

Abstract
Inflammation is often applied broadly to human disease. Despite its general familiarity, inflammation is highly complex. There are numerous injurious, immune and infectious determinants, functional elements and signaling pathways, ranging from genetic to epigenetic, environmental, racial, molecular and cellular that participate in disease onset and progression, phenotypic heterogeneity, and treatment selection and response. In addition, inflammation can be tissue and organ specific, adding a layer of complexity to achieving a detailed and translatable understanding of its role in health and disease. The following review takes a close look at inflammation in the context of two common heart diseases, hypertrophic cardiomyopathy and hypertensive cardiomyopathy.
Keywords: Tissue-specific inflammation, Von Willebrand Factor, neutrophil-derived extracellular traps, hypertrophic cardiomyopathy, hypertensive cardiomyopathy
Introduction
Inflammation is an area of significant interest and importance in human cardiovascular health and disease. There are several reasons that include, but are not limited to the following: first, inflammation is a dynamic and complex genetic, epigenetic and cellular process involved with many cardiovascular diseases, disorders and conditions; and second, targeted therapies can be selected and tested to determine their impact on measurable processes and tissue phenotypes. Our interest in tissue-level inflammation in hypertrophic cardiomyopathy (HCM) in adults and early hypertensive cardiomyopathy (HTCM) in youth is the foundation for the following review focused on the potential role of inflammation, Von Willebrand Factor (VWF) and leukocyte-derived extracellular traps (ETs) in these common cardiomyopathies associated with significant morbidity, mortality and health care expenditures worldwide1.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a common condition affecting one of every 500 adults. It is transmitted as an autosomal dominant trait and caused primarily by mutations in genes encoding sarcomere proteins; however, mutations 2 in genes involved in calcium signaling or development of the myocardial cytoskeleton can also cause HCM. Mutations in the cMyBP-C gene (cMyBP-C3) are associated with ~34% of all HCM cases 3 and affect ~60 million people worldwide 4. While many cases are heritable, spontaneous mutations also occur. There is considerable phenotypic variability, ranging from asymmetric septal hypertrophy and isolated apical hypertrophy to moderate left ventricular dilation and end-stage dilation 5. Sudden cardiac death is a feared and devastating complication that occurs most often in patients with ventricular fibrosis. Indeed, even small areas of fibrosis increase the risk substantially 6,7,8.
Hypertensive Cardiomyopathy
Systemic hypertension, most often referred to as essential hypertension, is the most common acquired cardiovascular risk factor in the world-affecting from one-half to two-thirds of adults greater than 40 years of age9. Essential hypertension can also begin during youth 10–12. Regardless of age of onset, elevated blood over time can cause target organ damage that includes concentric left ventricular hypertrophy and impaired diastolic relaxation collectively also known as hypertensive cardiomyopathy (HTCM) 13. Similar to HCM, HTCM is characterized by ventricular remodeling and myocardial fibrosis on cardiac MRI 14. Coronary microvascular disease also occurs in both conditions 15. The extent of fibrosis and its diffuse pattern, as well as heightened global longitudinal strain tends to be greater in HCM than HTCM, as does the heterogeneity of phenotypic expression 16.
Von Willebrand Factor: Fundamental Constructs and Biology
Von Willebrand Factor (VWF) is a large and complex glycoprotein ranging in size from 600,000 to 20 million Da that participates actively in platelet adhesion to either injured or disrupted vascular surfaces, platelet activation and platelet aggregation, particularly under high shear-stress conditions 17. VWF also possesses a wide variety of other biological properties that will be summarized in subsequent sections.
The VWF gene is located on the short arm p of chromosome 12 (12p13.2) with 52 exons that span 178kbp. Genetic regulation of plasma and vesicular VWF is complex and influenced by several non-VWF quantitative trait loci (reviewed in 18. Analysis of the VWF sequence can also be complicated by a non-coding partial VWF pseudogene located on chromosome 22q11.2, which shares 98% sequence homology with exons 23-34 of VWF 19.
Following its synthesis within bone marrow megakarocytes and endothelial cells (EC), VWF is packaged in EC Weibel-Palade Bodies (WPB) and platelet α-granules and released employing several distinct pathways. The first represents a constitutive pathway linked directly to synthesis. This applies primarily to vascular EC. The second is a regulated pathway that responds to EC or platelet stimulation by histamine, leukotriene D4, platelet-activating factor, vascular permeability factor, the terminal component of complement, epinephrine, fluid mechanical forces, blood vessel dynamic forces, factor VIIa, thrombin and fibrin 20. In addition, WPB rapidly translocate to the cell surface of vascular EC following activation 21. The size, complexity, function and redundancy of VWF, coupled with its secretion and activation underscore its biological and teleological relevance in human health and disease.
VWF associates with the luminal surface of EC by vitronectin receptors and constituents of the WPB itself22 (summarized in a subsequent section). The nature (or stimulus) of release also carries important functional ramifications. For example, thrombin stimulates the appearance of high-molecular weight multimers (HMWM) of VWF with increased functionality e.g. platelet binding and aggregation capacity among others 23.
Neutrophil Extracellular Traps: Pathogenesis, Structure and Function in Human Disease
In response to strong stimulation, neutrophils, and to a lesser degree monocytes and eosinophils, release extracellular traps (ETs), consisting of DNA and histones in a process known as NETosis. The process involves histone (H) citrullination (Cit) by peptidylarginine deiminase (PAD)-4, chromatin unwinding, breakdown of nuclear membranes and cytolysis 24,25. There is also a vital or non-lytic NETosis, wherein nuclear materials (DNA and histones) are released without cellular suicide 26, 27. Tissue-level inflammation and NETs have been detected in humans with acute myocarditis and in animal models of autoimmune myocarditis 28. The bioactive proteins released from NETs that cause tissue injury include: MPO (myeloperoxidase), NE (neutrophil elastase), matrix metalloproteinase (MMP)-9, high-mobility group box (HMGB)-1, proteinase (PTN)-3 and properdin 29–31. To the best of our knowledge, the presence and functionality of NETs in HCM have not been investigated previously. The available information suggests that a heightened sterile inflammatory response is necessary, but in the absence of NETs may not be sufficient to cause tissue injury, fibrosis and ventricular remodeling.
NET Architecture
Pires et.al 32 investigated the nanoscale properties of NETs following vascular injury. They combined fluorescence and atomic force microscopy and identified branching filament networks arranged in an organized porous structure with openings of 0.03+/− 0.04 μm2. Topological profiles were up to 3.0 ± 1.0 nm in height, supporting a “beads on a string” model of nucleosome and chromatin strands. Typical branch lengths of 153+/− 103 nm appeared as rigid rods and height profiles of naked DNA and NETs of 1.2 +/− 0.5 nm were indicative of extensive DNA super-coiling throughout the NETs. The existence of DNA duplexes was supported by force spectroscopy with the occurrence of force plateaus that ranged from ~65 pN to 300 pN. The findings suggested that NETs function as microscopic mechanical sieves, with elastic properties that stem from their DNA-protein composition and size. We hypothesize that these central features may be particularly important and determine their ability to interact with other cells, including myocytes, fibroblasts and platelets in diseases and conditions like HCM and HTCM (Fig. 1).
Figure 1:
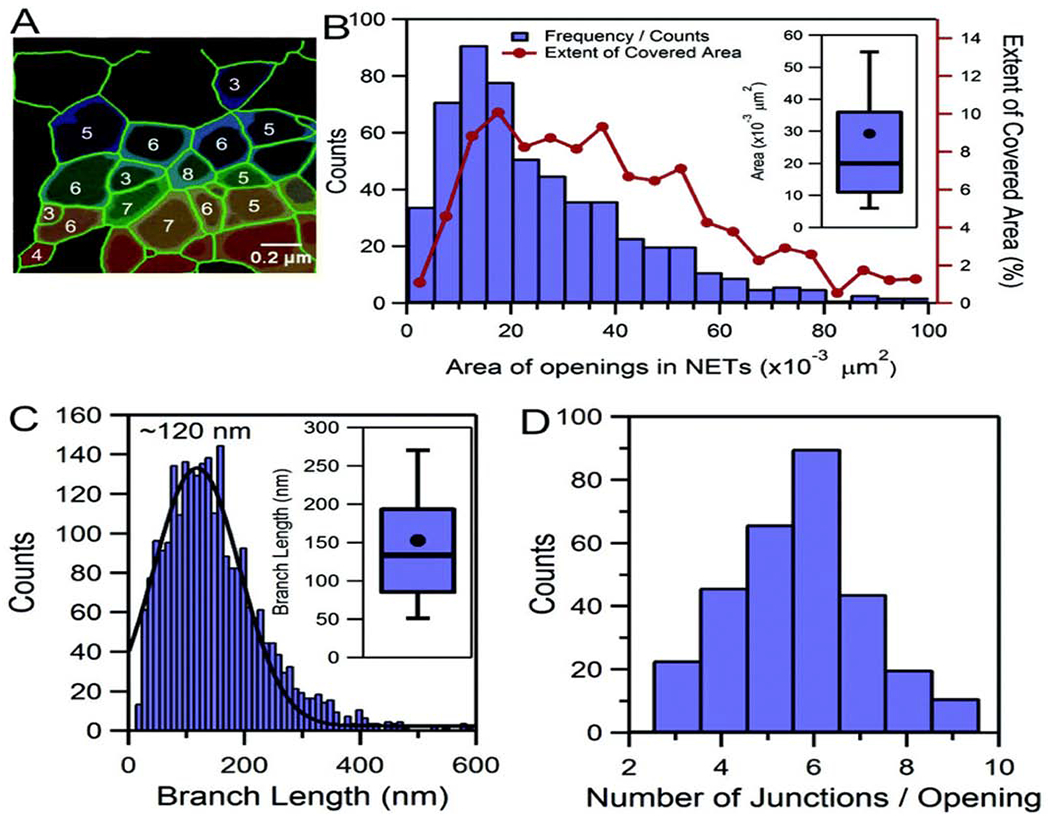
Filaments that only partially resemble NETs, exhibiting several orders of branching, but otherwise not forming a porous matrix (A). More advanced states of NET decomposition, in which DNA appears to exhibit a much more relaxed conformation (B). NETs treated with thermally inactivated trypsin (C). In partially digested NETs, the average branch length was 278 ± 189 (s.d.) nm, indicating that proteins within NETs participate in regulating branch-length and architecture. From Pires RH. Nanoscale 2016; 8: 14193-14202. With permission.
VWF in Hypertrophic Cardiomyopathy and Hypertensive Cardiomyopathy
Von Willebrand factor levels are elevated in patients with HCM 33 and correlate with progressive disease and major comorbidities. The same is true among patients with HTCM and end-organ damage 34. The responsible mechanisms and potential impact on cardiovascular disease progression are summarized below:
Vascular and Endocardial Injury
The early stages of HCM and HTCM are characterized by heightened endocardial and vascular wall stress that causes altered pulse wave velocity, EC injury and increased permeability 35,33,36–38. Vascular EC under stress exhibit changes in adhesion receptors for leukocytes and platelets. Endothelial cell WPB maturation, secretion of adhesive proteins, local environmental responses and exocytosis are altered substantially 39. In turn, VWF kinetics are impacted, leading to increased surface concentrations and availability for platelet, leukocyte and leukocyte-derived extracellular trap (ET) (primarily of neutrophil origin or NETs) binding to injured or altered EC and endocardial cells.
Weibel-Palade Body Mobilization
Weibel-Palade bodies are endothelial cell-derived elongated organelles that contain VWF and numerous functionally active proteins 40. Their unique structure and architecture is due to the arrangement of VWF as tubules and, based on local environmental factors, strings of varied length 22, 41, 42. In the presence of vascular and tissue injury, WPBs themselves can be organized and exocytosed in an elongated state, highlighting the dynamic nature and overall structural-functional plasticity of these organelles (Fig. 2).
Figure 2:
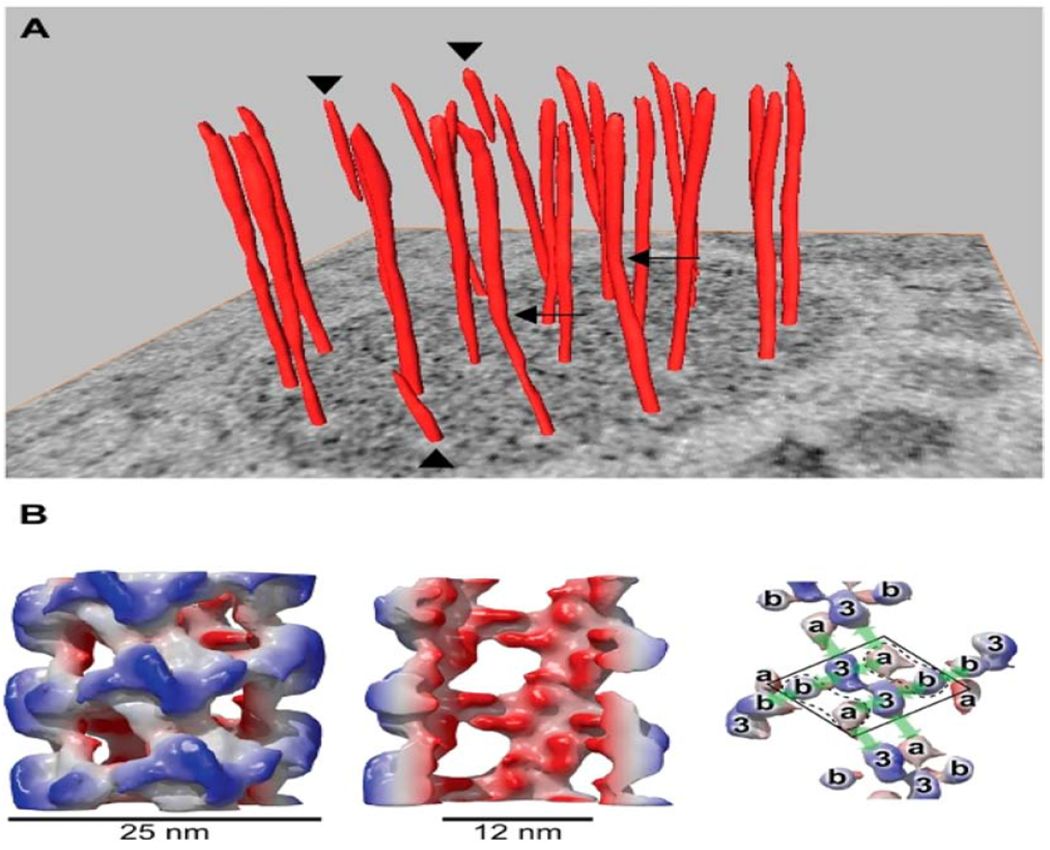
Helical arrangement of VWF in tubular striations of WPB. (A) orderly twisting of the tubules within WPBs is well-illustrated on a tomographic slice. Several tubules (see arrowheads) stop halfway into the WPB and others (arrows) display kinks. In panel B, a reconstruction of VWF tubules is shown with an outside diameter of 25nm (left), cutaway view of the internal diameter of 12nm (middle) and different domains within the helix (right). From Valentijn KM et al. Blood 2011; 119: 5033-5043. With permission. VWF (Von Willebrand Factor), WPB (Weibel-Palade Bodies).
In a two-step process, histones and DNA have been shown to promote WPB release from vascular endothelial cells and the former induces the release of VWF from WPB as well 43,44. The ability of histones to stimulate WPB release is caspase-, Ca 2+ - and charge-dependent. Peripheral blood levels of WPB-released proteins, including VWF correlate with both the degree and duration of inflammation 43.
Endothelial cell activation causes a rapid exocytosis of WPB 45. In addition to the release of VWF, WPB can release a host of adhesion molecules, micro-vesicles and proteins involved directly with maintaining vascular tone, vascular permeability, membrane/protein transport and Ca2+- ATPase signaling 46. Mechanical stretch is associated with inflammation and the exocytosis of WPB through vascular endothelial growth factor (VEGF) receptor-2 signaling pathways 47. Vascular stretch in a hypertensive mouse model was shown to cause a rapid release of VWF and interleukins (IL) 48,49.
Tissue-Specific Inflammation and VWF
Acute States
Platelets play a pivotal role in the recruitment of neutrophils to sites of inflammation as well as their subsequent trans-endothelial migration. VWF may be a key determinant of this event. Anti-VWF antibodies have been shown to stabilize the endothelial barrier 50. The interaction of platelets and neutrophils, to include NET formation, occurs through several signaling pathways independent of platelet aggregation and thrombosis (reviewed in Pitchford) 51. Accumulation of platelets and VWF within microvessels is a unifying step for EC cell activation, impaired vascular integrity, leukocyte recruitment, trans-endothelial migration, tissue inflammation and target organ injury 52 (see subsequent section).
While platelets dissociate from leukocytes during trans-endothelial migration in high shear stress conditions, platelet-leukocyte complexes can remain intact under low mechanical stress 53. Inflammation and its widely varied triggers provoke the formation of ultra-large VWF fibers, which are immobilized on the EC surface and transform to highly adhesive strings under shear conditions 54.
Not all neutrophils release NETs. The available evidence suggest that 20-25% of neutrophils have the capacity to release NETs and those that do are characterized by low density (low density granulocytes) (LDGs) found to co-localize with mononuclear cells 55. LDGs and NETs found within the circulation and tissues of patients with inflammatory myopathies correlate with disease activity and the degree of muscle damage. In addition, LDGs isolated from the peripheral blood display a condition-specific gene signature and have heightened NET formation capabilities 56. An ability of sera from inflammatory diseases to stimulate NET formation in neutrophils obtained from healthy controls supports the premise of tissue and condition-specific inflammatory responses 57.
Tissue-specific inflammation may be directly influenced by tissue-specific EC and vascular beds that typically possess a genetic signature identical to the surrounding tissues 58. In heart failure with reduced ejection fraction, inflammation tends not to be a dominant histological feature and more often reflects concomitant metabolic inflammatory stimuli than a disease-related sterile inflammation as is often the case with HCM and HTCM {reviewed in Paulus)59. Tissue-specific NET release is an area of interest and ongoing investigation for our group.
The hemodynamic-mechanical stress that characterizes HCM and HTCM leads to impaired autophagy 60. Consequently, there is an accumulation of damaged organelles, proteins and DNA that trigger danger-associated molecular patterns (DAMPs), NETs, alarmins and mitochondrial DNA that activate inflammatory pathways 61. Self-renewal of resident cardiac macrophages regulate the accumulation of these injurious molecular and protein materials and limits remodeling following myocardial injury62.
Biomarkers of myocardial remodeling, fibrosis and endothelial cell dysfunction are increased among proteins with HCM and HTCM 63, correlate with cMRI-determined left ventricular mass and inversely correlate with global myocardial perfusion 64.
In a murine heterozygous myosin-binding protein C (cMyBP-C3) mutation model subjected to thoracic aorta constriction (TAC), members of our group showed that cardiomyocytes undergo marked hypertrophy and have impaired force generation, higher Ca2+ sensitivity, and blunted length-dependent increase in force generation. RNA sequencing revealed several differentially regulated genes between cMyBP-C3 heterozygotes and wild-type mice, including regulators of inflammation (e.g. heat shock protein β−1 and tumor necrosis factor), hypertrophic response, myocardial fibrosis and ventricular remodeling. 65. Employing a well-established homozygous cMyBP-C3 mutation model, the group also observed that upregulation of inflammatory pathways and evidence of proinflammatory M1 macrophages on immunofluorescence staining of tissues persisted over time as the phenotype was transitioning from a hypertrophic state to a dilated cardiomyopathy 66. There are several initial triggers for inflammation in HCM, however, the available data suggest that oxidative stress stemming from disorganized sarcomere structure and contractile dyssynchrony are pivotal 67 early contributors 68.
Our group performed RNA-sequencing on peripheral blood samples obtained from adolescents with either hypertension or normal blood pressure 67, 69. In adolescents with elevated blood pressure and increased left ventricular mass by transthoracic echocardiography compared to those with normal blood pressure and normal left ventricular mass, there were 270 differentially expressed genes (unpublished data).
Employing a gene ontology database, differentially expressed gene clusters in several biological pathways, including inflammation, EC activation, systemic blood pressure regulation, vascular remodeling and regulation of apoptosis were identified in participants with elevated blood pressure and increased left ventricular mass. A KEGG pathway analysis revealed upregulation in TGF-β, relaxin and tumor necrosis factor (TNF)-a signaling and mitophagy. There was concomitant upregulation in miRNA involved with cardiovascular remodeling (JAK-STAT, TNF-α, TGF-β and Wnt), fibrosis (fibroblast growth factor receptor) and blood pressure (adrenergic signaling in cardiomyocytes and nitric-oxide synthase) (unpublished data).
In support of the scientific premise that sarcomere abnormalities as observed in HCM with accompanying myocyte dyssynchrony lead to oxidative stress, tissue inflammation and fibrosis are several observations: first, there is a reciprocal augmentation of myosin contractility in response to a step-wise loss of cMyBP-C3 70 and, second, mavacamten, a myosin modulator that reduces steady-state ATPase activity by inhibiting the rate of phosphate release of β-cardiac myosin-S1 and stabilizing an autoinhibited state of two-headed cardiac myosin 71, suppresses the development of ventricular hypertrophy, cardiomyocyte disarray, and myocardial fibrosis and attenuates hypertrophic, inflammatory and fibrosis gene expression in mice harboring heterozygous human mutations in the myosin heavy chain 72.
Chronic States
Levels of VWF antigen predict clinical events among patients with chronic heart failure 73 independently of other well-known risk factors. A relationship between reduced ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity, responsible for cleavage of VWF HMWM, and heart failure has also been reported 74.
A traditional view of inflammation places neutrophils at the forefront of acute inflammation, but down-plays their participation in chronic inflammatory states. The study of NETs and NETosis has changed this long-held paradigm significantly as remnants of leukocytes continue to play important roles in chronic inflammation, tissue injury and fibrosis after the parent cells are no longer present histologically 75.
Neutrophils are released into the blood in response to a wide variety of stimuli and circulate for 6-8 hours. By contrast, once they undergo endothelial cell transmigration and enter surrounding tissues, neutrophils can survive for days in duration 76. In tissues, NETs occupy a space several-fold larger than the cells from which they were derived and cause tissue injury. Additional injury can occur if the concentration of NETs is high and clearance mechanisms are not fully operational. The bioactive proteins released from NETs that are responsible for tissue injury include: MPO (myeloperoxidase), NE (neutrophil elastase), matrix metalloproteinase (MMP)-9, high-mobility group box (HMGB)-1, proteinase (PTN)-3 and properdin 29.
While NET formation is most robust in acute inflammatory states, chronic inflammation is also characterized by the presence of functional NETs. Several well-known examples include chronic obstructive pulmonary disease, cystic fibrosis and systemic lupus erythematosus 77. The same may be true in HCM and HTCM where ongoing hemodynamic-tensile wall stress, oxidative stress, and tissue ischemia from impaired perfusion-energetics, small-vessel vasculopathy and reduced capillary density for the existing ventricular mass are operative (see subsequent section). This is an area of ongoing research in our laboratory.
Platelet Intercellular Transfer
An important question when contemplating tissue-specific inflammation is the portal of entry from the intravascular to the extravascular space. As summarized previously, trans-endothelial migration of leukocytes plays an important role. In addition, platelets contain a major source of growth factors and inflammatory mediators that can also participate.
Platelets contain functional RNA that can be transferred to other cells in a process referred to as horizontal transfer (reviewed in Clancy) 78. The transfer of platelet cytosolic RNA to nucleated cells increases protein translation and biological effects at the vascular level and, if the recipient cell undergoes trans-endothelial migration, at the tissue level 79. An example of the functional signature of platelet horizontal transfer and its biological and pathological effects is hepatocyte proliferation that follows platelet internalization by hepatocytes and functional transfer of RNA in non-alcoholic fatty liver disease 80. Platelet micro-vesicles are also an important source of RNA that can be transferred to a variety of cells, including neutrophils, T lymphocytes, monocytes, macrophages and smooth muscle cells (reviewed in Edelstein)81.
VWF-NET Interactions
As summarized previously, VWF associates with the luminal surface of EC and is activated by shear stress, inflammatory cells and secreted proteins 40.
VWF Anchors NETs to the Vessel Wall and to Inflamed Tissues
NETs “trap” and kill bacteria 82, but, as discussed previously they can also injure host tissues. In animal models of methicillin-resistant Staphylococcus Aureus bacteremia, neutrophils release NETs into the liver vasculature, where they remain anchored to the vascular wall by VWF and display significant neutrophil elastase proteolytic activity 83. Similar processes have been described in the heart. For example, ischemia-reperfusion injury of the myocardium causes an increase in plasma nucleosomes, but in addition, there is abundant neutrophil infiltration at the tissue level and citrullinated histone H3 at the site of injury. 84, 85. We posit that targeting VWF-mediated leukocyte recruitment or NET adhesion may represent novel therapeutic strategies to reduce cardiac injury in HCM and HTCM.
Potential VWF Binding Site(s)
In studies performed by Grassle et.al 54 both functionalized surfaces and intact cell layers of human umbilical vein EC were perfused with isolated, protein-free DNA or leukocytes from whole blood at distinct shear rates. Isolated DNA and DNA released by stimulated leukocytes was able to bind to shear-activated, but not inactivated VWF. As previously described, VWF is known to mediate leukocyte adhesion to the vessel wall and facilitate extravasation into the perivascular space, a process that is augmented by DNA released from leukocytes and likely involved in tissue-level injury 50. The VWF A1 domain is believed to be the binding site for NETs-specifically for DNA (Fig. 3) 86.
Figure 3:
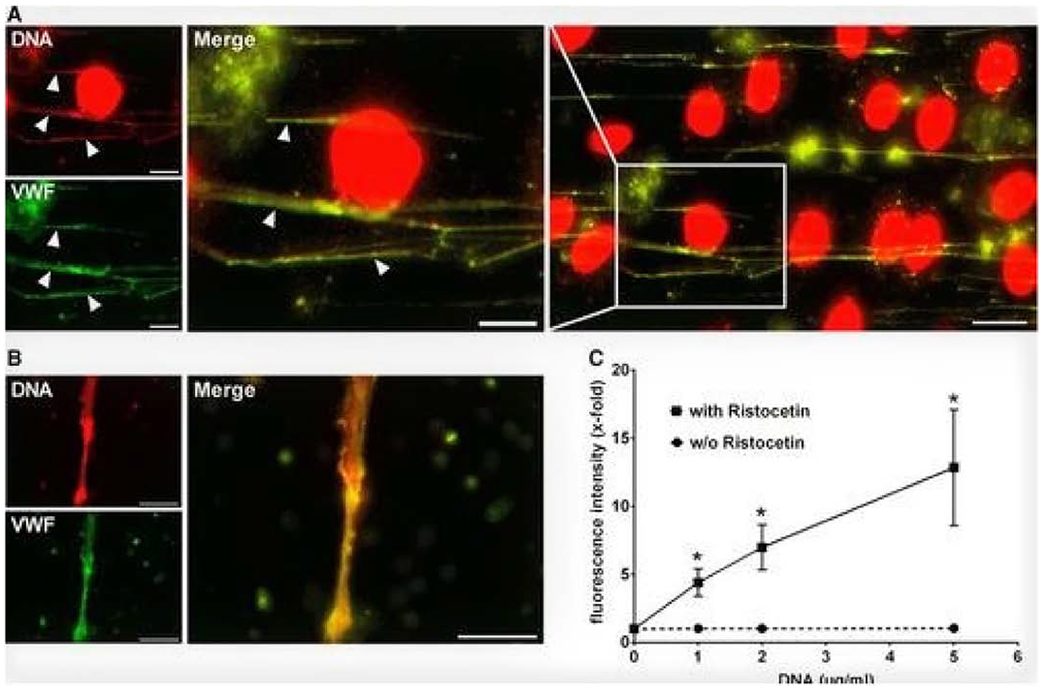
DNA binds to Von Willebrand Factor (VWF) under shear conditions. Panel A. DNA is localized with VWF strings (arrowheads) (Green represents VWF and red DNA); Panel B. Isolated DNA is perfused over VWF at 2 dyne/cm2. Binding only occurs with activated VWF. Panel C. Biding of DNA to VWF is Ristocetin-dependent. From Grassle S et al. Arter Thromb Vasc Biol 2014:34:1382-1389. With permission. Von Willebrand Factor (VWF).
NET Clearance and Regulation
While rapidly evolving, the available data suggest that NETs have an early protective effect following acute injury, particularly from infectious causes. This might not be the case in all tissues and in the setting of fungal infections 87. Emerging data also suggest that the regulation of NET formation and clearance is equally protective and necessary to prevent compounding injurious effects from an initial injury and an excessive inflammatory response. For example, NETs that are not removed from inflamed tissues or from the circulation can be cytotoxic or trigger an autoimmune response 88. DNase is one mechanism for NET degradation, but in physiologic amount’s is not sufficient for complete regulation. Human monocyte-derived macrophages and dendritic cells 89 are able to engulf NETs in a cytochalasin D-dependent manner. Moreover, recombinant C1q, found in human plasma, opsonizes NETS and facilitates their clearance. Once within macrophages, NETs are degraded in lysosomes. The regulation of NETs within tissues requires both normal macrophage activity and DNase, but ADAMTS-13 activity is important for the degradation of VWF and thus the ligand that allows binding of any residual NETs to site(s) of injury. We are currently investigating whether failure at any step in the process could lead to persistent tissue injury, inflammation and fibrosis in HCM and HTCM.
The modulation of cardiac macrophages is increasingly recognized for its importance in chronic disease, including heart failure. Tissue-resident and recruited monocyte-derived macrophages subsets play a particularly important role in HCM, HTCM and ischemia-related cardiac remodeling 90. Phenotypic expressions of disease are influenced by differing macrophage populations. For example, M1 macrophages, in general, are programmed to regulate inflammation while M2 macrophages play a pivotal role in reparative processes 91. Members of our group have previously shown in a murine model of homozygous cMyBP-C3 mutation that upregulation of inflammatory pathways and evidence of proinflammatory M1 macrophages on immunofluorescence staining of tissues persisted over time as the phenotype was transitioning from a hypertrophic state to a dilated cardiomyopathy 66.
VWF and Histone Interactions
The composition of NETs includes a high concentration of histones 92. Analysis of VWF modulators and the marked charge asymmetry of amino acid sequences within the A1 domain has led to an electrostatic model for VWF modulation. Both VWF and the 39/34-kDa VWF fragment bind strongly to histones 92. Histone binding to VWF neither activates nor inhibits VWF binding to platelets; however, in complex with DNA or platelets, attachment to the vessel wall facilitates its injurious effects. In addition, VWF derived from EC and platelets can trigger NET formation and subsequently bind to histones forming platelet-NET attachments. Histones activate platelets through toll-like receptor (TLR)-dependent mechanisms to generate the release of polyphosphates 93, which, in turn, increase both tissue-level inflammation and tissue injury.
VWF and Small Vessel Disease
Patients with HCM and HTCM commonly have microvascular disease and dysfunction referred to as small vessel vasculopathy 94,95. In many instances, the overall perfusion network of microvessels is insufficient for the burden of myocardial mass, while in others there is attendant dysfunction with impaired endothelium-dependent and-independent regulation of vessel tone 96, 97
Pathological studies have shown that intra-mural arteries undergo typical changes observed during vascular injury (fibromuscular dysplasia), which include myofibroblast transdifferentiation and activation, medial layer hypertrophy, smooth muscle cell intimal infiltration and elaboration of extracellular matrix, and breakdown of the internal elastic membrane 98. Studies using human subendocardial biopsies have shown that HTCM is associated with precapillary arteriole smooth muscle hyperplasia, whereas HCM hearts are characterized by a decreased density of such vessels without significant changes in the medial layer content 99. This difference may be one of many factors that explain the phenotypic variations in the site(s) and extent of myocardial hypertrophy between the conditions.
The investigation of myocardial remodeling in HCM and HTCM requires an in-depth understanding of early microvascular remodeling, dysfunction and thrombosis 100. The current evidence in humans indicates an early remodeling process with increases in circulating cathepsin S and endostatin that, in turn, correlate with diastolic dysfunction. Evidence of transcriptional changes in the pre-hypertrophic stage have been described in a thick filament mouse model of HCM with upregulation of TGFB1, CTGF, and periostin and also downregulation of SERCA2a, phospholamban, and sarcolipin—markers of fibrosis and cellular remodeling, respectively 101.
Early changes in myocardial energetics in HCM and HTCM may play an important role in the developing phenotype. Because diastole is crucial for myocardial perfusion, the subendocardium is prone to ischemia with increased flow velocities required to supply this layer. As discussed in a prior section, increased flow velocities with high shear stress mediates VWF release from WPB and flow-induced arterial remodeling. The extensive collateral network of the coronary microvasculature tends to mask the effects of vascular remodeling found dispersed throughout the myocardium in HCM and HTCM 102.
Betoni et. al investigated 20 patients with congenital thrombotic thrombocytopenic purpura (cTTP) who are unable to cleave VWF multimers because of a genetic ADAMTS13 deficiency 103. Using assays of ex vivo serum-induced C3 and C5b-9 deposits on EC, the group documented that in cTTP, complement is activated via the alternative pathway on the cell surface. The abnormality was corrected by restoring ADAMTS13 activity in cTTP serum, which prevented VWF multimer accumulation on EC. The same effect was observed with administration of an anti-VWF Ab. In mechanistic studies the group found that VWF interacted with C3b through its three type A domains and initiated activation of the alternative pathway, although assembly of active C5 convertase and formation of the terminal complement products C5a and C5b-9 occurred solely on the VWF-A2 domain. They showed that in ADAMTS13 deficiency, VWF-mediated formation of terminal complement products, particularly C5a, altered normal EC properties and induced microvascular thrombosis in a perfusion system.
Zheng et.al employed engineered microvessels to determine the pathological responses to EC activation 104. They found that endothelial-secreted VWF was able to assemble into thick bundles or complex meshes, depending on the vessel geometry and flow characteristics. Assembly was most robust in vessels of diameter ≤300 μm subjected to high shear stress, strong flow acceleration and with sharp turns. VWF bundles and webs bound platelets, leukocytes and erythrocytes, obstructed blood flow and also caused erythrocyte fragmentation.
Microvascular Thrombosis, Interstitial Fibrosis and Ventricular Remodeling
In the hearts of patients with HCM and HTCM, NET-mediated microvascular thrombosis may contribute to inflammation, further NET formation, tissue fibrosis and ventricular remodeling 84. Microvascular thrombosis 105 has been reported in several animal models of cardiomyopathy and typically occurs in the presence of active myocardial inflammation. This can be either infectious or non-infectious (sterile) in nature. The role of VWF in sterile inflammation is of particular interest to our group. Witch et.al described a profound inflammatory response and microvascular dysfunction in a mouse model of HCM employing TAC that led to fibrotic remodeling and cardiac failure. The down-stream effects were prevented by pre-TAC administration of rhADAMTS13. Early after the induction of pressure overload under rhADAMTS13 pre-treatment, there was less EC-associated VWF, fewer platelet aggregates, and decreased TGF-β1 levels than in vehicle-treated mice. There was also significant preservation of cardiac function and decreased fibrotic remodeling with rhADAMTS13 administration 106. The findings suggest that in the setting of pressure-overload-induced cardiomyopathy and associated sterile inflammation, decreased VWF-mediated recruitment of platelets, a major source of activated TGFβ1, and preservation of microvascular perfusion substantially attenuate ventricular fibrosis and remodeling.
Human in vitro Heart Tissue Models for Investigation
The ability to model human organs and specific diseases, disorders and conditions is vital to our understanding and advancements in both diagnosis and treatment. Human pluripotent stem cells (hiPSC), derived from patients with HCM and HTCM can be differentiated into cardiomyocytes to fabricate 3-dimensional cardiac organoids. They can be decorated with vascular channels or vascular organoids can concomitantly be differentiated via mesoderm induction of hiPSC aggregates composed of endothelial cells and pericytes to form a human vascular system for experimentation 107, 108. The complementary application of in silica modeling 109–111, systems-based pharmacology 112 and platelet-like particles113 or nanovesicles 114 could also offer a means to more fully and accurately capture the complexity and dynamic nature of HCM and HTCM from molecular, biological and physiological perspectives (reviewed in Becker 1). Gene manipulation of the cells, to include in the precardiac development stage of the first and second heart fields may provide insights 115. Similarly, iPSCs from PAD deficient mice116developed to spheroids/oranoids under proinflammatory and profibrotic conditions may provide a model in which to test therapeutic agents. We have developed human cardiac organoids in our laboratory for the investigation of HCM and HTCM.
Concluding Thoughts and Future Directions
Acute and chronic inflammation are common in cardiovascular disease, including cardiomyopathies such as HCM and HTCM. Changes in cardiomyocyte structure, intra- and extra-cardiac myocardial hemodynamic forces, microvascular dysfunction and capillary density-myocardial mass mismatches with impaired perfusion and cyclic ischemia-reperfusion injury predominate the pathobiology landscape. Sterile inflammation, through several well-characterized mechanisms triggers NET formation with inherent cytotoxic, pro-inflammatory and pro-fibrotic effects. VWF is a pivotal participant in each step to include NET binding and both its localized (vascular and tissue-level) and systemic detrimental effects.
The collective response to acute and chronic inflammation, coupled with impaired macrophage-and dendritic cell-mediated NET and DAMP protein regulation may represent key determinants of the phenotypic expression of HCM and HTCM. Heart and vascular organoids are available and, coupled with computer-based modeling and systems-based pharmacology could provide important mechanistic information. We believe that a more in-depth understanding of the roles of VWF, tissue-level inflammation and NETs would advance the field and, most importantly, benefit patients affected by these common heritable and acquired diseases.
Highlights for Review.
The pathogenesis and natural history of hypertrophic cardiomyopathy and hypertensive cardiomyopathy are considered to be distinct and highly complex. While they share some phenotypic similarities, the potential role of inflammation in these common and life-threatening heart diseases has not been defined fully. We provide an overview and offer a theoretical construct for the potential contributions of Von Willebrand factor and neutrophil extracellular traps.
ACKNOWLEDGEM ENTS
Dr. Sadayappan has received support from National Institutes of Health grants R01 HL130356, R56 HL139680, R01 AR067279, R01 HL105826 and R01 HL143490; American Heart Association 2019 Institutional Undergraduate Student (19UFEL34380251) and transformation (19TPA34830084) awards; and MyoKardia, AstraZeneca, Merck and Amgen.
Dr. Becker has received support from National Institutes of Health grants R01-HL065222, U54-HL112307 and American Heart Association 14FRN24110000.
Dr. Owens has received support from National Institutes of Health grants 5R01-HL141404-02, F31-HL143987-01, R01-HL141404-S2A; American Heart Association 191 PLO134760319, 19 TPA34910086.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: Dr. Sadayappan provided consulting and collaborative research studies to the Leducq Foundation, Red Saree Inc., Greater Cincinnati Tamil Sangam, MyoKardia, Merck and Amgen, but such work is unrelated to the content of this manuscript. No other disclosures are reported.
Dr. Becker serves on scientific advisory boards for: Janssen and DSMB Committees for Ionis Pharmaceuticals, Akcea Therapeutics and Novartis.
Dr. Owens receives research support from Bayer AG.
References
- 1.Becker RC, Owens AP 3rd and Sadayappan S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. Journal of thrombosis and thrombolysis. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen K, Roche S, Donal E, Odent S, Eicher JC, Faivre L, Millat G, Salgado D, Desvignes Jp , Lavoute C, Haentjens J, Consolino E, Janin A, Cerino M, Reant P, Rooryck C, Charron P, Richard P, Casalta AC, Michel N, Magdinier F, Beroud C, Levy N and Habib G. Whole Exome Sequencing Reveals a Large Genetic Heterogeneity and Revisits the Causes of Hypertrophic Cardiomyopathy. Circulation Genomic and precision medicine. 2019;12:e002500. [DOI] [PubMed] [Google Scholar]
- 3.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG and Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. The New England journal of medicine. 2008;358:1899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG and Seidman Ce. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. The New England journal of medicine. 1998;338:1248–57. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Maron MS, Maron BA and Loscalzo J. Moving Beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week. Journal of the American College of Cardiology. 2019;73:1978–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lorenzini M, Anastasiou Z, O’Mahony C, Guttman OP, Gimeno JR, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Garcia-Pavia P, Limongelli G, Pavlou M and Elliott PM. Mortality Among Referral Patients With Hypertrophic Cardiomyopathy vs the General European Population. JAMA cardiology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He D, Ye M, Zhang L and Jiang B. Prognostic significance of late gadolinium enhancement on cardiac magnetic resonance in patients with hypertrophic cardiomyopathy. Heart & lung : the journal of critical care. 2018;47:122–126. [DOI] [PubMed] [Google Scholar]
- 8.Marian AJ and Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circulation research. 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han M, Chen Q, Liu L, Li Q, Ren Y, Zhao Y, Liu D, Zhang D, Liu F, Chen X, Cheng C, Guo C, Zhou Q, Tian G, Qie R, Huang S, Wu X, Liu Y, Li H, Sun X, Lu J, Hu D and Zhang M. Stage 1 hypertension by the 2017 American College of Cardiology/American Heart Association hypertension guidelines and risk of cardiovascular disease events: systematic review, metaanalysis, and estimation of population etiologic fraction of prospective cohort studies. Journal of hypertension. 2020;38:573–578. [DOI] [PubMed] [Google Scholar]
- 10.Mendizabal B, Khoury P, Woo JG and Urbina EM. Racial Differences in the Influence of Risk Factors in Childhood on Left Ventricular Mass in Young Adulthood. The Journal of pediatrics. 2020;217:152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urbina EM, Khoury PR, Bazzano L, Burns TL, Daniels S, Dwyer T, Hu T, Jacobs DR Jr., Juonala M, Prineas R, Raitakari O, Steinberger J, Venn A, Woo JG and Sinaiko A. Relation of Blood Pressure in Childhood to Self-Reported Hypertension in Adulthood. Hypertension (Dallas, Tex: 1979). 2019;73:1224–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamdani G, Flynn JT, Becker RC, Daniels SR, Falkner B, Hanevold CD, Ingelfinger JR, Lande MB, Martin LJ, Meyers KE, Mitsnefes M, Rosner B, Samuels JA and Urbina EM. Prediction of Ambulatory Hypertension Based on Clinic Blood Pressure Percentile in Adolescents. Hypertension (Dallas, Tex : 1979). 2018;72:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tadic M, Cuspidi C, Plein S, Milivojevic IG, Wang DW, Grassi G and Mancia G. Comprehensive assessment of hypertensive heart disease: cardiac magnetic resonance in focus. Heart failure reviews. 2020. [DOI] [PubMed] [Google Scholar]
- 14.Nwabuo CC and Vasan RS. Pathophysiology of Hypertensive Heart Disease: Beyond Left Ventricular Hypertrophy. Current hypertension reports. 2020;22:11. [DOI] [PubMed] [Google Scholar]
- 15.Konst RE, Guzik TJ, Kaski JC, Maas A and Elias-Smale SE. The pathogenic role of coronary microvascular dysfunction in the setting of other cardiac or systemic conditions. Cardiovasc Res. 2020;116:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neisius U, Myerson L, Fahmy AS, Nakamori S, El-Rewaidy H, Joshi G, Duan C, Manning WJ and Nezafat R. Cardiovascular magnetic resonance feature tracking strain analysis for discrimination between hypertensive heart disease and hypertrophic cardiomyopathy. PloS one. 2019;14:e0221061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggeri ZM. Role of von Willebrand factor in platelet thrombus formation. Ann Med. 2000;32 Suppl 1:2–9. [PubMed] [Google Scholar]
- 18.Zolkova J, Sokol J, Simurda T, Vadelova L, Snahnicanova Z, Loderer D, Dobrotova M, Ivankova J, Skornova I, Lasabova Z, Kubisz P and Stasko J. Genetic Background of von Willebrand Disease: History, Current State, and Future Perspectives. Seminars in thrombosis and hemostasis. 2019. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad F, Kannan M, Biswas A and Saxena R. Impact of 789Ala/Ala genotype on quantitative type of von Willebrand disease. Ann Hematol. 2009;88:479–83. [DOI] [PubMed] [Google Scholar]
- 20.Huisman B, Hoore M, Gompper G and Fedosov DA. Modeling the cleavage of von Willebrand factor by ADAMTS13 protease in shear flow. Med Eng Phys. 2017;48:14–22. [DOI] [PubMed] [Google Scholar]
- 21.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. [DOI] [PubMed] [Google Scholar]
- 22.Sharda AV, Harrison J, Wilkie AR, Fang C, Mendez LM, Italiano JE and Flaumenhaft RC. VWF Exocytosis and Biogenesis of Weibel Palade Bodies in Endothelial Cells Are Differentially Controlled By Interactions between Bloc-2 and the Exocyst Complex. Blood. 2019;134:8. [Google Scholar]
- 23.Trabold K, Makhoul S, Gambaryan S, van Ryn J, Walter U and Jurk K. The Direct Thrombin Inhibitors Dabigatran and Lepirudin Inhibit GPIbalpha-Mediated Platelet Aggregation. Thrombosis and haemostasis. 2019;119:916–929. [DOI] [PubMed] [Google Scholar]
- 24.Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA and Wagner DD. Neutrophil extracellular traps promote deep vein thrombosis in mice. Journal of thrombosis and haemostasis : JTH. 2012;10:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thalin C, Daleskog M, Goransson SP, Schatzberg D, Lasselin J, Laska AC, Kallner A, Helleday T, Wallen H and Demers M. Validation of an enzyme-linked immunosorbent assay for the quantification of citrullinated histone H3 as a marker for neutrophil extracellular traps in human plasma. Immunologic research. 2017;65:706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J and Kubes P. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nature medicine. 2012;18:1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH and Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature medicine. 2007;13:463–9. [DOI] [PubMed] [Google Scholar]
- 28.Weckbach LT, Grabmaier U, Uhl A, Gess S, Boehm F, Zehrer A, Pick R, Salvermoser M, Czermak T, Pircher J, Sorrelle N, Migliorini M, Strickland DK, Klingel K, Brinkmann V, Abu Abed U, Eriksson U, Massberg S, Brunner S and Walzog B. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. The Journal of experimental medicine. 2019;216:350–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Euler M and Hoffmann MH. The double-edged role of neutrophil extracellular traps in inflammation. Biochemical Society transactions. 2019. [DOI] [PubMed] [Google Scholar]
- 30.Szatmary P, Huang W, Criddle D, Tepikin A and Sutton R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med. 2018;22:4617–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meegan JE, Yang X, Beard RS Jr., Jannaway M, Chatterjee V, Taylor-Clark TE and Yuan SY. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem Biophys Res Commun. 2018;503:1498–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pires RH, Felix SB and Delcea M. The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nanoscale. 2016;8:14193–202. [DOI] [PubMed] [Google Scholar]
- 33.Cambronero F, Vilchez JA, Garcia-Honrubia A, Ruiz-Espejo F, Moreno V, Hernandez-Romero D, Bonacasa B, Gonzalez-Conejero R, de la Morena G, Martinez P, Climent V, Valdes M and Marin F. Plasma levels of von Willebrand factor are increased in patients with hypertrophic cardiomyopathy. Thrombosis research. 2010;126:e46–50. [DOI] [PubMed] [Google Scholar]
- 34.Spencer CG, Gurney D, Blann AD, Beevers DG and Lip GY. Von Willebrand factor, soluble P-selectin, and target organ damage in hypertension: a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT). Hypertension (Dallas, Tex : 1979). 2002;40:61–6. [DOI] [PubMed] [Google Scholar]
- 35.Urbina EM, Kimball TR, Khoury PR, Daniels SR and Dolan LM. Increased arterial stiffness is found in adolescents with obesity or obesity-related type 2 diabetes mellitus. Journal of hypertension. 2010;28:1692–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alman AC, Talton JW, Wadwa RP, Urbina EM, Dolan LM, Hamman RF, D’Agostino RB Jr., Marcovina SM and Dabelea DM. Inflammation, adiposity, and progression of arterial stiffness in adolescents with type 1 diabetes: The SEARCH CVD Study. Journal of diabetes and its complications. 2018;32:995–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendizabal B, Urbina EM, Becker R, Daniels SR, Falkner BE, Hamdani G, Hanevold CD, Hooper SR, Ingelfinger JR, Lande M, Martin LJ, Meyers K, Mitsnefes M, Rosner B, Samuels JA and Flynn JT. SHIP-AHOY (Study of High Blood Pressure in Pediatrics: Adult Hypertension Onset in Youth). Hypertension (Dallas, Tex : 1979). 2018;72:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khoury M and Urbina EM. Cardiac and Vascular Target Organ Damage in Pediatric Hypertension. Frontiers in pediatrics. 2018;6:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holthenrich A, Drexler HCA, Chehab T, Nass J and Gerke V. Proximity proteomics of endothelial Weibel-Palade bodies identifies novel regulator of von-Willebrand factor secretion. Blood. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valentijn KM, Sadler JE, Valentijn JA, Voorberg J and Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCormack JJ, Harrison-Lavoie KJ and Cutler DF. Human endothelial cells size-select their secretory granules for exocytosis to modulate their functional output. Journal of thrombosis and haemostasis : JTH. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holthenrich A, Drexler HCA, Chehab T, Nass J and Gerke V. Proximity proteomics of endothelial Weibel-Palade bodies identifies novel regulator of von Willebrand factor secretion. Blood. 2019;134:979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michels A, Albanez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, James PD, Swystun LL and Lillicrap D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. Journal of thrombosis and haemostasis : JTH. 2016;14:2274–2286. [DOI] [PubMed] [Google Scholar]
- 44.Michels A, Swystun LL, Mewburn J, Albanez S and Lillicrap D. Investigating von Willebrand Factor Pathophysiology Using a Flow Chamber Model of von Willebrand Factor-platelet String Formation. Journal of visualized experiments : JoVE. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holthenrich A and Gerke V. Regulation of von-Willebrand Factor Secretion from Endothelial Cells by the Annexin A2-S100A10 Complex. International journal of molecular sciences. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chatterjee V, Yang X, Ma Y, Cha B, Meegan JE, Wu M and Yuan SY. Endothelial microvesicles carrying Src-rich cargo impair adherens junction integrity and cytoskeleton homeostasis. Cardiovasc Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong Y, Hu Z, Han X, Jiang B, Zhang R, Zhang X, Lu Y, Geng C, Li W, He Y, Huo Y, Shibuya M and Luo J. Hypertensive stretch regulates endothelial exocytosis of Weibel-Palade bodies through VEGF receptor 2 signaling pathways. Cell research. 2013;23:820–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand CM, Guzik TJ and Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114:1547–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao W, Zhang D, Li Q, Liu Y, Jing S, Cui J, Xu W, Li S, Liu J and Yu B. Biomechanical Stretch Induces Inflammation, Proliferation, and Migration by Activating NFAT5 in Arterial Smooth Muscle Cells. Inflammation. 2017;40:2129–2136. [DOI] [PubMed] [Google Scholar]
- 50.Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, Schneider SW, Jones C, Nieswandt B, Wild MK and Vestweber D. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–4719. [DOI] [PubMed] [Google Scholar]
- 51.Pitchford S, Pan D and Welch HC. Platelets in neutrophil recruitment to sites of inflammation. Current opinion in hematology. 2017;24:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM and Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. 2010;115:3632–9. [DOI] [PubMed] [Google Scholar]
- 53.van Gils JM, da Costa Martins PA, Mol A, Hordijk PL and Zwaginga JJ. Transendothelial migration drives dissociation of plateletmonocyte complexes. Thrombosis and haemostasis. 2008;100:271–9. [PubMed] [Google Scholar]
- 54.Grassle S, Huck V, Pappelbaum KI, Gorzelanny C, Aponte-Santamaria C, Baldauf C, Grater F, Schneppenheim R, Obser T and Schneider SW. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler Thromb Vasc Biol. 2014;34:1382–9. [DOI] [PubMed] [Google Scholar]
- 55.Yipp BG and Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–94. [DOI] [PubMed] [Google Scholar]
- 56.Seto NL, Torres-Ruiz JJ, Carmona-Rivera C, Pinal-Fernandez I, Pak K, Purmalek MM, Hosono Y, Fernandes-Cerqueira C, Gowda PC, Arnett N, Gorbach A, Benveniste O, Gomez-Martin D, Selva-O’Callaghan A, Milisenda JC, Grau-Junyent JM, Christopher-Stine L, Miller FW, Lundberg IE, Kahlenberg JM, Schiffenbauer AI, Mammen AL, Rider LG and Kaplan MJ. Neutrophil dysregulation is pathogenic in idiopathic inflammatory myopathies. JCI Insight. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mistry P, Carmona-Rivera C, Ombrello AK, Hoffmann P, Seto NL, Jones A, Stone DL, Naz F, Carlucci P, Dell’Orso S, Gutierrez-Cruz G, Sun HW, Kastner DL, Aksentijevich I and Kaplan MJ. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Annals of the rheumatic diseases. 2018;77:1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, Dai Y, Malik AB and Rehman J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paulus WJ and Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. Journal of the American College of Cardiology. 2013;62:263–71. [DOI] [PubMed] [Google Scholar]
- 60.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T and Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol. 2012;107:235. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Q, Kang R, Zeh HJ, 3rd, Lotze MT and Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9:451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS and Epelman S. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nature immunology. 2019;20:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanaani A and Fuisz A. Cardiac Magnetic Resonance for Diagnosis and Risk Stratification. Cardiology clinics. 2019;37:27–33. [DOI] [PubMed] [Google Scholar]
- 64.Fernlund E, Gyllenhammar T, Jablonowski R, Carlsson M, Larsson A, Arnlov J and Liuba P. Serum Biomarkers of Myocardial Remodeling and Coronary Dysfunction in Early Stages of Hypertrophic Cardiomyopathy in the Young. Pediatric cardiology. 2017;38:853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barefield D, Kumar M, Gorham J, Seidman JG, Seidman CE, de Tombe PP and Sadayappan S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J Mol Cell Cardiol. 2015;79:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lynch TLt, Ismahil MA, Jegga AG, Zilliox MJ, Troidl C, Prabhu SD and Sadayappan S. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J Mol Cell Cardiol. 2017;102:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamdani G, Flynn JT, Daniels S, Falkner B, Hanevold C, Ingelfinger J, Lande MB, Martin LJ, Meyers KE, Mitsnefes M, Rosner B, Samuels J and Urbina EM. Ambulatory blood pressure monitoring tolerability and blood pressure status in adolescents: the SHIP AHOY study. Blood pressure monitoring. 2019;24:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lynch TLt, Sivaguru M, Velayutham M, Cardounel AJ, Michels M, Barefield D, Govindan S, dos Remedios C, van der Velden J and Sadayappan S. Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation. Oxid Med Cell Longev. 2015;2015:424751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Urbina EM. Genome-wide association and Mendelian randomization study of NT-proBNP in patients with acute coronary syndrome. Hypertension (Dallas, Tex : 1979). 2016;25:1447–56. [DOI] [PubMed] [Google Scholar]
- 70.Toepfer CN, Wakimoto H, Garfinkel AC, McDonough B, Liao D, Jiang J, Tai AC, Gorham JM, Lunde IG, Lun M, Lynch TLt, McNamara JW, Sadayappan S, Redwood CS, Watkins HC, Seidman JG and Seidman CE. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Science translational medicine. 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rohde JA, Roopnarine O, Thomas DD and Muretta JM. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E7486–e7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG and Seidman CE. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (New York, NY). 2016;351:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gombos T, Mako V, Cervenak L, Papassotiriou J, Kunde J, Harsfalvi J, Forhecz Z, Pozsonyi Z, Borgulya G, Janoskuti L and Prohaszka Z. Levels of von Willebrand factor antigen and von Willebrand factor cleaving protease (ADAMTS13) activity predict clinical events in chronic heart failure. Thrombosis and haemostasis. 2009;102:573–80. [DOI] [PubMed] [Google Scholar]
- 74.Akyol O, Akyol S and Chen CH. Update on ADAMTS13 and VWF in cardiovascular and hematological disorders. Clinica chimica acta; international journal of clinical chemistry. 2016;463:109–118. [DOI] [PubMed] [Google Scholar]
- 75.Frangou E, Vassilopoulos D, Boletis J and Boumpas DT. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmunity reviews. 2019;18:751–760. [DOI] [PubMed] [Google Scholar]
- 76.Abu Abed U and Brinkmann V. Immunofluorescence Labelling of Human and Murine Neutrophil Extracellular Traps in Paraffin-Embedded Tissue. Journal of visualized experiments : JoVE. 2019. [DOI] [PubMed] [Google Scholar]
- 77.Castanheira FVS and Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133:2178–2185. [DOI] [PubMed] [Google Scholar]
- 78.Clancy L, Beaulieu LM, Tanriverdi K and Freedman JE. The role of RNA uptake in platelet heterogeneity. Thrombosis and haemostasis. 2017;117:948–961. [DOI] [PubMed] [Google Scholar]
- 79.Risitano A, Beaulieu LM, Vitseva O and Freedman JE. Platelets and platelet-like particles mediate intercellular RNA transfer. Blood. 2012;119:6288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kirschbaum M, Karimian G, Adelmeijer J, Giepmans BN, Porte RJ and Lisman T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood. 2015;126:798–806. [DOI] [PubMed] [Google Scholar]
- 81.Edelstein LC. The role of platelet microvesicles in intercellular communication. Platelets. 2017;28:222–227. [DOI] [PubMed] [Google Scholar]
- 82.Brinkmann V Neutrophil Extracellular Traps in the Second Decade. Journal of innate immunity. 2018;10:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G and Kubes P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y and Wagner DD. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123:141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, Libby P, Goldhaber SZ, Mitchell RN and Wagner DD. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. Journal of thrombosis and haemostasis : JTH. 2014;12:860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kawecki C, Lenting PJ and Denis CV. von Willebrand factor and inflammation. Journal of thrombosis and haemostasis : JTH. 2017;15:1285–1294. [DOI] [PubMed] [Google Scholar]
- 87.Alflen A, Aranda Lopez P, Hartmann AK, Maxeiner J, Bosmann M, Sharma A, Platten J, Ries F, Beckert H, Ruf W and Radsak MP. Neutrophil extracellular traps impair fungal clearance in a mouse model of invasive pulmonary aspergillosis. Immunobiology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Farrera C and Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. Journal of immunology (Baltimore, Md: 1950). 2013;191:2647–56. [DOI] [PubMed] [Google Scholar]
- 89.Lazzaretto B and Fadeel B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. Journal of immunology (Baltimore, Md : 1950). 2019;203:2276–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rhee AJ and Lavine KJ. New Approaches to Target Inflammation in Heart Failure: Harnessing Insights from Studies of Immune Cell Diversity. Annual review of physiology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D and Lavine KJ. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circulation research. 2019;124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ward CM, Tetaz TJ, Andrews RK and Berndt MC. Binding of the von Willebrand factor A1 domain to histone. Thrombosis research. 1997;86:469–77. [DOI] [PubMed] [Google Scholar]
- 93.Ammollo CT, Semeraro F, Xu J, Esmon NL and Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. Journal of thrombosis and haemostasis : JTH. 2011. ;9:1795–803. [DOI] [PubMed] [Google Scholar]
- 94.Moreno-Moral A, Mancini M, D’Amati G, Camici P and Petretto E. Transcriptional network analysis for the regulation of left ventricular hypertrophy and microvascular remodeling. Journal of cardiovascular translational research. 2013;6:931–44. [DOI] [PubMed] [Google Scholar]
- 95.Camaioni C, Knott KD, Augusto JB, Seraphim A, Rosmini S, Ricci F, Boubertakh R, Xue H, Hughes R, Captur G, Lopes LR, Brown LAE, Manisty C, Petersen SE, Plein S, Kellman P, Mohiddin SA and Moon JC. Inline perfusion mapping provides insights into the disease mechanism in hypertrophic cardiomyopathy. Heart. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Villa AD, Sammut E, Zarinabad N, Carr-White G, Lee J, Bettencourt N, Razavi R, Nagel E and Chiribiri A. Microvascular ischemia in hypertrophic cardiomyopathy: new insights from high-resolution combined quantification of perfusion and late gadolinium enhancement. Journal of cardiovascular magnetic resonance : official journal of the Society for Cardiovascular Magnetic Resonance. 2016;18:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bravo PE, Di Carli MF and Dorbala S. Role of PET to evaluate coronary microvascular dysfunction in non-ischemic cardiomyopathies. Heart failure reviews. 2017;22:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marszalek RJ, John Solaro R and Wolska BM. Coronary arterial vasculature in the pathophysiology of hypertrophic cardiomyopathy. Pflugers Archiv : European journal of physiology. 2019;471:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mundhenke M, Schwartzkopff B and Strauer BE. Structural analysis of arteriolar and myocardial remodelling in the subendocardial region of patients with hypertensive heart disease and hypertrophic cardiomyopathy. Virchows Archiv : an international journal of pathology. 1997;431:265–73. [DOI] [PubMed] [Google Scholar]
- 100.Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, Gonzalez A, Colan SD, Seidman JG, Diez J and Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. The New England journal of medicine. 2010;363:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim JB, Porreca GJ, Song L, Greenway SC, Gorham JM, Church GM, Seidman CE and Seidman JG. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science (New York, NY). 2007;316:1481–4. [DOI] [PubMed] [Google Scholar]
- 102.Pries AR and Secomb TW. Control of blood vessel structure: insights from theoretical models. American journal of physiology Heart and circulatory physiology. 2005;288:H1010–5. [DOI] [PubMed] [Google Scholar]
- 103.Bettoni G, Palla R, Valsecchi C, Consonni D, Lotta LA, Trisolini SM, Mancini I, Musallam KM, Rosendaal FR and Peyvandi F. ADAMTS-13 activity and autoantibodies classes and subclasses as prognostic predictors in acquired thrombotic thrombocytopenic purpura. Journal of thrombosis and haemostasis : JTH. 2012;10:1556–65. [DOI] [PubMed] [Google Scholar]
- 104.Zheng Y, Chen J and Lopez JA. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015;6:7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oehmcke S, Morgelin M and Herwald H. Activation of the human contact system on neutrophil extracellular traps. Journal of innate immunity. 2009;1:225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Witsch T, Martinod K, Sorvillo N, Portier I, De Meyer SF and Wagner DD. Recombinant Human ADAMTS13 Treatment Improves Myocardial Remodeling and Functionality After Pressure Overload Injury in Mice. Journal of the American Heart Association. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, Taubenschmid J, Hammerle M, Esk C, Bagley JA, Lindenhofer D, Chen G, Boehm M, Agu CA, Yang F, Fu B, Zuber J, Knoblich JA, Kerjaschki D and Penninger JM. Human blood vessel organoids as a model of diabetic vasculopathy. Nature. 2019;565:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wimmer RA, Leopoldi A, Aichinger M, Kerjaschki D and Penninger JM. Generation of blood vessel organoids from human pluripotent stem cells. Nat Protoc. 2019;14:3082–3100. [DOI] [PubMed] [Google Scholar]
- 109.Denardo SJ, Denardo BC, Carpinone PL, Dean WT, New DM, Estrada LE, Green CL, Yock PG and Karunasiri G. Validated model of platelet slip at stenosis and device surfaces. Platelets. 2019:1–10. [DOI] [PubMed] [Google Scholar]
- 110.Mishra A and Ashraf MZ. Using Artificial Intelligence to Manage Thrombosis Research, Diagnosis, and Clinical Management. Seminars in thrombosis and hemostasis. 2019. [DOI] [PubMed] [Google Scholar]
- 111.Belyaev AV, Dunster JL, Gibbins JM, Panteleev MA and Volpert V. Modeling thrombosis in silico: Frontiers, challenges, unresolved problems and milestones. Physics of life reviews. 2018;26-27:57–95. [DOI] [PubMed] [Google Scholar]
- 112.Elgart V, Lin JR and Loscalzo J. Determinants of drug-target interactions at the single cell level. PLoS computational biology. 2018;14:e1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nandi S, Sproul EP, Nellenbach K, Erb M, Gaffney L, Freytes DO and Brown AC. Platelet-like particles dynamically stiffen fibrin matrices and improve wound healing outcomes. Biomaterials science. 2019;7:669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jung H, Kang YY and Mok H. Platelet-derived nanovesicles for hemostasis without release of pro-inflammatory cytokines. Biomaterials science. 2019;7:856–859. [DOI] [PubMed] [Google Scholar]
- 115.Andersen P, Tampakakis E, Jimenez DV, Kannan S, Miyamoto M, Shin HK, Saberi A, Murphy S, Sulistio E, Chelko SP and Kwon C. Precardiac organoids form two heart fields via Bmp/Wnt signaling. Nat Commun. 2018;9:3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wong SL and Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2018:fj201800691R. [DOI] [PMC free article] [PubMed] [Google Scholar]