

1. Summary
The spreadsheet file reported herein provides centroid data, descriptive of deuterium uptake, for the Fab Fragment of NISTmAb (PDB: 5K8A) reference material, as measured by the bottom-up hydrogen-deuterium exchange mass spectrometry (HDX-MS) method. The protein sample was incubated in deuterium-rich solutions under uniform pH and salt concentrations between 3.6 °C and 25.4 °C for seven intervals ranging over (0 to 14,400) s plus a ∞pseudo s control. The deuterium content of peptic peptide fragments were measured by mass spectrometry. [1–3] These data were reported by fifteen laboratories, which conducted the measurements using orbitrap and Q-TOF mass spectrometers. The cohort reported ≈ 78,900 centroids for 430 proteolytic peptide sequences of the heavy and light chains of NISTmAb, providing nearly 100 % coverage. In addition, some groups reported ≈ 10,900 centroid measurements for 77 peptide sequences of the Fc fragment. The instrumentation and physical and chemical conditions under which these data were acquired are documented.
2. Data Specifications
| NIST Operating Unit(s) | Material Measurement Laboratory |
| Format | xlsx |
| Instrument | Orbitrap and Q-TOF mass spectrometers |
| Spatial or Temporal Elements | N/A |
| Data Dictionary | If applicable, provide URL of data dictionary |
| Accessibility | All datasets submitted to Journal of Research of NIST are publicly available. |
| License | https://www.nist.gov/open/license |
3. Methods
3.1. Bottom-up HDX-MS measurements
These centroid data were obtained using the “bottom-up” HDX-MS experiment (Fig. 1). The first step of the HDX-MS measurement process involves immersion of the protein sample into buffered solution environments (deuterium fraction, ) at temperature THDX = (3.6 to 25.4) °C and pD 7.48, which induces D for H exchange (Fig. 1A). After the period tHDX the solution is transferred into a cold acidic aqueous solution at Tquench ≈ 0 °C and pH ≈ 2.5 (Fig. 1B). This quench solution contains a chaotrope and reducing agent [4] that denatures the protein and reduces disulfide bonds. In this solution environment (), amide sites of the denatured protein exchange H for D near their minimum chemical rates [5]. NIST supplied the HDX-MS kit that provided buffers and reagents used during the first two steps (ref. yellow/gray box of Fig. 1). The kit assured uniform pH, salt concentration, and reducing power during these steps. However, due to the individual requirements of each laboratory apparatus, the incubation solution (Fig. 1A) differs in both protein concentration and .
Fig. 1.
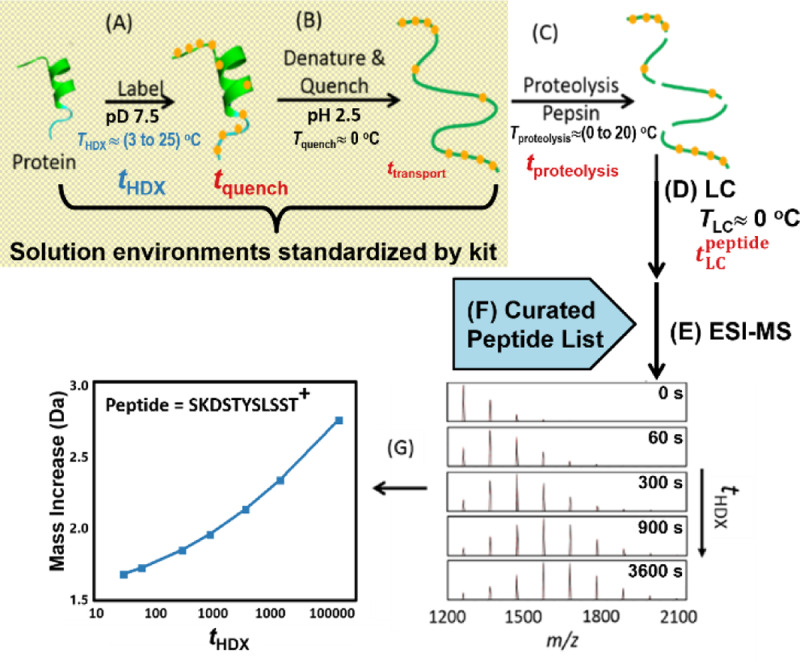
The “bottom-up” HDX-MS experiment broken into steps controlled by the NIST HDX-MS reagent kit (steps A and B in the yellow/gray box) and by each participating laboratory (steps C through G). H for D back-exchange occurs during time intervals listed in red.
Subsequently, the protein solution passes into the fluidic system comprising a column containing an immobilized protease (usually pepsin) and reverse-phase chromatography components (Fig. 1C). The solution undergoes proteolysis for an interval, tproteolysis = (4 to 240) s at a chosen temperature, Tproteolysis = (0 to 20) °C, and the resulting effluent passes through a guard column at TLC ≈ 0 °C that traps proteolytic peptides (Fig. 1D). To optimize the mass spectrometer sensitivity, many laboratories wash out salts by flowing additional loading solution through the guard column for twash = (30 to 180) s. Reverse-phase chromatography, conducted at TLC ≈ 0 °C, releases and separates peptides, which elute from the analytical column at (Fig. 1D). Electrospray ionization mass spectrometry (ESI-MS) detects the peptide ions (Fig. 1E).
Some laboratories use a variation of this procedure, where after D2O incubation has completed, the protein sample is quenched and flash frozen at −196 °C. Subsequently, the sample is thawed and analyzed using a workflow similar to that depicted in Figs. 1C through 1G.
The average mass change of each selected peptide (Fig. 1F) is calculated and plotted as a function of tHDX (Fig. 1G). Since side chains generally have more rapid exchange rates than backbone amides at pH 2.5 and ≈ 0 °C, [5, 6] the deuterium labels at these positions are equilibrated back to the natural protic isotope abundance during digestion and separation. This simplifies data analyses since each amino acid (except proline) can have a maximum of one deuterium label.
Separate experiments, equivalent to the HDX-MS experiment for tHDX = 0 s, determine the initial curated list of peptides (Fig. 1F) that associates each chromatographic peak with mass spectra. To improve the veracity of peptide identifications, the operator observes MS/MS data for the eluting peptides. With reference to the known sequence of fab fragment of NISTmAb [2] peptide ion identification software analyzes these data and proposes an amino acid sequence, charge state (z), and confidence rank for each peptide ion. The list of retention times and sequences becomes the filter through which peptide ions are selected for HDX-MS analyses. However, the curation process continues throughout the data analysis. Curation of the list assures that the kinetics behavior of sequences adheres to EX2 behavior, that the D-uptake kinetics match the sequence assignments, that the LC retention times remain stable, and that the peptides exhibit adequate intensity to support reliable centroid determinations and remain free of interfering ion signals. These requirements may disqualify many identified peptides from use in the HDX-MS analysis. Thus, the peptides listed on each spreadsheet may represent only a fraction of the peptide sets found by investigators during the proteomics discovery process. The discarded peptides are not reported.
Each laboratory conducted proteomics studies on the Fab fragment of NISTmAb (PDB: 5K8A) [1–3] and performed three HDX–MS runs. Here, the term “run” is defined by the procedures and capabilities of the laboratory equipment. Practically speaking, the term denotes approximately one day of HDX-MS measurements. Each “run” may include several replicant measurements, termed “reps”. Across the participating laboratories, each run comprised between one and five HDX-MS reps at each time point, tHDX. The database contains anonymized copies of the data spreadsheets and auxiliary information.
3.2. Reagents and Materials1
All reagents used to prepare samples and supplied with the HDX-MS kits originated from the same chemical lots. D2O (99.96 mole % D) was acquired from Cambridge Isotopes (Andover, MA). Sodium phosphate dihydrate, sodium phosphate monohydrate, and sodium chloride were purchased from Sigma-Aldrich (St. Louis, MO). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 98 % guanidine hydrochloride (GdmCl) were acquired from Thermo Scientific (Thermo Fisher Scientific, Pittsburgh, PA).
The test protein for the HDX-MS interlaboratory comparison was the Fab fragment that is enzymatically cleaved from Candidate RM 8670 (Lot #5F1b), which has the same structure as the Fab of the NIST IgG1κ reference material, NISTmAb RM8671. [1–3] Material used for this study contained a small fraction of free Fc fragment. Triple-angle light scattering data indicated that the Fab fragment of NISTmAb sample was in a monomeric state [2, 3]. Testing showed that the protein is unaltered by the mechanical shocks associated with shipping [2].
Samples of Fab fragment were used to prepare Fab-H2O and Fab-D2O buffered stock solutions. Fab-D2O solution was prepared by resuspending lyophilized Fab fragment of NISTmAb in buffered, 99.96 % D2O. Separate HDX-MS experiments revealed negligible differences between freshly prepared Fab-H2O and Fab solutions prepared by resuspending lyophilized Fab fragment of NISTmAb in buffered H2O (data not shown). The Fab-H2O and Fab-D2O solutions were dispensed through a 0.2 μm filter into 1 mL glass vials.
3.3. HDX-MS Kit
The NIST HDX-MS standard reagent kit comprising a padded box and 26 vials contained all solutions and materials necessary for conducting proteomics studies on Fab samples; for conducting three runs of HDX-MS kinetics studies involving immersion of Fab in buffered D2O for selected durations, tHDX (Fig. 1A); and for quenching the H/D exchange process, denaturing the protein, and reducing disulfide bonds during the analysis process (Fig. 1B).
Each kit contained a glass vial holding ≈200 μL Fab–H2O and a glass vial containing ≈200 μL Fab–D2O, respectively. Each kit contained one 4 mL glass vial and one 2.5 mL plastic vial of H2O dilution buffer solution, comprising 20 mmol/L sodium phosphate buffer and 150 mmol/L sodium chloride in deionized H2O (pH 7.50 ± 0.02). Each kit contained three 4 mL glass vials and one 2.5 mL plastic vial of D2O exchange buffer solution, comprising 20 mmol/L sodium phosphate buffer and 150 mmol/L sodium chloride in 99.96 % D2O (pDcorrected 7.48 ± 0.02).
Each kit contained four 4 mL glass vials of quench buffer solution, comprising 8 mol/L guanidine-HCl and 0.4 mol/L sodium phosphate in H2O (pH 3.1 ± 0.02). To assure the preparation of solutions with uniform disulfide bond reducing potential, the kits contained four samples of dry ≈0.70 g tris(2-carboxyethyl) phosphine hydrochloride (TCEP). Each TCEP sample (Pierce TCEP; Thermo Fisher Scientific, Pittsburgh, PA) was doublesealed within a 1.5 mL Eppendorf tube inside a 5 mL glass vial.
3.4. pH Measurements
During preparation of solutions provided in the HDX-MS kit, pH measurements were conducted with a Thermo Scientific Model Orion 3-Star Benchtop pH meter (Thermo Fisher Scientific, Pittsburgh, PA) coupled to a Fisherbrand double junction refillable glass pH Electrode (Fisher Scientific Catalog# 13-620-223A, Thermo Fisher Scientific, Pittsburgh, PA). The meter was calibrated using a point-by-point method with four point calibration solutions, pH 1.68, 4.01, 7.00, 10.01 (Oakton Instruments, Vernon Hills, IL).
3.5. Intact Mass Analysis of Fab fragment of NISTmAb
Intact mass analyses were conducted by direct infusion of native and perdeuterated Fab fragment of NISTmAb into a Thermo Fisher Scientific LTQ Orbitrap Elite (Thermo Fisher Scientific Inc., San Jose, CA) and an Agilent 6545 Q-TOF (Agilent Technologies, Santa Clara, CA). For these instruments, MagTran 1.03 software (Amgen Inc., Thousand Oaks, CA) [7] and BioConfirm 8.0 (Agilent Technologies, Santa Clara, CA), respectively, deconvoluted the resulting spectra. The 1 221.990 637 m/z ion of the HP-1221 calibration standard (Agilent Technologies, Santa Clara, CA) served as the reference mass for Q-TOF measurements. A native Fab control was treated similarly to perdeuterated Fab by incubating at 37 °C for 96 hours in 99.96 % D2O. Deconvolution of the [M+nH+]n+ mass envelopes of the Fab-H2O sample yielded a molecular mass of [M] = 47,628 (± 2) g/mol. This mass is in good accord with the theoretical mass of Fab-H2O, [M] = 47,628 g/mol and a previous measurement of [M] = 47,628 (±5) g/mol with a Q-TOF mass spectrometer. [2] Calculation of the theoretical mass accounts for the N-terminal pyroglutamate residue.
Prior to conducting measurements on Fab-D2O the mass spectrometer sample handling systems were preconditioned with 99.96 % D2O. Using the same mass spectrometers and infusion procedures, deconvolution of the [M+nD+]n+ data for Fab-D2O yielded a molecular mass of [M] = 48,206 (± 2) g/mol. The theoretical mass of Fab-D2O is 48,390 g/mol. [8] The difference indicates that Fab-D2O control samples contain (184 ± 2) protons. These protons may indicate that certain portions of the 412 amide sites of Fab-D2O are essentially inert to H/D exchange. Alternately, the protons may reside mainly among the 346 amino acid side-group sites that are available to rapid H/D exchange with H2O within the MS electrospray source. The intact molecule ESI-MS data do not resolve this question of site-occupancy type. This uncertainty of the amide occupancy type within the finished Fab-D2O control directs us to assume that 〈m (∞pseudo)〉peptide ≤ 〈m (∞)〉peptide.
3.6. Onsite Sample and Control Preparation
The kit was shipped to participants via an overnight delivery service in an insulated box. Dry gel packets maintained the package contents at +4 °C and had sufficient capacity to accommodate transits of at least 48 h. Upon receipt of a package, each participant ascertained that the internal temperature was near +4 °C. (Replacement kits were issued to participants upon request.) Using H2O dilution buffer solution or D2O exchange buffer solution, as appropriate, laboratories promptly diluted the Fab stock solutions to the concentration suitable for use in HDX-MS studies. These vials were stored at −80 °C until needed.
Onsite preparation included additional finishing of the Fab-D2O control sample to assure that the deuterium content corresponded to tHDX ≈ ∞pseudo material. At each participating laboratory the Fab-D2O sample was diluted with 99.96 % D2O buffered solution to the concentration suitable for the incumbent HDX-MS apparatus. The sample was then incubated at 37 °C for 96 h prior to its centroid measurement. Although this procedure does not assure that the amide sites, (NH)k, contain 99.96 % D, it does assure that all (NH)k sites contain maximum deuterium, as dictated by structural properties.
3.7. Summary of the Data
The centroid data and additional documentation for each laboratory (numbered #X = 1 to 15) is reported on four distinct Worksheets within the Excel Workbook:
- Lab#X A-Centroids. The A-centroid data sheet from each laboratory lists the centroid mass observed at time tHDX = 0 s, 30 s, 60 s, 300 s, 900 s, 3,600 s, 14,400 s and ∞pseudo. Each centroid is computed by integrating the m/z spectrum of the peptide isotopic envelope:
where z is the ion charge, n is the number of isotopic m/z features in the mass spectrum of the ion, (m/z)i is the measured mass to charge ratio, Ii is the intensity of the ith ion feature, and is the proton mass. [9] Lab#X B-Questionnaire. This data sheet details the solution conditions of the proteomics and HDX-MS experiments and a description of the experimental protocol, including a list of protease, guard, analytical columns, chromatography gradients, and ion source conditions.
Lab#X B-Supplemental. This data sheet reports the temperature of the D2O incubation solution and the uncertainty or variation of the solution temperature measurement.
Lab#X C-Instruments. This sheet lists the make and model of chromatograph, mass spectrometer, sample-handling system (i.e. robot), the search engine used to identify peptides, and the software used to conduct the HDX-MS analyses.
4. Impact
These data have use for the development and testing of HDX-MS analysis software. The original use of these spreadsheets was for a determination of the reproducibility of bottom-up HDX-MS. [10, 11] The data comprise the most comprehensive collection of HDX-MS measurements for an antibody fragment reported by a cohort of laboratories. The laboratories have measured the same sample using the same reagents. Protein concentrations and D2O fraction varied in the incubation solutions, but the salt concentrations were uniform. The study spans multiple laboratories, diverse instrumentation, and different software. These attributes are documented.
Acknowledgments
JWH at NIST wishes to acknowledge Dr. Dean Ripple for his advice on procedures and legal hurdles, Ms. Tsega Solomon for her assistance with experiments, and Ms. Natalie McDonald for assistance with the composition analysis software and data archiving. IK, ESG, and KWA of NIST and IBBR acknowledge the support of National Academy of Science’s National Research Council postdoctoral fellowships. JWH also thanks Dr. Yoshitomo Hamuro for insightful discussions. RY-CH and GC of Bristol-Myers Squibb acknowledge Dr. Adrienne Tymiak and Dr. Bruce Car for their support of this project. AE and EH at Centro de Investigación Lilly, S.A. acknowledge Mr. Sergio Cano for technical assistance. HMZ, BW, and JZ at Genentech, Inc. acknowledge Dr. Yung-Hsiang Kao and Dr. John Stults for their support for this project. XL and RP of MedImmune LLC acknowledge Dr. Qing (Paula) Lei and Dr. Michael Washabaugh for their support for this study. DDW at University of Kansas acknowledges Agilent Technologies for an equipment loan. DD and PLW acknowledge support of University of Maryland Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014).
Funding Sources
The NIST project (design, test reagents, data analysis, and manuscript preparation) was funded by the NIST Biomanufacturing Program. CS was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH), Award Number U54 GM094586. GSA was supported by grants from the Singapore Ministry of Education Academic Research Fund Tier 3 (No. MOE2012-T3-1-008). DD and PW declare that work at their institution was supported in part by the University of Maryland Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014). MP and JDL declare that work conducted at their institution was supported by National Institutes of Health (grants AI068730 and AI030040).
About the NIST authors:
Jeffrey W. Hudgens is a Research Chemist in the Biomolecular Measurement Division at NIST. During part of this project Elyssia S. Gallagher, Ioannis Karageorgos, and Kyle W. Anderson were National Institute of Standards and Technology/National Research Council Postdoctoral Associates in the Biomolecular Measurement Division at NIST. Elyssia S. Gallagher is now an Assistant Professor of Chemistry and Biochemistry at Baylor University (Waco, TX). Ioannis Karageorgos and Kyle W. Anderson are currently Research Chemists in the Biomolecular Measurement Division at NIST. The non-NIST authors are scientists at the institutions and companies listed in the byline. The National Institute of Standards and Technology is an agency of the U.S. Department of Commerce.
Footnotes
Certain commercial equipment, instruments, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
5. References
- [1].Marino JP, Brinson RG, Hudgens JW, Ladner JE, Gallagher DT, Gallagher ES, Arbogast LW, Huang RYC (2015) Emerging Technologies To Assess the Higher Order Structure of Monoclonal Antibodies State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization, Vol 3: Defining the Next Generation of Analytical and Biophysical Techniques, ACS Symposium Series, eds Schiel JE, Davis DL, Borisov OV (American Chemical Society, Washington, DC: ), Vol. 1202, pp 17–43. [Google Scholar]
- [2].Karageorgos I, Gallagher ES, Galvin C, Gallagher DT, Hudgens JW (2017) Biophysical characterization and structure of the Fab fragment from the NIST reference antibody, RM 8671. Biologicals 50:27–34. 10.1016/j.biologicals.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gallagher DT, Karageorgos I, Hudgens JW, Galvin CV (2018) Data on crystal organization in the structure of the Fab fragment from the NIST reference antibody, RM 8671. Data in Brief 16:29–36. 10.1016/j.dib.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang HM, McLoughlin SM, Frausto SD, Tang HL, Emmettt MR, Marshall AG (2010) Simultaneous Reduction and Digestion of Proteins with Disulfide Bonds for Hydrogen/Deuterium Exchange Monitored by Mass Spectrometry. Analytical Chemistry 82(4):1450–1454. 10.1021/ac902550n [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bai YW, Milne JS, Mayne L, Englander SW (1993) Primary Structure Effects on Peptide Group Hydrogen-Exchange. Proteins-Structure Function and Genetics 17(1):75–86. 10.1002/prot.340170110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Engen JR, Wales TE (2015) Analytical Aspects of Hydrogen Exchange Mass Spectrometry Annual Review of Analytical Chemistry, Vol 8, Annual Review of Analytical Chemistry, eds Cooks RG & Pemberton JE (Annual Reviews, Palo Alto: ), Vol. 8, pp 127–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang Z, Marshall AG (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. Journal of The American Society for Mass Spectrometry 9(3):225–233. 10.1016/S1044-0305(97)00284-5 [DOI] [PubMed] [Google Scholar]
- [8].Kavan D, Man P (2011) MSTools - Web based application for visualization and presentation of HXMS data. International Journal of Mass Spectrometry. 302:53–58. 10.1016/j.ijms.2010.07.030 [DOI] [Google Scholar]
- [9].Weis DD ed (2016) Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications (John Wiley & Sons, Ltd., Chichester: ), 1st Ed. [Google Scholar]
- [10].Hudgens JW, Gallagher ES, Karageorgos I, Anderson KW, Filliben JJ, Huang RY-C, Chen G, Bou-Assaf GM, Espada A, Chalmers MJ, Harguindey E, Zhang H-M, Walters BT, Zhang J, Venable J, Steckler C, Park I, Brock A, Lu X, Pandey R, Chandramohan A, Anand GS, Nirudodhi SN, Sperry JB, Rouse JC, Carroll JA, Rand KD, Leurs U, Weis DD, Al-Naqshabandi MA, Hageman TS, Deredge D, Wintrode PL, Papanastasiou M, Lambris JD, Li S, Urata S (2019) Interlaboratory Comparison of Hydrogen-Deuterium Exchange Mass Spectrometry Measurements of the Fab fragment of NIST-mAb. Analytical Chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bureau International des Poids et Mesures (BIPM) (2012) JGCM 200 2012 International vocabulary of metrology – Basic and general concepts and associated terms (VIM) Joint Committee for Guides in Metrology 2012 (Bureau International des Poids et Mesures, Cedex, France: ). Available at http://www.bipm.org/en/publications/guides/vim.html [Google Scholar]