

To the Editor:
Lymphangioleiomyomatosis (LAM) is a rare, slowly progressive pulmonary disease causing cystic lung destruction and respiratory failure. It affects predominantly premenopausal women, and rarely men. It can occur as a sporadic condition (sporadic LAM) or in association with tuberous sclerosis complex (TSC) [1]. LAM is caused by biallelic inactivation of the tumour suppressor gene TSC2 in LAM cells, which leads to hyperactivation of mammalian target of rapamycin complex (mTORC)1, resulting in anabolism and LAM cell proliferation [2]. Sirolimus and everolimus, mTORC1 allosteric inhibitors, have been shown to retard progression of LAM [3].
Histopathologically, LAM is characterised by the presence of nodules containing spindle-shaped LAM cells scattered throughout the lung that express smooth muscle and melanocytic markers, including smooth muscle α-actin and HMB45, respectively. Similar cells proliferate on the luminal surface of the thoracic duct and other lymphatic channels, from which they break off as clusters, and are transported through the lymphatic and then venous circulation, and then the pulmonary artery, to the lung [4].
~30% of women with sporadic LAM have renal angiomyolipoma, which are benign tumours of the kidney composed of vascular, smooth muscle and fat cells. Genetic analysis of microdissected LAM lesions from the lung and kidney angiomyolipoma of the same patient have shown identical TSC2 mutations, indicating that they are clonal neoplasms with a common cell of origin [5].
Although high-resolution chest computed tomography shows the hallmark bilateral thin-walled cysts in patients with LAM, diagnostic uncertainty for such patients is not uncommon. Increased serum vascular endothelial growth factor (VEGF)-D levels (>800 pg·mL−1) are common in LAM and can be used to facilitate diagnosis. However, lung biopsy is often required for definitive diagnosis [6]. Therefore, new biomarkers for LAM are of great interest.
Plasma cell-free DNA (cfDNA) is double-stranded, short (median size 150–200 base pairs in length) and thought to be derived from apoptotic or necrotic cells [7]. In patients with cancer, plasma cfDNA often contains DNA derived from cancer cells and cfDNA analysis is now commonly performed in multiple cancer types to detect mutations that enable targeted therapy, such as epidermal growth factor receptor mutations for lung adenocarcinoma [8, 9]. Given the model of LAM pathogenesis with universal involvement of the lymphatics and release of LAM cell clumps into the circulation, we hypothesised that LAM cells might die in the circulation to some degree and contribute to the pool of plasma cfDNA. Thus, analysis of cfDNA in LAM patients might identify TSC2 mutations, and such findings might serve as both a diagnostic and prognostic marker in LAM.
We collected peripheral blood plasma from 61 sporadic LAM patients, who did not meet diagnostic criteria for TSC, using EDTA tubes (for rapid processing) or in Streck tubes (for shipment overnight). Plasma was isolated and cfDNA extracted by standard methods. Buffy coats were used for genomic DNA isolation. On average, we obtained 10 ng cfDNA per millilitre of plasma with median of 7 ng·mL−1 and range of 2–74 ng·mL−1. Targeted capture for 50 mammalian target of rapamycin pathway genes, including the entire nonrepetitive genomic extent of TSC2, was performed [10]. The mean coverage was 1060× (median, 1107×, range 318–1788×). We used a custom pipeline to identify low-frequency variants of TSC1 and TSC2 [10–12]. Candidate pathogenic single-nucleotide variants were identified based on presence in at least three reads, with at least one read in each orientation, and allele frequency (AF) ⩾0.5% [10].
Candidate insertions and deletions were identified based on presence in at least two reads and AF ⩾0.2%. Candidate variants were reviewed extensively, including using Integrative Genomics Viewer (Broad Institute, San Diego, CA, USA) and functional assessments in silico. All candidate variants were independently validated using amplicon massively parallel sequencing (MPS). For this purpose, primers flanking mutation sites were designed and PCR amplification was performed on all available samples, including additional cfDNA samples and blood genomic DNA. Two normal control DNA samples for each amplicon were included.
The 61 female sporadic LAM patients who were screened had median age at diagnosis of 41 years (range 23–73 years) (figure 1a) with median duration of disease of 4 years (range 1–28 years). 26 (43%) patients had angiomyolipoma and these tumours were bilateral in four (7%). 27 (44%) patients had experienced pneumothorax; three (5%), chylothorax; one (2%), haemothorax; and two (3%), pleural effusion. 12 (20%) had retroperitoneal lymphangioleiomyoma. Pulmonary function tests (PFT) interpreted according to standard criteria [13] indicated that 14 (23%) patients had severe, 16 (26%) moderate and eight (13%) mild loss of lung function; 22 (36%) had normal lung function (for one (2%) patient, PFT information was not available). Seven (11%), all with severe PFT, were being treated with rapalogues at the time of analysis. Serum VEGF-D was assessed in 55 (90%) out of 61 patients, with a median of 755 pg·mL−1 (range 247–5616 pg·mL−1) (figure 1b). Testing was performed either as a College of American Pathologists/Clinical Laboratory Improvement Amendments-compliant test (Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA) or in a research setting using the Quantikine Human VEGF-D Immunoassay (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions, while blinded to other clinical information and the genetic analyses.
FIGURE 1.
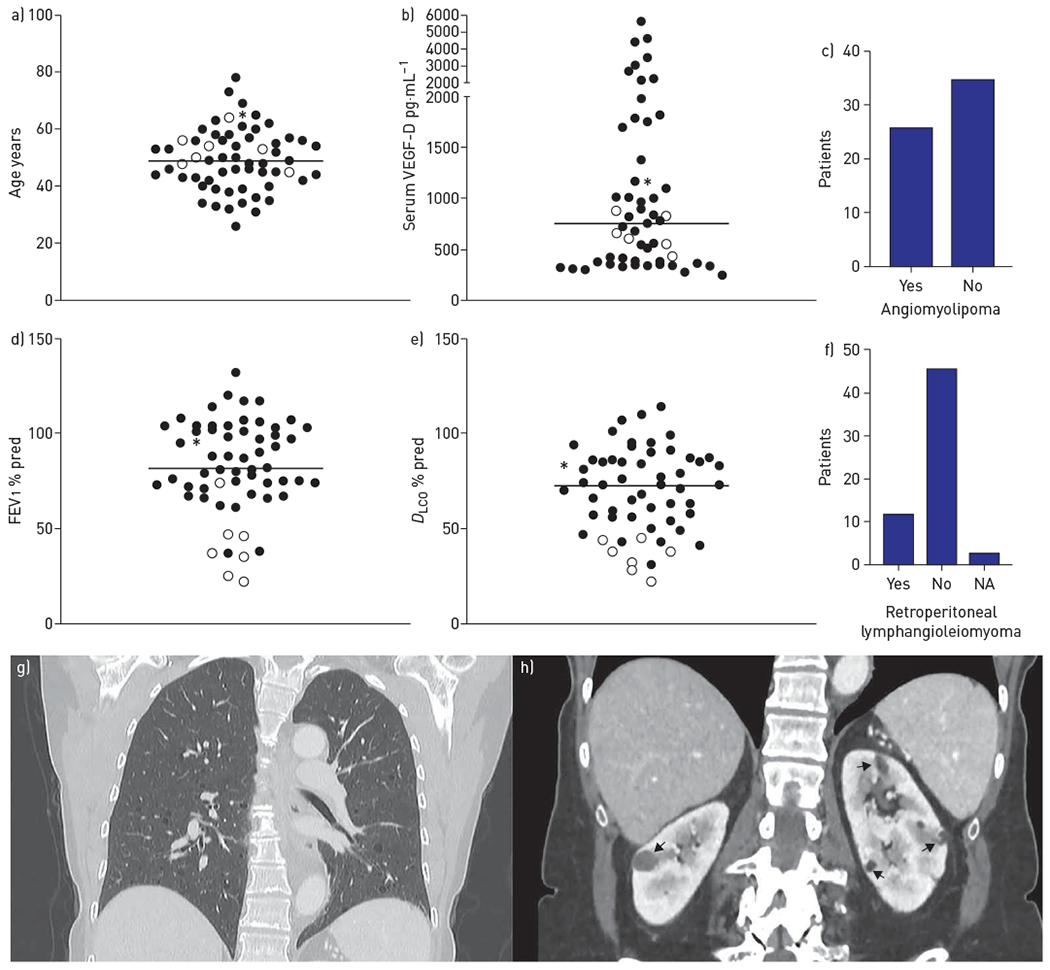
Clinical characteristics of lymphangioleiomyomatosis (LAM) cohort and patient with generalised TSC2 mosaicism. a) Current age distribution for 61 LAM patients (median 49 years). b) Serum vascular endothelial growth factor (VEGF)-D concentrations for 55 out of 61 LAM patients (median 755 ng·μL−1). c) Number of patients with angiomyolipoma. d) Forced expiratory volume in 1 s (FEV1) for 60 of 61 LAM patients (median 82% pred). e) Diffusing capacity of the lung for carbon monoxide (DLCO) for 45 of 61 LAM patients (median 76% pred). f) Number of patients with retroperitoneal lymphangioleiomyoma. a, b, d and e) Median is indicated by the horizontal line. Empty circles correspond to individuals treated with rapamycin. g) Chest and h) abdominal computed tomography images (coronal view) of mosaic patient demonstrating lung cysts (g) and bilateral kidney angiomyolipoma (arrows in h). NA: not available. *: patient with generalised TSC2 mosaicism.
A single pathogenic variant of TSC2 (c.2251C>T, p.Arg751*) at AF 1.83% was identified in one LAM cfDNA sample. There were no genetic findings in any of the other cfDNA samples. Validation by amplicon MPS of multiple DNA samples from the same patient, including those derived from blood, normal skin biopsy, saliva, urine and a second plasma cfDNA, showed that the p.Arg751* variant was present in all samples with a median allele frequency of 2.13% ranging from 1.51% (saliva) to 3.12% (blood), indicating that the patient had generalised, low-level mosaicism for this TSC2 mutation. Following this finding, this patient was seen again clinically, and had a thorough detailed review of all TSC manifestations including skin examination by an expert TSC dermatologist (T.N. Darling) and review of all past scans, including brain magnetic resonance imaging. The patient had a bilateral angiomyolipomas (the largest 1.8×1.3 cm) (figure 1h), retroperitoneal nodules (left iliac 1.2 cm) and 10 sclerotic bone lesions. She had an elevated serum VEGF-D level (1143 pg·mL−1) and normal PFT results. There was no other evidence of TSC in this individual.
These findings indicate that plasma cfDNA analysis in LAM does not detect TSC2 mutations at significant frequency, at least using the methods utilised here, in which a minimum mutant AF ⩾0.5% can be detected, an approach that has been robust in multiple past studies of TSC mosaicism [11, 12, 14]. Future studies with a more sensitive assay might increase TSC2 variant identification in LAM cfDNA.
Nonetheless, we have identified one LAM subject who did not meet TSC diagnostic criteria, who had generalised mosaicism for a pathogenic TSC2 mutation that has previously been identified in at least 36 TSC individuals (www.lovd.nl). A variable phenotype, with some individuals not meeting TSC diagnostic criteria, has been reported for several rare TSC2 missense mutations. However, to our knowledge, this is the first patient identified with mosaicism for a strongly inactivating TSC gene mutation who did not meet diagnostic criteria for TSC. It is notable that this patient had bilateral angiomyolipoma, one of only four in this cohort, and multiple sclerotic bone lesions, suggesting that these clinical findings may be predictive of mosaicism for a TSC2 mutation in sporadic LAM. This possibility requires further investigation.
Acknowledgements:
We would like to thank Karthik Karnik and Edward Kwiatkowski (Brigham and Women’s Hospital, Boston, MA, USA) for their help with bioinformatic analysis.
Support statement: This was work was supported by the The Engles Family LAM/TS Research Fund, National Institutes of Health (NIH)/NHLBI U01 HL131022, the LAM Foundation (L.R. Young), and the Intramural Research Program, NIH/NHLBI. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: B. Ogórek has nothing to disclose. L Hamieh has nothing to disclose. K. Lasseter has nothing to disclose. S. Bagwe has nothing to disclose. T. Machado has nothing to disclose. C. Herranz-Ors has nothing to disclose. A.R. Thorner has nothing to disclose. A. Nag has nothing to disclose. P. Gulleman has nothing to disclose. K. Giannikou has nothing to disclose. L.R. Young reports grants from The LAM Foundation during the conduct of the study. She has served a paediatric advisory board member for Boehringer Ingelheim and has received payment for authorship from UpToDate, outside the submitted work. In addition, she has a patent “VEGF-D in diagnosis of LAM” licensed to Cincinnati Children’s (no personal royalties). M.À. Pujana reports grants from Roche Pharma outside the submitted work. T.N. Darling has nothing to disclose. S. El-Chemaly has nothing to disclose. J. Moss has nothing to disclose. E.P. Henske has nothing to disclose. D.J. Kwiatkowski has nothing to disclose.
References
- 1.Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet 2018; 178: 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwiatkowski DJ, Manning BD. Molecular basis of giant cells in tuberous sclerosis complex. N Engl J Med 2014; 371: 778–780. [DOI] [PubMed] [Google Scholar]
- 3.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011; 364: 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumasaka T, Seyama K, Mitani K, et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol 2005; 29: 1356–1366. [DOI] [PubMed] [Google Scholar]
- 5.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 2000; 97: 6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta N, Finlay GA, Kotloff RM, et al. Lymphangioleiomyomatosis diagnosis and management: high-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An official American Thoracic Society/Japanese Respiratory Society clinical practice guideline. Am J Respir Crit Care Med 2017; 196: 1337–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heitzer E, Haque IS, Roberts CES, et al. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 2019; 20: 71–88. [DOI] [PubMed] [Google Scholar]
- 8.Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017; 8: 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwapisz D The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med 2017; 5: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bongaarts A, Giannikou K, Reinten RJ, et al. Subependymal giant cell astrocytomas in tuberous sclerosis complex have consistent TSC1/TSC2 biallelic inactivation, and no BRAF mutations. Oncotarget 2017; 8: 95516–95529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyburczy ME, Dies KA, Glass J, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet 2015; 11: e1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyburczy ME, Wang JA, Li S, et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet 2014; 23: 2023–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005; 26: 948–968. [DOI] [PubMed] [Google Scholar]
- 14.Han MK, Tyburczy ME, Darling TN, et al. Apparent sporadic lymphangioleiomyomatosis in a man as a result of extreme mosaicism for a TSC2 mutation. Ann Am Thorac Soc 2017; 14: 1227–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]