

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of pathologic immune activation, often associated with genetic defects of lymphocyte cytotoxicity. Though a distinctive constellation of features has been described for HLH, diagnosis remains challenging as patients have diverse presentations associated with a variety of triggers. We propose two concepts to clarify how HLH is diagnosed and treated: within the broader syndrome of HLH, “HLH disease” should be distinguished from “HLH disease mimics” and HLH subtypes should be categorized by specific etiologic associations, not the ambiguous dichotomy of “primary” and “secondary.” We provide expert-based advice regarding the diagnosis and initiation of treatment for patients with HLH, rooted in improved understanding of its pathophysiology.
Keywords: hematology, hemophagocytic lymphohistiocytosis, immunology
1 |. INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) has been recognized since the middle of the 20th century as both a familial disorder and an apparently sporadic disorder associated with a variety of environmental triggers including infection, malignancy, and rheumatologic disorders. Despite clinical and scientific advances, uncertainty remains about how to conceptualize and recognize HLH. Due to the complexity of diagnostic criteria and similarity to other inflammatory disorders, diagnosis is often delayed and misdiagnosis remains a significant concern. Members of the HLH committee from the North American Consortium for Histiocytic Disorders have compiled this review focusing on the pitfalls of HLH diagnosis, based on a review of the medical literature and the collective expertise and experience consulting with treating physicians nationally and internationally. This review will focus on four areas: clarifying concepts of HLH, describing the diversity of presentations, providing practical advice regarding diagnosis, and providing a brief overview of initial considerations for the treatment of HLH.
1.1 |. What is HLH?
1.1.1 |. HLH is a syndrome encompassing diverse genetic and environmental contributions
The first report of what we now recognize as familial HLH (F-HLH) in 1952 described two siblings who developed fever and hepatosplenomegaly at 9 weeks of age.1 Subsequently, familial clusters of children with similar phenotypes were observed, as well as apparently sporadic cases of a similar syndrome were seen in the context of severe infection, rheumatologic disorders, or malignancy.2–6 In 1999, defects of PRF1 (encoding perforin) were discovered as the first inherited gene defect underlying F-HLH.7 Before this discovery, a committee from the Histiocyte Society developed consensus enrollment criteria for the HLH-94 study to capture the HLH disease phenotype: fever, splenomegaly, cytopenias, increased triglycerides/decreased fibrinogen, and hemophagocytosis.8 Enrollment criteria for the subsequent HLH2004 trial were revised to incorporate ongoing discovery of the genetic basis of F-HLH and development of specialized immune studies.9 Over the last decade, these enrollment criteria have become the de facto criteria for defining/diagnosing HLH, though their sensitivity and specificity remain unknown and an updated, evidence-based international consensus is needed. The details of these criteria are listed in Table 1, along with explanation/commentary on their individual meaning.
TABLE 1.
Diagnostic criteria used for the HLH2004 trial and relevance for clinical diagnosis
| HLH2004 entry criteria | Comment |
|---|---|
| A. Molecular diagnosis consistent with HLH: Pathologic mutations of PRF1, UNC13D, STXBP2, Rab27a, STX11, SH2D1A, or XIAP or | In a patient with known genetic defects, treatment before full development of HLH may be appropriate, but genetic studies usually just help to define HLH recurrence risk, not the presence of an active disease state. |
| B. Five of the eight criteria listed below are fulfilled | |
| 1. Fever ≥38.3°C | Nearly universal in untreated HLH. |
| 2. Splenomegaly | While splenomegaly and hepatomegaly are very common in HLH, adenopathy is not. |
| 3. Cytopenias (affecting at least two of three lineages in the peripheral blood): Hemoglobin <9 g/dL (in infants <4 weeks: hemoglobin <10 g/dL) Platelets <100 × 103/mL Neutrophils < 1 × 103/mL | Cytopenias are ubiquitous in HLH. Lack of cytopenias should make one doubt a diagnosis of HLH, except in the special case of isolated, CNS-only disease. |
| 4. Hypertriglyceridemia (>265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL) | Low fibrinogen in the context of inflammation is paradoxical and one of the more distinctive features of HLH. |
| 5. Hemophagocytosis in bone marrow or spleen or lymph nodes or liver | Not specific to HLH, or essential for the diagnosis, but helpful as a disease marker. Of note, it is often not evident early after disease onset. |
| 6. Low or absent NK-cell activity | More modern and robust assays measuring perforin levels and its degranulation should replace this assay for reliable diagnosis of HLH. This assay is not specific for primary HLH. |
| 7. Ferritin >500 ng/mL | Most patients have much higher levels than this threshold suggests. |
| 8. Elevated Soluble CD25 (soluble IL-2 receptor alpha) | As HLH is a T-cell driven disease, this assay is extremely informative for diagnosis and response to therapy. See Section 3 for more information. |
1.1.2 |. Experimental studies have defined the unique pathophysiology of familial HLH
Understandably, the diversity of clinical presentations of HLH has led to confusion. For F-HLH at least, experimental studies in animal models have demonstrated that HLH is caused by excessive activation of CD8+ T cells, which are otherwise triggered appropriately by infection.10,11 The lack of normal cytotoxic function deregulates antigen presentation, leading to excessive T-cell activation.12–14 Several key concepts are apparent from these studies:
The immune response itself, not the infectious (or other environmental) trigger, drives disease pathology.
Unlike many other immune disorders, immune activation, not self-reactivity (or autoimmunity), is the cause of disease pathology.
While macrophages are clearly involved in disease development, T cells (specifically CD8+ T cells) are key upstream drivers of HLH disease.15
Across multiple genetic models, interferon gamma (IFN-γ) is a key mediator of disease development and is likely the principle connector between activated T cells and activation of macrophages.16–19
Since the discovery of defects in PRF1, mutations in multiple other genes (UNC13D, STX11, STXBP2, RAB27A, LYST) affecting perforin delivery have been identified as causes of F-HLH. All these defects appear to share the common pathophysiology described above in experimental contexts. Defects in other genes related to effector T-cell function (SH2D1A, ITK, CD27) are also known to cause HLH, and at least in the case of SH2D1A, appear to cause it in a similar fashion as perforin-related defects. With a less well defined and perhaps distinctive immune physiology, are cases of HLH associated with inflammasome activation, as seen with genetic defects in XIAP or NLRC4, and systemic onset juvenile arthritis (soJIA). A recent whole exome–based study has expanded the list of potentially HLH-associated genes considerably to include other genes associated with immune function and regulation, albeit with less clear causality.20 However, the overall theme of all these genetic findings is that HLH is caused by defective immune regulation leading to excessive activation of T cells and macrophages.
1.2 |. HLH: A pattern or a disease?
HLH is an inflammatory syndrome with a distinctive clinical pattern including unusual features: cytopenias, splenomegaly, low fibrinogen, extreme elevations of inflammatory markers, a (sometimes) protracted course, and liver or brain injury. The presence of this pattern suggests the abnormal immune physiology described above and the need for immune suppression as the key therapeutic intervention. But it is clear that not all patients who display this pattern have underlying immune dysregulation, and not all would benefit from immune suppression. However, the HLH literature has been ambiguous about how to conceptualize HLH, and confusion remains about when to make this diagnosis and initiate HLH-specific therapies.
To clarify thinking and terminology regarding HLH, we favor the concept that patients displaying sufficient HLH diagnostic features should be considered to be fulfilling the “HLH syndrome,” while only a subset of these patients, those in whom this distinctive immune dysregulation is the core problem and who are likely to benefit from immune suppression, should be considered to have “HLH disease.” Disorders leading to the HLH pattern, but which are not likely to benefit from immune suppression, should therefore be viewed as “HLH disease mimics.” For example, compare two neonates with fever, cytopenias, coagulopathy, and elevated sCD25/ ferritin. While both may fulfill HLH criteria (display the “HLH syndrome”), the one with biallelic perforin mutations and no apparent infection clearly has “HLH disease,” while the other one with disseminated HSV infection (for which there is no general evidence of immune dysregulation or benefit from immune suppression) should be considered to have a condition which is an “HLH disease mimic.” This dichotomy does not preclude anti-inflammatory treatments in the latter group (as illustrated in this context21), but does clarify diagnostic and therapeutic priorities. Thus, diagnosis of HLH disease is based on recognizing the HLH pattern and careful exclusion of HLH disease mimics. This concept is illustrated in Figure 1.
FIGURE 1.
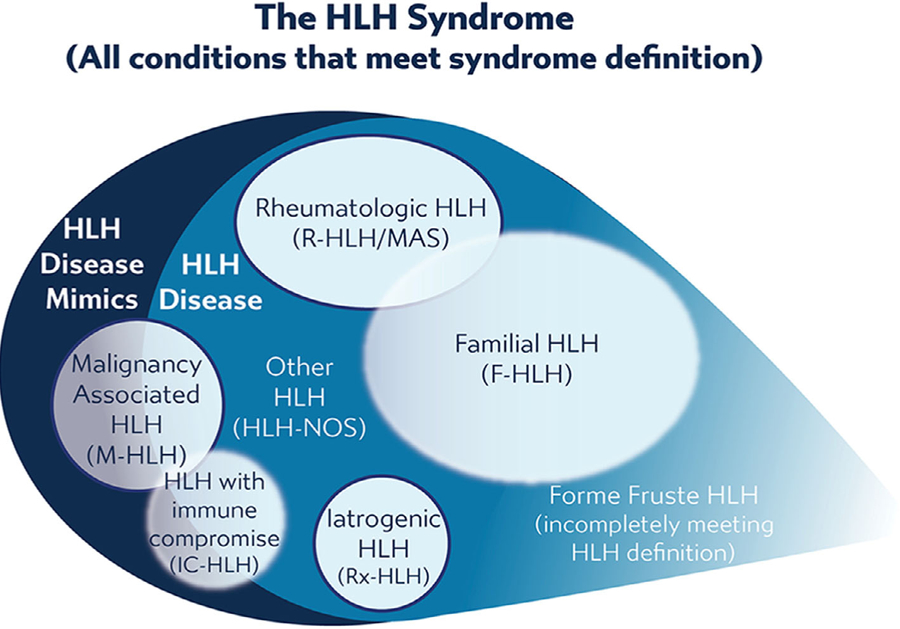
Categorizing HLH
Note. The HLH syndrome includes all conditions meeting consensus diagnostic criteria. This syndrome includes conditions that would benefit from HLH-directed immunosuppressive therapies, which are termed “HLH disease,” and those conditions that would not benefit from such therapy or require entirely different treatments, termed as “HLH disease mimics.” HLH disease includes recognizable subgroups: familial HLH with clear genetic etiology, HLH associated with malignancy, HLH associated with rheumatologic conditions (also called MAS), HLH observed after immune activating therapies (iatrogenic HLH, also called cytokine release syndrome), HLH associated with immune compromise (either primary immune deficiency or treatment-related immune suppression), and HLH not associated with other specific conditions. Recognition of these subcategories is valuable as this may alter treatment, though some categories overlap with each other or have indistinct borders (as indicated above), and may include examples of both HLH disease and HLH disease mimics. Use of these category-specific terms is preferred over the historical terms of “primary” and “secondary” because the older concepts are ambiguous due to increasing understanding of genetic complexity, involvement of infection in triggering multiple distinct variations of HLH, and varied application. Incomplete, forme fruste episodes of HLH (similar but not completely fulfilling diagnostic criteria) are also well recognized in patients with familial HLH and may occur in others, such as “mild MAS” in patients with soJIA.
Why is this concept valuable? HLH typically requires prompt recognition and treatment, often before all data can be gathered. However, this aggressive stance should be balanced by a cautious and careful exclusion of conditions that look like HLH disease but would not benefit from (or even be harmed by) immune suppression and may require different specific treatments. Undiagnosed malignancy is especially problematic as HLH-directed therapy often obscures diagnosis. Simply identifying the pattern of HLH is not sufficient for prudent application of HLH-directed therapies.
1.2.1 |. HLH disease has a range of clinical phenotypes that fit poorly into traditional categories
Historically, a dichotomy between familial and apparently nonfamilial patients has been noted. Familial patients are often very young and may not have an obvious infectious trigger. Apparently, nonfamilial patients are typically older and usually have a clear environmental trigger: infection, malignancy, or rheumatologic disorders. Accordingly, patients with HLH are often split into two groups, “primary” and “secondary,” but with increasing recognition of the clinical diversity of HLH, these labels are increasing ambiguous and misleading. We see at least four reasons for this. First, this dichotomy oversimplifies the complex reality of genetic causality in HLH. A widening spectrum of genetic lesions associated with HLH is now recognized, in which the most severe ones inevitably lead to HLH at an early age (mutation as destiny)22 and milder ones are associated with older ages of onset and stronger environmental triggers (mutation as a risk factor).23,24 Single allele mutations, typically thought of as conferring only carrier status, are frequently seen in patients with HLH associated with rheumatologic disorders or malignancy.25–28 Thus, genetic contribution/risk does not fit neatly into the primary/secondary dichotomy. Second, because this historical classification does not recognize the differences between the HLH pattern and HLH disease, it may lead physicians to miss HLH disease mimics. Furthermore, lumping essentially all non-F-HLH into secondary HLH implies a similarity of pathophysiology and treatment that is unwarranted and may lead to suboptimal or inappropriate therapy. Third, this dichotomy is ambiguous, particularly with regard to infection, and may lead to confusion. Many patients with clear F-HLH experience disease triggered by (secondary to) infections,29 while infection-associated cases occurring in older patients may be associated with varying or indeterminate genetic (primary) lesions. Fourth, unlike the concept of HLH disease and mimics, this dichotomy is not helpful when choosing initial therapy for HLH. As historically defined, both patient groups experience significant morbidity and mortality during the first several weeks of disease, and overall outcomes were not influenced by the presence of known genetic lesions in the HLH-94 and HLH-2004 studies.8,29
For maximal clarity, we favor designating subgroups of HLH by their specific etiologic associations and also specifying whether an individual patient is experiencing HLH disease or a mimic thereof. Thus, cases with clear genetic etiology should be referred to as “familial” HLH (F-HLH), and those seen with malignant or rheumatologic disorders as associated with those conditions (M-HLH or R-HLH, respectively). Other notable subgroups (discussed below in more detail) of HLH disease include HLH associated with immune compromise (immune deficiency or immunosuppression, IC-HLH) and iatrogenic HLH (Rx-HLH, secondary to immune activating therapies). All other cases of HLH disease (including those with negative or ambiguous genetic findings, with or without infectious triggers) should simply be referred to as “HLH disease-NOS,” without reference to “primary” or “secondary.” Notably, as infection may act as a trigger for HLH in any subcategory (except Rx-HLH), it is not distinguishable as a distinct category or cause. Finally, some patients develop a recognizable but incomplete or “forme fruste” HLH syndrome. While we do not wish to broaden the HLH syndrome indefinitely, this forme fruste is important to recognize as some patients with F-HLH or R-HLH may experience repeated, often self-limited episodes of this partial HLH, before developing definitive disease. Such a history should be a strong indicator for genetic workup/careful monitoring, and may justify treatment. Figure 1 illustrates the diversity of HLH that can be precisely described in this fashion.
2 |. TYPICAL AND UNUSUAL HLH PRESENTATION PHENOTYPES
2.1 |. Infant with Fever
While “classic” F-HLH may present at almost any age, it most commonly presents in infants under 1 year of age, and neonatal or in utero presentations are well described.30 This “typical” patient with HLH usually presents with fever and is then noted to have cytopenias and splenomegaly. Further investigation reveals an illappearing patient, sometimes with a critical sepsis-like appearance. Marrow biopsy is often obtained to evaluate for leukemia and may (variably) reveal hemophagocytosis, while not displaying malignancy. Though infection and malignancy must be carefully considered, a triggering infection may not be identified.31 While these patients are well described by current diagnostic criteria, their most striking features are typically the degree of cytopenias, hypofibrinogenemia (in the face of inflammation), and hepatosplenomegaly. This patient population is capable of sudden, severe, and unpredictable worsening, necessitating rapid diagnosis and aggressive intervention.
2.2 |. HLH with isolated CNS involvement
Patients with F-HLH present with central nervous system (CNS) involvement about one-third of the time with symptoms including seizures, meningismus, signs of cranial nerve involvement, ataxia, dysarthria, and encephalopathy.32,33 However, patients may also rarely present with isolated CNS inflammation, without complete or even any systemic signs of HLH. Though rare, this isolated disease is an important forme fruste of HLH, which often presents insidiously with patients experiencing substantial delays in diagnosis.34 Cerebrospinal fluid analysis shows pleocytosis and hyperproteinemia, and brain biopsy reveals T-cell and histiocytic infiltrates. MRI findings in isolated or non-isolated CNS HLH typically include increased signal in T2-weighted images of widespread areas, with symmetric periventricular white matter hyperintensity, meningeal enhancement, diffusion restriction, and may progress to include necrosis.35–37 Age of onset of isolated CNS HLH varies greatly, but is most often seen in children over a year of age, as younger children will usually have accompanying systemic HLH.
2.3 |. Acute liver failure
Patients with F-HLH can present with acute liver failure (ALF), sometimes requiring liver transplantation.38 Significant overlap exists between patients with HLH, who present with ALF, compared to those with ALF of unknown etiology, but patients with HLH are more likely to have fever, splenomegaly, highly elevated ferritin, hemophagocytosis, hypofibrinogenemia, and CNS inflammation.39 However, sCD25 does not appear to be specific for HLH in this context, as approximately one-third of patients with ALF (most without HLH) have elevations, though higher levels were associated with increased mortality.40,41 Additionally, ferritin may not be a reliable marker in this patient population as dying hepatocytes may release it.
2.4 |. Older children with infection
Children over the age of 1 year with HLH typically have definable triggers, most commonly viral infection. Epstein-Barr virus (EBV) is the most frequently identified infection triggering HLH, followed by cytomegalovirus (CMV), though numerous other viral, bacterial, parasitic, and fungal infections have been reported.6,42,43 Though underlying genetic mutations may not be found in patients with EBV-HLH, disease is often responsive to standard treatments and rituximab may be a helpful adjunct, as it may decrease viral burden.44 EBV-associated HLH is usually seen with primary infection, though some patients may present with HLH (or HLH recurrence) associated with chronic active EBV infection.45 These patients typically have predominant EBV infection of T cells (less often NK cells), instead of B cells. Overall, clinical presentations in older patients are similar to those in younger patients, though they may vary widely in severity. While patients presenting with relatively milder acuity may only require corticosteroids or intravenous immune globulin, aggressive treatment may be appropriate, as early initiation of etoposide is associated with improved survival in severe EBV-HLH.46–48 Ferritin values are consistently high in older patients, and sCD25 is always elevated, though often not to the degree seen in infants.
2.5 |. Sepsis-like presentations
Patients with HLH (pediatric and adult) stand out as some of the most critically ill patients in the ICU.49–51 While HLH can be very difficult to discern from typical sepsis or multiple organ dysfunction syndrome (MODS), there are often clues that the severity of inflammation may be more than expected for critical illness and further investigation for possible HLH needs to be considered. These patients tend to be persistently febrile with significant cytopenias, especially thrombocytopenia, and require multiple transfusions.49,51 Hypofibrinogenemia may be helpful to discern HLH from sepsis, though disseminated intravascular coagulation (DIC) may lead to a similar appearance.9,52 Hepatosplenomegaly often seen in HLH but not in sepsis can aid in the diagnosis. If present, hemophagocytosis in tissue and bone marrow can be suggestive of HLH, but hemophagocytosis can be found in critically ill patients with no other HLH features.53,54 Beyond patients with clear HLH disease, there is a growing body of evidence to support a macrophage activation syndrome (MAS) like phenotype in some patients with sepsis.55–58 Unfortunately, there are multiple definitions that have been used for MAS-like sepsis, but in general these patients are described as having MODS characterized by hyperferritinemia, hepatobiliary dysfunction, and DIC with or without bone marrow failure.55–58 Whether these patients may benefit from immunosuppressive therapy is an active area of investigation.
2.6 |. Rheumatologic HLH (R-HLH) or macrophage activation syndrome
MAS is the term most commonly used to refer to an HLH or HLH-like syndrome occurring in the context of rheumatologic disorders (R-HLH). While MAS and HLH are very similar and should be viewed as the same disease, there are notable differences in presentation.59–61 Consensus criteria for recognizing MAS in the context of soJIA have been developed, and in general, these patients with MAS are older than patients with F-HLH and they present with substantially higher platelet and neutrophil counts as well as higher fibrinogen levels.62 These laboratory indices are typically elevated in patients with JIA, so “falling” to near-normal levels is often a substantial change indicating a more severe process than would ordinarily be implied by such values. The frequency of MAS is ∼10% of patients with soJIA, but 30–40% may have an incomplete presentation of MAS.62,63 MAS in adults is most commonly seen with adult-onset Still’s disease (AOSD), reported in ∼15% of AOSD cases.64,65 MAS has also been reported in systemic lupus (estimated incidence of MAS in 4% of cases), dermatomyositis, Kawasaki’s disease, and rheumatoid arthritis.66,67 Obviously, a pre-existing diagnosis of a rheumatologic disorder would define R-HLH, but unexplained rash, recurrent fever, arthralgia/arthritis in an undiagnosed patient presenting with HLH may suggest R-HLH.
2.7 |. HLH in the context of malignancy (M-HLH)
The association of HLH with malignancy (M-HLH) has been recognized for decades and has been reviewed elsewhere.68 Patients may present with the clinical syndrome of HLH associated with undiagnosed underlying malignancy (new onset), or they may develop HLH during treatment for known malignancy (on therapy), usually in the context of infection. With new onset M-HLH, this HLH syndrome is typically an HLH disease mimic, as the disease features appear to be driven directly by the cancer, and antineoplastic (instead of anti-HLH) treatment should be the priority, though pathophysiology is not well defined and therapy of malignancy may overlap with HLH treatment. “On therapy” M-HLH should most often be considered to be a manifestation of HLH due to infection in the context of immune compromise (IC-HLH, see next), though it may rarely be associated with occult relapse.
Lymphoma deserves special mention as it is the most common malignancy associated with HLH at its initial presentation. Because of the difficulty of distinguishing lymphoma from F-HLH or R-HLH, thorough imaging and aggressive biopsy, often guided by PET-CT, should be pursued or at least considered before starting corticosteroids and other therapies that may obscure diagnosis.69 Malignancy may present with HLH at all ages (including infancy), but is increasingly likely at older ages, and is associated with perhaps a majority of cases in adults.66,68,70 When considering M-HLH, it is important to note that the presence of EBV viremia does not rule out malignancy (including B- or T-cell lymphomas) and sCD25 may be disproportionately elevated compared to other features of HLH in patients with occult lymphoma.71,72
2.8 |. HLH in the context of immune compromise (IC-HLH)
HLH has been reported to occur in a variety of patients who are immune compromised, including those receiving chemotherapy or those with primary immune deficiency (PID). As noted above, HLH may occur in patients with malignancy who have often received chemotherapy for an extended period of time and who typically have a recent triggering infection. Similarly, a number of children (and adults) with inflammatory bowel disease (IBD), typically treated with azathioprine or mercaptopurine have been reported to develop HLH after primary infection with EBV or CMV.73 In both contexts, pathophysiology is not well understood and while it may be related to the underlying disease, HLH appears to be a dysregulated response to infection which is related to immunosuppression. It is difficult to generalize about whether IC-HLH may benefit from significant immune suppression. For patients with IBD, withdrawal of mercaptopurine, treatment of infection, supportive care, and moderate dose corticosteroids often suffice. “On therapy” M-HLH does not appear to respond well to any treatment, though corticosteroids ± etoposide are usually tried. Thus, IC-HLH ambiguously straddles the categories of HLH disease and HLH disease mimics.
A variety of PIDs have been reported to present with HLH74,75: severe combined immunodeficiency (SCID), Omenns syndrome, severe DiGeorge syndrome, Wiskott-Aldrich syndrome, chronic granulomatous disease (CGD), X-linked agammaglobulinemia, and autoimmune lymphoproliferative syndrome. Patients with PID and HLH often have unresolved, severe infections. Patients with SCID most often have viral infections, while those with CGD present with bacterial infections. Thus, the presence of HLH associated with unusual or unusually severe infection should suggest undiagnosed immune deficiency. For patients with SCID and infection, immunosuppressive therapy is generally not helpful and this condition should be considered to be a mimic of HLH disease. HLH in patients with CGD is less clear, though typical treatment for HLH beyond corticosteroids is usually not indicated. Thus, the constellation of HLH symptoms in the context of PID should usually be considered a mimic of HLH disease, though some patients may require immunosuppressive therapy.
2.9 |. HLH in the context of immune activating therapies (Rx-HLH)
The HLH syndrome develops in some patients receiving immune activating therapies such as T-cell engaging antibodies, CAR T cells, or immune checkpoint inhibitors. In this context, this syndrome is usually referred to as cytokine release syndrome (CRS), though its pathophysiology appears quite similar to F-HLH and should be recognized as a variant of HLH, which we term as Rx-HLH. Diagnosis and treatment for CRS has been extensively reviewed elsewhere.
3 |. PRACTICAL APPROACH AND ADVICE FOR THE DIAGNOSIS OF HLH
3.1 |. Essential workup for patients with suspected HLH disease
Initial symptoms of HLH are nonspecific and may overlap with other inflammatory or hematopoietic diseases. The initial workup is two-pronged and aims (a) to establish the diagnosis of HLH promptly and (b) to identify mimickers of HLH disease (if present), or potentially treatable underlying HLH disease triggers. Figure 2 displays a diagnostic algorithm for consideration of HLH in newly presenting patients. Table 2 provides additional details regarding the utility of specialized and general diagnostic assays relevant to HLH. It should be emphasized that since T-cell activation is central to HLH pathogenesis, elevated sCD25 should always be observed in untreated HLH.72,76,77 If not elevated, then one should doubt a diagnosis of HLH. Similarly, though not as well established because HLH appears to be largely driven by IFN-γ, elevations of CXCL9 (a sensitive indicator of IFN-γ bioactivity) should be seen in untreated cases of HLH disease (MJ and CA, unpublished data).60,78,79 Additionally, elevated expression of granzyme B in NK cells has been shown to be similarly ubiquitous in HLH; normal expression levels would suggest that HLH is not the correct diagnosis.80 Ferritin levels above 10 000 ng/mL appear to be relatively specific for HLH but are not very sensitive.81 While specialized immunologic testing may facilitate diagnosis, if a diagnosis can be made without them then treatment should not be delayed pending these results. Likewise, treatment should not be delayed for assessment of CNS involvement, though this should always be conducted (once a lumbar puncture may be safely performed).
FIGURE 2.
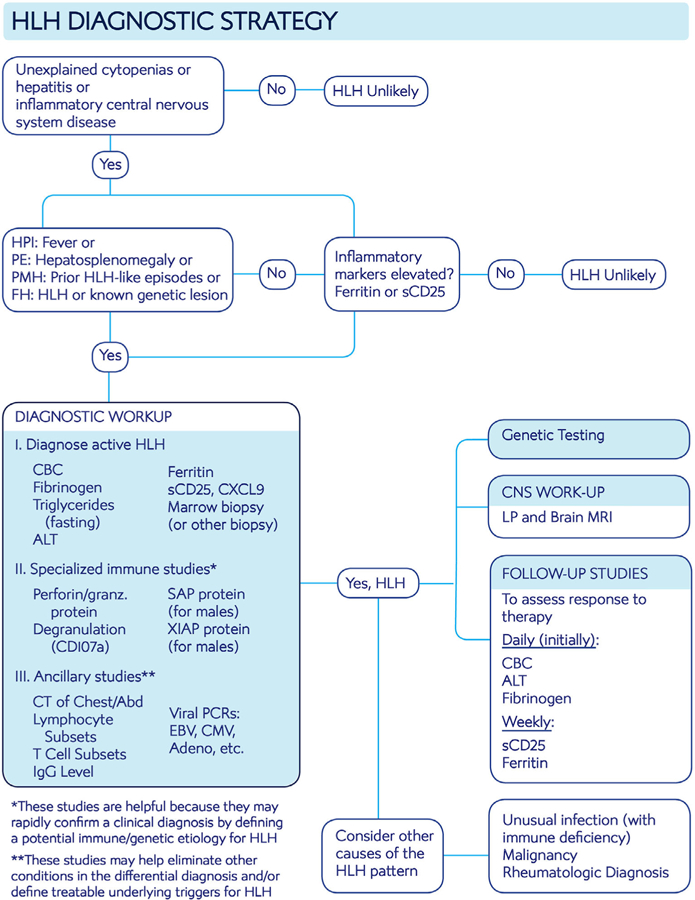
Algorithm for diagnostic workup of HLH
Note. Suggested assessment for patients suspected of having HLH. See Tables 1 and 2 for further explanation/context and Figure S1 for standard treatment roadmap with monitoring strategy.
TABLE 2.
Utility of various diagnostic assays
| Study | Rationale |
|---|---|
| Common laboratory studies | |
| 1. CBC, hepatic panel, fibrinogen, triglycerides 2. Ferritin 3. Cultures, viral PCRs: EBV, CMV, adenovirus |
1. Essential to define typical features of HLH. Though not part of diagnostic criteria, hepatitis is extremely common in HLH. 2. Usually (but not always) very elevated 3. Search for triggering infection |
| Specialized laboratory studies | |
| 1. Markers of immune activation: sCD25, granzyme B expression, CXCL9 2. IL-18 level 3. Measurement of proteins affected in familial HLH: perforin, SAP, XIAP 4. Functional studies: lymphocyte degranulation (CD107a mobilization), NK cell function 5. Lymphocyte subsets, T-cell subsets, immune globulin levels 6. Genetic testing (multigene panel or whole exome) |
1. Essentially always elevated in untreated HLH; very sensitive, but not specific. sCD25, and perhaps CXCL9, are also useful for monitoring response to therapy 2. Elevated in HLH associated with inflammasome-opathies (XIAP, NLRC4, soJIA) 3. Helpful to quickly confirm a suspected familial HLH diagnosis 4. May help to fulfill diagnostic criteria. Degranulation is the preferred functional assay over NK cell function.102 5. Screening studies for PID 6. Essential for defining HLH recurrence risk |
| Imaging | |
| Body cavity CT’s (chest/abd/pelvis) Consider PET-CT if suspicion of lymphoma MRI of brain |
Should be routinely performed to help rule out malignancy, or unusual infections if trigger is unknown (CXR/abdominal ultrasound may suffice) PET-CT very helpful for guiding biopsy if concern for lymphoma Brain MRI complements LP for CNS assessment |
| Tissue sampling | |
| Bone marrow biopsy Lumbar puncture Other tissue biopsies as appropriate: liver, lymph node, masses |
Essential to rule out malignancy, identify hemophagocytosis, and identify CNS involvement |
3.2 |. Differential diagnosis: All patients
3.2.1 |. Infections
Many infections may trigger HLH. Search for common HLH associated viruses by polymerase chain reaction (not serology) should be performed, as concurrent disseminated infection, not prior exposure, is the principal concern. Atypical infections that may lead to cytopenias, elevations of inflammatory markers, and other features of HLH include visceral leishmaniasis, atypical/tuberculous mycobacteria, histoplasmosis, ehrlichia, bartonella and Brucella species, disseminated adenovirus, and disseminated herpes simplex. In most cases, these infections should be considered as mimics of HLH disease as direct treatment of the infection is preferable to HLH-directed immune suppression, and the HLH syndrome does not appear to have a largely immune-mediated etiology in these patients.
3.2.2 |. Other hematologic disorders
Hematologic diseases that may present with a clear pattern of HLH include Langerhans cell histiocytosis involving the marrow and/or visceral organs; multicentric Castleman disease, especially the thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly variant (TAFRO). In both cases, therapy should be directed toward the underlying disorder, ±additional corticosteroids.
3.2.3 |. Drug reactions
In particular, drug rash with eosinophilia and systemic symptoms (DRESS) may present as HLH, but should be treated by withdrawal of the offending agent, ±corticosteroids.
3.2.4 |. Storage and metabolic disorders
Patients with storage diseases including Wolman’s disease (infantile lysosomal acid lipase deficiency), Gaucher’s, and other disorders have been reported to fulfill HLH diagnostic criteria.82–85 In most cases, these disorders should be considered as mimics of HLH disease, as they develop features of HLH (eg, splenomegaly, cytopenias) due to processes not involving immune hyperactivation. However, inflammation in these patients remains poorly studied and there is evidence of abnormal immune regulation in some contexts.86,87 Similarly, patients with metabolic disorders including lysinuric protein intolerance (LPI) and others have also been reported to develop HLH.88,89 Though these disorders should be also be considered as mimics of HLH disease, as they require different, specific treatment, the inflammatory features of LPI in particular are notable and may benefit from immune suppression.90
3.2.5 |. Differential diagnosis: Special situations
Newborns:
The differential in newborns with fulminant liver failure includes gestational alloimmune liver disease (GALD), also called neonatal hemochromatosis. While patients with GALD present with hyperferritinemia and coagulopathy, hemophagocytosis, and other signs of inflammation are absent and extrahepatic siderosis is typically present.91 As mentioned above, disseminated HSV presenting with the syndrome of HLH should be viewed as a mimicker of HLH disease, and immune suppression may be more harmful than beneficial to patients.
Isolated CNS disease:
Differential diagnosis includes autoimmune disseminated encephalomyelitis (ADEM), CNS vasculitis, multiple sclerosis, Rasmussen’s encephalitis, FIRES (febrile infection–related epilepsy) syndrome, and acute necrotizing encephalopathy (ANE), or interferonopathies.33 Rasmussen’s encephalitis is usually recognized based on single hemispheric involvement; ANE may be distinguished from isolated CNS HLH due to its typical presentation with symmetric thalamic necrosis and an absence of extensive white matter changes; and interferonopathies may present with cerebral calcifications.92 FIRES remains poorly defined and in some cases may actually represent undiagnosed HLH.93
3.2.6 |. Practical examples of when to not diagnose HLH disease
In our experience, certain clinical features may suggest mimics of HLH disease:
Despite the common presence of splenomegaly, prominent or abnormal lymphadenopathy is not typically seen in HLH. This finding strongly suggests lymphoma, Castleman disease, or unusual infection such as HIV, histoplasmosis or mycobacterial disease.69 Similarly, leukocytosis is not typical of HLH (except in R-HLH/MAS) and should prompt a search for an alternative diagnosis.
An extremely elevated sCD25 (>10- to 20-fold above normal) in a noninfantile patient suggests undiagnosed lymphoma, especially when ferritin is not similarly elevated.69,94
The early occurrence of isolated and aggressive “CNS relapse” during treatment of systemic HLH may suggest an undiagnosed malignant disorder relapsing in the CNS.
A normal or only modestly elevated sCD25, despite extremely elevated ferritin, suggests disseminated infection in the context of PID, especially in an infant.74
4 |. YOU HAVE DIAGNOSED HLH, NOW WHAT?
4.1 |. Prompt and aggressive initiation of treatment may be life-saving
Currently, standard of care for HLH should be considered to be treatment with etoposide and dexamethasone, as per a slightly modified HLH-94 protocol, which has been reviewed elsewhere.95 Figure S1 displays a roadmap describing this treatment. In general, immediate treatment of HLH is warranted once a diagnosis is made, while it is important to rule out HLH disease mimics or malignancies before starting therapy in order to avoid inappropriate treatment and/or obscuring the underlying diagnosis. The risk of patients deteriorating during prolonged diagnostic evaluation must be balanced against these concerns, especially in the face of limited data. If treatment is initiated before a firm diagnosis is established, the uncertainty of the diagnosis should be revisited once the patient is stable. Though aggressive treatment is needed for most patients, initial treatment with dexamethasone alone with close inpatient monitoring may be appropriate before starting etoposide in patients who are not infants and not severely ill.
4.2 |. Regular monitoring for response to therapy and intercurrent infection are essential
Because etoposide is myelosuppressive, it may be difficult to ascertain whether a patient is responding to therapy based on assessment of peripheral blood counts. Regular monitoring of immune activation, via measurement of sCD25, is essential to gauge success of therapy and need for treatment reintensification or alternative salvage approaches. Ferritin tends to lag sCD25 and may not fully normalize for many months in some patients despite resolution of HLH. Patients with HLH frequently reactivate as steroids are weaned (typically around week 6 of the HLH94 protocol), so close clinical and regular laboratory monitoring should continue as long as patients are receiving treatment. Salvage therapy for HLH has been reviewed in recent years.96 To date, most published experience has been with alemtuzumab, but emapalumab, an IFN-γ blocking monoclonal antibody, was recently approved for refractory or recurrent HLH, and is the first drug ever approved by the US Food and Drug Administration for the treatment of HLH.97 Based on the importance of IFN-γ signaling in HLH, ruxolitinib is being investigated currently in several trials (NCT03795909, NCT02400463, NCT03533790).98–100
4.3 |. Plan ahead for potential hematopoietic transplantation
Allogeneic hematopoietic transplantation is performed to prevent potentially fatal HLH disease recurrence. Patients with clear family histories and/or genetic etiologies for HLH, as well as those with significant CNS involvement or who have recurrent disease, should proceed to BMT as soon as disease is reasonably well controlled. Relapsing HLH, however does not always reflect an underlying genetic predisposition; sometimes it may indicate undiagnosed underlying infection or malignancy rather than a genetic etiology. Since genetic risks are usually not known when patients present, HLA typing and preliminary donor searches should be performed early after diagnosis. Sibling donors should be screened for HLH-associated genetic lesions. Decisions regarding whether or not to proceed to transplantation hinge upon an assessment of HLH recurrence risk, balanced against the risks of transplantation.
5 |. SUMMARY
Without prompt diagnosis and treatment, the natural history of HLH is almost uniformly fatal.101 Due to the rarity, diversity, and complexity of the HLH syndrome, diagnosis is difficult and historically was often delayed. Rising awareness of HLH in recent years has improved recognition, but also increased the risk of misdiagnosis and inappropriate treatment of HLH-mimicking conditions. Evolving understanding of HLH pathophysiology is beginning to alter therapy for this disease (eg, recent FDA approval of emapalumab for second line treatment of HLH) and will alter how it is conceptualized and diagnosed. In this review, we put forth a pragmatic framework to conceptualize HLH rooted in scientific understanding. As a disease not defined by histologic criteria, recognition of clinical patterns is essential for diagnosis. Yet, because of the nonspecific nature of the clinical symptoms, the differential diagnoses of other inflammatory conditions must be considered to avoid unnecessary or harmful immune suppression induced by HLH therapy. In “HLH disease,” immune dysregulation is the central problem for patients and therapeutic immune suppression is the priority. In contrast, other therapies should be the priority for patients displaying the pattern of HLH, which is caused by processes only mimicking the sort of immune dysregulation typified by F-HLH. Furthermore, HLH disease should be categorized into specific disease contexts instead of the historical dichotomy of “primary” and “secondary,” which can be ambiguous and confusing for treatment decisions. The distinction of “HLH disease” and “HLH disease mimics” within the broader syndrome of HLH and more precise categorization of HLH disease subtypes will aid treating physicians in considering all relevant diagnostic considerations when confronting the mystery of HLH.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank St. Baldrick’s Foundation for generous support of NACHO and Liam’s Lighthouse Foundation for support (to MJ) for efforts better define HLH.
Funding information St. Baldrick’s Foundation, Grant/Award Number: 318937
Abbreviations:
- ALF
acute liver failure
- ANE
acute necrotizing encephalopathy
- AOSD
adult-onset Still’s disease
- CGD
chronic granulomatous disease
- CMV
cytomegalovirus
- CNS
central nervous system
- DIC
disseminated intravascular coagulation
- DRESS
drug rash with eosinophilia and systemic symptoms
- EBV
Epstein-Barr virus
- F-HLH
familial HLH
- FIRES
febrile infection–related epilepsy
- HLH
hemophagocytic lymphohistiocytosis
- IBD
inflammatory bowel disease
- IC-HLH
HLH associated with immune compromise
- IFN-γ
interferon gamma
- MAS
macrophage activation syndrome
- M-HLH or R-HLH
malignant or rheumatologic disorders as associated with those conditions
- MODS
multiple organ dysfunction syndrome
- PID
primary immune deficiencies
- Rx-HLH
iatrogenic HLH
- SCID
severe combined immunodeficiency
- soJIA
systemic onset juvenile arthritis
- TAFRO
thrombocytopenia, anasarca, myelofibrosis, renal dysfunction and organomegaly
Footnotes
CONFLICT OF INTEREST
Michael B. Jordan is a consultant and advisory board member for Novimmune, Sobi. Kim E. Nichols received research funding from Incyte. Ashish Kumar is a consultant for Novimmune, Sobi. Michael Henry is a consultant for Sobi. Michelle L. Hermiston is an external advisory board member for Novartis. Carl E. Allen is a consultant and advisory board member for Novimmune, Sobi.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27(136):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ladisch S, Poplack DG, Holiman B, Blaese RM. Immunodeficiency in familial erythrophagocytic lymphohistiocytosis. Lancet. 1978;1(8064):581–583. [DOI] [PubMed] [Google Scholar]
- 3.Jaffe ES, Costa J, Fauci AS, Cossman J, Tsokos M. Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med. 1983;75(5):741–749. [DOI] [PubMed] [Google Scholar]
- 4.Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–566. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg J, Nezelof C. Lymphohistiocytosis: a multi-factorial syndrome of macrophagic activation clinico-pathological study of 38 cases. Hematol Oncol. 1986;4(4):275–289. [DOI] [PubMed] [Google Scholar]
- 6.Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44(3):993–1002. [DOI] [PubMed] [Google Scholar]
- 7.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. [DOI] [PubMed] [Google Scholar]
- 8.Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. [DOI] [PubMed] [Google Scholar]
- 10.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): cD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–743. [DOI] [PubMed] [Google Scholar]
- 11.Pachlopnik Schmid J, Ho CH, Chretien F, et al. Neutralization of IFN gamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1(2):112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118(3):618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terrell CE, Jordan MB. Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood. 2013;122(15):2618–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. 2013;121(26):5184–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoller EE, Lykens JE, Terrell CE, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med. 2011;208(6):1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pham NL, Badovinac VP, Harty JT. Epitope specificity of memory CD8+ T cells dictates vaccination-induced mortality in LCMV-infected perforin-deficient mice. Eur J Immunol. 2012;42(6):1488–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sepulveda FE, Garrigue A, Maschalidi S, et al. Polygenic mutations in the cytotoxicity pathway increase susceptibility to develop HLH immunopathology in mice. Blood. 2016;127(17):2113–2121. [DOI] [PubMed] [Google Scholar]
- 18.Kogl T, Muller J, Jessen B, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: t-cell exhaustion limits fatal disease. Blood. 2013;121(4):604–613. [DOI] [PubMed] [Google Scholar]
- 19.Jessen B, Kogl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. 2013;4: 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chinn IK, Eckstein OS, Peckham-Gregory EC, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. 2018:132:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeba S, Hasegawa S, Shimomura M, et al. Successful Treatment of corticosteroid with antiviral therapy for a neonatal liver failure with disseminated herpes simplex virus infection. AJP Rep. 2015;5(2):e089–e092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27(1):62–68. [DOI] [PubMed] [Google Scholar]
- 23.Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13–4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9–15. [DOI] [PubMed] [Google Scholar]
- 25.Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13–4 polymorphisms. Arthritis Rheum. 2008;58(9):2892–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufman KM, Linghu B, Szustakowski JD, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014;66(12):3486–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciambotti B, Mussolin L, d’Amore ES, et al. Monoallelic mutations of the perforin gene may represent a predisposing factor to childhood anaplastic large cell lymphoma. J Pediatr Hematol Oncol. 2014;36(6):e359–e365. [DOI] [PubMed] [Google Scholar]
- 28.Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192(5):755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergsten E, Horne A, Arico M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah AJ, Kapoor N, Cooper RM, et al. Pre- and post-natal treatment of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;52(1):139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heeg M, Ammann S, Klemann C, et al. Is an infectious trigger always required for primary hemophagocytic lymphohistiocytosis? Lessons from in utero and neonatal disease. Pediatr Blood Cancer. 2018;65(11):e27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horne A, Wickstrom R, Jordan MB, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis. Curr Treat Options Neurol. 2017;19(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benson LA, Li H, Henderson LA, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. 2019;6(3):e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997;89(3):794–800. [PubMed] [Google Scholar]
- 36.Lounder DT, Khandelwal P, Chandra S, et al. Incidence and outcomes of central nervous system hemophagocytic lymphohistiocytosis relapse after reduced-intensity conditioning hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(5):857–860. [DOI] [PubMed] [Google Scholar]
- 37.Rego I, Severino M, Micalizzi C, et al. Neuroradiologic findings and follow-up with magnetic resonance imaging of the genetic forms of haemophagocytic lymphohistiocytosis with CNS involvement. Pediatr Blood Cancer. 2012;58(5):810–814. [DOI] [PubMed] [Google Scholar]
- 38.de Kerguenec C, Hillaire S, Molinie V, et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96(3):852–857. [DOI] [PubMed] [Google Scholar]
- 39.Ryu JM, Kim KM, Oh SH, et al. Differential clinical characteristics of acute liver failure caused by hemophagocytic lymphohistiocytosis in children. Pediatr Int. 2013;55(6):748–752. [DOI] [PubMed] [Google Scholar]
- 40.Bucuvalas J, Filipovich L, Yazigi N, et al. Immunophenotype predicts outcome in pediatric acute liver failure. J Pediatr Gastroenterol Nutr. 2013;56(3):311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DiPaola F, Grimley M, Bucuvalas J. Pediatric acute liver failure and immune dysregulation. J Pediatr. 2014;164(2):407–409. [DOI] [PubMed] [Google Scholar]
- 42.Marsh RA, Vaughn G, Kim MO, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116(26):5824–5831. [DOI] [PubMed] [Google Scholar]
- 43.Xu XJ, Wang HS, Ju XL, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer. 2017;64(4):e26264 10.1002/pbc.26264 [DOI] [PubMed] [Google Scholar]
- 44.Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29(8):569–573. [DOI] [PubMed] [Google Scholar]
- 45.Imashuku S Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44(3):259–272. [DOI] [PubMed] [Google Scholar]
- 46.Shiraishi A, Ohga S, Doi T, et al. Treatment choice of immunotherapy or further chemotherapy for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;59(2):265–270. [DOI] [PubMed] [Google Scholar]
- 47.Arca M, Fardet L, Galicier L, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015;168(1):63–68. [DOI] [PubMed] [Google Scholar]
- 48.Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19(10): 2665–2673. [DOI] [PubMed] [Google Scholar]
- 49.Leow EH, Soh SY, Tan AM, Mok YH, Chan MY, Lee JH. Critically ill children with hemophagocytic lymphohistiocytosis: a case series of 14 patients. J Pediatr Hematol Oncol. 2017;39(6):e303–e306. [DOI] [PubMed] [Google Scholar]
- 50.Rajagopala S, Singh N, Agarwal R, Gupta D, Das R. Severe hemophagocytic lymphohistiocytosis in adults-experience from an intensive care unit from North India. Indian J Crit Care Med. 2012;16(4):198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buyse S, Teixeira L, Galicier L, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med. 2010;36(10):1695–1702. [DOI] [PubMed] [Google Scholar]
- 52.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2):95–109. [DOI] [PubMed] [Google Scholar]
- 53.Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients–a postmortem clinicopathologic analysis. Crit Care Med. 2004;32(6): 1316–1321. [DOI] [PubMed] [Google Scholar]
- 54.Stephan F, Thioliere B, Verdy E, Tulliez M. Role of hemophagocytic histiocytosis in the etiology of thrombocytopenia in patients with sepsis syndrome or septic shock. Clin Infect Dis. 1997;25(5):1159–1164. [DOI] [PubMed] [Google Scholar]
- 55.Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyriazopoulou E, Leventogiannis K, Norrby-Teglund A, et al. Macrophage activation-like syndrome: an immunological entity associated with rapid progression to death in sepsis. BMC Med. 2017;15(1):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carcillo JA, Podd B, Simon DW. From febrile pancytopenia to hemophagocytic lymphohistiocytosis-associated organ dysfunction. Intensive Care Med. 2017;43(12):1853–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carcillo JA, Sward K, Halstead ES, et al. A systemic inflammation mortality risk assessment contingency table for severe sepsis. Pediatr Crit Care Med. 2017;18(2):143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2010;49(3):441–449. [DOI] [PubMed] [Google Scholar]
- 60.Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-gamma and interferon-gamma-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–172. [DOI] [PubMed] [Google Scholar]
- 61.Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–971. [DOI] [PubMed] [Google Scholar]
- 62.Minoia F, Bovis F, Davi S, et al. Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr. 2017;189:72–78. [DOI] [PubMed] [Google Scholar]
- 63.Ravelli A, Davi S, Minoia F, Martini A, Cron RQ. Macrophage activation syndrome. Hematol Oncol Clin North Am. 2015;29(5):927–941. [DOI] [PubMed] [Google Scholar]
- 64.Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014;33(3):305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lenert A, Yao Q. Macrophage activation syndrome complicating adult onset Still’s disease: a single center case series and comparison with literature. Semin Arthritis Rheum. 2016;45(6):711–716. [DOI] [PubMed] [Google Scholar]
- 66.Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. [DOI] [PubMed] [Google Scholar]
- 67.Ravelli A, Minoia F, Davi S, et al. Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against rheumatism/American college of rheumatology/paediatric rheumatology international trials organisation collaborative initiative. Ann Rheum Dis. 2016;75(3):481–489. [DOI] [PubMed] [Google Scholar]
- 68.Lehmberg K, Sprekels B, Nichols KE, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170(4):539–549. [DOI] [PubMed] [Google Scholar]
- 69.Gurunathan A, Boucher AA, Mark M, et al. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018;65(12): e27400. [DOI] [PubMed] [Google Scholar]
- 70.Machaczka M, Vaktnas J, Klimkowska M, Hagglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–619. [DOI] [PubMed] [Google Scholar]
- 71.Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol. 2012;91(1):63–71. [DOI] [PubMed] [Google Scholar]
- 72.Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fitzgerald MP, Armstrong L, Hague R, Russell RK. A case of EBV driven haemophagocytic lymphohistiocytosis complicating a teenage Crohn’s disease patient on azathioprine, successfully treated with rituximab. J Crohns Colitis. 2013;7(4):314–317. [DOI] [PubMed] [Google Scholar]
- 74.Bode SF, Ammann S, Al-Herz W, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schultz KA, Neglia JP, Smith AR, Ochs HD, Torgerson TR, Kumar A. Familial hemophagocytic lymphohistiocytosis in two brothers with X-linked agammaglobulinemia. Pediatr Blood Cancer. 2008;51(2):293–295. [DOI] [PubMed] [Google Scholar]
- 76.Lin M, Park S, Hayden A, et al. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: a systematic scoping review. Ann Hematol. 2017;96(8):1241–1251. [DOI] [PubMed] [Google Scholar]
- 77.Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015;62(2):346–352. [DOI] [PubMed] [Google Scholar]
- 78.Buatois V, Chatel L, Cons L, et al. Use of a mouse model to identify a blood biomarker for IFN gamma activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res. 2017;180:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prencipe G, Caiello I, Pascarella A, et al. Neutralization of IFN-gamma reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol. 2018;141(4):1439–1449. [DOI] [PubMed] [Google Scholar]
- 80.Mellor-Heineke S, Villanueva J, Jordan MB, et al. Elevated granzyme B in cytotoxic lymphocytes is a signature of immune activation in hemophagocytic lymphohistiocytosis. Front Immunol. 2013;4: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1227–1235. [DOI] [PubMed] [Google Scholar]
- 82.Rabah F, Al-Hashmi N, Beshlawi I. Wolman’s disease with secondary hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. 2014;31(6):576–578. [DOI] [PubMed] [Google Scholar]
- 83.Karaman S, Urganci N, Kutluk G, Cetinkaya F. Niemann - Pick disease associated with hemophagocytic syndrome. Turk J Haematol. 2010;27(4):303–307. [DOI] [PubMed] [Google Scholar]
- 84.Duzenli Kar Y, Ozdemir ZC, Kiral E, Kilic Yildirim G, Dinleyici EC, Bor O. Hemophagocytic lymphohystiocytosis associated with type Ia glycogen storage disease. J Pediatr Hematol Oncol. 2019;41(4):e260–e262. [DOI] [PubMed] [Google Scholar]
- 85.Gholamreza B, Ghasem MA. Hemophagocytic lymphohistiocytosis syndrome associated with Gaucher disease type 2. Turk J Haematol. 2014;31(3):307–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sotiropoulos C, Theodorou G, Repa C, et al. Severe impairment of regulatory T-cells and Th1-lymphocyte polarization in patients with Gaucher disease. JIMD Rep. 2015;18:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aflaki E, Borger DK, Grey RJ, et al. Efferocytosis is impaired in Gaucher macrophages. Haematologica. 2017;102(4):656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duval M, Fenneteau O, Doireau V, et al. Intermittent hemophagocytic lymphohistiocytosis is a regular feature of lysinuric protein intolerance. J Pediatr. 1999;134(2):236–239. [DOI] [PubMed] [Google Scholar]
- 89.Gokce M, Unal O, Hismi B, et al. Secondary hemophagocytosis in 3 patients with organic acidemia involving propionate metabolism. Pediatr Hematol Oncol. 2012;29(1):92–98. [DOI] [PubMed] [Google Scholar]
- 90.Bader-Meunier B, Parez N, Muller S. Treatment of hemophagocytic lymphohistiocytosis with cyclosporin A and steroids in a boy with lysinuric protein intolerance. J Pediatr. 2000;136(1):134. [DOI] [PubMed] [Google Scholar]
- 91.Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol. 2013;3(4):313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neilson DE. The interplay of infection and genetics in acute necrotizing encephalopathy. Curr Opin Pediatr. 2010;22(6):751–757. [DOI] [PubMed] [Google Scholar]
- 93.Farias-Moeller R, LaFrance-Corey R, Bartolini L, et al. Fueling the FIRES: hemophagocytic lymphohistiocytosis in febrile infection-related epilepsy syndrome. Epilepsia. 2018;59(9):1753–1763. [DOI] [PubMed] [Google Scholar]
- 94.Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014;93(5):821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508–1517. [DOI] [PubMed] [Google Scholar]
- 96.Marsh RA, Jordan MB, Talano JA, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: a review of the published experience. Pediatr Blood Cancer. 2017;64(4):e26308 10.1002/pbc.26308 [DOI] [PubMed] [Google Scholar]
- 97.Al-Salama ZT. Emapalumab: first global approval. Drugs. 2019;79(1): 99–103. [DOI] [PubMed] [Google Scholar]
- 98.Broglie L, Pommert L, Rao S, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. 2017;1(19):1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang JS, Wang YN, Wu L, Lai WY, Wang Z. [Refractory/relapsed hemophagocytic lymphohistiocytosis treated with ruxolitinib: three cases report and literatures review]. Zhonghua Xue Ye Xue Za Zhi. 2019;40(1):73–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sin JH, Zangardi ML. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: first case report. Hematol Oncol Stem Cell Ther. 2017. 10.1016/j.hemonc.2017.07.002. [DOI] [PubMed]
- 101.Janka GE. Familial hemophagocytic lymphohistiocytosis. European Journal of Pediatrics. 1983;140(3):221–230. [DOI] [PubMed] [Google Scholar]
- 102.Rubin TS, Zhang K, Gifford C, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. 2017;129(22):2993–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.