

Abstract
Macrophages are critical mediators of tissue homeostasis, with tumors distorting this proclivity to stimulate proliferation, angiogenesis, and metastasis. This had led to an interest in targeting macrophages in cancer, and preclinical studies have demonstrated efficacy across therapeutic modalities and tumor types. Much of the observed efficacy can be traced to the suppressive capacity of macrophages, driven by microenvironmental cues such as hypoxia and fibrosis. As a result, tumor macrophages display an ability to suppress T cell recruitment and function as well as regulate other aspects of tumor immunity. With the increasing impact of cancer immunotherapy, macrophage targeting is now being evaluated in this context. Here we will discuss the results of clinical trials and the future of combinatorial immunotherapy.
Keywords: tumor-associated macrophages, immunotherapy, checkpoint blockade, chemotherapy, tumor microenvironment
Introduction
The presence of tumor-associated macrophages (TAMs) is generally associated with a poor prognosis in solid tumors. This has been shown in studies performed on individual tumor types using traditional immunohistochemistry techniques to quantify cellular density1,2 and in more recent analyses that infer the presence of macrophages across malignancies using gene expression profiles3. These findings are consistent with the established role of macrophages in promoting multiple aspects of tumorigenesis in experimental models, from initiation through to angiogenesis and systemic dissemination4,5. Most relevant for patients, TAMs are known to suppress responses to standard-of-care therapeutics, including chemotherapy, irradiation, and angiogenic inhibitors6–9. Although this includes direct regulation of survival and cell death pathways in tumor cells10,11, in vivo modeling indicates that improved efficacy following macrophage depletion is often dependent upon enhanced recruitment or function of cytotoxic CD8+ T cells6. Perhaps not surprisingly, macrophage antagonists demonstrate combinatorial efficacy when combined with immunotherapy, including checkpoint blockade12. Clinical trials examining these combinations are now ongoing. In this Review, we will discuss how macrophages are induced into becoming immunosuppressive, the mechanisms by which they suppress anti-tumor immunity, and how this information is being utilized to develop therapeutics and design clinical trials.
Factors Regulating Macrophage Function
Macrophages are not a single cell population with a defined phenotype and biological activity, but rather a diverse collection of cell types with a wide range of functional roles in homeostatic and pathological conditions. This diversity of cellular activities is regulated by input from three distinct elements: developmental origin, tissue of residence, and acute microenvironmental cues (Figure 1). The diversity of macrophage functions is regulated in turn by the integration of the epigenetic memory of these cells and their plasticity to respond to new cues13–16. The extent to which macrophages regulate tumor growth is therefore critically linked to properties of the tumor itself. This includes a role for malignant cell-derived factors such as CSF1 and CCL2 in promoting macrophage recruitment; however, the elements within the tumor microenvironment (TME) and tumor immune microenvironment (TIME), such as fibrosis, hypoxia, nutrient availability, and lymphocyte-derived factors, appear to most dramatically shift macrophage phenotypes (Figure 2). Prior to discussing these factors, it is important to note that most of the available data are contextualized within the binary M1/M2 polarization system. Thus, macrophages have traditionally been considered anti-tumorigenic when they express high levels of tumor necrosis factor (TNF), inducible nitric oxide synthase (iNOS) or MHC class II molecules, and pro-tumorigenic when they express high levels of arginase-1 (ARG1), IL-10, CD163, CD204, or CD20617. Changes to any of these markers were then used to conclude that macrophage repolarization has occurred. However, it is now clear that macrophage activation states consist of a continuum of phenotypes, and the use of markers to delineate their functional role within the tumor is circumspect18. In the following sections we will therefore highlight studies that demonstrate a change in macrophage phenotype and function in vivo. Although not discussed here, it should also be acknowledged that factors such as anatomical location, pathological or molecular cancer subtype, and even the specific microenvironmental niche occupied by the cell likely contribute to inter- and intra-tumoral macrophage heterogeneity (Box 1). Thus the role of macrophages in cancer types can differ considerably6, and there may even be an underappreciated level of variability between individual patients.
Figure 1. Macrophage origin and polarization state.
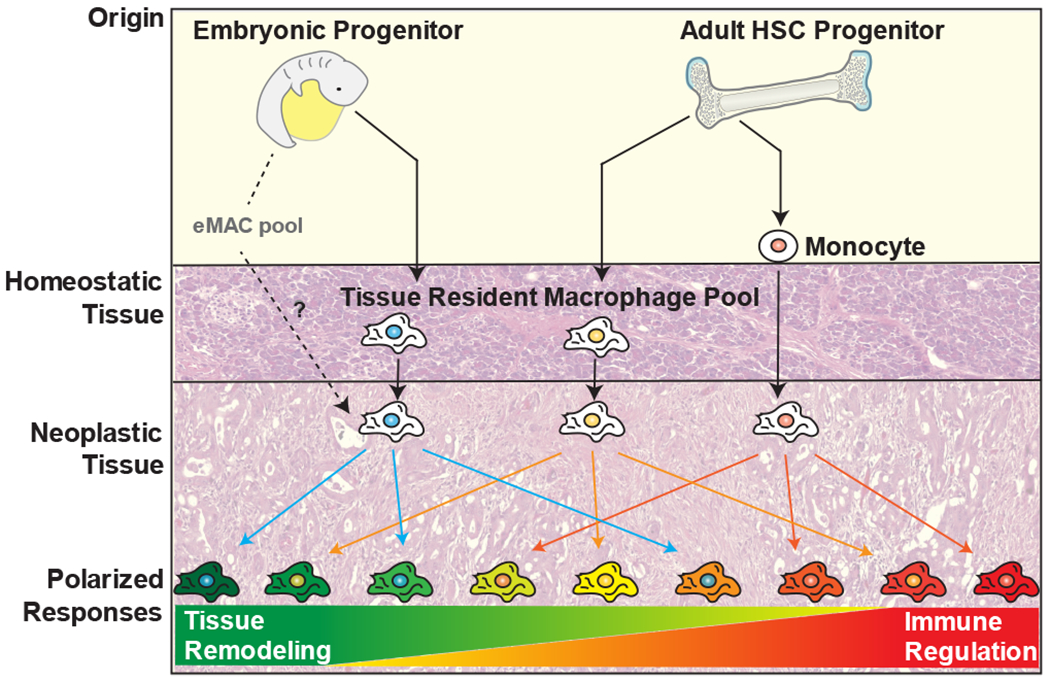
Tissue macrophages are derived from embryonic or adult progenitor cells under homeostatic conditions, with the relative contribution of these populations varying by tissue. Monocyte-derived cells also contribute to the macrophage population in some tissues, but are mostly associated with a response to inflammatory conditions, including cancer. The combination of their developmental origin and tissue of residence is thought to fine-tune their eventual response to polarizing stimuli.
Figure 2. Direct and indirect regulation of tumor immunity by TAMs.
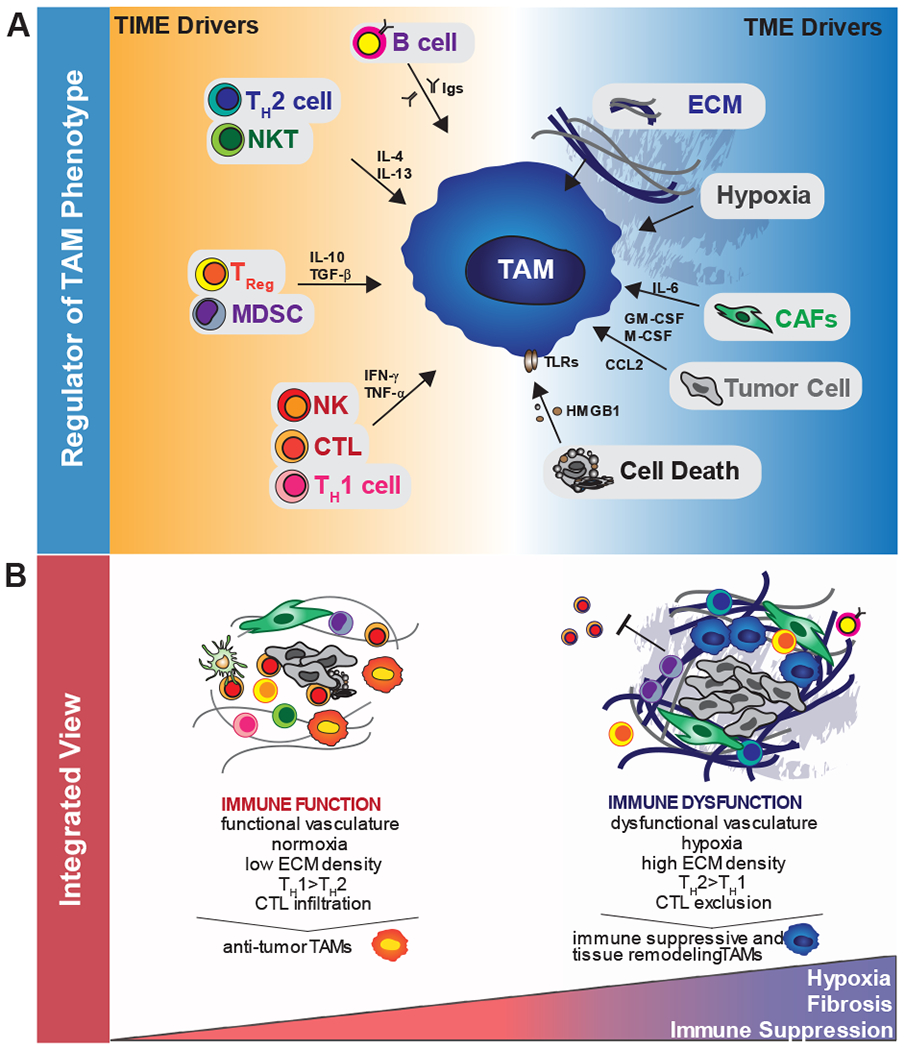
A) TAMs can directly inhibit T cell responses through three distinct mechanisms. These include checkpoint engagement via expression of molecules such as PD-L1, production of inhibitory cytokines such as IL-10, and through their metabolic activities, including depletion of metabolites and production of reactive oxygen species (ROS). B) TAMs also inhibit T cell responses indirectly by controlling the immune microenvironment. This includes recruitment of immunosuppressive populations (e.g Tregs) or by inhibiting stimulatory populations (e.g. cDCs). TAMs also blunt T cell recruitment via regulation of vascular structure, and through their ability to exclude T cells from intratumoral regions via regulation of the extracellular matrix (ECM) and the chemokine milieu.
Box 1. Tissue localization and macrophage heterogeneity.
Most of the drivers of macrophage phenotype occur within distinct and heterogeneous microscopic areas, or niches. As reviewed elsewhere209,210, these niches exist in both primary tumors and disseminated sites, and can include areas defined by their relative proximity to cancer cell invasive fronts, tumor cell nests, fibrotic stroma, functional vasculature, or even the presence of tertiary lymphoid structures. These parameters lead to the classification of distinct macrophage populations (e.g. perivascular or hypoxic macrophages) that are better defined by their functionality within these niches than their expression of surface markers or activation state. As a consequence, it is likely that macrophages within these unique niches differentially regulate T cell function. For example, perivascular macrophages are expected to control vascular structure and T cell infiltration into the tumor parenchyma186, while hypoxic macrophages might have a more specialized role in governing T cell infiltration into tumor beds39. Additionally, these niches may be shaped by cancer therapy, for example, by immunotherapy increasing the frequency of tertiary lymphoid structures, or by vascular directed-therapy reshaping perivascular and hypoxic niches. Certainly multiple cytotoxic and targeted therapies are known to alter the phenotype of tumor macrophages as a whole7. Thus, while there are consistent drivers of macrophage function, the cumulative impact of these is highly dependent upon the microenvironmental niche occupied by the cells within the malignant tissue, in addition to the macroenviroment of the tumor and tissue type of residence.
Macrophage origin.
Macrophages arise from three distinct developmental pathways (Figure 1). A large proportion of tissue-resident macrophages are now recognized as originating from embryonic precursors that seed tissues in the prenatal and perinatal periods. This occurs in at least two functional waves, with macrophage precursor cells from fetal yolk sac or fetal liver progenitors16,19. These precursors seed distant tissue and give rise to locally proliferating, self-maintained tissue-resident macrophages that can persist into adulthood16,19–22. For some tissues, such as the colon, these embryonic macrophages are rapidly replaced by monocytes derived from hematopoietic stem cells (HSCs) after birth. However, for other macrophage subsets, such as microglia, their sole origin appears to be embryonic, with little contribution from HSCs under homeostatic conditions23,24. Still other tissues contain macrophages with a mixed origin, including the pancreas, breast, and lung25–28. The relevance of this remains to be determined, though it is known that tissue macrophages of both embryonic and HSC origin assume epigenetically-regulated programs indicative of their residence in these tissues (e.g., brain, liver, and lung) to drive specific phenotypes, especially those related to metabolism and interferon responsiveness28–30.
In tumors, however, several recent studies have suggested that embryonic-derived macrophages may have distinct phenotypes and functions compared to their monocyte-derived counterparts28,31,32. TAMs are usually thought to predominantly derive from circulating monocytes33,34, but up to 50% of the macrophages in murine models of brain, lung and pancreatic cancer were found to derive from tissue-resident populations28,31,32. Within these tumors the TAMs of HSC origin have elevated expression of genes involved in immunosuppressive networks and antigen presentation31,32,35. In contrast, embryonic-derived TAM gene sets are enriched for tissue remodeling and wound healing. These data suggest that tissue and origin-specific programs can fine tune macrophage responses in ways that may have significant impact on tumor immunity; however, the rules for how these programs might integrate with macrophage polarizing cues are largely unknown. Additionally, while the origins of macrophages have been mapped in multiple animal models, our ability to interrogate these populations in human tissues is limited. Thus, one challenge for the field going forward will be to determine the impact of these findings in human cancers, perhaps with an approach such as single cell RNA-sequencing to permit an evaluation of macrophage heterogeneity and origin.
Metabolism.
Tissue hypoxia impacts macrophages in two ways. First, hypoxia can induce the production of key monocyte recruitment factors including CCL2, CCL5, CXCL12, CSF-1, and VEGF by tumor cells and the stroma. Once recruited into hypoxic regions, the receptors for several of these factors are downregulated, effectively locking TAMs in hypoxic microenvironments36. Second, macrophages directly sense hypoxic conditions via hypoxia-inducible factors (HIFs): the absence of HIF-1α leads to reduced arginase-1 expression and immunosuppressive activity in vitro37, and the absence of HIF-2α reduces macrophage infiltration and cytokine production38. In both cases, myeloid-specific loss of these factors significantly delays tumor progression in autochthonous tumor models37,38. Notably, the localization of TAMs in hypoxic regions is critical to the generation of an immunosuppressive phenotype in vivo, because myeloid-specific loss of neuropilin-1, which excludes macrophages from hypoxic areas, promotes anti-tumor immunity39. However, it should be noted that the roles of neuropilin-1 and its ligand, SEMA3A, remain controversial40, and that HIF-1α can also be stabilized by the presence of lactic acid41.
Irrespective of tissue hypoxia, aerobic glycolysis within tumors can limit glucose availability and promote the accumulation of organic acids. Do these factors also impact macrophage function? Macrophages stimulated with lipopolysaccharide (LPS) and/or IFNγ display enhanced glucose uptake and aerobic glycolysis42, and consistent with the important role for this metabolic shift, increasing glucose transport promotes expression of reactive oxygen species43. This is in contrast to IL-4-stimulated macrophages, which show preferential oxidative phosphorylation and fatty acid oxidation42, and are therefore not impacted by impaired glycolysis44. Presumably low glucose availability will thus favor TAMs to adopt a pro-tumor polarization state, but this has not been formally demonstrated. However, because a metabolic shift does control macrophage function, interfering with the genetic regulators of this process can blunt the pro-tumor bioactivity of TAMs and reduce tumor growth in animal models45–47.
As mentioned above, lactic acid promotes Vegf and Arg1 expression by macrophages in a Hif1a-depenent manner41. This is not true for lactate salt41, indicative of a role for acidic pH in either promoting the activity of monocarboxylate transporters48 and/or acidic pH directly regulating macrophage polarization. In support of the second scenario, a recent study found that a pH of 6.1 was sufficient to promote expression of Arg1, Vegfa, and Hif1a by unstimulated macrophages in vitro49, and similar observations have been made at pH 6.8 during stimulation with IL-450. Increasing the pH within tumors similarly reduces expression of Arg1 by TAMs50. How macrophages sense pH at a molecular level is somewhat vague, but activation appears to be mediated by G protein-coupled receptors and production of cAMP51 leading to expression of the transcription factor ICER (inducible cyclic AMP early repressor)49. Importantly, mice with myeloid-specific deficiency of ICER resist the growth of highly glycolytic tumors49.
Fibrosis.
Desmoplasia is a hallmark of many solid tumors, with pancreatic cancer representing one extreme end of the spectrum. Fibrotic stroma has the potential to shape the TAM phenotype through direct effects of its components, like activated fibroblasts, changes in the extracellular matrix (ECM), or indirect effects on factors such as oxygen and nutrient availability. Cancer-associated fibroblasts (CAFs) are perhaps the most relevant component of fibrosis because these cells overexpress numerous pro-inflammatory cytokines (e.g., CCL2, CCL3, CCL5, IL-6, GM-CSF, CSF-1, VEGF, and CXCL8) with the potential to regulate recruitment, differentiation, and activation of TAMs52–56. In particular, CAFs have been reported to impair the maturation of macrophages, locking recruited monocytes in an immature, suppressive state. This is possibly due to high levels of IL-6 production, especially in pancreatic CAFs, which can induce STAT3 phosphorylation and prevent macrophage differentiation57–59. In addition, IL-6 production by endothelial cells has been shown to promote M2-like polarization and tumor growth in a glioblastoma model60, and TAMs themselves produce IL-6 in multiple other model systems33,61,62. The source of these polarizing cytokines may therefore vary considerably across tumor types or even within microenvironments of the tumor. Adding to this complexity is the diversity of CAF subsets and their differential potential to alter immune function63,64. Thus, although CAFs are assumed to be important regulators of TAM function, their role remains poorly defined in vivo.
The extensive ECM associated with fibrosis can also impact macrophages in several ways. Periostin has been shown to promote TAM recruitment via integrin binding65, while collagen promotes an M2-like phenotype66–68, with the increased presence of collagen I acting as a reservoir for secreted factors, such as transforming growth factor β (TGFβ), or directly activating inhibitory receptors such as leukocyte-associated immunoglobulin-like receptor 1 (LAIR1)69. In addition to the concentration of ECM components, their macromolecular structure, including biophysical and mechanical properties, can direct macrophage function within tumors. Collagen crosslinking and matrix stiffness are properties of solid tumors that have direct effects on macrophage differentiation, motility, and phenotype70–72. This is largely mediated by β1 integrin clustering and hyperactivation of focal adhesion kinases (FAK), and genetic or pharmacological loss of FAK signaling reduces tumor fibrosis73 and the number of M2-like TAMs74,75, leading to improved anti-tumor immunity and response to immune checkpoint blockade74.
Other ECM components known to regulate macrophage activation include hyaluronan, versican, and tenascin, mostly through altered forms of these molecules being recognized as damage-associated molecular patterns (DAMPs) by Toll-like receptor 2 (TLR2) or TLR476. Versican acts via TLR2 to increase expression of receptors for IL-6 and IL-10 and sensitizes cells to these cytokines77. In tumor models, versican has been suggested to promote metastasis via macrophage activation78. Meanwhile, hyaluronan production by fibroblasts or keratinocytes promotes macrophage recruitment79,80, possibly through its role in providing a scaffold for ECM proteoglycans and enhancing chemokine retention. Hyaluronan has also been shown to alter macrophage activation in vitro through CD44 or TLR2/TLR4, depending on the state of the cells and the molecular weight of the hyaluronan81.
Cellular debris.
Cell death is prevalent within tumors, particularly regions of hypoxia, and is significantly induced by anti-cancer therapies. Whereas the release of intracellular DAMPs can promote tumor immunity through activation of dendritic cells (DCs)82, the chronic stimulation of macrophages induces negative regulatory mechanisms to dampen inflammation. Thus, although the release of HMGB1 in response to chemotherapy promotes immunity via TLR4, it can also drive IL-10 expression in TAMs via the receptor for advanced glycation end products83. Whether this is true for other DAMPs in tumors is unknown, but even the expression of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF by macrophages can promote tumor progression6. Macrophage recognition of apoptotic cells is also well-known to suppress their activation potential84, with activation of the MerTK receptor elevating expression of immunosuppressive factors such as TGFβ, IL-10, and arginase-185,86. Consistent with this observation, loss of MerTK in the stromal compartment leads to macrophage repolarization and T cell-dependent growth restraint86, as well as a prolonged response to radiation therapy87. Similarly, the deletion of protein S in tumor cells, which mediates the recognition of phosphatidylserine-presenting membranes by MerTK/Tyro3, increases iNOS expression and leukocyte infiltration88. It remains possible that activation of select pattern recognition receptors may shift macrophages towards an anti-tumor phenotype, especially when using agonists therapeutically. However, it generally appears that recognition of cellular and extracellular debris by macrophages is detrimental in terms of tumor growth.
Lymphocytes.
Most lymphocyte subpopulations can be identified within the tumor stroma, with the composition and density varying by tissue, molecular subtype, and individual patient. In addition to directly impacting tumor growth, many of these populations have been shown to shape the phenotype of macrophages in some fashion89 (Figure 2). IFNγ can be produced by CD8+ CTLs, T helper 1 (TH1) cells and natural killer (NK) cells and primes macrophages towards a classically-activated phenotype, increasing macrophage antigen presentation and pro-inflammatory cytokine production, and direct tumor cell killing90. Similarly, expression of CD40 ligand (CD40L) by T cells can activate monocytes via CD40 to increase their expression of MHC class II, iNOS, and TNF. This induction of an anti-tumor phenotype is needed during acute T cell responses following therapeutic intervention91,92, and could presumably promote a cycle of anti-tumor immunity if maintained.
Unfortunately, many lymphocyte-derived factors engage the tumor-promoting activities of TAMs. This includes production of IL-4 and IL-13 by TH2-polarized CD4+ T cells, which enhance epidermal growth factor expression by TAMs to foster cancer cell metastasis93, as well as the suppressive activity of TAMs to blunt CD8+ T cell responses to chemo- and radiation therapy94. It also includes a role of FOXP3+ regulatory T (Treg) cells in driving TAMs towards an immunosuppressive phenotype marked by production of IL-10 and expression of B7-H495,96. Although the available data suggest that neutrophils are the major effector cells associated with IL-17-driven responses97, IL-17 produced by TH17 or γδ T cells can increase monocyte recruitment and macrophage activation, and this could possibly lead to immune tolerance when macrophages engulf recruited neutrophils undergoing apoptosis in the tissue98–100. Finally, the loss of B cells in murine models inhibits tumor progression, and in squamous cell carcinoma this has been traced to the pathogenic production of autoantibodies, which act via Fcγ receptor signaling to promote macrophage-dependent angiogenesis and tumor progression101,102. A similar phenotype has been observed in pancreatic tumors, and in both cases, B cell depletion or inhibition of Fcγ receptor signaling (Btk, Syk, and PI3Kγ) relieves macrophage-mediated T cell dysfunction, leading to delayed progression or improved responses to chemotherapy and PD1 blockade103–106.
Regulation of T cell function by TAMs
Macrophages are widely acknowledged as one of the central suppressive populations within tumors and depleting these cells can unleash T cell responses under several therapeutic conditions6–8. Underlying this functional role are molecular mechanisms that range from nutrient depletion to recruitment of Treg cells, although the extent to which these mechanisms are involved in any particular tumor is less well defined. Here we will describe what is known about the ability of macrophages to directly or indirectly suppress T cells responses within tumors, as well as discuss several theoretical concepts that may be applicable to cancer (Figure 3).
Figure 3. Cell type versus integrated views on drivers of TAM phenotype.
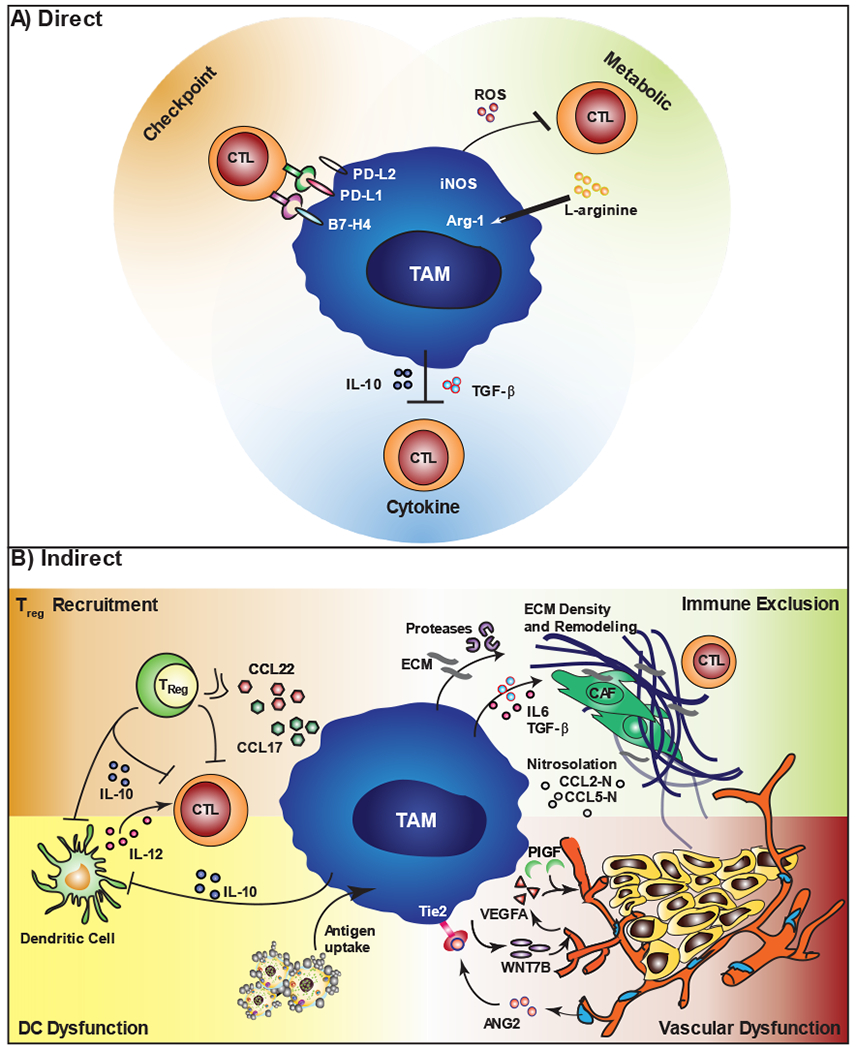
A) TAM phenotype is driven by a combination of the tumor microenvironment (TME) and the tumor immune microenvironment (TIME). On the left, responses by adaptive and innate immune cells provide cytokines and other factors that regulate macrophage bioactivities. On the right, properties of the tumor microenvironment like hypoxia, fibrosis and cellular stress also tailor the phenotype of TAMs. B) Both immune and non-immune related factors integrate to drive functional or dysfunctional anti-tumor immunity. On the left, the presence of a robust adaptive immune response is concomitant with limited tissue pathology and macrophages programmed to drive inflammation. On the right, tumor hypoxia and fibrosis are integrated with high CAF and immunosuppressive cell infiltration, and macrophages are programmed to drive immune suppression and tissue remodelling, leading to CTL exclusion and/or suppression.
Direct regulation of T cells by TAMs.
Numerous studies have shown that TAMs suppress naïve T cell proliferation ex vivo, indicating that macrophages can directly suppress T cell function33,37,107–109. This is often thought to relate to the ability of murine tumor macrophages to metabolize L-arginine via expression of arginase-1110, a common marker of M2-like polarization in murine macrophages, and L-arginine is necessary for T cell fitness and anti-tumor activity111. However, neither arginase-1 inhibition nor Arg-1 deficiency impact macrophage suppressive capacity in vitro109,112. Instead a secondary role for arginase-1 is only observed when inducible nitric oxide synthase (iNOS/NOS2) is inhibited or absent33,109,112. These observations may be a byproduct of the supraphysiological concentration of L-arginine in cell culture medium, because tumor macrophages can deplete L-arginine and prevent recovery of CD3ε expression by T cells following stimulation113. Inhibition of arginase-1 in vivo also reduces the growth of tumors only in immune competent mice113, and Arg1-deficiency in macrophages improves responses to adoptive cell transfer therapy (ACT)112. However, it remains unclear if these in vivo observations are the result of direct immune suppression, the effects of polyamines on tumor cell proliferation114, or enhanced production of NO112.
The importance of iNOS expression by tumor macrophages is less opaque, at least ex vivo33,109,112. Nos2-deficient tumor myeloid cells lose significant suppressive capacity in co-culture assays112 and iNOS inhibition restores T cell proliferation in the presence of macrophages33. In addition to potential direct effects of NO on T cells115, this may be due to secondary production of peroxynitrites, which can prevent the interaction of the T cell receptor with MHC through nitration of either protein116–118. In vivo, scavenging peroxynitrites with uric acid improves T cell activation and enhances responses to a tumor vaccine118, although whether this is attributed to expression by macrophages, other myeloid subsets, or the combination may vary by the tumor model. Based on these observations, it is surprising that iNOS expression by myeloid cells has also been implicated in promoting a T cell response92,112. Nos2-deficiency diminishes the efficacy of ACT112, and at least in the context of low-dose irradiation, this is due to iNOS-expressing macrophages inducing vascular cell adhesion protein (VCAM)-1 expression by the tumor endothelium, leading to enhanced recruitment of adoptively-transferred CD8+ T cells92. Thus, the impact of iNOS expression by macrophages may be highly context dependent, promoting or hindering T cell responses under different therapeutic conditions.
Despite the results in murine models, L-arginine metabolism by macrophages has not been implicated in regulating T cell responses in humans; arginase-1 is expressed by human granulocytes instead of macrophages95,119,120, and even combining arginase-1 and iNOS inhibitors fails to blunt macrophage suppressive activity95. Instead, the available data points to expression of immune checkpoint ligands as being necessary for suppression. Programmed cell death ligand 1 (PD-L1) expression by CD14+ or CD68+ macrophages is observed in multiple cancer tissues, including hepatocellular carcinoma, melanoma, and ovarian cancer121,122, and positive correlations have been noted specifically between PD-L1 expression by macrophages and response to programmed cell death 1 (PD-1) blockade121. Induced expression of PD-L1 on monocytes also suppresses activation of tumor-specific T cells in vitro, and in vivo following adoptive transfer122. Similarly, PD-L1 blockade depends upon target expression by CD11b+ myeloid cells in murine models121,123. However, it should be noted that PD-L1 expression by macrophages has not been established as an independent predictor of response, and the relative contribution of PD-L1 expression by tumor cells or the various stromal populations is likely to be highly variable between patients.
The other immune checkpoint ligand that has been identified as functionally important in human macrophages is B7-H4 (also known as B7S1). Although the receptor for B7-H4 is unknown, B7-H4 expressing cells and B7-H4-Ig fusion proteins suppress IL-2 production and T cell proliferation124. The fusion protein has also been used to identify expression of the “receptor” in T lymphocytes within healthy livers and hepatocellular carcinomas125. Importantly, B7-H4 is preferentially expressed by CD14+ macrophages within ovarian and liver tumors, and inducing B7-H4 expression on human monocytes/macrophages confers suppressive capacity in vitro95,125. Blocking B7-H4 also suppresses the growth of subcutaneously implanted tumors in mice by reducing CD8+ T cell exhaustion125; however, other groups have found that B7-H4 promotes or has no impact on tumor immunity in murine models of breast and prostate cancer, respectively126,127. These differences may relate to the specifics of the tumor model examined, especially as B7-H4 expression is regulated by cytokines common in the tumor microenvironment such as IL-6, IL-10, and IFN-γ95,127.
Several other molecules expressed by macrophages potentially have direct suppressive effects on tumor-infiltrating T cells. For example, tumor macrophages can be an important source of IL-10108, and IL-10 is known to suppress CD8+ T cell stimulation by increasing N-glycan branching, thereby reducing co-localization of CD8 protein with the T cell receptor128. This process requires the presence of galectin-3, and interfering with this association restores IFN-γ expression by CD8+ T cells from human ovarian ascites129. As macrophages can be an important source of galectin-3 in inflamed tissues130, tumor macrophages may even regulate both aspects of this suppressive pathway. Macrophages actually express a variety of C-type and I-type lectins, which could represent an additional layer of regulation via protein-carbohydrate interactions. This has not been extensively evaluated, but it is worth noting that the mannose receptor, CD206, which is highly expressed by tumor macrophages, can impair the cytotoxicity of CD8+ T cells by suppressing CD45 phosphatase activity131. Given the importance of carbohydrate modifications in T cell signaling, lectin expression by macrophages may have an unappreciated role in controlling anti-tumor immunity.
Indirect regulation of T cells by TAMs.
Extending from their function in maintaining tissue homeostasis, macrophages are important regulators of multiple aspects of the tumor microenvironment, in particular the structure of the vasculature and extracellular matrix6. Do tumor macrophages regulate T cell recruitment? Several studies have reported that inhibiting macrophage recruitment by targeting the CSF1–CSF1R pathway improves T cell infiltration, including during chemotherapy or high-dose irradiation12,61,94,107,108. Similar results are observed when blocking macrophage recruitment through CCR2 inhibition61. Assuming the increase in T cell numbers is not due to changes in cell death or proliferation, the mechanism(s) underlying these observations is unclear.
One possibility could be through regulation of vascular adhesion molecules, but iNOS-expressing macrophages actually promote VCAM1 expression during low-dose irradiation92, and while perivascular macrophages regulate vascular structure through expression of VEGFA132–134, this would be expected to enhance vessel leakiness and expression of adhesion molecules. Another possible mechanism by which macrophages could suppress T cell recruitment is through production of peroxynitrites and the subsequent nitration of CCL2 or CCL5135,136. Nitration of CCL2 specifically has been shown to reduce accumulation of T cells within subcutaneous tumors137. This appears to be due to low CCL2-responsiveness by T cells, resulting in nitration preventing chemotaxis of human and mouse T cells without impacting monocyte migration137. Although this does not explain the increased T cell recruitment following CCR2 inhibition61, it is possible that nitration of CCL5 has a similar effect, especially given its more important role in promoting T cell recruitment into tumours138.
Rather than recruitment, it may be that macrophages are involved in restricting the intratumoral localization of T cells. For example, inhibiting reactive nitrogen species can increase the number of CD8+ T cells within tumors without impacting their frequency in the surrounding stroma137. Increasing the degree of fibrosis would be another mechanism by which macrophages could shield tumors from T cell infiltration. This has been shown in pancreatic cancer with macrophage production of granulin promoting the accumulation of myofibroblasts139,140. Two recent studies have also reported that TGFβ signaling acts to exclude T cells from human and murine tumors141,142. Tumor macrophages are one of many cells known to produce TGFβ133,108, and are not a major source of TGFβ2 or TGFβ3141; however, it is possible that macrophages can regulate desmoplasia through expression of matrix metalloproteinases and activation of latent TGFβ143. Human monocytes and macrophages even display an enhanced ability to activate TGFβ through their expression of integrin αvβ8144. Alternatively, because tumor macrophages are chemotactic for CD8+ T cells ex vivo103 and have been described to highly express the T cell attracting chemokine Cxcl1033,145,146, it is conceivable that they could retain T cells within the stroma or perivascular regions of the tumor. Macrophages and T cells are predominantly found within these regions, and at least one study has shown that CSF1R inhibition increases T cell motility and localization in tumor islets147.
Macrophages have also been shown to promote Treg cell recruitment to human ovarian carcinomas via CCL22 production148. In addition to, presumably, suppressing cytotoxic T cell responses, Treg cells from ovarian ascites stimulate IL-6 and IL-10 expression by macrophages and increase B7-H4 expression in an autocrine fashion in vitro95,96. This ability of macrophages to suppress T cell responses via regulation of intermediate cell types has also been described in mammary carcinoma models. In this case, macrophage production of IL-10 suppresses IL-12 expression by CD103+ conventional dendritic tumor cells (cDC), resulting in a diminished CD8+ T cell response during chemotherapy108. Together, these data highlight macrophages as central drivers of the immunosuppressive tumor microenvironment through their ability to regulate the recruitment and the function of multiple immune subtypes.
Phagocytosis and antigen presentation.
Consistent with their name, several studies have confirmed macrophages are the main phagocytic population within tumors145,146,149. However, tumor macrophages do not express CCR7 and are unable to migrate into the draining lymph nodes149,150. They also display relatively limited ability to activate or restimulate CD8+ T cells146. What impact could phagocytosis by macrophages have on the induction of anti-tumor immunity? First, it is possible that macrophages redirect antigens away from cDCs, an observation that might not be apparent in experiments conducted using highly expressed model antigens (e.g., ovalbumin). Second, clearance of apoptotic cells and debris might be expected to reduce the presence of alarmins or DAMPs. Third, tissue-resident macrophages are known to suppress their own activation in response to apoptotic cell phagocytosis84. This includes phagocytic pathways designed to avoid activation of cytosolic sensors such as stimulator of interferon genes (STING)151,152, as well as the secretion of factors that suppress the activation of neighboring cells153. One of these secreted factors, insulin-like growth factor-1, has already been shown to promote the regrowth of gliomas following CSF1R inhibition154. Any of these mechanisms, or a combination of them, could theoretically reduce the capacity of the CD103+ cDC subset to transport tumor antigens to the draining lymph node and initiate a de novo CD8+ T cell response149,150.
Alternatively, it has recently been described that embryonically-derived tissue-resident macrophages are present within tumors, and that these cells retain a distinct phenotype from their monocyte-derived macrophage counterparts31,32. A unique property of many tissue-resident macrophages is expression of CD169/Siglec-1155, which has been shown to selectively bind to CD8α+ cDCs and promote antigen transfer within secondary lymphoid organs156. Whether CD169+ macrophages are present in tumors and transfer antigen to the equivalent CD103+ cDC subset is unknown. This pathway also appears irrelevant for soluble or Fc receptor-mediated uptake of antigens by cDCs156, but could be involved in the transfer of cellular antigens, particularly during early tumor progression when tissue-resident macrophages might be expected to dominate the microenvironment. Interestingly, it has been demonstrated that tissue-resident CD169+ macrophages in the spleen and lymph node capture extracellular vesicles157, a process that restricts entrance into the lymph node cortex and prevents their interaction with B cells157,158. In tumor models, the absence of CD169+ macrophages increases production of immunoglobulin158, which subsequently drives the polarization of tumor macrophages via activating Fcγ receptors to increase neoplastic progression, tumor growth, and resistance to chemotherapy101–103.
Therapeutic targeting of TAMs
Given the importance of TAMs in regulating tumor immunity, there has been considerable interest in therapeutic strategies that target macrophages, which can be roughly divided into those that either deplete or alter TAM protumoral activities. As preclinical evidence largely supports combinatorial approaches being necessary to observe efficacy6, these strategies are currently in evaluation either to augment tumor immunity during standard chemotherapy or radiation therapy, or in combination with T cell-directed immunotherapy (Table 1). Herein, we will discuss the potential strengths and weaknesses of these approaches, and suggest future directions for clinical trials.
Table 1.
Immuno-oncology Combinations
| TAM Targeted Agent | Immune Modulator | Clinical Trial(s) |
|---|---|---|
| CSF1/CSF1R agonists | PD-1 or PD-L1 antagonists |
NCT02554812* NCT02452424-n NCT02777710* NCT02880371* NCT03238027* NCT02526017-n NCT02829723* NCT02323191* NCT03158272* NCT02713529-n |
| CCR2/5-antagonists | PD-1 or PD-L1 antagonists |
NCT02723006-n NCT03184870* |
| CXCR4 antagonist | PD-L1 antagonists |
NCT02737072-n NCT03193190* NCT02907099* NCT03154827* NCT03337698* NCT02823405* |
| Ang2/Tie2 | PD-1 antagonists | NCT03239145* |
| CD40-agonists | PD-1 or PD-L1 antagonists |
NCT03123783* NCT02304393* |
| CD47 (SIRPα-Fc) | PD-1 or PD-L1 antagonists |
NCT03530683* NCT02890368* NCT03013218* |
| PI3Kγ/δ inhibitors | PD-1 or PD-L1 antagonists |
NCT03471351* NCT02637531* |
| Multimodality Immuno-oncology Combinations | ||
| TAM Targeted Agent | Immune Modulator | Clinical Trial(s) |
| CSF1R | Chemotherapy + PD1/PDL1 |
NCT02323191* NCT03336216* |
| CSF1R | PD-1 antagonist + SBRT | NCT03431948* |
| CSF1R | PD-1 antagonist + CD40 agonist | NCT03502330* |
| CSF1R | GVAX + PD-1 antagonist | NCT03153410* |
| CCR2/5 inhibition | Chemotherapy + PD-1 antagonist |
NCT03184870* NCT03496662* |
trial currently open and active
trial closed
Targeting TAM recruitment and survival.
One strategy to deplete TAMs is to cut off their replenishment by circulating inflammatory monocytes. Circulating monocytes are highly dependent on CCL2–CCR2 signaling for their mobilization from the bone marrow and recruitment into inflammatory sites; thus, CCR2 inhibition keeps monocytes in the bone marrow, resulting in depleted pools of circulating cells and reduced numbers of TAMs in primary and metastatic sites159–164. In preclinical models, CCL2/CCR2 blockade can improve the efficacy of chemotherapy, radiation therapy, and immunotherapy61,159,160,165–167. Several CCR2 blockade combination clinical trials are therefore ongoing. One early study in pancreatic cancer showed a >40% increase in responsiveness to chemotherapy when CCR2 inhibitors were combined with the chemotherapy regimen, FOLFIRNOX159. Biomarker analysis in this study also showed that combination therapy was associated with increased T cell infiltration. Similar observations have been made in some of the preclinical models, facilitating further combinations with checkpoint immunotherapy. Clinical testing of this triple combination, i.e., CCR2 inhibition, chemotherapy, and checkpoint blockade, are now ongoing. Although CCR2 plays a dominant role in macrophage recruitment, important considerations for combined efficacy include rapid compensation by granulocytes and lack of efficacy in impacting resident TAMs populations31,168. It has also been observed that cessation of CCL2/CCR2 blockade leads to release of the monocytes previously trapped within the bone marrow, and this can exacerbate metastasis in a murine model of breast cancer169. These parameters will be critical to consider in the design of future clinical trials.
The CSF1–CSF1R axis has also been heavily investigated in preclinical models. In most tumors, inhibition of CSF1–CSF1R signaling leads to the apoptotic death of a significant portion of TAMs, the outlier being glioma models where compensatory activity by GM-CSF leads to TAM survival and repolarization10. Independent of the mechanism of action, in a significant array of animal models, CSF1R inhibition improves T cell responses in combination with radiation or chemotherapeutic agents61,107,170–173. Additionally, CSF1–CSF1R blockade improves the efficacy of the diversity of immunotherapeutic modalities, including CD40 agonists, PD-1, or CTLA-4 antagonists, and adoptive T cell therapy12,147,174–176. The sum of these studies has led to a number of clinical trials combining CSF-1/CSF-1R inhibitors with immune checkpoint blockade. In a promising study in pancreatic cancer patients, which do not traditionally respond to immunotherapy, it has been reported that some patient responses were observed with the combination of CSF-1R and PD-1 antagonists177, and these studies are now moving forward to a multi-arm Phase II clinical trial. Despite the breadth and consistency of these findings, it is perhaps not surprising that compensatory resistance pathways have already been defined that ultimately limit the durability of the response to CSF-1R inhibitors154,178,179. The other major barrier to translation may be that TAM depletion coincides with loss of tissue-resident populations important for maintaining homeostasis180. Although elevated liver enzymes appear to be a byproduct of reduced clearance by Kupffer cells, and not evidence of liver toxicity, grade 3 adverse events may limit the utility of CSF-1R antagonists in combination studies181.
Other pathways involved in macrophage recruitment include the CXCL12/CXCR4 and angiopoietin-2/Tie2 axes. In glioma and breast models, radiation, chemotherapy, and vascular disruption have been shown to increase CXCL12 expression and promote CXCR4-dependent macrophage repopulation and treatment resistance133,182–184. Intriguingly, it appears that CXCL12 acts by preferentially recruiting Tie2+ macrophages, a population that is strongly associated with the vasculature and is important for tumor angiogenesis185. Thus, depleting Tie2+ macrophages improves vascular disruption184, neutralizing angiopoietin-2 improves responses to VEGFA blockade186, and inhibiting Tie2 blocks chemotherapy-induced Tie2+ TAM recruitment in breast models and leads to decreased metastasis187,188. Though the impact of Tie2+ TAMs on tumor immunity and the potential for immunotherapy combinations are unclear, it has been found that dual neutralization of angiopoietin-2 and VEGFA promotes T cell infiltration, and that efficacy is completely dependent upon CD8+ T cells186. CXCR4 inhibition also renders pancreatic cancer models more responsive to checkpoint blockade189, although whether this is related to macrophages is unclear.
Targeting TAM activation
An intrinsic downside to depleting TAMs is the loss of their latent immune stimulatory role as the primary phagocyte and professional antigen-presenting cell within tumors. Reprogramming or ‘repolarizing’ TAMs towards an anti-tumor phenotype could therefore prove a more efficacious—if potentially more toxic—approach to augmenting other forms of immunotherapy. One of the most productive approaches to date has been the use of an agonist CD40 antibody190, which shows combinatorial efficacy in pancreatic cancer with gemcitabine191 and gemcitabine/nab-paclitaxel192. As CD40 is expressed by DCs, the relative contribution of TAM versus DC activation is unclear, but critically, enhanced responses to PD-1 and CTLA-4 antagonists have been observed193. Clinical trials combining a CD40 agonist with chemotherapy, immunotherapy, and angiogenic inhibitors are currently ongoing. Intriguingly, efficacy has also been described when combining CD40 agonists with various CSF-1R-targeted agents. This effect occurs in systems where the number of TAMs is not impacted by CSF-1R inhibition176,194, but also when TAM depletion is effective195. In either case, it appears that blocking CSF-1R sensitizes residual TAMs to reprogramming by anti-CD40 and promotes enhanced T cell-dependent immunity.
Epigenetic reprogramming of macrophages through inhibition of histone deacetylases (HDAC) can also elicit a T cell supportive role. Specifically, in mammary tumor models, a selective HDAC IIa inhibitor induces anti-tumor macrophage phenotypes that support T cell responses and increase responses to chemotherapy and immune checkpoint blockade196. Pan-HDAC inhibitors are already being tested with PD-1 antagonists, and it is possible that macrophage reprogramming could have a significant role in mediating therapeutic efficacy. An alternative approach to reprogramming, which has been evaluated extensively in preclinical models, involves targeting the major pathways that drive the immunosuppressive function of TAMs, including IL-4, IL-13, and immunoglobulins6. However, although clinical agents for these pathways exist, the interest in using them in solid malignancies to promote immunity has been minimal. An exception to this has been the targeting of PI3Kγ, which is activated in TAMs downstream of multiple pathways, including the Fcγ receptor. Activation of PI3Kγ signaling in macrophages has been shown to drive TAM immunosuppressive activities in models of lung, melanoma, and pancreatic cancer106,197–200. In animal models, pharmacological inhibition of PI3Kγ results in macrophage reprograming and augmentation of T cell responses as a single agent105, and in combination with T cell checkpoint blockade106,198,200.
Future directions
Given the myriad of roles for TAMs, therapeutically targeting one specific function of these cells may prove difficult. Success has been observed in multiple xenograft models by blocking CD47, a membrane bound protein that interacts with SIRPα on TAMs to inhibit phagocytosis201. However, it should be noted that SIRPα is also expressed on CD11b+ DCs, and in syngeneic models, the ability of CD47 blockade to promote an immune response is dependent upon cytosolic sensing of tumor DNA by DCs202,203. Although this does not negate the approach, the degree to which efficacy is macrophage-dependent is unclear. Intriguingly, recent data have suggested that PD-L1/PD-1 signaling on TAMs impairs their phagocytic capacity and anti-tumor phenotype204,205, suggesting a second point of synergy for CD47/PD1 combinations. In addition, macrophages have been shown to uptake anti-PD-1 antibodies through their Fcγ receptors, thereby limiting efficacy, in animal models206. The binding of antibodies to macrophage Fc receptors has been shown to regulate a number of therapeutic responses in vivo, and is a critical aspect of antibody drug design207. The importance of macrophages in regulating these responses also needs to be considered when designing clinical studies, especially when incorporating CSF-1R or CCR2 inhibitors that could limit therapeutic efficacy.
The sum of pre-clinical animal and correlative human studies suggest that targeting TAMs could significantly improve the efficacy of conventional and immunotherapeutics. However, in spite of the clinical interest and a few suggestive early trial outcomes159,177, the optimum therapeutic approach has yet to be identified. This may be partially due to a lack of data on key clinical parameters that may determine the success or failure of combinatorial therapeutic approaches in humans. For example, while several cancer types, such as ovarian, pancreas, and mesothelioma, have relatively high macrophage infiltration, it is unknown if these reflect patient populations that will see maximal benefit. Other questions include what type of approach (i.e. depletion or reprogramming) should be employed, and whether the best therapeutic modalities might be cancer type dependent. In addition, if TAM antagonists are being used to overcome resistance to immunotherapy, then more clinical data correlating macrophage infiltration or phenotype with patient outcomes is needed to guide patient selection. Finally, it will be critical to determine how exactly to employ these therapeutic combinations, including data driven considerations of dosing strategies and sequencing to minimize potential toxicities and/or maximize immune stimulatory properties. For example, anti-CD40 antibody demonstrated toxicity in mice when given prior to gemcitabine in a pancreatic tumor model208. The challenge will be to empirically design trials rather than base them upon historical doses, clinical practicality, or financial considerations. In spite of these challenges, there remains significant potential to harness macrophage biology to improve outcomes for cancer patients.
Acknowledgements
The laboratory of D.G.D. is supported by funding from National Cancer Institute, including P50CA196510, R01CA177670, R01CA203890, P30CA091842 Supplement-15S3, as well as The Mary Kay Foundation. The laboratory of B.R. is supported by funding from the National Institute of Health (R00CA185325), Florida Department of Health Bankhead-Coley Cancer Research Program (8BC02) and the Florida Breast Cancer Foundation. The authors thank members of their laboratories for helpful discussion.
Glossary Terms:
- Tumor microenvironment
The tumor microenvironment (TME) is the cellular and acellular components in which malignant cells reside. These include surrounding blood vessels, immune cells, fibroblasts, extracellular matrix components, extracellular signaling molecules such as chemokines, cytokines, and growth factors, as well as metabolic regulators such as oxygen.
- Tumor immune microenvironment
The tumor immune microenvironment (TIME) is the components of the tumor microenvironment represented by leukocytes or their derived factors.
- Desmoplasia
Cancer associated desmoplasia is the growth and expansion of fibrous and/or connective tissue surrounding the malignant cells. Desmoplasia may occur around a growing neoplasm and consists of expansion of the non-malignant cellular components, such as activated fibroblasts beyond the norms of the homeostatic tissue levels.
References
- 1.Zhang QW et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PloS one 7, e50946, doi: 10.1371/journal.pone.0050946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komohara Y, Jinushi M & Takeya M Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer science 105, 1–8, doi: 10.1111/cas.12314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945, doi: 10.1038/nm.3909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canli O et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer cell 32, 869–883 e865, doi: 10.1016/j.ccell.2017.11.004 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Qian BZ & Pollard JW Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51, doi: 10.1016/j.cell.2010.03.014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruffell B & Coussens LM Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 27, 462–472, doi: 10.1016/j.ccell.2015.02.015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffelt SB & de Visser KE Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends in immunology 36, 198–216, doi: 10.1016/j.it.2015.02.006 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Engblom C, Pfirschke C & Pittet MJ The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447–462, doi: 10.1038/nrc.2016.54 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61, doi: 10.1016/j.immuni.2014.06.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pyonteck SM et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19, 1264–1272, doi: 10.1038/nm.3337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson OC, Kim H, Quail DF, Foley EA & Joyce JA Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell Rep 19, 101–113, doi: 10.1016/j.celrep.2017.03.038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74, 5057–5069, doi: 10.1158/0008-5472.CAN-13-3723 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnardel J & Guilliams M Developmental control of macrophage function. Curr Opin Immunol 50, 64–74, doi: 10.1016/j.coi.2017.12.001 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Epelman S, Lavine KJ & Randolph GJ Origin and functions of tissue macrophages. Immunity 41, 21–35, doi: 10.1016/j.immuni.2014.06.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavin Y, Mortha A, Rahman A & Merad M Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol 15, 731–744, doi: 10.1038/nri3920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginhoux F & Guilliams M Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 44, 439–449, doi: 10.1016/j.immuni.2016.02.024 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Mantovani A, Sozzani S, Locati M, Allavena P & Sica A Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23, 549–555 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20, doi: 10.1016/j.immuni.2014.06.008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mass E et al. Specification of tissue-resident macrophages during organogenesis. Science 353, doi: 10.1126/science.aaf4238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto D et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804, doi: 10.1016/j.immuni.2013.04.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulz C et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90, doi: 10.1126/science.1219179 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Yona S et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91, doi: 10.1016/j.immuni.2012.12.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoeffel G et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678, doi: 10.1016/j.immuni.2015.03.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginhoux F et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845, doi: 10.1126/science.1194637 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calderon B et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med 212, 1497–1512, doi: 10.1084/jem.20150496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Epelman S et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104, doi: 10.1016/j.immuni.2013.11.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbings SL et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126, 1357–1366, doi: 10.1182/blood-2015-01-624809 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loyher PL et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med 215, 2536–2553, doi: 10.1084/jem.20180534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavin Y et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326, doi: 10.1016/j.cell.2014.11.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gosselin D et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340, doi: 10.1016/j.cell.2014.11.023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Y et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 47, 323–338, doi: 10.1016/j.immuni.2017.07.014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowman RL et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 17, 2445–2459, doi: 10.1016/j.celrep.2016.10.052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Movahedi K et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70, 5728–5739, doi: 10.1158/0008-5472.CAN-09-4672 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Strachan DC et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology 2, e26968, doi: 10.4161/onci.26968 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res 77, 2266–2278, doi: 10.1158/0008-5472.CAN-16-2310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henze AT & Mazzone M The impact of hypoxia on tumor-associated macrophages. J Clin Invest 126, 3672–3679, doi: 10.1172/JCI84427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doedens AL et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res 70, 7465–7475, doi: 10.1158/0008-5472.CAN-10-1439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imtiyaz HZ et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120, 2699–2714, doi: 10.1172/JCI39506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casazza A et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24, 695–709, doi: 10.1016/j.ccr.2013.11.007 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Wallerius M et al. Guidance Molecule SEMA3A Restricts Tumor Growth by Differentially Regulating the Proliferation of Tumor-Associated Macrophages. Cancer research 76, 3166–3178, doi: 10.1158/0008-5472.CAN-15-2596 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563, doi: 10.1038/nature13490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geeraerts X, Bolli E, Fendt SM & Van Ginderachter JA Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front Immunol 8, 289, doi: 10.3389/fimmu.2017.00289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freemerman AJ et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289, 7884–7896, doi: 10.1074/jbc.M113.522037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang F et al. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab 28, 463–475 e464, doi: 10.1016/j.cmet.2018.08.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang SC et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 45, 817–830, doi: 10.1016/j.immuni.2016.09.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wenes M et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab 24, 701–715, doi: 10.1016/j.cmet.2016.09.008 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Penny HL et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 5, e1191731, doi: 10.1080/2162402X.2016.1191731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones RS & Morris ME Monocarboxylate Transporters: Therapeutic Targets and Prognostic Factors in Disease. Clin Pharmacol Ther 100, 454–463, doi: 10.1002/cpt.418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohn T et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol 19, 1319–1329, doi: 10.1038/s41590-018-0226-8 (2018). [DOI] [PubMed] [Google Scholar]
- 50.El-Kenawi A et al. Acidity promotes tumor progression by altering macrophage phenotype in prostate cancer. bioRxiv, doi: 10.1101/478420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radu CG, Nijagal A, McLaughlin J, Wang L & Witte ON Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proceedings of the National Academy of Sciences of the United States of America 102, 1632–1637, doi: 10.1073/pnas.0409415102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalluri R The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598, doi: 10.1038/nrc.2016.73 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Chomarat P, Banchereau J, Davoust J & Palucka AK IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nature immunology 1, 510–514, doi: 10.1038/82763 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Wu MH et al. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 1306–1316, doi: 10.1158/1078-0432.CCR-10-1824 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Torres S et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 6006–6019, doi: 10.1158/1078-0432.CCR-13-1130 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Mathew E et al. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 18, 142–151, doi: 10.1016/j.neo.2016.01.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JH et al. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochemical and biophysical research communications 423, 60–66, doi: 10.1016/j.bbrc.2012.05.081 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Mace TA et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer research 73, 3007–3018, doi: 10.1158/0008-5472.CAN-12-4601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar V et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 44, 303–315, doi: 10.1016/j.immuni.2016.01.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat Commun 9, 559, doi: 10.1038/s41467-018-03050-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitchem JB et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73, 1128–1141, doi: 10.1158/0008-5472.CAN-12-2731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song L et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest 119, 1524–1536, doi:37869 [pii] 10.1172/JCI37869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Costa A et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 33, 463–479 e410, doi: 10.1016/j.ccell.2018.01.011 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Givel AM et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun 9, 1056, doi: 10.1038/s41467-018-03348-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou W et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17, 170–182, doi: 10.1038/ncb3090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pickup MW, Mouw JK & Weaver VM The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15, 1243–1253, doi: 10.15252/embr.201439246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stahl M et al. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One 8, e81382, doi: 10.1371/journal.pone.0081382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wesley RB 2nd, Meng X, Godin D & Galis ZS Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol 18, 432–440 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Meyaard L The inhibitory collagen receptor LAIR-1 (CD305). J Leukocyte Biol 83, 799–803, doi: 10.1189/jlb.0907609 (2008). [DOI] [PubMed] [Google Scholar]
- 70.McWhorter FY, Davis CT & Liu WF Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci 72, 1303–1316, doi: 10.1007/s00018-014-1796-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I & Le Cabec V Matrix Architecture Dictates Three-Dimensional Migration Modes of Human Macrophages: Differential Involvement of Proteases and Podosome-Like Structures. Journal of immunology 184, 1049–1061, doi: 10.4049/jimmunol.0902223 (2010). [DOI] [PubMed] [Google Scholar]
- 72.McWhorter FY, Davis CT & Liu WF Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci 72, 1303–1316, doi: 10.1007/s00018-014-1796-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sulzmaier FJ, Jean C & Schlaepfer DD FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14, 598–610, doi: 10.1038/nrc3792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang H et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860, doi: 10.1038/nm.4123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laklai H et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med, doi: 10.1038/nm.4082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sorokin L The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10, 712–723, doi: 10.1038/nri2852 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Tang M et al. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep 13, 2851–2864, doi: 10.1016/j.celrep.2015.11.053 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Kim S et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106, doi: 10.1038/nature07623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kobayashi N et al. Hyaluronan Deficiency in Tumor Stroma Impairs Macrophage Trafficking and Tumor Neovascularization. Cancer research 70, 7073–7083, doi: 10.1158/0008-5472.CAN-09-4687 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Jameson JM, Cauvi G, Sharp LL, Witherden DA & Havran WL Gammadelta T cell-induced hyaluronan production by epithelial cells regulates inflammation. The Journal of experimental medicine 201, 1269–1279, doi: 10.1084/jem.20042057 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee-Sayer SS et al. The where, when, how, and why of hyaluronan binding by immune cells. Front Immunol 6, 150, doi: 10.3389/fimmu.2015.00150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kroemer G, Galluzzi L, Kepp O & Zitvogel L Immunogenic cell death in cancer therapy. Annu Rev Immunol 31, 51–72, doi: 10.1146/annurev-immunol-032712-100008 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Huber R et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci Rep 6, 29914, doi: 10.1038/srep29914 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roberts AW et al. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 47, 913–927 e916, doi: 10.1016/j.immuni.2017.10.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Graham DK, DeRyckere D, Davies KD & Earp HS The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 14, 769–785, doi: 10.1038/nrc3847 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Cook RS et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest 123, 3231–3242, doi: 10.1172/JCI67655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crittenden MR et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 7, 78653–78666, doi: 10.18632/oncotarget.11823 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ubil E et al. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J Clin Invest 128, 2356–2369, doi: 10.1172/JCI97354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruffell B, Affara NI & Coussens LM Differential macrophage programming in the tumor microenvironment. Trends in immunology 33, 119–126, doi: 10.1016/j.it.2011.12.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Biswas SK & Mantovani A Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11, 889–896, doi:ni.1937 [pii] 10.1038/ni.1937 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Marigo I et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer cell 30, 377–390, doi: 10.1016/j.ccell.2016.08.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klug F et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24, 589–602, doi: 10.1016/j.ccr.2013.09.014 (2013). [DOI] [PubMed] [Google Scholar]
- 93.DeNardo DG et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102, doi: 10.1016/j.ccr.2009.06.018 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shiao SL et al. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res 3, 518–525, doi: 10.1158/2326-6066.CIR-14-0232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kryczek I et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med 203, 871–881, doi: 10.1084/jem.20050930 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kryczek I et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res 67, 8900–8905, doi: 10.1158/0008-5472.CAN-07-1866 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Coffelt SB et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature, doi: 10.1038/nature14282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shahrara S, Pickens SR, Dorfleutner A & Pope RM IL-17 induces monocyte migration in rheumatoid arthritis. J Immunol 182, 3884–3891, doi: 10.4049/jimmunol.0802246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jovanovic DV et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol 160, 3513–3521 (1998). [PubMed] [Google Scholar]
- 100.Greenlee-Wacker MC Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 273, 357–370, doi: 10.1111/imr.12453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andreu P et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 17, 121–134, doi: 10.1016/j.ccr.2009.12.019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Visser KE, Korets LV & Coussens LM De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7, 411–423, doi: 10.1016/j.ccr.2005.04.014 (2005). [DOI] [PubMed] [Google Scholar]
- 103.Affara NI et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 25, 809–821, doi: 10.1016/j.ccr.2014.04.026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gunderson AJ et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov 6, 270–285, doi: 10.1158/2159-8290.CD-15-0827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaneda MM et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov 6, 870–885, doi: 10.1158/2159-8290.CD-15-1346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaneda MM et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442, doi: 10.1038/nature19834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.DeNardo DG et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1, 54–67, doi: 10.1158/2159-8274.CD-10-0028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ruffell B et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kusmartsev S & Gabrilovich DI STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol 174, 4880–4891 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Gabrilovich DI & Nagaraj S Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9, 162–174, doi: 10.1038/nri2506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Geiger R et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842 e813, doi: 10.1016/j.cell.2016.09.031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marigo I et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 30, 377–390, doi: 10.1016/j.ccell.2016.08.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodriguez PC et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64, 5839–5849, doi: 10.1158/0008-5472.CAN-04-0465 (2004). [DOI] [PubMed] [Google Scholar]
- 114.Chang CI, Liao JC & Kuo L Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res 61, 1100–1106 (2001). [PubMed] [Google Scholar]
- 115.Bronte V & Zanovello P Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 5, 641–654, doi: 10.1038/nri1668 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Lu T et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest 121, 4015–4029, doi: 10.1172/JCI45862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu T & Gabrilovich DI Molecular pathways: tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin Cancer Res 18, 4877–4882, doi: 10.1158/1078-0432.CCR-11-2939 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nagaraj S et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13, 828–835, doi: 10.1038/nm1609 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zea AH et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res 65, 3044–3048, doi: 10.1158/0008-5472.CAN-04-4505 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Munder M et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 105, 2549–2556, doi: 10.1182/blood-2004-07-2521 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Lin H et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 805–815, doi: 10.1172/JCI96113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kuang DM et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med 206, 1327–1337, doi: 10.1084/jem.20082173 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tang H et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 128, 580–588, doi: 10.1172/JCI96061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ceeraz S, Nowak EC & Noelle RJ B7 family checkpoint regulators in immune regulation and disease. Trends in immunology 34, 556–563, doi: 10.1016/j.it.2013.07.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li J et al. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells. Immunity, doi: 10.1016/j.immuni.2018.03.018 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Kreymborg K et al. Ablation of B7-H3 but Not B7-H4 Results in Highly Increased Tumor Burden in a Murine Model of Spontaneous Prostate Cancer. Cancer Immunol Res 3, 849–854, doi: 10.1158/2326-6066.CIR-15-0100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rahbar R et al. B7-H4 expression by nonhematopoietic cells in the tumor microenvironment promotes antitumor immunity. Cancer Immunol Res 3, 184–195, doi: 10.1158/2326-6066.CIR-14-0113 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Smith LK et al. Interleukin-10 Directly Inhibits CD8(+) T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 48, 299–312 e295, doi: 10.1016/j.immuni.2018.01.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Demotte N et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity 28, 414–424, doi: 10.1016/j.immuni.2008.01.011 (2008). [DOI] [PubMed] [Google Scholar]
- 130.Henderson NC et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 172, 288–298, doi: 10.2353/ajpath.2008.070726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schuette V et al. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proceedings of the National Academy of Sciences of the United States of America 113, 10649–10654, doi: 10.1073/pnas.1605885113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stockmann C et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456, 814–818, doi: 10.1038/nature07445 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hughes R et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res 75, 3479–3491, doi: 10.1158/0008-5472.CAN-14-3587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Harney AS et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov 5, 932–943, doi: 10.1158/2159-8290.CD-15-0012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sato E, Simpson KL, Grisham MB, Koyama S & Robbins RA Effects of reactive oxygen and nitrogen metabolites on RANTES- and IL-5-induced eosinophil chemotactic activity in vitro. Am J Pathol 155, 591–598, doi: 10.1016/S0002-9440(10)65154-1 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sato E, Simpson KL, Grisham MB, Koyama S & Robbins RA Effects of reactive oxygen and nitrogen metabolites on MCP-1-induced monocyte chemotactic activity in vitro. Am J Physiol 277, L543–549 (1999). [DOI] [PubMed] [Google Scholar]
- 137.Molon B et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 208, 1949–1962, doi: 10.1084/jem.20101956 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C & Mami-Chouaib F Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res 72, 6325–6332, doi: 10.1158/0008-5472.CAN-12-2027 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Nielsen SR et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol 18, 549–560, doi: 10.1038/ncb3340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Quaranta V et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res 78, 4253–4269, doi: 10.1158/0008-5472.CAN-17-3876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tauriello DVF et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543, doi: 10.1038/nature25492 (2018). [DOI] [PubMed] [Google Scholar]
- 142.Mariathasan S et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548, doi: 10.1038/nature25501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kessenbrock K, Plaks V & Werb Z Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67, doi: 10.1016/j.cell.2010.03.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kelly A et al. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J Exp Med 215, 2725–2736, doi: 10.1084/jem.20171491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.de Mingo Pulido A et al. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 33, 60–74 e66, doi: 10.1016/j.ccell.2017.11.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Broz ML et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652, doi: 10.1016/j.ccell.2014.09.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Peranzoni E et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proceedings of the National Academy of Sciences of the United States of America 115, E4041–E4050, doi: 10.1073/pnas.1720948115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Curiel TJ et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10, 942–949, doi: 10.1038/nm1093 (2004). [DOI] [PubMed] [Google Scholar]
- 149.Salmon H et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938, doi: 10.1016/j.immuni.2016.03.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roberts EW et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336, doi: 10.1016/j.ccell.2016.06.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cunha LD et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175, 429–441 e416, doi: 10.1016/j.cell.2018.08.061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ahn J, Xia T, Rabasa Capote A, Betancourt D & Barber GN Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 33, 862–873 e865, doi: 10.1016/j.ccell.2018.03.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Han CZ et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature 539, 570–574, doi: 10.1038/nature20141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Quail DF et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018, doi: 10.1126/science.aad3018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gupta P et al. Tissue-Resident CD169(+) Macrophages Form a Crucial Front Line against Plasmodium Infection. Cell Rep 16, 1749–1761, doi: 10.1016/j.celrep.2016.07.010 (2016). [DOI] [PubMed] [Google Scholar]
- 156.van Dinther D et al. Functional CD169 on Macrophages Mediates Interaction with Dendritic Cells for CD8(+) T Cell Cross-Priming. Cell Rep 22, 1484–1495, doi: 10.1016/j.celrep.2018.01.021 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Saunderson SC, Dunn AC, Crocker PR & McLellan AD CD169 mediates the capture of exosomes in spleen and lymph node. Blood 123, 208–216, doi: 10.1182/blood-2013-03-489732 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pucci F et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 352, 242–246, doi: 10.1126/science.aaf1328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nywening TM et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol, doi: 10.1016/S1470-2045(16)00078-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sanford DE et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: a Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-13-0525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao L et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 57, 829–839, doi: 10.1002/hep.26094 (2013). [DOI] [PubMed] [Google Scholar]
- 162.Qian BZ et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225, doi: 10.1038/nature10138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lim SY, Yuzhalin AE, Gordon-Weeks AN & Muschel RJ Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710, doi: 10.18632/oncotarget.7376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhang J, Patel L & Pienta KJ CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev 21, 41–48, doi:S1359-6101(09)00116-6 [pii] 10.1016/j.cytogfr.2009.11.009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Connolly KA et al. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 7, 86522–86535, doi: 10.18632/oncotarget.13287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kalbasi A et al. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin Cancer Res 23, 137–148, doi: 10.1158/1078-0432.CCR-16-0870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fridlender ZG et al. CCL2 blockade augments cancer immunotherapy. Cancer research 70, 109–118, doi: 10.1158/0008-5472.CAN-09-2326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nywening TM et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut, doi: 10.1136/gutjnl-2017-313738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bonapace L et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133, doi: 10.1038/nature13862 (2014). [DOI] [PubMed] [Google Scholar]
- 170.Ruffell B et al. Macrophage IL10 Blocks CD8+ T Cell-Dependent Responces to Chemotherapy by Supressing IL12 Expression in Intratumoral Dendritic Cells. Cancer Cell 26, 623–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xu J et al. Abrogating the protumorigenic influences of tumor-infiltrating myeloid cells byCSF1R signaling blockade improves the efficacy of radiotherapy in prostatecancer. Cancer Res, doi: 10.1158/0008-5472.CAN-12-3981 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Seifert L et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 150, 1659–1672 e1655, doi: 10.1053/j.gastro.2016.02.070 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang Q et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 32, 176–183, doi: 10.1038/leu.2017.193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Neubert NJ et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Science translational medicine 10, doi: 10.1126/scitranslmed.aan3311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mok S et al. Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res, doi: 10.1158/0008-5472.CAN-13-1816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wiehagen KR et al. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol Res 5, 1109–1121, doi: 10.1158/2326-6066.CIR-17-0258 (2017). [DOI] [PubMed] [Google Scholar]
- 177.Wainberg ZA et al. First-in-Human Phase 1 Dose Escalation and Expansion of a Novel Combination, Anti–CSF-1 Receptor (cabiralizumab) Plus Anti–PD-1 (nivolumab), in Patients With Advanced Solid Tumors. SITC-Meeting Abstract, O42 (2017). [Google Scholar]
- 178.Kumar V et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 32, 654–668 e655, doi: 10.1016/j.ccell.2017.10.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nywening T et al. Targeting tumor associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut In-Press (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ries CH et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer cell 25, 846–859, doi: 10.1016/j.ccr.2014.05.016 (2014). [DOI] [PubMed] [Google Scholar]
- 181.Cannarile MA et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 5, 53, doi: 10.1186/s40425-017-0257-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sanchez-Martin L et al. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 117, 88–97, doi: 10.1182/blood-2009-12-258186 (2011). [DOI] [PubMed] [Google Scholar]
- 183.Wang SC, Yu CF, Hong JH, Tsai CS & Chiang CS Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS One 8, e69182, doi: 10.1371/journal.pone.0069182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Welford AF et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest 121, 1969–1973, doi: 10.1172/JCI44562 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mazzieri R et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19, 512–526, doi: 10.1016/j.ccr.2011.02.005 (2011). [DOI] [PubMed] [Google Scholar]
- 186.Schmittnaegel M et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 9, doi: 10.1126/scitranslmed.aak9670 (2017). [DOI] [PubMed] [Google Scholar]
- 187.Harney AS et al. The Selective Tie2 Inhibitor Rebastinib Blocks Recruitment and Function of Tie2(Hi) Macrophages in Breast Cancer and Pancreatic Neuroendocrine Tumors. Mol Cancer Ther 16, 2486–2501, doi: 10.1158/1535-7163.MCT-17-0241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Karagiannis GS et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Science translational medicine 9, doi: 10.1126/scitranslmed.aan0026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Feig C et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217, doi: 10.1073/pnas.1320318110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vonderheide RH The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 33, 563–569, doi: 10.1016/j.ccell.2018.03.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Beatty GL et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616, doi: 10.1126/science.1198443 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Byrne KT & Vonderheide RH CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep 15, 2719–2732, doi: 10.1016/j.celrep.2016.05.058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Winograd R et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 3, 399–411, doi: 10.1158/2326-6066.CIR-14-0215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Perry CJ et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. The Journal of experimental medicine 215, 877–893, doi: 10.1084/jem.20171435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hoves S et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med 215, 859–876, doi: 10.1084/jem.20171440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guerriero JL et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432, doi: 10.1038/nature21409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Schmid MC et al. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS One 8, e60226, doi: 10.1371/journal.pone.0060226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sai J et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin Cancer Res 23, 3371–3384, doi: 10.1158/1078-0432.CCR-16-2142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Foubert P, Kaneda MM & Varner JA PI3Kgamma Activates Integrin alpha4 and Promotes Immune Suppressive Myeloid Cell Polarization during Tumor Progression. Cancer Immunol Res 5, 957–968, doi: 10.1158/2326-6066.CIR-17-0143 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.De Henau O et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447, doi: 10.1038/nature20554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Willingham SB et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 109, 6662–6667, doi: 10.1073/pnas.1121623109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Liu X et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nature medicine 21, 1209–1215, doi: 10.1038/nm.3931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Xu MM et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363–373 e365, doi: 10.1016/j.immuni.2017.07.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gordon SR et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499, doi: 10.1038/nature22396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hartley GP, Chow L, Ammons DT, Wheat WH & Dow SW Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res 6, 1260–1273, doi: 10.1158/2326-6066.CIR-17-0537 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Arlauckas SP et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9, doi: 10.1126/scitranslmed.aal3604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.DiLillo DJ & Ravetch JV Fc-Receptor Interactions Regulate Both Cytotoxic and Immunomodulatory Therapeutic Antibody Effector Functions. Cancer Immunol Res 3, 704–713, doi: 10.1158/2326-6066.CIR-15-0120 (2015). [DOI] [PubMed] [Google Scholar]
- 208.Byrne KT, Leisenring NH, Bajor DL & Vonderheide RH CSF-1R-Dependent Lethal Hepatotoxicity When Agonistic CD40 Antibody Is Given before but Not after Chemotherapy. J Immunol 197, 179–187, doi: 10.4049/jimmunol.1600146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kitamura T, Qian BZ & Pollard JW Immune cell promotion of metastasis. Nat Rev Immunol 15, 73–86, doi: 10.1038/nri3789 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yang M, McKay D, Pollard JW & Lewis CE Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res 78, 5492–5503, doi: 10.1158/0008-5472.CAN-18-1367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]