.), NIEHS Pilot Grant ES009089 (to S.-H.K.), Ministry of Science and Technology in Taiwan [grants MOST 104-2314-B-002-076-MY3, MOST 107-2321-B-002-020, MOST 108-2321-B-002-011, and MOST 108-2321-002-059-MY2 (to M.-K.P.)], National Taiwan University Hospital [grants 105-N3227 and 108-039 (to M.-K.P.)], and Yun-Lin branch of the hospital [grant NTUHYL104.N007 (to M.-K.P.)].
Footnotes
Competing interests:
The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Louis ED, Ferreira JJ, How common is the most common adult movement disorder? Update on the worldwide prevalence of essential tremor. Mov. Disord 25, 534–541 (2010). [DOI] [PubMed] [Google Scholar]
- 2.McAuley JH, Marsden CD, Physiological and pathological tremors and rhythmic central motor control. Brain 123 (Pt. 8), 1545–1567 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Kuhlenbäumer G, Hopfner F, Deuschl G, Genetics of essential tremor: Meta-analysis and review. Neurology 82, 1000–1007 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Thier S, Lorenz D, Nothnagel M, Poremba C, Papengut F, Appenzeller S, Paschen S, Hofschulte F, Hussl AC, Hering S, Poewe W, Asmus F, Gasser T, Schöls L, Christensen K, Nebel A, Schreiber S, Klebe S, Deuschl G, Kuhlenbäumer G, Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology 79, 243–248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefansson H, Steinberg S, Petursson H, Gustafsson O, Gudjonsdottir IH, Jonsdottir GA, Palsson ST, Jonsson T, Saemundsdottir J, Bjornsdottir G, Bottcher Y, Thorlacius T, Haubenberger D, Zimprich A, Auff E, Hotzy C, Testa CM, Miyatake LA, Rosen AR, Kristleifsson K, Rye D, Asmus F, Schöls L, Dichgans M, Jakobsson F, Benedikz J, Thorsteinsdottir U, Gulcher J, Kong A, Stefansson K, Variant in the sequence of the LINGO1 gene confers risk of essential tremor. Nat. Genet 41, 277–279 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller SH, Girard SL, Hopfner F, Merner ND, Bourassa CV, Lorenz D, Clark LN, Tittmann L, Soto-Ortolaza AI, Klebe S, Hallett M, Schneider SA, Hodgkinson CA, Lieb W, Wszolek ZK, Pendziwiat M, Lorenzo-Betancor O, Poewe W, Ortega-Cubero S, Seppi K, Rajput A, Hussl A, Rajput AH, Berg D, Dion PA, Wurster I, Shulman JM, Srulijes K, Haubenberger D, Pastor P, Vilarino-Guell C, Postuma RB, Bernard G, Ladwig KH, Dupre N, Jankovic J, Strauch K, Panisset M, Winkelmann J, Testa CM, Reischl E, Zeuner KE, Ross OA, Arzberger T, Chouinard S, Deuschl G, Louis ED, Kuhlenbaumer G, Rouleau GA, Genome-wide association study in essential tremor identifies three new loci. Brain 139, 3163–3169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louis ED, Zheng W, Jurewicz EC, Watner D, Chen J, Factor-Litvak P, Parides M, Elevation of blood β-carboline alkaloids in essential tremor. Neurology 59, 1940–1944 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louis ED, Zheng W, Applegate L, Shi L, Factor-Litvak P, Blood harmane concentrations and dietary protein consumption in essential tremor. Neurology 65, 391–396 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louis ED, Jiang W, Pellegrino KM, Rios E, Factor-Litvak P, Henchcliffe C, Zheng W, Elevated blood harmane (1-methyl-9H-pyrido[3,4-b]indole) concentrations in essential tremor. Neurotoxicology 29, 294–300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis ED, Environmental epidemiology of essential tremor. Neuroepidemiology 31, 139–149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopfner F, Helmich RC, The etiology of essential tremor: Genes versus environment. Parkinsonism Relat. Disord 46 (suppl. 1), S92–S96 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Pan M-K, Ni C-L, Wu Y-C, Li Y-S, Kuo S-H, Animal models of tremor: Relevance to human tremor disorders. Tremor Other Hyperkinet. Mov. (N Y) 8, 587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahn S, The 200-year journey of Parkinson disease: Reflecting on the past and looking towards the future. Parkinsonism Relat. Disord 46 (suppl. 1), S1–S5 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Klingelhoefer L, Reichmann H, Pathogenesis of Parkinson disease—The gut–brain axis and environmental factors. Nat. Rev. Neurol 11, 625–636 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Kuo S-H, Lin C-Y, Wang J, Sims PA, Pan M-K, Liou J.-y., Lee D, Tate WJ, Kelly GC, Louis ED, Faust PL, Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol. 133, 121–138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin C-Y, Louis ED, Faust PL, Koeppen AH, Vonsattel J-PG, Kuo S-H, Abnormal climbing fibre-Purkinje cell synaptic connections in the essential tremor cerebellum. Brain 137, 3149–3159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyazaki T, Yamasaki M, Takeuchi T, Sakimura K, Mishina M, Watanabe M, Ablation of glutamate receptor GluRδ2 in adult Purkinje cells causes multiple innervation of climbing fibers by inducing aberrant invasion to parallel fiber innervation territory. J. Neurosci 30, 15196–15209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee D, Gan S-R, Faust PL, Louis ED, Kuo S-H, Climbing fiber-Purkinje cell synaptic pathology across essential tremor subtypes. Parkinsonism Relat. Disord 51, 24–29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishina M, Uemura T, Yasumura M, Yoshida T, Molecular mechanism of parallel fiber-Purkinje cell synapse formation. Front. Neural Circuits 6, 90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe M, Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. Tohoku J. Exp. Med 214, 175–190 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Lou J-S, Jankovic J, Essential tremor: Clinical correlates in 350 patients. Neurology 41, 234–238 (1991). [DOI] [PubMed] [Google Scholar]
- 22.Zesiewicz TA, Kuo S-H, Essential tremor. BMJ Clin. Evid 2015, 1206 (2015). [PMC free article] [PubMed] [Google Scholar]
- 23.Elias WJ, Shah BB, Tremor. JAMA 311, 948–954 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Piper RC, Slot JW, Li G, Stahl PD, James DE, Recombinant Sindbis virus as an expression system for cell biology. Methods Cell Biol 43 (Pt. A), 55–78 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Lalouette A, Lohof A, Sotelo C, Guénet J-L, Mariani J, Neurobiological effects of a null mutation depend on genetic context: Comparison between two hotfoot alleles of the delta-2 ionotropic glutamate receptor. Neuroscience 105, 443–455 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Lu H, Yang B, Jaeger D, Cerebellar nuclei neurons show only small excitatory responses to optogenetic olivary stimulation in transgenic mice: In vivo and in vitro studies. Front. Neural Circuits 10, 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathy A, Ho SSN, Davie JT, Duguid IC, Clark BA, Häusser M, Encoding of oscillations by axonal bursts in inferior olive neurons. Neuron 62, 388–399 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugihara I, Shinoda Y, Molecular, topographic, and functional organization of the cerebellar nuclei: Analysis by three-dimensional mapping of the olivonuclear projection and aldolase C labeling. J. Neurosci 27, 9696–9710 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin JY, Sann SB, Zhou K, Nabavi S, Proulx CD, Malinow R, Jin Y, Tsien RY, Optogenetic inhibition of synaptic release with chromophore-assisted light inactivation (CALI). Neuron 79, 241–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahn M, Prigge M, Ron S, Levy R, Yizhar O, Biophysical constraints of optogenetic inhibition at presynaptic terminals. Nat. Neurosci 19, 554–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoogland TM, De Gruijl JR, Witter L, Canto CB, De Zeeuw CI, Role of synchronous activation of cerebellar Purkinje cell ensembles in multi-joint movement control. Curr. Biol 25, 1157–1165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukamel EA, Nimmerjahn A, Schnitzer MJ, Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welsh JP, Lang EJ, Suglhara I, Llinás R, Dynamic organization of motor control within the olivocerebellar system. Nature 374, 453–457 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Ozden I, Sullivan MR, Lee HM, Wang SS-H, Reliable coding emerges from coactivation of climbing fibers in microbands of cerebellar Purkinje neurons. J. Neurosci 29, 10463–10473 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang EJ, Apps R, Bengtsson F, Cerminara NL, De Zeeuw CI, Ebner TJ, Heck DH, Jaeger D, Jorntell H, Kawato M, Otis TS, Ozyildirim O, Popa LS, Reeves AM, Schweighofer N, Sugihara I, Xiao J, The roles of the olivocerebellar pathway in motor learning and motor control. A consensus paper. Cerebellum 16, 230–252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.di Biase L, Brittain J-S, Shah SA, Pedrosa DJ, Cagnan H, Mathy A, Chen CC, Martín-Rodríguez JF, Mir P, Timmerman L, Schwingenschuh P, Bhatia K, Di Lazzaro V, Brown P, Tremor stability index: A new tool for differential diagnosis in tremor syndromes. Brain 140, 1977–1986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marty B, Wens V, Bourguignon M, Naeije G, Goldman S, Jousmäki V, De Tiège X, Neuromagnetic cerebellar activity entrains to the kinematics of executed finger movements. Cerebellum 17, 531–539 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Cao L, Veniero D, Thut G, Gross J, Role of the cerebellum in adaptation to delayed action effects. Curr. Biol 27, 2442–2451.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrojo Ruiz M, Maess B, Altenmüller E, Curio G, Nikulin VV, Cingulate and cerebellar beta oscillations are engaged in the acquisition of auditory-motor sequences. Hum. Brain Mapp 38, 5161–5179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin C-H, Tierney TM, Holmes N, Boto E, Leggett J, Bestmann S, Bowtell R, Brookes MJ, Barnes GR, Miall RC, Using optically pumped magnetometers to measure magnetoencephalographic signals in the human cerebellum. J. Physiol 597, 4309–4324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hills LB, Masri A, Konno K, Kakegawa W, Lam A-TN, Lim-Melia E, Chandy N, Hill RS, Partlow JN, Al-Saffar M, Nasir R, Stoler JM, Barkovich AJ, Watanabe M, Yuzaki M, Mochida GH, Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology 81, 1378–1386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louis ED, Hernandez N, Chen KP, Naranjo KV, Park J, Clark LN, Ottman R, Familial aggregation of the cerebellar signs in familial essential tremor. Tremor Other Hyperkinet. Mov. (N Y) 7, 439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim J-Y, Jafar-Nejad P, di Ronza A, Kang H, Sayegh LS, Cooper TA, Orr HT, Sillitoe RV, Zoghbi HY, Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell 160, 1087–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bondar VV, Adamski CJ, Onur TS, Tan Q, Wang L, Diaz-Garcia J, Park J, Orr HT, Botas J, Zoghbi HY, PAK1 regulates ATXN1 levels providing an opportunity to modify its toxicity in spinocerebellar ataxia type 1. Hum. Mol. Genet 27, 2863–2873 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halje P, Brys I, Mariman JJ, da Cunha C, Fuentes R, Petersson P, Oscillations in cortico-basal ganglia circuits: Implications for Parkinson’s disease and other neurologic and psychiatric conditions. J. Neurophysiol 122, 203–231 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Gatev P, Darbin O, Wichmann T, Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov. Disord 21, 1566–1577 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Orekhova EV, Stroganova TA, Prokofyev AO, Nygren G, Gillberg C, Elam M, Sensory gating in young children with autism: Relation to age, IQ, and EEG gamma oscillations. Neurosci. Lett 434, 218–223 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Barstein J, Ethridge LE, Mosconi MW, Takarae Y, Sweeney JA, Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord 5, 24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Machado C, Estévez M, Leisman G, Melillo R, Rodríguez R, DeFina P, Hernández A, Pérez-Nellar J, Naranjo R, Chinchilla M, Garófalo N, Vargas J, Beltrán C, QEEG spectral and coherence assessment of autistic children in three different experimental conditions. J. Autism Dev. Disord 45, 406–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Diessen E, Senders J, Jansen FE, Boersma M, Bruining H, Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur. Arch. Psychiatry Clin. Neurosci 265, 537–540 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Orekhova EV, Stroganova TA, Nygren G, Tsetlin MM, Posikera IN, Gillberg C, Elam M, Excess of high frequency electroencephalogram oscillations in boys with autism. Biol. Psychiatry 62, 1022–1029 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Sinha S, McGovern RA, Sheth SA, Deep brain stimulation for severe autism: From pathophysiology to procedure. Neurosurg. Focus 38, E3 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Sturm V, Fricke O, Bührle CP, Lenartz D, Maarouf M, Treuer H, Mai JK, Lehmkuhl G, DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: A case report and hypothesis on the pathogenesis of the disorder. Front. Hum. Neurosci 6, 341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park HR, Kim IH, Kang H, Lee DS, Kim B-N, Kim DG, Paek SH, Nucleus accumbens deep brain stimulation for a patient with self-injurious behavior and autism spectrum disorder: Functional and structural changes of the brain: Report of a case and review of literature. Acta Neurochir. 159, 137–143 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Tang G, Gudsnuk K, Kuo S-H, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D, Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowling H, Klann E, Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron 83, 994–996 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M, Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Groth JD, Sahin M, High frequency synchrony in the cerebellar cortex during goal directed movements. Front. Syst. Neurosci 9, 98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartmann MJ, Bower JM, Oscillatory activity in the cerebellar hemispheres of unrestrained rats. J. Neurophysiol 80, 1598–1604 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Pellerin J-P, Lamarre Y, Local field potential oscillations in primate cerebellar cortex during voluntary movement. J. Neurophysiol 78, 3502–3507 (1997). [DOI] [PubMed] [Google Scholar]
- 61.de Solages C, Szapiro G, Brunel N, Hakim V, Isope P, Buisseret P, Rousseau C, Barbour B, Léna C, High-frequency organization and synchrony of activity in the purkinje cell layer of the cerebellum. Neuron 58, 775–788 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Servais L, Cheron G, Purkinje cell rhythmicity and synchronicity during modulation of fast cerebellar oscillation. Neuroscience 134, 1247–1259 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Jarius S, Wildemann B, ‘Medusa head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J. Neuroinflammation 12, 167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Louis ED, Faust PL, Vonsattel J-PG, Honig LS, Rajput A, Robinson CA, Rajput A, Pahwa R, Lyons KE, Ross GW, Borden S, Moskowitz CB, Lawton A, Hernandez N, Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain 130, 3297–3307 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Pan M-K, Kuo S-H, Tai C-H, Liou J-Y, Pei J-C, Chang C-Y, Wang Y-M, Liu W-C, Wang T-R, Lai W-S, Kuo C-C, Neuronal firing patterns outweigh circuitry oscillations in parkinsonian motor control. J. Clin. Invest 126, 4516–4526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan M-K, Tai C-H, Liu W-C, Pei J-C, Lai W-S, Kuo C-C, Deranged NMDAergic cortico-subthalamic transmission underlies parkinsonian motor deficits. J. Clin. Invest 124, 4629–4641 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kakegawa W, Miyazaki T, Kohda K, Matsuda K, Emi K, Motohashi J, Watanabe M, Yuzaki M, The N-terminal domain of GluD2 (GluRδ2) recruits presynaptic terminals and regulates synaptogenesis in the cerebellum in vivo. J. Neurosci 29, 5738–5748 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. mGluR1 expression in the postmortem ET cerebellum.
Fig. S2. The amino acid alignment of the protein products of WT and hotfoot17J GluRδ2 cDNA.
Fig. S3. Quantification of cellular and subcellular expression of GluR?2 in WT and Grid2dupE3 cerebellum.
Fig. S4. Suppression of mouse tremor by ET medications.
Fig. S5. Regression of CF synapses after rescue of GluRδ2 expression.
Fig. S6. Tremor in Grid2dupE3 mice receiving intracerebellar injection of control SINV.
Fig. S7. Tremor in hotfoot4J mice.
Fig. S8. Cryoinjury of mouse cerebellum by dry ice.
Fig. S9. Frequency profiles of tremor in Grid2dupE3 mice by IO silencing.
Fig. S10. Spike-phase coupling between IO spikes and tremor phases.
Fig. S11. Spike-phase coupling between IO spikes and cerebellar LFPs.
Fig. S12. Suppression of cerebellar oscillations and tremor by IO silencing.
Fig. S13. Optogenetic stimulation of PC outputs at 10 Hz.
Fig. S14. Optogenetic stimulation of PC outputs by nonactivating green light.
Fig. S15. Signals and characteristics of cerebellar EEG leads and nearby muscle leads.
Fig. S16. Signal comparison between cerebellar EEG leads and nearby muscle leads in patients with ET.
Fig. S17. Occipital alpha rhythm and cerebellar oscillations in a patient with ET.
Fig. S18. CF synaptic pruning deficits, cerebellar oscillations, and tremor.
Table S1. Clinical and pathological features of postmortem cerebellum in patients with ET and control subjects.
Table S2. Demographic data of patients with ET and control subjects for the EEG study.
Table S3. Demographic data of patients with ET and control subjects for cerebellar oscillation index.
Movie S1. Tremor characteristics in WT versus Grid2dupE3 mice.
Movie S2. Tremor modulation by dry ice-mediated cerebellar lesioning.
Movie S3. Tremor modulation by optogenetic inhibition of cerebellar outputs.
Movie S4. Tremor modulation by synaptic inhibition of CFs.
Movie S5. Tremor induction by PC stimulation in a WT mouse.
Data file S1. Raw data (provided as separate excel file).