

Abstract
Defining the biologically active structures of proteins in their cellular environments remains challenging for proteins with multiple conformations and functions, where only a minor conformer might be associated with a given function. Here, we use deep mutational scanning to probe the structure and dynamics of α-synuclein, a protein known to adopt disordered, helical, and amyloid conformations. We examined the effects of 2,600 single-residue substitutions on the ability of intracellularly expressed α-synuclein to slow the growth of yeast. Computational analysis of the data showed that the conformation responsible for this phenotype is a long, uninterrupted, amphiphilic helix with increasing dynamics toward the C terminus. Deep mutational scanning can therefore determine biologically active conformations in cellular environments, even for a highly dynamic multi-conformational protein.
INTRODUCTION
The three-dimensional structures of proteins are critical determinants of biological activity, but identifying the conformations responsible for activity in vivo remains a formidable challenge in structural biology. This is especially pertinent for intrinsically disordered proteins, which adopt a wide variety of conformational states, many of which are poorly defined. The cellular environment plays a critical role in shaping the conformational landscape of intrinsically disordered proteins; however, many conventional approaches for structure determination are impractical or impossible to apply within living cells with sufficient resolution to discriminate between states. Moreover, activity might be driven by a small structural subpopulation, complicating detection, and techniques such as X-ray crystallography or electron microscopy that are optimized for examining specific structures can overlook contributions from poorly ordered conformational states. Importantly, most structural biological approaches do not directly report the activity of a protein, which must instead be inferred from additional experiments, making it challenging to determine which conformational state drives activity. It is therefore desirable to infer the structure of a protein directly from its activity in vivo.
The functional significance of different protein structural models can be tested by engineered mutations that selectively perturb specific conformations. More broadly, functionally significant features and interactions can be inferred from sequence conservation and covariation analyses, enabling structural modeling1–4. Nevertheless, natural sequences sample the sequence–activity landscape sparsely and balance a variety of selective pressures that are challenging to deconvolute. As a complement to analysis of natural sequences, deep mutational scanning (DMS) has emerged as a powerful approach to systematically map the sequence–activity landscapes of proteins under controlled experimental conditions5. In DMS, a library of protein missense variants is screened for relative activity in competitive selection; by applying modest selective pressures, the relative effect of many mutations can be quantified using deep sequencing. DMS has typically been applied to probe structure and function of proteins of known three-dimensional structures, and it has recently been extended to identify residues that are important for the activities of intrinsically disordered proteins6,7.
Here, we ask whether it can be used to identify the three-dimensional structure and dynamics of the active state of a multi-conformational intrinsically disordered protein. Because structures obtained by DMS are based on the activity of the protein, this approach should allow identification of conformational states associated with specific activities8,9. In particular, we leverage the comprehensive sequence landscapes generated by DMS to generate a high-resolution model for the structure and dynamics of the conformational state of α-synuclein that slows the growth of yeast, which highlights underappreciated biophysical features of this protein with functional implications.
RESULTS
Structural discrimination by mutational scanning
α-Synuclein has been proposed to adopt a large number of conformational states that are difficult to characterize, particularly within a cellular environment. These structures include intrinsically disordered populations10,11, poorly structured and/or helix-rich oligomers12,13, a variety of membrane-bound helical states14–16, and diverse amyloid-like conformations with distinct cross-β structures and disordered regions17–19 (Fig. 1a). Many of these structures remain poorly defined and it remains unclear which states are populated in living cells. Moreover, it remains unclear which structural states drive different activities, each of which might be associated with a minor structural subpopulation.
Fig. 1 |. Conformational landscape of α-synuclein.
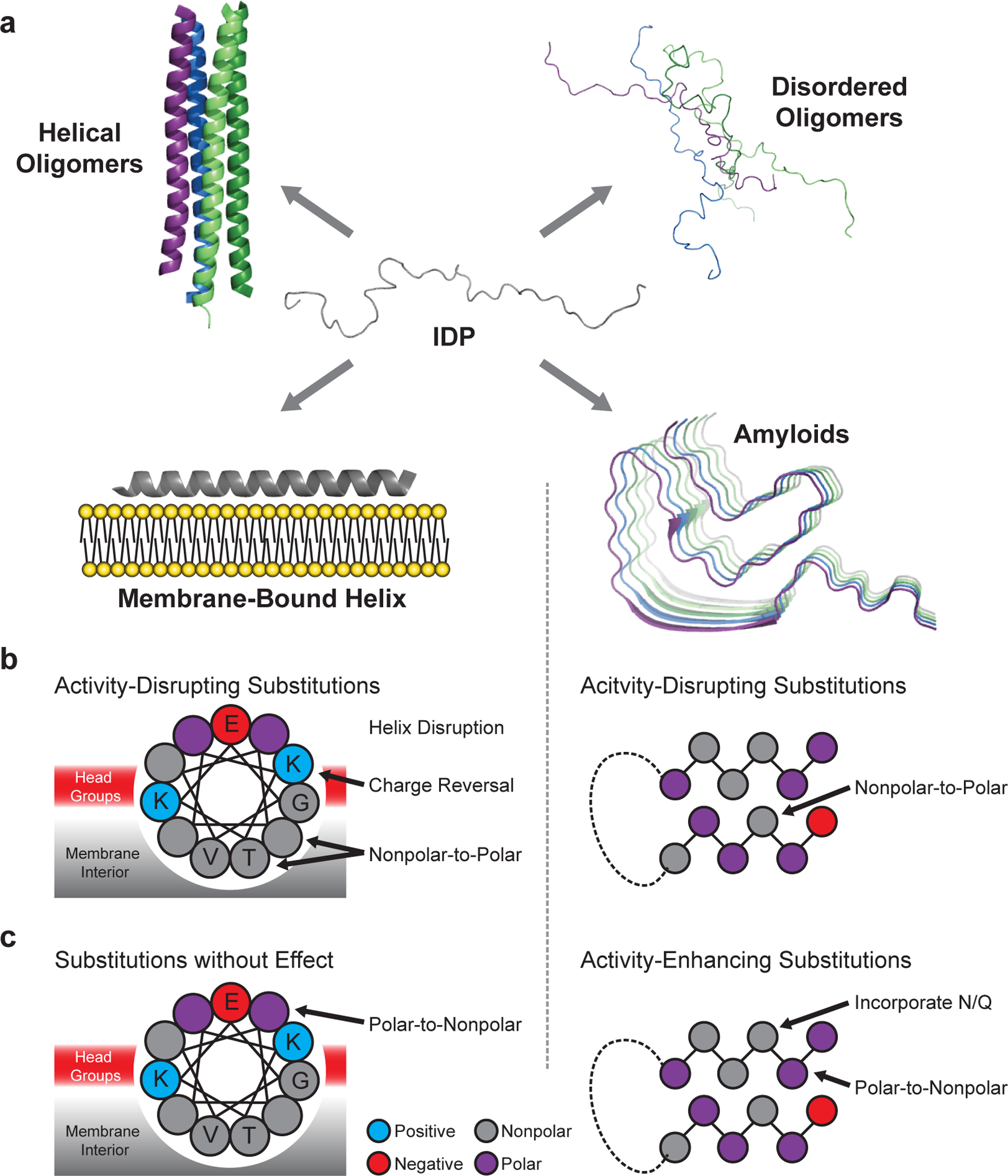
a, α-Synuclein adopts intrinsically disordered conformations10,11, poorly structured and/or helix-rich oligomers12,13, a variety of membrane-bound helical states14–16, and amyloid-like conformations with distinct cross-β structures and disordered regions17–19. Amino acid substitutions are predicted to differentially perturb each structure, allowing for model discrimination by mutational scanning. b, Substitutions predicted to disrupt the activity of the membrane-bound helix (left) and the amyloid (right) structures. c, Substitutions predicted to enhance or have little effect on activity. Mutations affecting other possible active states are not shown, but discussed below.
We anticipated that each structural state of α-synuclein would have differential sensitivity to specific mutations (Fig. 1b–c), and that this differential sensitivity would enable DMS to determine which structure(s) drive a particular activity. For example, if the amphiphilic helix (Fig. 1a, membrane-bound helix model) drives activity, then substitutions could reduce activity by reducing favorable interactions with the membrane surface or by disrupting α-helix formation through incorporation of helix-breaking residues such as proline (Fig. 1b). Based on the WT sequence, α-synuclein is predicted to form attractive electrostatic and hydrophobic interactions with lipid membranes. Thus, nonpolar-to-polar substitutions of the membrane-contacting face should reduce activity if activity is driven by a membrane-binding helix (Fig. 1b). Also, because membrane-contacting residues alternate with solvent-exposed residues, the mutational landscape should feature periodicity consistent with the α-helix, approximately 3.6 residues per turn.
If, instead, amyloid formation drives the activity of α-synuclein (Fig. 1a, amyloid model), a distinct sequence landscape should be observed. Core residues of the amyloid should be sensitive to nonpolar-to-polar substitutions or changes in residue size that disrupt packing (Fig. 1b). Well-known amyloid-promoting residues such as nonpolar amino acids, which promote aggregation, or Asn/Gln substitutions, which can form hydrogen-bonded ladders in amyloid fibers20, should promote activity (Fig. 1c). Moreover, if amyloid formation drives activity, the effect of mutations should be well-described by amyloid-predictive software such as TANGO21. Finally, because amyloid fibers are enriched in β-sheet, disruptive mutations might recur every other residue, corresponding to residues facing to the same side of the sheet.
Sequence–activity landscape of α-synuclein
Among the various activities reported for α-synuclein, we focused on conformations that contribute to its cellular toxicity. We sought a tractable model system in which a detrimental α-synuclein-dependent phenotype arises spontaneously upon the induction of α-synuclein expression, as opposed to systems that depend on templating with preformed amyloids to bias the conformational landscape. We therefore undertook these studies in yeast, a well-validated cellular model for α-synuclein activity. Expression of α-synuclein in yeast recapitulates several critical features of α-synuclein behavior, including membrane-binding, vesicle clustering, aggregation, and dose-dependent toxicity22. Moreover, the aggregates observed in yeast resemble those found in humans; both consist of α-synuclein-enriched clusters of membranous structures23,24. Importantly, these features develop in the course of hours, and one can easily grow millions of yeast cells to enable the analysis of a complete library. Finally, although not all α-synuclein conformations or features of α-synuclein toxicity in humans are recapitulated in any in vitro eukaryotic cell culture system, genetic25–28 and chemical modifiers29,30 of α-synuclein toxicity have been discovered by screening in yeast and subsequently validated in neuron and animal models26,27,31. Thus, the yeast system provides a well calibrated model system in which to assess the viability of DMS for probing the structural basis of important cellular phenotypes.
We constructed a barcoded, single-variant library that exhaustively samples substitutions of each amino acid at every position of α-synuclein, ultimately containing 98% of the 2,660 possible α-synuclein single point mutants, and screened this library for growth phenotypes in yeast. Expression of α-synuclein in yeast slows growth in a dose-dependent manner that can be modified by point mutations22,32, providing the basis on which to identify substitutions that disrupt the toxic conformation(s); variants with reduced toxicity become enriched in the library over generations of cell division. Therefore, we can define a fitness score (Methods) based on changes in the relative proportion of each variant in the library over time, which reflects their relative toxicity. The screen was performed in quadruplicate, with excellent reproducibility between replicates (Supplementary Fig. 1). In addition, by sorting a population of yeast cells expressing each α-synuclein variant fused to GFP, we observed that, with few exceptions, nontoxic variants have higher fluorescence on average than toxic variants (Supplementary Fig. 2). Thus, decreases in toxicity are not generally due to decreases in expression level.
The phenotypic landscape of α-synuclein (Fig. 2a) has two prominent features that point to an α-helix as the toxic conformation in yeast. First, proline residues mitigate the slow-growth phenotype throughout the first ~90 residues (Fig. 2b), suggesting helix formation across this region. Second, substitutions that disrupt toxicity recur every 3.67 ± 0.01 residues (95% CI) along the first 90 residues of the protein (Figs. 2b, 3a), which is consistent with the periodicity of the α-helix. The remarkable length of the predicted helix allowed for accurate determination of the period, which remains uninterrupted over ~90 residues.
Fig. 2 |. Fitness scores of α-synuclein point mutants.
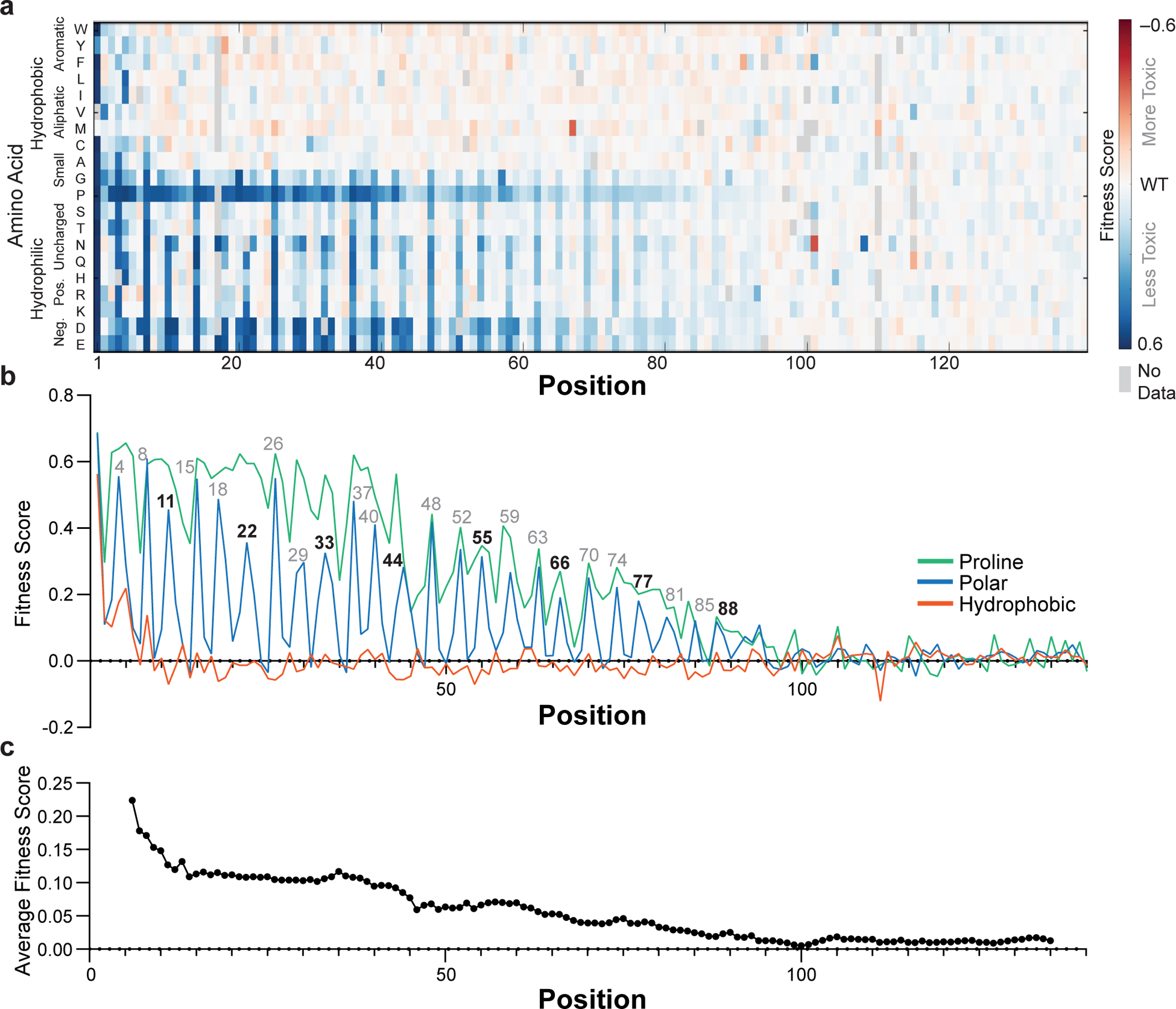
a, Fitness scores (defined as the slope of the line describing change in log-transformed variant frequencies over time) for expression of α-synuclein point mutants in yeast. b, Average fitness scores of mutants with hydrophobic (W, Y, F, L, I, V, M, C, A), polar (S, T, N, Q, H, R, K, D, E), or proline residues. c, Fitness score averaged over an 11-residue window. Experiments were performed four independent times with similar results (Supplementary Fig. 1) and the average of all four replicates is shown.
Closer inspection strongly supports a membrane-bound α-helix. α-Synuclein’s helix-forming region has seven tandemly repeated 11-residue repeats believed to mediate membrane binding, and this very precise 11-residue periodicity is also observed in the mutational profile, which shows peaks at 11-residue multiples of positions 4, 8 and 11, throughout the entire 90-residue sequence (see residue numbers over peaks in Fig. 2b). The mean effects of substitutions at each position along these repeats are shown in Fig. 3b, and the results are precisely as one would expect for the membrane-helix model. Nonpolar-to-polar substitutions have the largest effect at positions believed to penetrate most deeply into the membrane. Substitutions that increased the hydrophobicity tended to slightly increase the toxicity, but only when applied to residues that face the membrane interior or headgroup region. In the headgroup region, mutations of Lys residues, which are expected to interact with anionic lipid headgroups, to acidic residues strongly decreased toxicity. The importance of conserved lysine residues for toxicity also argues against ordered water-soluble helical bundles as drivers of toxicity in yeast, since the alignment of lysine residues on either face of the helix should destabilize helix-helix association. Hydrophobic packing of helical interfaces should also be sensitive to amino acid size, which we do not observe, again excluding contributions from discrete helical bundles. One interesting substitution pattern was observed at position 5, which places a Gly residue in the headgroup region, where substitutions of Gly to any other residue except Pro, Asp, or Glu slightly increased toxicity. We believe this effect is a result of increasing the helix propensity, as Gly has the lowest helix-forming propensity, next to Pro. Together, the mutational analysis provides remarkably strong support for the membrane helix model.
Fig. 3 |. Mutational sensitivity varies by depth of membrane burial.
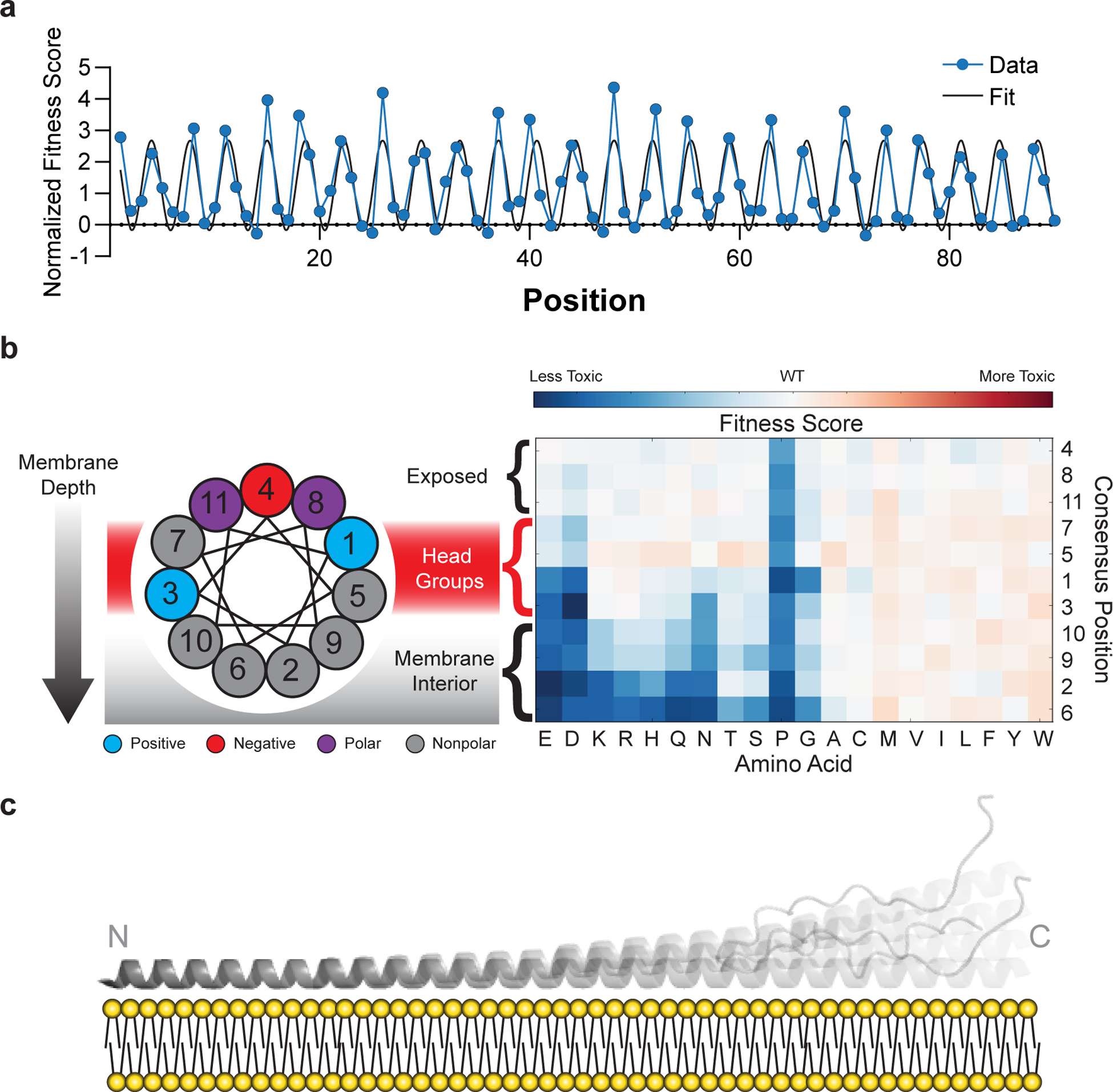
a, Average fitness scores of mutations to polar (S, T, N, Q, H, R, K, D, E) residues as a function of position, normalized against an 11-residue window average (Fig. 2C). A sinusoid fit describes the data as having a periodicity of 3.67 ± 0.01 residues (95% CI). b, Fitness scores were averaged over equivalent positions in each of the seven 11-residue repeating segments and ordered by predicted depth of membrane penetration. c, Structural model of α-synuclein as a single amphiphilic helix interacting with a lipid bilayer, demonstrating increased dynamics toward the C terminus, which is consistent with data from deep mutational scanning, EPR spectroscopy15, and NMR spectroscopy16,33.
The comprehensive sequence–toxicity landscape also excludes contributions from other structures. For example, toxic mutants have less predicted disorder than non-toxic mutants (Supplementary Fig. 3A), which argues against a fully disordered state being the toxic species. The data also argue against the amyloid model as a driver of toxicity. Polar-to-nonpolar substitutions do not generally increase toxicity (Fig. 2b), and we do not observe an increase in toxicity with increased N/Q content (Fig. 2a). The amyloid-forming region of α-synuclein spans from approximately residues 35 to 9517–19, but the region that is most sensitive to mutation spans from residues 1 to 90. Scores from the amyloid-predicting program TANGO are only 5% lower for nontoxic variants than for toxic variants (Supplementary Fig. 3B). Finally, the periodicity of the sequence landscape does not match that expected for β-sheet formation (see Supplementary Information for an extended discussion), demonstrating that the toxicity observed in yeast is not driven by even small or undetectable amounts of amyloid.
Dynamics in the membrane-bound helix
The structural details of our model are also supported by orthogonal biophysical measurements made in the presence of membrane mimetics. Specifically, the periodicity in our data (Fig. 3a) is remarkably consistent with the depth parameters measured by electron paramagnetic resonance (EPR) spectroscopy15. In addition, the gradual decrease in sensitivity to substitution across the protein (Fig. 2c) is consistent with increased dynamics that have been observed by solution- and solid-state nuclear magnetic resonance (NMR) spectroscopy in phospholipid bilayers16,33. In contrast, the strict periodicity and steady attenuation of the sensitivity argue against two very stable helices separated by a fully disordered region, as seen in a horseshoe-shaped model developed from NMR data collected in the presence of detergent micelles14, since we do not observe any regions that accommodate proline residues (Fig. 2b) or that deviate from the observed periodicity (Fig. 3a, Online Methods). We therefore conclude that the toxic structural state of α-synuclein in yeast most resembles models with an extended, 90-residue single helix with increased dynamics toward the C terminus (Fig. 3c)16,33.
Membrane binding is critical for toxicity
To test directly the membrane-binding helix hypothesis, we measured the membrane-binding affinity of twelve α-synuclein mutants by titrating purified α-synuclein with 50 nm phospholipid vesicles composed of a 3:1 mixture of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) and 1-palmitoyl-2-oleoylphosphatidylglycerol (POPG) and monitoring structural changes by circular dichroism (CD) spectroscopy at 222 nm; we performed these experiments at low protein concentrations (350 nM, see Methods) to minimize vesicle remodeling that occurs at higher protein concentrations34. We mutated two positions that are predicted to contact the membrane and two that do not (Supplementary Fig. 4); at each position, we substituted the WT residue for a hydrophobic residue (Leu), a polar residue (Asn), or Pro. Pro substitutions reduced membrane binding at all four positions, manifested by a shift to the right in the binding isotherm (Fig. 4a–d). Pro substitution was less disruptive at G31 than other positions, likely because glycine also disrupts helices. For positions predicted to contact the membrane, Asn and Pro similarly reduced membrane binding capacity, relative to WT and the Leu mutant (Fig. 4b/d). In contrast, at positions that do not contact the membrane, nonpolar-to-polar substitutions had little effect on membrane binding (Fig. 4a/c), consistent with their weak effects on toxicity (Fig. 2a).
Fig. 4 |. Mutations that disrupt toxicity perturb formation of the amphiphilic helix.
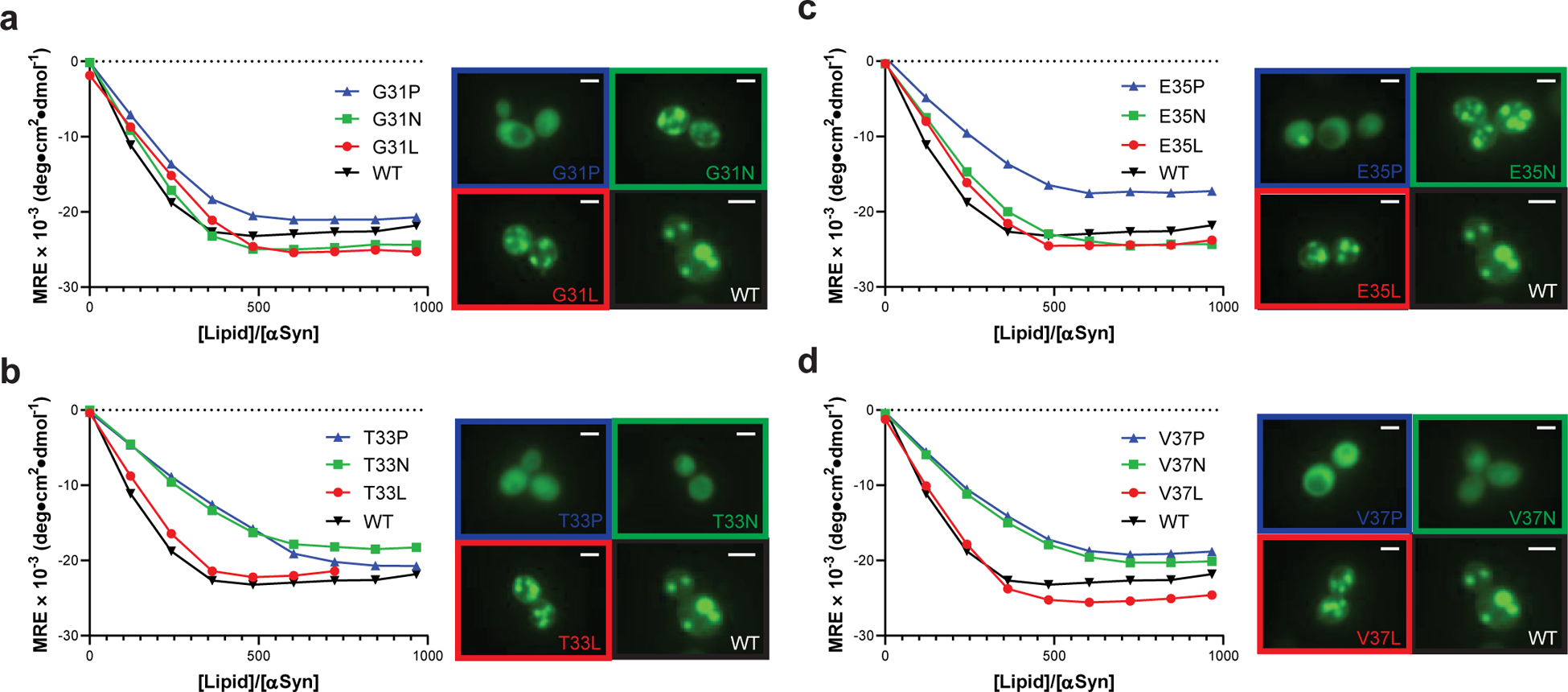
Membrane binding of α-synuclein point mutants measured by CD titration of purified α-synuclein with lipid vesicles ([α-synuclein] = 350 nM) or fluorescence microscopy of α-synuclein variants expressed in yeast. 2 μm scale bars are shown. Data are shown for variants with substitutions at (a) G31, (b) T33, (c) E35, or (d) V37. Experiments were performed three independent times with similar results, and representative examples are shown.
We observed similar trends when we used fluorescence microscopy to determine the membrane affinity of the same mutants expressed in yeast. Mutants with hydrophobic residues contacting the membrane induced ubiquitous inclusions (Fig. 4a–d), which, like Lewy bodies in humans24, have previously been shown to consist of clustered membranes in yeast23. Substitution with proline disrupted inclusion formation and resulted in diffuse cytosolic localization, while polar residues only disrupted inclusions when incorporated on the membrane binding face. Mutants that did not form inclusions were uniformly less toxic (Fig. 2a), supporting our hypothesis that membrane binding is critical for toxicity in yeast.
Our results are broadly consistent with previous measurements of the membrane-binding capacity of α-synuclein mutants35; proline substitutions, in particular, consistently reduce membrane binding. The correlation between membrane binding and yeast toxicity suggests that the former is a critical feature of α-synuclein pathology, although additional mechanisms likely operate in humans. Our results are also consistent with previous observations that genes modifying α-synuclein toxicity are enriched in vesicle-trafficking pathways25. At sufficiently high concentrations, binding of α-synuclein to lipid membranes likely interferes with normal membrane remodeling and vesicle trafficking36, which can be rescued by overexpression of signaling proteins that stimulate vesicle transfer26.
Sequence features underlying dynamics
The phenotypic landscape highlights unique and related features of α-synuclein structure: a long, contiguous helix with 3.67 residue periodicity and progressive C terminal dynamics. The N-terminal region of the α-synuclein sequence contains seven 11-residue repeats that mediate membrane binding. Similar sequence motifs are present in apolipoproteins, where they likewise serve to mediate lipid binding. However, in apolipoproteins, these 11-residue segments are often separated by proline residues or other disruptions to the amphiphilic conformation, which allow helix distortion and hairpin formation37. In α-synuclein, in contrast, the 11-residue segments are concatenated without proline residues, and moreover, proline substitutions reduce the toxicity of α-synuclein (Fig. 2b). Without breaks between repeats, concatenation of the 11-residue repeats creates a long, continuous hydrophobic face along the extended helix.
In an idealized α-helix with 3.60 residues per turn, the hydrophobic face will gradually wrap around the helix (Fig. 5a, Methods). Upon membrane binding, the protein can either distort the helical conformation to maintain a favorable match of the hydrophobic residues with the membrane surface38, which would create strain due to overwinding, or the protein might increasingly engage in dynamic excursions from the membrane surface or from an ideal helical conformation near the C-terminal region (Fig. 3c). Conserved glycine residues within this region likely facilitate both the distortion and dynamics associated with membrane binding. The strain imparted by direct concatenation of the 11-residue membrane-binding repeats therefore provides a molecular mechanism that might promote the increased dynamics observed at the C-terminal end of the amphiphilic helix.
Fig. 5 |. Topology of α-synuclein–membrane interactions.
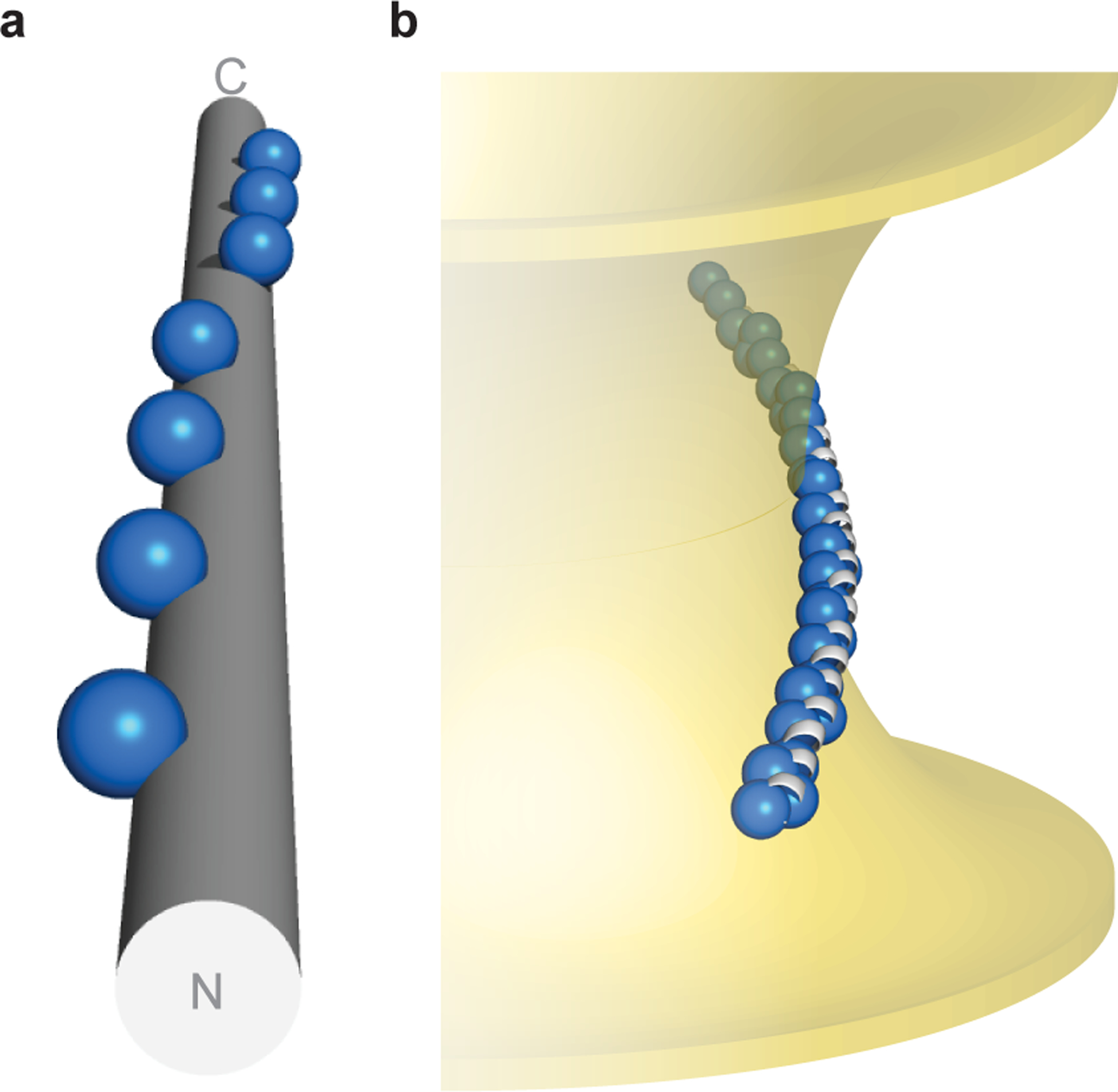
a, α-Synuclein’s hydrophobic residues (repeat position 2 shown as spheres) wind around an idealized α-helix ([φ, ψ] = [–57°, −47°], 3.60 residues/turn). b, Hypothetical model of α-synuclein interacting with membranes of negative Gaussian curvature, which is consistent with both NMR spectroscopy in the presence of anionic membranes of native-like composition16,39, as well as functional data in neurons45. Membrane-contacting residues (repeat positions 2, 6, 9, 10) shown as blue spheres.
Functional implications of membrane binding
The greater dynamics at the C terminus of the membrane-binding helix should allow partial dissociation of α-synuclein from the membrane and formation of additional interactions. Once dissociated, the dynamic region could interact with other nearby membranes, thereby tethering them together. This is consistent with the observation that variants with increased dynamics (as determined by NMR) are more effective at vesicle clustering39, a likely feature of α-synuclein’s normal36 and pathologic26 activities.
Alternatively, once partially dissociated from the membrane, the C-terminal region of α-synuclein would be poised to aggregate. Membrane binding via the N terminus increases local concentration, facilitating self-association of the hydrophobic NAC region40. This is consistent with the propensity of lipid membranes to drive α-synuclein aggregation and amyloid formation41. Conversely, amyloids and other aggregates provide a high local concentration that could interfere with membrane integrity or remodeling if the membrane-binding regions are accessible. Indeed, α-synuclein oligomers with exposed N-terminal segments are highly toxic to neuronal models, whereas oligomers with sequestered N termini are not42.
As discussed above, the hydrophobic surface of α-synuclein would spiral around an ideal α-helix (Fig. 5a), creating tension when bound to either a flat membrane or a curved membrane with constant curvature. Interestingly, however, if the helix bound along a surface with negative Gaussian curvature, as would occur at vesicle buds and fusion pores, α-synuclein could bury its hydrophobic groups without overwinding (Fig. 5b), thereby relieving strain. Helix bending requires significantly less energy than twisting43, which might promote interaction with membranes of this topology. Indeed, α-synuclein has been previously observed to induce negative Gaussian curvature in simulation44, and was recently shown to stabilize fusion pores45. Twisting of the hydrophobic face around the helix might therefore mediate the native roles of α-synuclein in endo- and exocytosis46, and dysregulation of these activities might also contribute to toxicity.
DISCUSSION
By systematically exploring the sequence–toxicity landscape of α-synuclein, we were able to test and significantly refine alternative structural models for its toxicity. Although previous experiments in both yeast and neurons show that mutations that compromise membrane binding can also affect toxicity32,35, it was not possible to infer a specific structure from these relatively sparse data alone. Using the comprehensive sequence–toxicity landscape generated by DMS, we were able to define an active structural and dynamic model with single-residue resolution, and the features of this structure suggest molecular mechanisms for activity that unite several previous lines of inquiry. While phylogenetic analysis of WT synuclein sequences from different species had predicted the presence of 11-residue membrane-binding motifs that suggested a long helical model, the present comprehensive deep mutational scan identified a gradation in the sensitivity and hence likely dynamic character that was not apparent from analysis of natural sequences alone. Moreover, while magnetic resonance investigations had provided evidence for a helix in the membrane-bound state of α-synuclein, differences between the models were far from subtle – coming to different conclusions about the extension of the N-terminal helix depending on the precise method and membrane mimetic employed. Our work highlights the power of sequence–activity landscapes to reveal critical structural features of conformationally plastic proteins within living cells. Furthermore, intrinsically disordered proteins respond to various conditions and stimuli within the cellular environment. By screening variants under various cellular stresses, DMS offers a convenient method to determine how protein conformation responds to changes in the cellular environment, a problem difficult to approach by other methods. Finally, this work provides a prototype for investigation of other intrinsically disordered proteins involved in neurodegenerative disorders and other diseases. While this study was conducted in yeast, it is increasingly possible to conduct similar studies in mammalian cells that more closely represent the human cell types of interest47,48. Although technically more challenging, obtaining highly residue-by-residue structural, functional, and dynamic information in highly relevant cell types is now clearly a feasible and important endeavor.
ONLINE METHODS
dummy
Overview of Library Construction
In order to examine the relative toxicity of α-synuclein variants, we designed a barcoded DNA library to encode each of the 2,660 possible missense mutations of the protein. Double-stranded protein-coding DNA was produced commercially by solid-phase oligonucleotide synthesis and microfluidic assembly. This DNA library was cloned into a yeast vector under control of an inducible promoter. We then appended random 26-nucleotide barcodes downstream of the stop codon. These barcodes serve two purposes: (1) they simplify quantification of relative fitness by decreasing the necessary read length from 420 bp to less than 50 bp, which is easily accessible by Illumina sequencing, and (2) they provide a source of internal redundancy, since the fitness of each variant can be estimated from the change in frequency of ~11 independent barcodes.
Library Cloning
A pooled double-stranded DNA library containing the sequences of 2,660 α-synuclein mutants was generously provided by Twist Bioscience and cloned into pYES2-αSyn-EGFP, which was generously provided by the laboratory of V. M.-Y. Lee. Briefly, the α-synuclein library was amplified using primers specific to overhangs outside of the α-synuclein sequence. The product was purified by gel extraction and used as a megaprimer for amplification of the pYES-αSyn-EGFP plasmid by the EZClone method (Agilent). Following DpnI digestion, products were column purified and transformed into TG1 cells (Invitrogen) by electroporation, which produced 2.5M transformants. The transformed culture was grown overnight, and plasmids were extracted by miniprep. A GFP sequence containing an N-terminal flexible linker was amplified with primers that append a random 26 nt barcode immediately after the stop codon. The product was purified by gel extraction and used as a megaprimer for amplification of the plasmid extracted from transformed TG1 cells using the EZClone method. Transformation into TG1 cells by electroporation produced 10M transformants. The transformed culture was grown overnight, and plasmids were extracted by miniprep. The resulting plasmid pool was then transformed into DH5α cells to produce 60,000 transformants. The transformed culture was grown overnight, and plasmids were extracted by miniprep. The resulting plasmid pool was subjected to deep sequencing (see below) and transformed into W303 by the LiAc/PEG/ssDNA method49, producing 21M transformants. Transformed cultures were grown overnight, back-diluted, and returned to log-phase growth. From this culture, glycerol stocks were prepared in SCD-Ura with 25% glycerol, each containing ~500M cells. Plating of these cultures after one freeze-thaw cycle yielded approximately 100M cells, ensuring appropriate representation of the library.
Deep Sequencing
To link α-synuclein variants to specific barcode sequences, plasmids isolated from DH5α cells were amplified by PCR to isolate the α-synuclein–GFP–barcode DNA sequence using primers that append Illumina adapter sequences. This product was gel purified and sequenced on an Illumina MiSeq using custom Read 1, Index Read 1, and Read 2 primers with 305, 20, and 205 cycles, respectively. Read 1 and Read 2 sequence α-synuclein and the index read sequenced the barcode. Plasmids isolated from yeast were amplified by PCR to isolate barcode DNA using primers that append Illumina adapter sequences, a unique index to distinguish each replicate and time point, and the standard index sequencing primer binding site. Products were gel purified, pooled, and sequenced on the HiSeq 4000 in SE50 mode using a custom Read 1 primer to sequence the barcode.
Barcode Association
Clusters with a greater than 5% chance of at least 1 error in the barcode sequence, judged by quality scores, were rejected from further analysis. Full-length α-synuclein sequences were constructed by merging paired-end reads. Non-overlapping regions were taken as the base call from the appropriate read. Bases in overlapping regions were taken as the consensus. In the event that the two reads disagreed, but one read indicated the WT base, the WT base was called. In the event that the two reads disagreed, and neither read indicated WT, the base call with the higher quality score was taken. Resulting merged reads were aligned against the synthesized sequences using Bowtie2, giving a set of barcode–variant pairs, one for each read. The list of variants mapping to each barcode was compiled across the entire set of reads, and accurate barcode–variant mappings were taken as those for which the most common mapping was at least twice as common as the next most common mapping. This workflow yielded a dictionary mapping 31,484 barcodes to 2,600 α-synuclein variants.
Yeast Library Selection by Outgrowth
Glycerol stocks of the transformed yeast library were thawed into SCD-Ura and grown overnight at 30°C with shaking. Cells were collected, washed, and resuspended in 50 mL SCR-Ura, after which the cultures were maintained in log phase for 12 hours. Cells (approximately 250M) were collected for t=0h timepoints by centrifugation and stored at −80°C. The remaining culture was diluted to OD 0.2 in 50mL SCR-Ura with 1% galactose to induce protein expression. After 12 hours, cells were collected as before and the culture was diluted to OD 0.2 in 50 mL SCR-Ura with 1% galactose maintain log-phase growth. Final aliquots were collected after 12 more hours of growth. The selection was performed in quadruplicate, starting with a new glycerol stock. Plasmids were isolated from each aliquot of cells as described previously50.
Determination of Fitness Scores
Barcode sequences with high-confidence mappings to protein variants were counted at each time point and converted to frequency to account for differences in read depth. Frequencies were log transformed to reflect the exponential growth of the yeast cultures. The log-transformed frequencies for each barcode were fit to a line over the three time points. The slopes for each barcode were averaged across replicates, and the average slopes for synonymous barcodes were averaged to determine a fitness score for each α-synuclein variant.
Yeast Library Selection by Cell Sorting
To estimate the relative expression level of each variant, glycerol stocks of the transformed yeast library were thawed into SCD-Ura and grown overnight at 30°C with shaking. Cells were collected, washed, and resuspended in 50 mL SCR-Ura, after which the cultures were maintained in log phase for 12 hours. The culture was then diluted to OD 0.2 in 50mL SCR-Ura with 1% galactose to induce protein expression. After 12 hours, cells were collected and sorted on a BD FACSAria II depending on GFP intensity. The distribution of GFP fluorescence values within the population was bimodal, corresponding to GFP-negative and GFP-positive populations. The GFP-positive population was sorted into three approximately equally sized bins, corresponding to low-, medium-, and high-fluorescence; the GFP negative population was collected separately. Plasmids were isolated from each aliquot of cells as described previously50.
Determination of Abundance Scores
Barcode sequences with high-confidence mappings to protein variants were counted in the low- and high-GFP populations to yield a ratio. This ratio was averaged over synonymous barcodes and then normalized by dividing by the corresponding ratio for the WT protein to yield an abundance score.
Mutant Protein Expression and Purification
Tag-free α-synuclein purification was adapted from previous methods51,52. The α-synuclein coding sequence was cloned into the pET28a vector between the NcoI and SacI restriction sites. Point mutations were introduced into the α-synuclein coding sequence by the QuikChange method (Stratagene). Plasmids were transformed into BL21(DE3) E. coli for expression. 5 mL LB cultures were grown to saturation overnight at 37°C with shaking and diluted to 100 mL LB the following morning. Once OD reached 0.8, IPTG was added to a final concentration of 0.5 mM and shaking continued at 37°C for 3 hours. Cells were harvested by centrifugation and resuspended in 10 mL osmotic shock buffer (20 mM Tris-HCl, 40% sucrose, 2 mM EDTA, pH 7.2). After 10 minutes at room temperature, the suspension was pelleted at 10k×g for 20 minutes and quickly resuspended in 10 mL cold water with 4 μL saturated MgCl2. After 3 minutes on ice, the suspension was pelleted at 10k×g for 20 minutes. The supernatant was collected, frozen, and lyophilized. Lyophilized extracts were dissolved in 2 mL 5% B in A (A = 0.1 % TFA in H2O; B = 0.1% TFA, 1% H2O in MeCN). After filtration through a 0.22 μm PVDF syringe filter, the solution was purified to homogeneity by HPLC using a semi-preparative C4 column (25 mm Vydac 214TP). Fractions containing α-synuclein were combined, frozen, and lyophilized.
CD Spectroscopy
Small unilamellar vesicles (SUVs) of 3:1 POPC:POPG were prepared by combining appropriate ratios of chloroform stock solutions (Avanti Polar Lipids), drying to a film, and resuspending to 25 mM lipid concentration in 20 mM phosphate buffer, pH 7.0. Tip sonication (Fisher Sonic Dismembrator Model 500, 20% power, 5 minutes, 2s on – 2s off) yielded SUVs with an average size of 50 ± 28 nm, as determined by dynamic light scattering. Lyophilized proteins were dissolved to 6 mg/mL in 20 mM phosphate buffer, pH 7.0. Proteins were subsequently diluted to 5 μg/mL in 3 mL 20 mM phosphate buffer, pH 7.0 with 150 mM NaCl in a 10 mm cuvette. CD data were collected on a Jasco J-810 (222 nm, 8s averaging, 2 nm bandwidth, triplicate measurements) following increasing addition of SUVs.
Fluorescence Microscopy
Point mutations were introduced into the α-synuclein coding region of pYES2-aSyn-GFP by the QuikChange method, and plasmids were transformed into W303 as described previously53. Individual transformants were inoculated into SCR-Ura and maintained in log phase growth for 12 hours before being diluted to OD 0.2 in SCR-Ura with 1% galactose. Cells were imaged 12 hours later on a Nikon Ti-E microscope with a Plan Apo 40x/0.95 Corr air objective. Excitation was achieved using a Sutter LS-2 xenon arc lamp and Semrock 5-band filter set for FITC (485/20ex, 525/30em). Images were recorded using a DS-Qi2 monochrome camera and NIS-Elements 4.30. Images were cropped and colored using ImageJ.
Statistical Support for Helix Formation
To estimate the probability that the observed periodicity could have arisen by chance, we took the average fitness score for substitution of polar amino acids at each position (Fig. 2b) and scrambled their order 10,000 times. For each shuffling, we computed the amplitude of the signal having 11/3 periodicity:
where
This procedure provided a distribution for the amplitude of 11/3 periodicity (which has the same units as fitness score) expected if the amino acids were randomly arranged. Based on the mean and standard deviation of this distribution (1.2 ± 0.6), the observed amplitude of the 11/3 periodicity for the real data (6.57) has a z-score of 8.34, which corresponds to a probability of 4.9 × 10−16 for a Gaussian distribution. The 11/3 periodicity in our data is therefore highly significant. Conversely, the observed magnitude of the β-sheet periodicity in the real data (0.66) has an associated z-score of −0.92, which corresponds to a probability of 0.41, indicating no significant β-sheet periodicity.
To estimate the probability that we erroneously concluded that there is no break in the helix, we estimated the probability that the observed effects would arise by chance. Specifically, we calculated the probability that helix-breaking proline residues would consistently disrupt the toxicity of the protein across the non-helical region observed when bound to SDS micelles. In the non-helical region of α-synuclein (approximately residues 100–140), 18% of proline substitutions result in a fitness score increase of at least 0.05, which is approximately 10% of the maximal effect we observe. Therefore, assuming that proline residues in non-helical segments have a 0.18 probability of significantly increasing fitness, we would expect that the probability that 9 non-helical residues (as identified from the micelle structure), would all cause a significant increase in fitness to be 0.189 or approximately 2 × 10−7. Even if we assume that the probability that a proline substitution would increase fitness is 0.5 (that is, increasing or decreasing fitness are equally likely), the probability that 9 residues would all cause an increase in fitness is 0.59 or 0.0019. Either way, it is unlikely that our data would be observed for a non-helical segment.
Calculation of Expected Phase Shift
The value of the helical periodicity seen in Fig. 3 (3.67 ± 0.01 residues) represents a slight structural mismatch with respect to the period of an ideal α-helix, which is approximately 3.6 residues. When propagated sufficiently long the repeat this mismatch could lead to two opposing forces. If the chain adopts an ideal helix, the hydrophobic residues will gradually rotate out of register, no longer maintaining favorable contacts with the membrane surface. For example, in an idealized α-helix, with 3.6 residue/turn periodicity (100° per residue), 11 residues will twist a total of 1100°, which is 20° over that expected for three turns (1080°). This would lead to a 100° rotation after just five consecutive repeats, and loss of energetically favorable membrane interactions. On the other hand, if the helix is slightly distorted from ideal, say to 3.64 residues/turn the extent of rotation would be less extreme (app. 50° after five repeats), and if the helix were fully distorted to 3.67 residues per turn the hydrophobic residues would remain fully in register. While it is energetically easy to bend an α-helix, the helical conformation is less easily twisted43. Thus, a protein with a long stretch of repeats can maintain favorable membrane interactions only at the expense of a slight distortion of the helical conformation. While this distortion is not expected to be large, the cost would increase with chain length, possibly contributing to chain length-dependent decrease sensitivity to mutation and the experimentally observed fraying of the helix over the same region16. By contrast, if the helix bound along a surface with negative Gaussian curvature, as would occur at vesicle buds and fusion pores, the membrane surface would gradually rotate around the bound helix. In this case, α-synuclein could bury its hydrophobic groups without overwinding (Fig. 5b), thereby relieving strain. In proteins such as apolipoproteins that have 11-residue repeats, the mismatch would have few structural consequences, as the number of direct repeats is modest, generally on the order of three, which does not lead to accumulated strain.
Material Availability
Requests for materials should be addressed to william.degrado@ucsf.edu.
Data Availability
Raw sequencing data is available at the NCBI Sequence Read Archive (PRJNA564806). The data plotted in Fig. 2–4 are supplied as supplemental information. Unprocessed images are available upon request.
Code Availability
Programs developed to analyze the data reported are available at github.com/rnewberry17.
Supplementary Material
Acknowledgements
We thank D. Larsen for technical assistance. We acknowledge students of the Integrated Program in Quantitative Biology at UCSF for contributions to our analytical approach and initial models of toxicity; their findings regarding α-synuclein DMS under different environmental conditions will be described in a manuscript currently in preparation. We thank the lab of H. El-Samad (University of California, San Francisco) for yeast strains, the lab of V. M-Y. Lee for plasmids (University of Pennsylvania), and Twist Bioscience for providing the dsDNA variant library in support of our educational efforts. This work was supported by grants from the NIH to M.K. (DP2 GM119139) and to W.F.D. (R35-122603, P01-AG002132, R01-GM117593) and by the UCSF Program in Breakthrough Biomedical Research, which is funded in part by the Sandler Foundation, through grants to M.K. R.W.N. was supported by NIH training grant T32-HL007731 and a UCSF Program in Breakthrough Biomedical Research Postdoc Independent Research Grant, which is funded in part by the Sandler Foundation. Flow cytometry was supported by grant P30-CA082103 (NIH).
Footnotes
Competing Interests
The authors declare no competing interests.
REFERENCES
- 1.Socolich M et al. Evolutionary information for specifying a protein fold. Nature 437, 512–518 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Morcos F et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U. S. A 108, E1293–E1301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marks DS et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One 6, e28766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth-Petroczy A et al. Structured States of Disordered Proteins from Genomic Sequences. Cell 167, 158–170 e112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fowler DM & Fields S Deep mutational scanning: a new style of protein science. Nat. Meth 11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolognesi B et al. The mutational lanscape of a prion-like domain. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray VE et al. Elucidating the molecular determinants of Aβ aggregation with deep mutational scanning. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmiedel JM & Lehner B Determining protein structures using deep mutagenesis. Nat. Genet (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rollins NJ et al. Inferring protein 3D structure from deep mutation scans. Nat. Genet (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinreb PH, Zhen W, Poon AW, Conway KA & Lansbury PT Jr. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 35, 13709–13715 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Theillet FX et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Conway KA et al. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U. S. A 97, 571–576 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartels T, Choi JG & Selkoe DJ α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulmer TS, Bax A, Cole NB & Nussbaum RL Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem 280, 9595–9603 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Jao CC, Hegde BG, Chen J, Haworth IS & Langen R Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U. S. A 105, 19666–19671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fusco G et al. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun 5, 3827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuttle MD et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol 23, 409–415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerrero-Ferreira R et al. Cryo-EM structure of alpha-synuclein fibrils. Elife 7, e36402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li B et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun 9, 3609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson R et al. Structure of the cross-β spine of amyloid-like fibrils. Nature 435, 773–778 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J & Serrano L Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotech 22, 1302–1306 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Outeiro TF & Lindquist S Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology. Science 302, 1772–1775 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soper JH et al. α-Synuclein-induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 1093–1103 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahmoradian SH et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci 22, 1099–1109 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL & Muchowski PJ Yeast Genes That Enhance the Toxicity of a Motant Huntingtin Fragment or α-Synuclein. Science 302, 1769–1772 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Cooper AA et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 313, 324–328 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gitler AD et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet 41, 308–315 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khurana V et al. Genome-Scale Networks Link Neurodegenerative Disease Genes to α-Synuclein through Specific Molecular Pathways. Cell Syst 4, 157–170.e114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tardiff DF et al. Yeast Reveal a “Druggable” Rsp5/Nedd4 Network that Ameliorates α-Synuclein Toxicity in Neurons. Science 342, 979–983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanning S et al. Lipidomic Analysis of α-Synuclein Nerotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 73, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung CY et al. Identification and Rescue of α-Synuclein Toxicity in Parkinson Patient-Derived Neurons. Science 342, 983–987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volles MJ & Lansbury PT Jr. Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol 366, 1510–1522 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodner CR, Dobson CM & Bax A Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol 390, 775–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fakhree MAA, Engelbertink SAJ, van Leijenhorst-Groener KA, Blum C & Claessens M Cooperation of Helix Insertion and Lateral Pressure to Remodel Membranes. Biomacromolecules 20, 1217–1223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burré J, Sharma M & Südhof TC Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci 32, 15227–15242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendor JT, Logan TP & Edwards RH The function of α-synuclein. Neuron 79, 1044–1066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segrest JP et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid Res 33, 141–166 (1992). [PubMed] [Google Scholar]
- 38.Bussell R & Eliezer D A Structural and Functional Role for 11-mer Repeats in α-Synuclein and Other Exchangeable Lipid Binding Proteins. J. Mol. Biol 329, 763–778 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Fusco G et al. Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun 7, 12563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snead D & Eliezer D Intrinsically disordered proteins in synaptic vesicle trafficking and release. J. Biol. Chem 294, 3325–3342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galvagnion C et al. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol 11, 229–234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fusco G et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Emberly EG, Mukhopadhyay R, Wingreen NS & Tang C Flexibility of α-Helices: Results of a Statistical Analysis of Database Protein Structures. J. Mol. Biol 327, 229–237 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Braun AR et al. α-Synuclein induces both positive mean curvature and negative Gaussian curvature in membranes. J. Am. Chem. Soc 134, 2613–2620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Logan T, Bendor J, Toupin C, Thorn K & Edwards RH α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci 20, 681–689 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lautenschläger J, Kaminski CF & Kaminski Schierle GS α-Synuclein - Regulator of Exocytosis, Endocytosis, or Both? Trends Cell Biol 27, 468–479 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Starita LM et al. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 200, 413–422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matreyek KA et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet 50, 874–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gietz RD & Schiestl RH Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 38–41 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Fowler DM, Stephany JJ & Fields S Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc 9, 2267–2284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kloepper KD, Woods WS, Winter KA, George JM & Rienstra CM Preparation of α-synuclein fibrils for solid-state NMR: expression, purification, and incubation of wild-type and mutant forms. Protein Expr. Purif 48, 112–117 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Huang C, Ren G, Zhou H & Wang CC A new method for purification of recombinant human α-synuclein in Escherichia coli. Protein Expr. Purif 42, 173–177 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Gietz RD & Schiestl RH High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 31–34 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data is available at the NCBI Sequence Read Archive (PRJNA564806). The data plotted in Fig. 2–4 are supplied as supplemental information. Unprocessed images are available upon request.