

Abstract
Oropharyngeal candidiasis (OPC, thrush) is an opportunistic infection caused by the commensal fungus Candida albicans. IL-17 and IL-22 are cytokines produced by Type 17 lymphocytes. Both cytokines mediate antifungal immunity yet activate quite distinct downstream signaling pathways. While much is now understood about how IL-17 promotes immunity in OPC, the activities of IL-22 are far less well delineated. We show that, despite having similar requirements for induction from Type 17 cells, IL-22 and IL-17 function non-redundantly during OPC. We find that the IL-22 and IL-17 receptors are required in anatomically distinct locations within the oral mucosa; loss of IL-22RA1 or STAT3 in the oral basal epithelial layer (BEL) causes susceptibility to OPC, whereas IL-17RA is needed in the suprabasal epithelial layer (SEL). Transcriptional profiling of the tongue linked IL-22/STAT3 to oral epithelial cell proliferation and survival, but also, unexpectedly, to driving an IL-17-specific gene signature. We show that IL-22 mediates regenerative signals on the BEL that replenish the IL-17RA-expressing SEL, thereby restoring the ability of the oral epithelium to respond to IL-17 and thus to mediate antifungal events. Consequently, IL-22 signaling in BEL ‘licenses’ IL-17 signaling in the oral mucosa, revealing spatially distinct yet cooperative activities of IL-22 and IL-17 in oral candidiasis.
One sentence summary
IL-22 signaling limits oral candidiasis via proliferation of basal epithelial cells and replenishment of an IL-17-responsive layer.
INTRODUCTION
Fungal infections are a serious threat to public health, but our understanding of immunity to fungi lags behind that of other microbes (1). Even today there are no licensed vaccines to any fungal microbes (2, 3). Oropharyngeal candidiasis (OPC, thrush) is an opportunistic infection of the oral mucosa caused by the commensal fungus Candida albicans. OPC occurs commonly in the settings of HIV/AIDS, head and neck cancer radiation treatment, immunosuppressive therapies, or the suboptimal immune responses in infants and the elderly (4, 5). OPC is also a characteristic infection of patients with gene mutations impairing the IL-17/Th17 pathway, such as STAT3, ACT1, IL17RA or IL17RC, among others (6–9). Consistently, whereas immunocompetent wild type (WT) mice are resistant to OPC, corticosteroid immunosuppression or loss-of-function mutations in the IL-17 receptor (Il17ra, Il17rc) or related genes (Act1, Il23, Il12b or Rorc) result in high susceptibility to OPC (10–14). However, peak oral fungal burdens in susceptible Il17ra−/− mice are still lower than in animals immunosuppressed with corticosteroids (12), indicating that signals in addition to IL-17 are needed for full protection in OPC.
Although IL-17 is the eponymous cytokine of Th17 cells and other ‘Type 17’ lymphocytes, IL-22 is also characteristic of these cells (15). Multiple studies indicate that IL-22, like IL-17, helps control oral candidiasis. For example, OPC occurs in patients with autoimmune polyendocrine syndrome type 1 (APS-1), a congenital autoimmune syndrome caused by mutations in AIRE and characterized by circulating auto-antibodies that neutralize not only IL-17A and IL-17F but also IL-22 (16–19). Mice with IL-22 impairments (via gene deficiency or antibody neutralization) are susceptible to OPC (12, 20, 21), and reduced IL-22 expression is associated with human chronic mucocutaneous candidiasis (CMC) (8). Nonetheless, relatively little is known about the mechanisms of antifungal immunity mediated by IL-22 in this setting or in other oral diseases.
The oral mucosa provides a vital physical barrier to limit pathogen invasion, yet mechanisms of oral mucosal immunity remain surprisingly under-studied, especially compared to other mucosal tissues such as the lung or gut. During OPC, oral epithelial cells (OECs) sense the transition of C. albicans from a benign yeast morphotype to a damaging, invasive hyphal state (22). This early recognition is mediated in part by EGFR family receptors and also involves sensing of oral tissue damage induced by a secreted fungal toxin, candidalysin (23–27). This OEC immunosurveillance response triggers production of IL-17 from lymphocytes (both innate and adaptive) via IL-1-dependent signals (28–30). IL-17 then activates essential antifungal responses in OECs, including myeloid and lymphoid chemoattractants and antimicrobial peptides (AMPs), particularly β defensin-3 (BD3) (5, 12, 31–33).
The oral mucosa is a stratified non-keratinizing tissue composed of distinct epithelial layers (34, 35). A proliferative basal epithelial layer (BEL) undergoes a program of differentiation that gives rise to the post-mitotic suprabasal epithelial layer (SEL). This differentiation process maintains the tissue and restores barrier immunity after infection or injury (35–37). Each layer is characterized by expression of specific pairs of cytokeratin filaments. Like most stratified epithelia, BEL expresses keratins-5 and −14 (K14, K5). However, the SEL expresses K4 and K13, which have a more restricted expression pattern. IL-17R signaling activity is generally restricted to non-hematopoietic cells (38, 39), and we previously demonstrated that IL-17RA in K13+ SEL cells is essential for immunity to OPC (31). Similar to the IL-17R, the IL-22 receptor is expressed mainly in non-hematopoietic cells and is implicated in gastric C. albicans infections (40), yet remarkably little is known about IL-22 signaling and function in the oral mucosa.
Here, we show that IL-22 functions non-redundantly with IL-17 to limit fungal infection. Although expression kinetics are similar between these cytokines, there were unexpected differences in cellular sources, downstream target gene expression and the nature and localization of the essential cytokine-responsive cells within the stratified oral epithelium. Transcriptomic analysis revealed that IL-22 signaling through STAT3 induces proliferation and survival of BEL cells. Moreover, IL-22-dependent signals are required for renewing the IL-17-responsive SEL. Hence, IL-22/STAT3 signaling ‘licenses’ the oral IL-17 signaling response, despite acting in a distinct epithelial layer.
RESULTS
IL-22 is non-redundant with IL-17 in mediating protection against OPC
To compare the roles of IL-22 and IL-17 in OPC, we tracked the time course of Il22 and Il17a mRNA expression in the oral mucosa (tongue) of wild-type (WT) immunocompetent mice following sublingual C. albicans infection (41, 42). Il22 and Il17a transcripts were not detectable at baseline but were induced contemporaneously at ~16–24 h post-infection (p.i.). Expression of both peaked at 48 h and returned to undetectable levels by 96 h (Fig. 1A). As previously observed (12), Il22−/− mice were susceptible to OPC, with fungal loads consistently averaging ~103 CFU/g of tongue tissue at 4–5 days p.i., whereas control WT mice fully cleared the infection by this time point (Fig. 1B). Fungal loads in Il22−/− mice were typically ~1/2 log lower than in mice with IL-17 signaling defects (Il17ra−/− or Act1−/−), also consistent with prior findings (Fig 1B) (12, 14, 43).
Figure 1. IL-22 protects against OPC non-redundantly with IL-17RA.
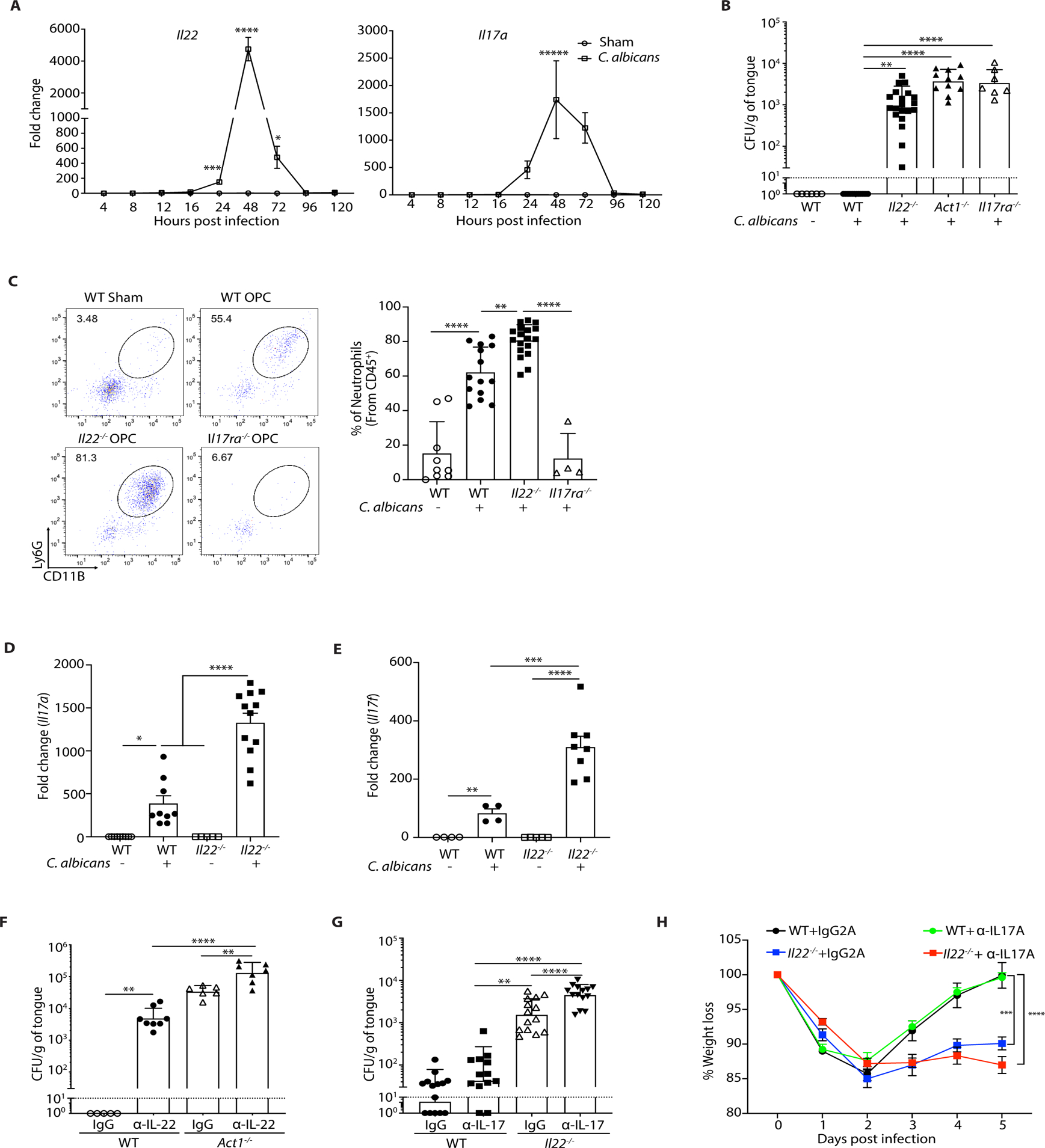
The indicated mice were sublingually inoculated with cotton ball-saturated PBS (Sham) or C. albicans (OPC). Each symbol represents one mouse. A. Total mRNA from tongue homogenates of infected WT mice was subjected to qPCR for Il22 and Il17a and normalized to Gapdh at each time point. Graphs show mean ± SEM. Data are pooled from 4–9 mice per group. B. Fungal burdens were determined by CFU enumeration on YPD/Amp agar at day 5 p.i. Graphs show geometric mean ± SD. Data were pooled from 3 independent experiments. Dashed line indicates limit of detection (~30 CFU/g). C. Tongue homogenates were prepared on day 2 p.i. Left: A representative FACS plot showing percent of CD11b+Ly6G+ neutrophils (gated on live, CD45+ cells). Right: Data from 3 independent experiments. D-E. Il17a and Il17f in total RNA from tongue on day 2 p.i. was assessed by qPCR relative to Gapdh. Graphs show mean ± SEM relative to Sham-infected WT mice. F. Mice were injected i.p. with anti-IL-22 or isotype control IgG (150 μg) on days −1, 0, 1, 2, and 3 relative to infection. CFU was assessed on day 4, pooled from 2 independent experiments. G. Mice were treated with anti-IL-17A or isotype control (IgG2a) (200 μg) injected i.p. on days −1, 1, and 3 relative to infection. CFU was assessed on day 5, pooled from 3 independent experiments. H. Weight loss was assessed daily, shown relative to day 0 in mice from panel F. Data were analyzed by ANOVA or Student’s t-test, with Mann-Whitney U test for fungal load analysis.
We next evaluated the impact of IL-22 on antifungal events in OPC. In line with previous data, Il17ra−/− mice exhibited impaired neutrophil recruitment to the tongue following infection, measured at day 2 (12). Unexpectedly, Il22−/− mice showed increased oral neutrophil frequencies and numbers (Fig. 1C, fig. S1A), despite no differences in fungal load compared to Il17ra−/− mice at this time point (fig. S1B). Il17a and Il17f mRNA were elevated in Il22−/− mice, which may explain this observation (Fig. 1D, E). Because mice are naïve to C. albicans, the IL-17 produced during a first encounter derives from innate immune cells (43–47). We previously showed that antigen-independent CD4+TCRαβ+ cells that express IL-17 [‘natural’ Th17 cells, nTh17 (48)] are required for effective immunity to OPC (29, 44, 49). These nTh17 cells undergo proliferative expansion during the first two days of C. albicans infection in an IL-1R-dependent manner (28, 29, 44). In Il22−/− mice, the frequencies of proliferating (Ki67+) CD4+TCRαβ+ cells were comparable to WT mice following infection (fig S1C), indicating that IL-22 does not regulate nTh17 proliferation.
In view of the many similar functions ascribed to IL-22 and IL-17 at mucosal surfaces (50), we asked whether IL-22 acts redundantly with IL-17 in OPC. Treatment with neutralizing Abs against IL-22 in Act1−/− mice (which are fully impaired for IL-17R signaling (51)) resulted in higher fungal burdens than isotype-treated controls (Fig. 1F). Similarly, blocking IL-17A in Il22−/− mice led to higher fungal burdens compared to isotype-treated controls (Fig. 1G). Weight loss was more pronounced in mice lacking IL-22 than in WT mice, but addition of anti-IL-17A Abs did not cause further weight loss (Fig 1H). Thus, IL-22 and IL-17 act cooperatively but non-redundantly to control OPC.
Determinants of IL-22 production in acute OPC
Conventional adaptive Th17 responses to C. albicans are triggered by sensing of β-glucans in the fungal cell wall via the Dectin-1/CARD9 signaling, which induces Th17-polarizing cytokines (IL-23 and IL-6) (52–58). Here, we assessed the essential triggers of IL-22 in innate responses to OPC. As expected, Il23r−/− mice showed impaired oral Il22 expression following C. albicans infection (Fig 2A), consistent with elevated fungal loads seen in Il23r−/− mice (12, 30). However, mice lacking Dectin-1 (Clec7a−/−) or CARD9 induced Il22 mRNA normally (Fig. 2A). In some settings, IL-22 is induced in Th17 cells by the aryl hydrocarbon receptor (AhR) or serum amyloid A (SAA) (59, 60). However, Ahr−/− mice did not exhibit an obvious deficit in Il22 induction and were resistant to OPC (fig. S2A, B). Similarly, SAA1/2-deficient mice were resistant to OPC and Il17a, Il17f and Il22 were induced normally in the oral mucosa (fig. S2C, D). Since these prototypical IL-22-inducing signals were dispensable for acute IL-22 induction, we next evaluated the role of candidalysin, a recently-described trigger of anti-Candida immunity. This fungal cytolytic peptide is generated by proteolytic cleavage of the fungal Ece1 protein (encoded by the ECE1 gene). Candidalysin is produced only by hyphae and is needed for optimal expression of IL-17 and proliferation of nTh17 cells after oral C. albicans challenge (29, 61). WT mice were infected with C. albicans strains lacking either the full ECE1 gene (ece1Δ/Δ), or just the candidalysin peptide (ClysΔ/Δ), or an ECE1 re-complemented strain (Rev). Infection with ece1Δ/Δ or ClysΔ/Δ caused reduced Il22 mRNA expression compared to Rev (Fig 2B). IL-22 protein levels were similarly affected, assessed using IL22TdTomato reporter mice (62) (fig. S2E). Notably, these strains showed similar fungal burdens at this time point (fig. S2F). Thus, IL-23 and candidalysin but not Dectin-1/CARD9, AhR or SAA1/2 are required for innate IL-22 production during OPC.
Figure 2. Determinants of IL-22 induction in acute OPC.
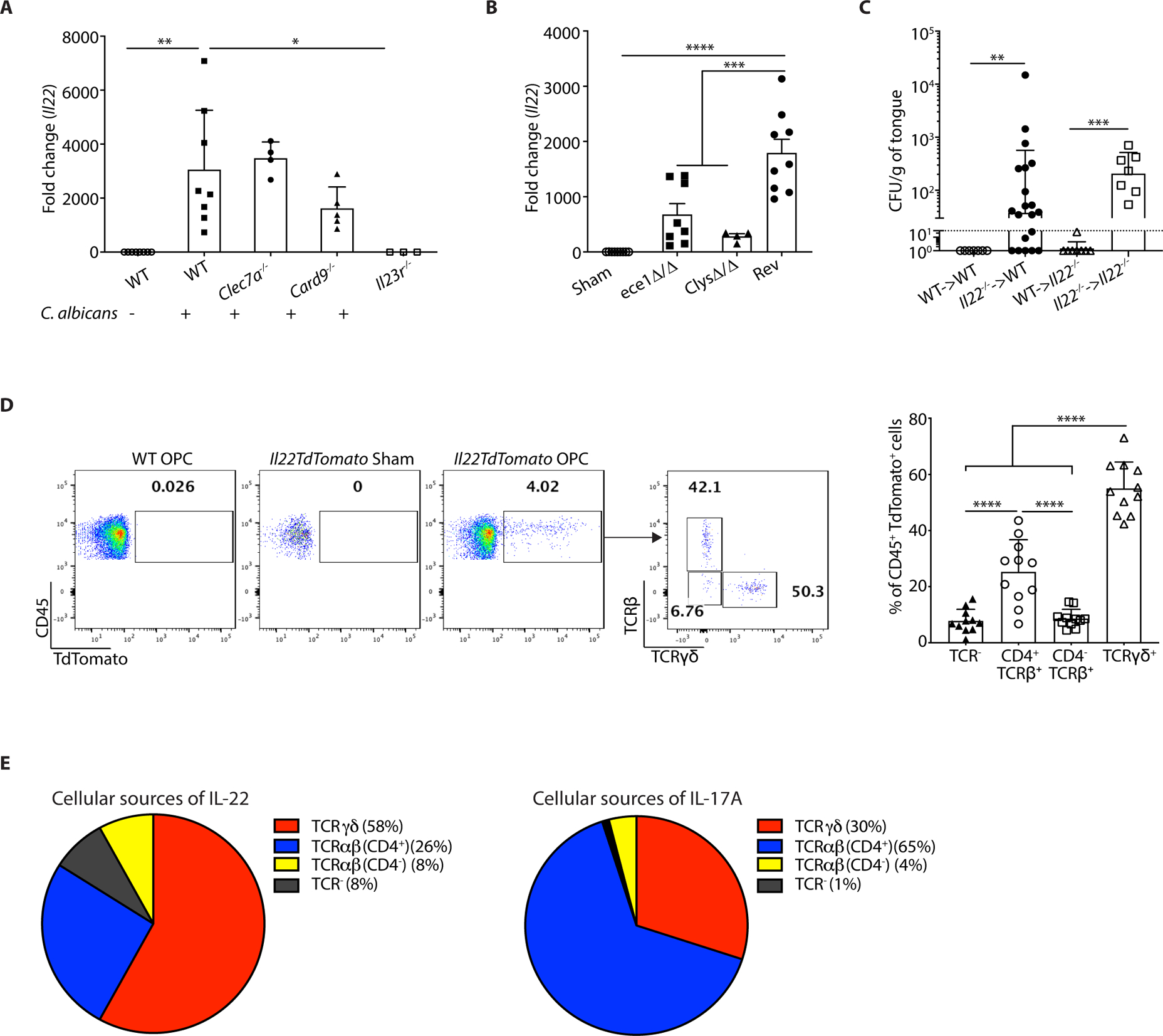
The indicated mice were subjected to OPC. Each symbol represents one mouse A. Tongues were harvested on day 2 p.i. and Il22 mRNA was assessed by qPCR, normalized to Gapdh. Graphs show mean ± SEM relative to sham-infected WT mice. B. WT mice were infected with the indicated C. albicans strains. Il22 mRNA in tongue on day 2 p.i. was assessed by qPCR, normalized to Gapdh. Data were pooled from 2 independent experiments. C. BM from indicated donors was transferred into irradiated recipients. After 6–9 weeks, mice were subjected to OPC and fungal burdens assessed on day 5 p.i. Data were pooled from 2 experiments. D. On day 2 p.i., tongue homogenates from IL22TdTomato mice were stained for the TCRβ and TCRγδ and gated on the live CD45+TdTomato+ population. Left: representative FACS plots. Right: Pooled results from 3 independent experiments. Each symbol represents data from 2 pooled tongues. Data were analyzed by ANOVA or Student’s t-test, with Mann-Whitney U test for fungal load analysis. E. Comparisons of the relative percentages of IL-22+ cells (left) or IL-17+ cells (data from (29)) isolated from tongues of mice 2 days p.i. and analyzed by flow cytometry.
We next delineated the oral cellular sources of IL-22 during OPC using bone marrow (BM) BM chimeras created from WT and Il22−/− mice. As expected, mice receiving Il22−/− BM regardless of host genotype developed OPC, indicating that IL-22 in hematopoietic cells was required for C. albicans clearance (Fig. 2C). We then analyzed tongue tissue from IL22TdTomato reporter mice at day 2 p.i., the time point when Il22 mRNA expression peaks (Fig 1A). There were almost no detectable CD45+TdTomato+ cells prior to infection, indicating that IL-22 is not expressed in the oral mucosa at baseline. After infection, however, a substantial population of reporter-positive cells was present (Fig. 2D). Of these, γδ-T cells constituted the dominant TdTomato+ population (58%), followed by CD4+TCRαβ+ cells (26%). A population of CD4−TCRαβ+ cells (8%) and of TCR-negative cells (8%) also expressed the reporter (Fig 2D). This expression pattern contrasts to some extent with that of IL-17, based on prior studies using an IL-17A fate tracking reporter mouse (63) (Fig 2E) (29, 44).
IL-22 signaling in the basal epithelial layer is required to control OPC
IL-22 signals via the IL-10R2 and IL-22RA1 subunits, and the latter is shared among several cytokines (64). Il22ra1−/− mice showed a similar, albeit not statistically identical, susceptibility to OPC compared to Il22−/− mice (Fig. 3A); the difference could be indicative of a contribution of other IL-22R-dependent cytokines, though this possibility was not pursued. By 14 days p.i., most Il22ra1−/− mice had cleared the infection (fig. S3A), which contrasts with Il17ra−/− mice that were previously shown to maintain oral fungal loads as long as 17 days p.i. (12, 31). To identify cell compartments requiring IL-22RA1 in OPC, we created reciprocal BM chimeras with WT and Il22ra1−/− mice. Regardless of BM source, WT recipients were resistant to OPC, whereas irradiated Il22ra1−/− recipients failed to clear C. albicans (Fig. 3B). Thus, IL-22RA1-mediated signaling in non-hematopoietic cells is required for effective antifungal immunity.
Figure 3. IL-22 signaling in the oral basal epithelial layer is required for protection against OPC.
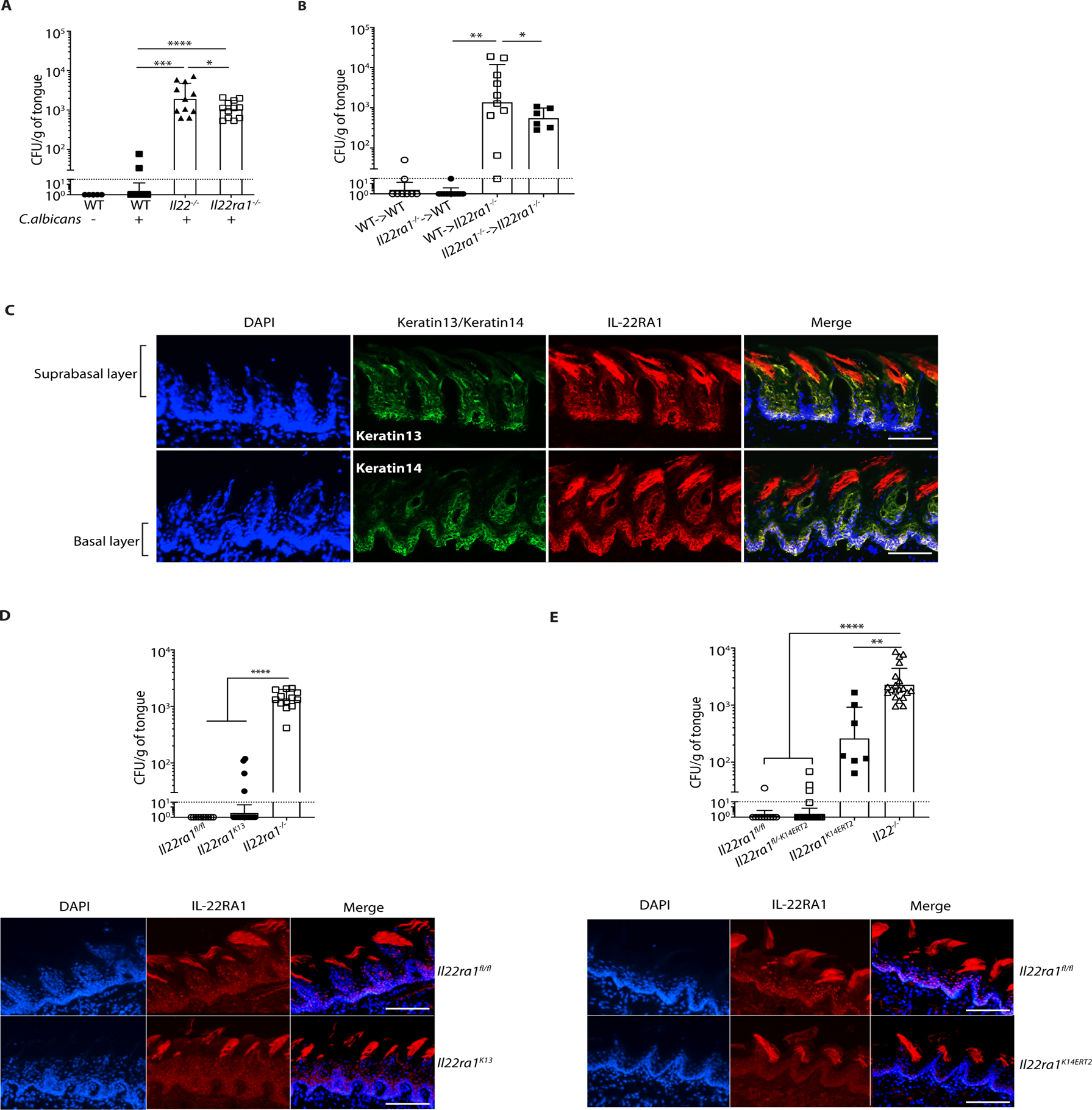
The indicated mice were subjected to OPC. A. Fungal burdens were assessed on day 5 p.i. Bars show geometric mean ± SD. Data were pooled from 2 independent experiments. B. BM from indicated donors was transferred into irradiated recipients. After 6–9 weeks, mice were subjected to OPC and fungal burdens assessed on day 5 p.i. Data were pooled from 2 experiments. C. Frozen sections from WT tongues were co-stained with DAPI and Abs against K13, K14 or IL-22RA1. Suprabasal and basal epithelial layers are indicated. Images are representative of a minimum of 3 sections. Size bar = 200 μm. D. Top: Fungal burdens were assessed on day 5 p.i. Data are pooled from 3 independent experiments. Bottom: IF staining of tongues from the indicated mice were co-stained with DAPI and α-IL-22RA1 Abs. Size bar = 200 μm. E. Top: All mice except Il22−/− were administered tamoxifen for 5 days prior to OPC, and fungal burden assessed on day 5 p.i. Bars show geometric mean ± SD. Bottom: Frozen sections from tongues from the indicated mice were co-stained with DAPI and α-IL-22RA1. Size bar = 200 μm. Data were pooled from 3 independent experiments and analyzed by ANOVA with Mann-Whitney U test.
Oral epithelial tissue is characterized by morphologically distinct expression of cytokeratins. Keratin 13 (K13) is expressed in differentiating SEL cells in the post-mitotic, terminally differentiated layer, which overlies the proliferative K14-expressing BEL (35). Consequently, K13+ epithelial cells make first contact with C. albicans during hyphal invasion and are highly subject to fungal-induced tissue damage. Moreover, this SEL layer is then sloughed and swallowed as part of the antimicrobial clearance response (34). The SEL is replenished by proliferation of the underlying basal K14+ cells which have stem-like properties, but how this is controlled in OPC is unclear. Immunofluorescent (IF) staining indicated that IL-22RA1 was prominent in the K14+ BEL, with some staining in K13+ SEL and papillae (Fig 3C). Isotype controls verified Ab specificity (fig. S3B, C).
To define the relative contributions of IL-22RA1 in oral epithelial cell subtypes, Il22ra1fl/fl mice were crossed to K13Cre or K14CreERT2 mice (31, 65). Conditional deletion of Il22ra1 in SEL and BEL was efficient, as verified by IF (Fig 3D, E). Il22ra1K13 mice were resistant to OPC (mean fungal load ~19), indicating that IL-22 signaling in SEL is dispensable for fungal control (Fig 3D). Remarkably, these results show that IL-22 signals in a spatially distinct manner from IL-17, since deletion of Il17ra in K13+ cells resulted in significant susceptibility to OPC (31). To delete IL-22RA1 in BEL, Il22ra1K14ERT2 mice and controls (Il22ra1fl/fl, Il22ra1fl/-K14ERT2, Il22ra1K14ERT2) were administered tamoxifen for 5 days and infected with C. albicans. In contrast to Il22ra1K13 mice, loss of Il22ra1 in K14+ cells led to markedly increased susceptibility to OPC (fungal load ~508), demonstrating that IL-22R signaling is commensurate with its expression profile in the BEL (Fig. 3E). Hepatic IL-22R is needed for pulmonary bacterial immunity (66), but mice with a liver-specific deletion of IL-22RA1 (Il22ra1Alb) were resistant to OPC (fig. S3D). Hence, these data show that IL-22RA1 signaling in the K14+BEL, but not the K13+SEL, is required for oral fungal control, and accordingly that IL-17 and IL-22 function in different epithelial compartments.
IL-22 activates STAT3 in the BEL to sustain epithelial proliferation and antifungal immunity
To determine the mechanisms by which IL-22 promotes immunity during OPC and whether this differs from IL-17-driven responses, we evaluated RNASeq profiles of total tongue mRNA from C. albicans-infected Il22−/−, II17ra−/− and WT mice. In keeping with the observation that IL-17 and IL-22 act non-redundantly, there were distinct gene sets induced by IL-22 (368 genes) versus IL-17RA (931 genes). There were also many overlapping genes controlled by both cytokines (215 genes) (Fig. 4A). As expected (15), Geneset Enrichment Analysis (GSEA) identified downregulation of IL-6/STAT3 gene sets in Il22−/− mice compared to Il17ra−/− and WT mice, with a normalized enrichment score (NES) of 1.4 (P<0.05) (Fig. 4B). Consistently, Ingenuity Pathway analysis identified STAT3 as a central upstream regulator that integrates the IL-22 and IL-17RA transcriptional networks (Fig. 4C). Within this network, STAT3 was connected to proliferation and apoptosis genes and to transcription factors that regulate inflammation such as NF-κB/IκBξ (Nfkbiz) and MAPK/AP-1 (Fig. 4C). In line with these bioinformatic predictions, IL-22 induced STAT3 phosphorylation and IκBξ expression in a human oral epithelial cell line (fig. S4A, B).
Figure 4. STAT3 in oral epithelial cells is required for protection against OPC.
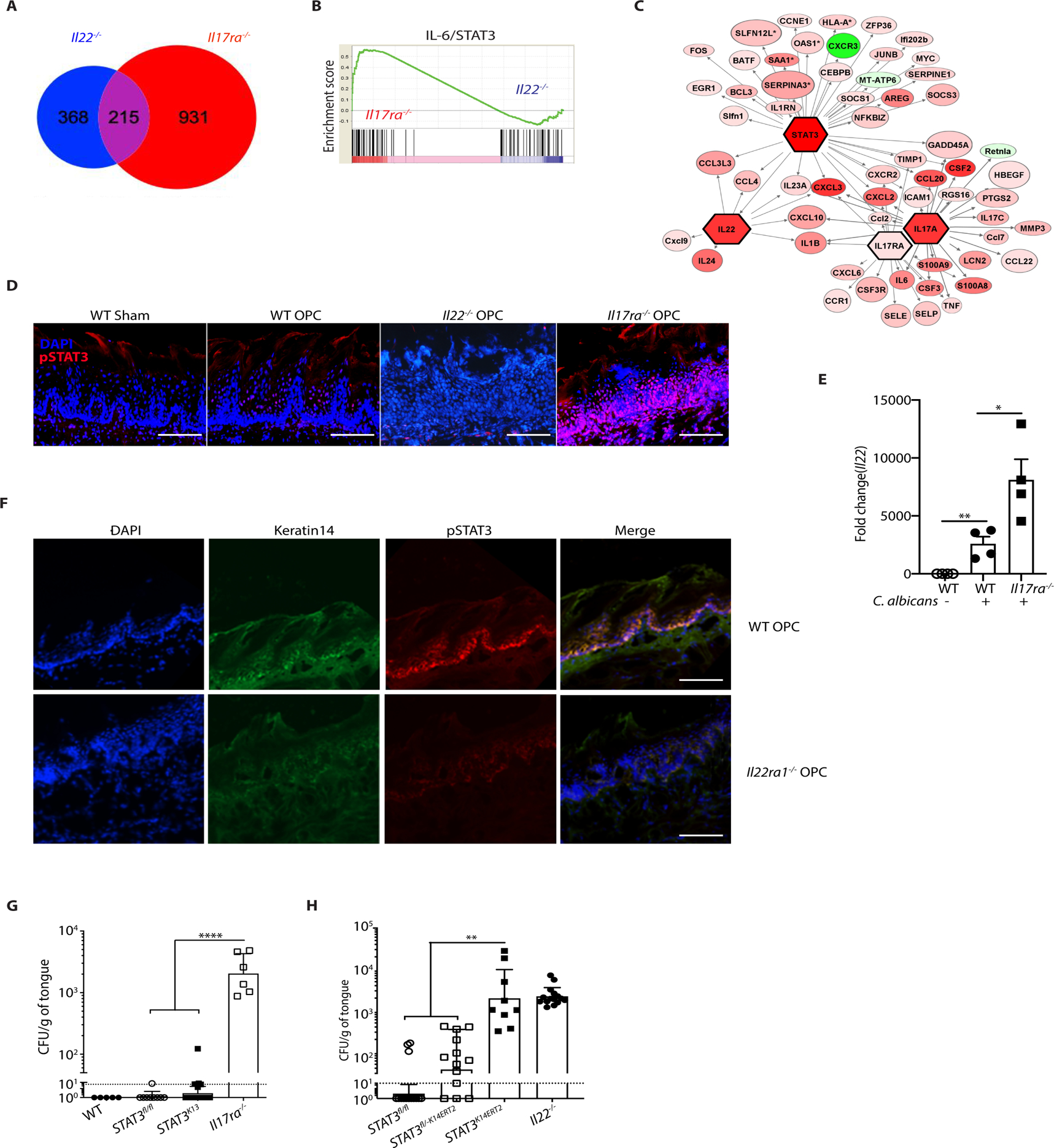
A. RNASeq was performed on whole tongue mRNA from WT, Il22−/− and Il17ra−/− mice subjected to OPC and harvested on day 1 p.i. Venn diagram of differentially regulated or overlapping genes in infected Il22−/− and Il17ra−/− compared to WT mice. 215 genes were regulated both by IL-22 and IL-17RA, whereas 368 genes are regulated only by IL-22, and 931 genes were regulated only by IL-17RA. B. GSEA enrichment of predicted IL-6/STAT3 gene sets in Il17ra−/− and Il22−/− mice from panel A. C. Ingenuity Pathway Analysis of RNASeq data from panel A, indicating that STAT3 is an upstream regulator integrating Il22- and Il17ra- driven transcriptional networks. D. IF staining of tongue frozen sections with DAPI and anti-pSTAT3 (Tyr705) in WT, Il22−/− and Il17ra−/− mice harvested 2 days p.i. Size bar = 200 μm. E. qPCR of Il22 in tongue mRNA from WT or Il17ra−/− mice at 2 days p.i. normalized to Gapdh F. IF staining of DAPI, pSTAT3 (Tyr705) and K14 in WT or Il22ra1−/− mice at 2 days p.i. Size bar = 200 μm. G. The indicated mice were subjected to OPC and fungal burden quantified at day 5 p.i.. Data are pooled from 3 experiments H. All mice except Il22−/− were administered tamoxifen for 5 d, subjected to OPC and fungal burden assessed on day 5 p.i. Bars show geometric mean ± SD. Data was pooled from 3 experiments. Data analyzed by ANOVA with Mann-Whitney U test.
In vivo, STAT3 phosphorylation was evident in the BEL after C. albicans infection, but markedly reduced in Il22−/− and Il22ra1−/− mice (Fig. 4D, F). Unexpectedly, STAT3 phosphorylation was increased in Il17ra−/− BEL during OPC, possibly due to elevated Il22 expression in these mice (Fig 4D, E). These results show that, even though numerous stimuli have potential to activate STAT3, the dominant STAT3 response during OPC is mediated by IL-22RA1-dependent cytokines. This finding also supports the idea that there may be defective IL-22-driven signaling in the pathogenesis of OPC associated with STAT3 mutations (e.g., Job’s syndrome). K14 staining intensity by IF was reduced in Il22ra1−/− mice compared to WT (Fig 4F). This was commensurate with RNASeq data that showed reduced Krt14 mRNA in Il22−/− mice (Fig. 4A, data not shown), a phenomenon also observed in skin (67). Additionally, we confirmed that mice lacking STAT3 in K14+ but not K13+ cells were susceptible to OPC (Fig 4G, H, fig. S4C). Together, these data demonstrate a central role of STAT3 in the oral epithelium that sustains antifungal immunity.
GSEA also revealed increased expression of mitotic spindle checkpoint genes and decreased expression of cell death-associated genes in WT versus Il22−/− mice (Fig. 5A), NES 1.4 (P<0.01) and 1.5 (P<0.003), respectively. Specifically, positive cell cycle regulatory genes (Stat3, Jun, Sphk1) were reduced and negative regulators of cell cycle (Cdkn1c, Ddit3, Ets1) were elevated (Fig 5B). Concordantly, Il22ra1−/− mice showed decreased IF staining of Ki67 in K14+ cells following C. albicans infection, indicating that IL-22 is a major driver of BEL proliferation (Fig. 5C). Consistently, there was a trend to decreased bromodeoxyuridine (BrdU) incorporation in Il22−/− K14+ BEL cells following oral C. albicans infection (fig. S5A). We next evaluated cell cycle progression in EpCAM+ epithelial cells from Il22−/− and WT tongues by BrdU and 7-aminoactinomycin D (7-AAD) staining. As shown, there were comparable proportions of cells in the Gap2/mitotic (G2/M) phase, fewer in the synthesis (S) phase and more in the G0/G1 phase (Fig. 5D, fig. S5A, B). GSEA also suggested enrichment of cell death-associated and DNA damage response genes (Fig 5A, fig. S5C). Indeed, Il22−/− mice had higher frequencies of active caspase 3+ and TUNEL+ epithelial (EpCAM+) cells following infection (Fig. 5E, F, G). Hence, IL-22 promotes BEL proliferation during OPC, while limiting accumulation of apoptotic cells.
Figure 5. IL-22 promotes cell survival and proliferation during OPC.
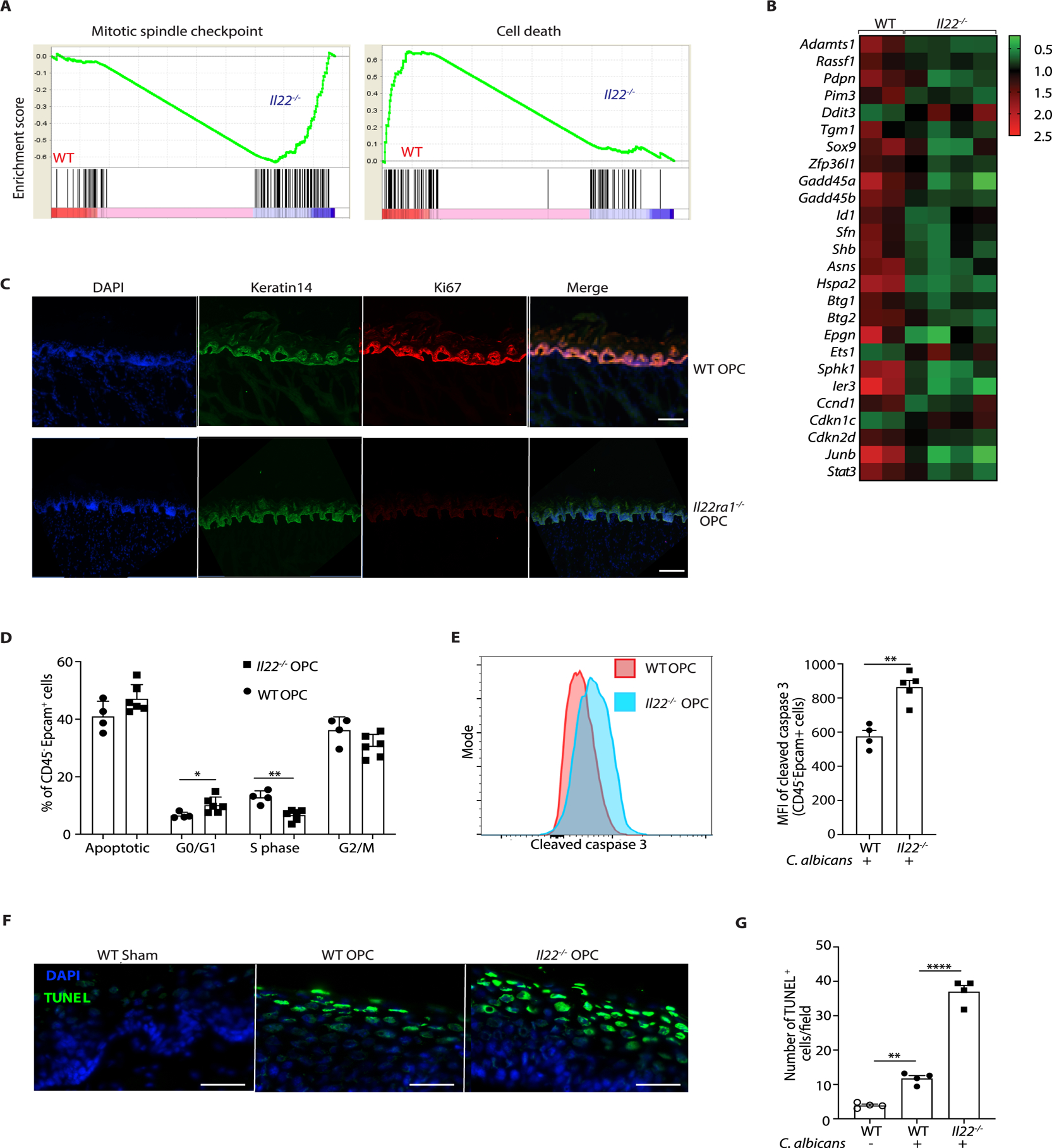
RNASeq was performed on tongue mRNA from WT, Il22−/− or Il17ra−/− mice, isolated 24 h p.i. A. GSEA of mitotic spindle checkpoint pathway and cell death pathway genes. Normalized enrichment score is shown on Y-axis. B. Heatmap of cell cycle pathway genes in global differential gene expression analysis (Partekflow) in WT or Il22−/− mice. C. IF staining of DAPI, Ki67 and K14 in WT and Il22ra1−/− mice at 2 days p.i. Data are representative of images from 2 mice per group. Size bar = 100 μm. D. BrdU was administered 24 h p.i., and tongues harvested on day 2 p.i. Cell cycle/apoptotic status of CD45−EpCAM+ cells was determined by bromodeoxyuridine (BrdU) and 7-aminoactinomycin (7AAD) staining. Data show mean ± SEM. E. Frequency of CD45−EpCAM+ cells staining positive for active (cleaved) caspase-3 in tongue homogenates at day 2 pi measured by flow cytometry. Left: Representative FACS plot. Right: Pooled data from 4 independent samples showing mean fluorescence intensity (MFI) of cleaved Caspase 3 within the CD45−EpCAM+ compartment. Data analyzed by Student’s t-test. F. DAPI and TUNEL staining of tongue sections from the indicated mice at day 2 p.i. Images are representative of 4 mice per group. Size bar = 200 μm. G. Quantification of TUNEL+ cells from panel. F. Data analyzed by ANOVA and post-hoc Tukey’s test.
IL-22 restores IL-17R expression and signaling in SEL to sustain antifungal immunity in OPC
In stratified epithelia such as the oral mucosa, proliferating BEL regenerate the post-mitotic SEL (35, 36). Consistently, genes implicated in tissue repair, keratinization and epithelial differentiation were impaired in Il22−/− mice compared to WT (Fig. 6A). Though IL-17RA signaling in K13+ SEL cells is critical for immunity to OPC (31), the factors that restore the SEL after sloughing have not been well defined. In view of the impaired proliferation of BEL in Il22−/− mice, we hypothesized that IL-22 might indirectly impact IL-17 signaling in OPC by restoring the IL-17RA-expressing SEL. Indeed, transcriptomic data and qPCR revealed that there was decreased Il17ra mRNA expression in Il22−/− tongue (Fig. 6B, C). IF staining revealed loss of IL-17RA in the SEL of WT mice after C. albicans infection, which was even more pronounced in Il22−/− mice (Fig. 6D). Flow cytometry also demonstrated that IL-17RA cell surface expression was reduced in WT and Il22−/− CD45−EpCAM+ oral epithelial cells following C. albicans infection, though the difference between WT and Il22−/− mice was modest (Fig. 6E). Consistent with reduced IL-17RA in oral epithelium, canonical IL-17 target genes associated with immunity to C. albicans were downregulated in Il22−/− mice during OPC, including the essential antimicrobial peptide β-defensin-3 (Defb3) (Fig 6F). Collectively, these data support a model in which IL-22 signaling promotes BEL-intrinsic signals that mediate anti-Candida immunity. Accordingly, IL-22 ‘licenses’ IL-17R signaling by renewing the SEL and thereby restoring the capacity of the tissue to respond to IL-17R, which is vital in controlling OPC (Fig 6G).
Figure 6. IL-22 licenses IL-17 signaling during OPC.
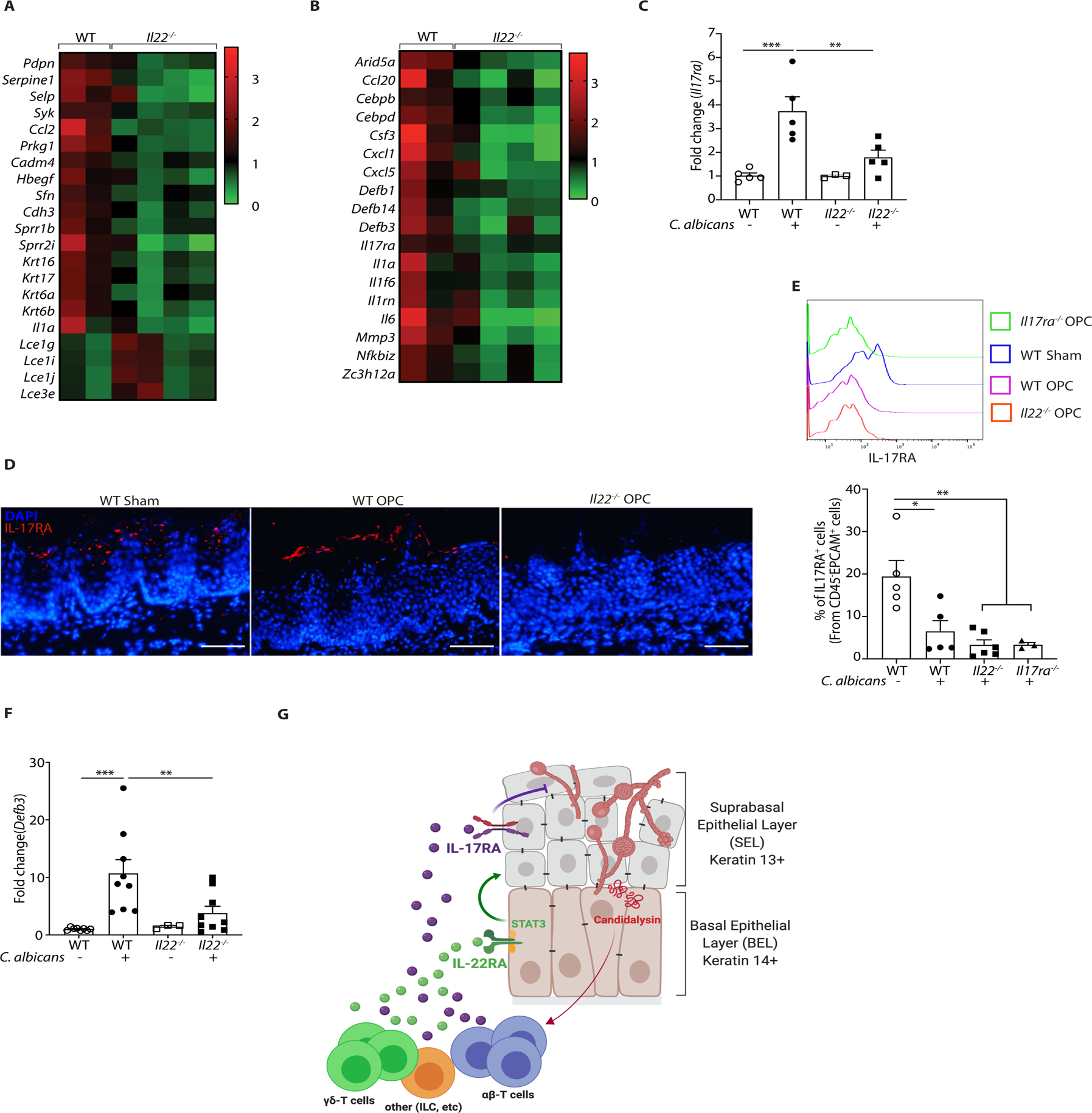
A. Heatmap of genes implicated in tissue repair, wound healing, keratinization and epithelial differentiation. B. Heatmap of IL-17 signature genes in OPC. C. qPCR of Il17ra expression normalized to Gapdh in tongue tissue from the indicated mice subjected to OPC and analyzed on day 2 p.i.. Data show mean ± SEM relative to sham-infected mice. Data analyzed by ANOVA or Student’s t-test. D. IF staining of IL-17RA and DAPI in the indicated mice on day 2 p.i. E. IL-17RA expression in CD45−EpCAM+ oral epithelial cells in WT or Il22−/− mice during OPC. Top: representative FACS histogram. Bottom: Pooled data from 2 independent experiments. Size bar = 200 μm. F. Expression of BD3 (Defb3) mRNA in tongue from WT or Il22−/− mice during OPC, normalized to Gapdh. Data analyzed by ANOVA or student’s t-test. G. Diagram of stratified oral epithelium during a first encounter with C. albicans. Fungal hyphae induces cellular damage and secrete the peptide candidalysin, which facilitates tissue invasion and activates innate IL-17- and IL-22-producing lymphocytes (see Refs (29, 61)). IL-17 was shown previously to act dominantly on K13+ cells of the suprabasal epithelial layer (SEL) (31). In contrast, IL-22/STAT3 promotes proliferation of the K14+ basal epithelial layer (BEL) that serves to restore the IL-17R-expressing SEL, thus maintaining IL-17-induced antifungal signals such as β-defensins and neutrophil responses that are required to mediate clearance of C. albicans. Diagram created on Biorender.com.
DISCUSSION
Although IL-22 and IL-17 are both produced by Type 17 cells, these cytokines are distinct in structure, receptors, and downstream signaling pathways (50). Even so, they are usually viewed interchangeably in the context of mucosal immunity. The protective function of IL-22 in oral candidiasis has been recognized for some time, yet its mechanisms of action are incompletely understood. In non-oral manifestations of C. albicans infection or colonization (systemic, vaginal, dermal or gastric), IL-22 is not necessarily protective, emphasizing that specific cytokine responses even to the same pathogen are influenced by tissue milieu (12, 40, 68–71).
While multiple families of CMC patients have been identified with mutations in IL-17R pathway genes (7, 72, 73), thus far no humans with genetic IL-22-deficiencies have been reported. Nonetheless, it does not necessarily follow that this cytokine is unimportant in humans, only that its contribution to host defense is likely to be more modest than that of IL-17, or possibly that IL-22 deficiency is not compatible with life for some other reason. The former would be analogous to the role of IL-17F. In mice, loss of IL-17F alone does not cause OPC, yet dual blockade of IL-17F and IL-17A significantly increases susceptibility (43, 51). In humans, a family with IL-17F mutations has been described with CMC, suggesting a role in mucosal candidiasis (73). AIRE-deficient humans have circulating neutralizing Abs against IL-17F, IL-22 and IL-17A, thought to help explain the CMC manifestations in these patients (9, 16–18). In most mammals, though not mice, Th17 cells also express IL-26, a member of the same cytokine family as IL-22, and this cytokine may be also a target of biologic therapy (74); however, there is no evidence for autoantibodies against IL-26 in AIRE deficiency nor have IL-26-deficient humans been described (16).
The mechanisms by which IL-17 and IL-22 act in OPC are divergent (50). Whereas loss of IL-17 impairs neutrophil recruitment (12, 31), IL-22 deficiency led to increased neutrophil tissue infiltration, presumably because Il17a is concomitantly elevated. These opposing activities on the neutrophil response may help to restrain excessive inflammation. Interestingly, the capacity of IL-17 to drive oral neutrophil recruitment in OPC is not observed by all who use this OPC model (28, 75), possibly reflecting altered microbiota or other distinctions among animal facilities. Hence, the events controlling oral mucosal immunity are complex and dynamic.
The immunology of the oral cavity is less well understood than other mucosae (34, 37). In part, this is due to technical challenges associated with isolating cells from oral mucosal sites and the paucity of tools available to interrogate cell types within this tissue (76). We show here that IL-22 and IL-17 are produced by and function in distinct oral cell subtypes in the setting of OPC. Unlike humans, mice do not harbor C. albicans as a commensal organism, and hence acute oral infection with C. albicans reflects an innate, not adaptive, immune response. In prior studies, IL-17 was shown to be produced by several innate lymphocyte subsets, including TCRαβ+ ‘natural’ Th17 cells (nTh17), which express CD4, have a diverse TCR repertoire, and are activated in a non-antigen-specific manner (29, 44). IL-17 is also detected to a lesser extent in γδ-T cells and, in some reports, ILC3 cells (29, 43, 44). Upon a recall encounter to C. albicans, mice generate conventional, antigen-specific Th17 cells that additionally contribute to the IL-17-producing pool, where they augment immunity to C. albicans (46, 47, 77). C. albicans-specific Th17 cells are abundant in humans, as C. albicans is encountered very early in life. Moreover, C. albicans-specific Th17 cells recognize and provide cross-reactive protection against other fungal species, which is likely why maintaining C. albicans as a commensal is evolutionarily advantageous (78–81).
We observed that γδ-T cells were the predominant oral source of IL-22 during OPC, followed by nTh17 cells and TCR-negative cell types. These results parallel observations made in skin upon C. albicans infection, where γδ-T cells are the major source of Type 17 cytokines (52). Since the sources of these cytokines were identified using reporter mice which may under-report, defining the relative differences in sources of IL-17 and IL-22 is worth pursuing in more detail (62, 63). Nonetheless, the induction requirements for IL-22 during OPC are remarkably similar to that of IL-17; namely, IL-23 and the fungal peptide candidalysin are crucial, whereas classical fungal PRRs such as Dectin-1, CARD9 or activators of conventional Th17 cells such as AhR or SAA1/2 are dispensable (29, 59, 60, 77). The finding that AhR was not essential to induce IL-22 or clear C. albicans from the mouth, although initially unexpected, is consistent with observations that AhR facilitates EGFR phosphorylation in OPC, a key step in fungal adhesion, endocytosis and invasion in OECs (25, 27, 82, 83).
IL-17 and IL-22 are typically depicted signaling on the same cell types to mediate mucosal immunity, which is the case in non-stratified epithelia such as gut or lung (50, 84). Hence, we did not anticipate that IL-17 and IL-22 would act upon spatially distinct cell types in the oral mucosa. To show this, we made use of a mouse that expresses Cre under control of the murine Krt13 proximal promoter, one of the first tools allowing relatively specific deletion in the oral mucosa (31). The K13Cre mouse deletes conditionally in the SEL of the tongue, buccal mucosa, esophagus and vaginal tract, with no detectable Cre activity in BEL of any tissue examined. Using this system, we found that IL-17RA acts predominantly within K13+ cells in the setting of OPC, with fungal burdens only slightly reduced compared to a full Il17ra−/− animal (31). In contrast, deletion of IL-22RA1 or STAT3 in K13+ SEL did not impact fungal clearance during OPC. GSEA of RNASeq data predicted a role for the IL-22RA1/STAT3 axis in tissue proliferation and repair during C. albicans infection. Indeed, IL-22R signaling, presumably through STAT3, was vital for proliferation and survival of the K14+BEL, consequently replenishing the SEL. Thus, IL-22 indirectly permits essential IL-17-driven antifungal events to occur by restoring the post-mitotic superficial layer where IL-17RA is expressed. This spatial stratification of the IL-22R versus the IL-17R enforces the specificity, diversification, and integration of cues that ensure oral epithelial integrity, restrain undue inflammation and promote antifungal immunity.
The oral mucosa is among the most resilient epithelial surfaces (85). By virtue of their location, superficial K13+ OECs are the first to make contact with C. albicans. In its non-invasive yeast (commensal) form, C. albicans causes no damage to the SEL, which was recently shown to be due the fact that this form of the fungus does not produce the pore-forming virulence factor candidalysin (61). Accordingly, there is insufficient expression of IL-1 or other DAMPs that would activate innate lymphocytes to produce IL-17 or IL-22. This creates an environment where benign commensal C. albicans colonization is favored. However, in conditions that are conducive to hypha formation and invasion into tissue, a different scenario ensues. As part of the response to fungal invasion, the damaged SEL is sloughed and swallowed, which helps to clear C. albicans. The resulting epithelial cell damage also triggers production of IL-1 and IL-36, which promote IL-17 and IL-23 expression, respectively (28–30, 86). IL-17R signaling in the SEL upregulates chemokines that recruit neutrophils as well as β-defensins that exert direct antifungal activity (12, 31, 33, 87). Hyphal invasion thus establishes an inflammatory milieu that is initiated by candidalysin-induced SEL damage, potentiated by IL-17- and candidalysin-induced effectors such as IL-1 and IL-36, and ultimately resolved upon clearance of the pathogen (29, 88).
IL-22 acts in many epithelial surfaces. Events in the oral epithelia are reminiscent of skin, where K14+ stem cells resupply superficial epithelial cells to maintain barrier integrity (36, 89). The skin also possesses ‘memory’ properties that accelerate tissue repair after future insults, though whether this occurs in the mouth is unknown (90). IL-22RA1+ epithelial cells in colonic epithelium maintain genome integrity and limit apoptotic cell accumulation during genotoxic stress (91). Similarly, IL-22 signaling in the BEL during OPC promotes replacement of damaged epithelial cells, prevents accumulation of inflammatory apoptotic cells, preserves genome integrity and, as shown here, helps maintain IL-17-driven antifungal activities.
IL-22 maintains the intestinal epithelial barrier during intestinal colonization of C. albicans (40, 92). Unlike the mouth, IL-17 is not protective in gut but rather promotes a tissue-destructive inflammatory cycle in response to C. albicans colonization (93). In fact, IL-17 signaling in gut is generally more reparative than inflammatory (94–98), which is thought to explain why anti-IL-17 biologic therapies failed in treatment trials of Crohn’s disease (99). Anti-IL-17 biologics are associated with low but statistically significant rates of OPC, though it is rare for patients to stop therapy for this reason (100). Anti-IL-22 antibodies are under evaluation for skin pathologies such as atopic dermatitis (101), but rates of C. albicans infections have not been reported. Our data predict that combinations of anti-IL-17 and anti-IL-22 could result in more severe mucosal candidiasis infections than either therapy alone.
GSEA data show that STAT3 is an integrating hub between the IL-22 and IL-17 signaling networks. The pathways inferred from bioinformatics data (proliferation, cell cycle analysis, and apoptosis) are commensurate with the known roles of IL-22 and STAT3 in epithelial proliferation and repair (64). Surprisingly, IL-22/STAT3 genesets and phosphorylation of STAT3 were enriched in the Il17ra−/− mice, even though expression of other STAT3-activating cytokines (e.g., IL-6 and G-CSF) were impaired in the absence of IL-17 signaling. This could be due to elevated Il22 expression in Il17ra−/− mice, but likely reflects overall perturbation of immune response networks inferred from GSEA that may involve STAT3. Susceptibility to CMC in Job’s syndrome patients with STAT3 mutations is attributed to impaired STAT3-dependent Th17 differentiation (9). However, in acute OPC in mice, STAT3 in CD4+ T cells is not required to control fungal loads (44). Thus, STAT3 likely modulates antifungal immunity beyond the hematopoietic compartment. Supporting this, we found that HIES patients exhibit impairments in salivary antifungal immunity, with reduced levels of salivary AMPs including β-defensins and histatins (102). STAT3 deficiency in lacrimal epithelial cells enhances apoptosis, causing a Sjögren’s syndrome-like phenotype (103). STAT3 also regulates metabolism in various settings to meet energy needs for cell proliferation, and STAT3-regulated functions in mitochondria are becoming increasingly appreciated (104–106). Hence, the IL-22/STAT3 axis coordinates antimicrobial immunity in a variety of environments.
In summary, these results reveal a deeper understanding of the antimicrobial defense functions of oral epithelial cells and a complex interplay between distinct cytokines of the Type 17 axis. It is clear that cells within the oral epithelium are not simply physical barriers, but are topographically structured sentinels that work in concert to dictate the outcome of oral C. albicans colonization, the most common fungal infection of humans (4, 107).
METHODS
Study design
The objective of this study was to delineate functions of IL-22 in immunity to OPC. We used a mouse model of C. albicans infection combined with knockout mouse and fungal strains to interrogate sources and activators of IL-22, genes induced in the oral mucosa, and cell-specific functions of this cytokine in vivo. Sample sizes were determined by power analyses from pilot or previously published data. Mice of both sexes were assigned randomly to experimental cohorts. Unless noted, experiments were done 2–3 independent times. Data from multiple experiments were pooled unless noted. Investigators were not blinded to groups. No data were excluded. Endpoints were selected based on prior kinetic studies (1–2 days for gene expression or IF staining in OPC, 4–5 days for fungal load enumeration).
Mice
IL-22TdTomato, Il22ra1fl/fl, K13Cre, Card9−/−, K14CreERT2, STAT3fl/fl, Saa1/2, and Il23r−/− mice were described (31, 62, 66, 108, 109). Il17ra−/− mice were from Amgen and Il22−/− mice were from Genentech. All mice are available by MTA. Other mice were from JAX or Taconic Farms. Mice were on the C57BL/6 background and housed in SPF conditions. Experiments were performed on age-matched mice (6–10 weeks) of both sexes. BM chimeras were generated by irradiation (900 rads) followed by i.v. injection of 106 femoral/tibial BM. Immune reconstitution after 6–9 weeks was verified by CD45.1/2 FACS staining. Experiments were performed in accordance with protocols approved by the University of Pittsburgh IACUC and NIAID and followed guidelines in the Guide for the Care and Use of Laboratory Animals of the NIH.
Oropharyngeal candidiasis (OPC) and fungal strains
OPC was induced by sublingual inoculation with C. albicans (strain CAF2–1 or SC5314)-saturated cotton balls for 75 min under anesthesia, as described (12, 41). Tongue homogenates were prepared on a GentleMACS homogenizer (Miltenyi Biotec) with C-tubes, and CFU was determined by serial dilution plating on YPD/Amp agar. Anti-IL-22 Abs (Genentech, clone 8E-11) or control IgG1 (Bio X Cell) (150 μg) were injected i.p. on days −1, 0, 1, 2, 3 relative to infection. Anti-IL-17A or IgG2a control Abs (200 μg) were from Janssen Research & Development LLC (110) and administered i.p. on days −1, 1, and 3. Mutant C. albicans lacking the full Ece1 protein(ece1Δ/Δ), the candidalysin peptide (ece1Δ/Δ+ECE1Δ184–279, herein called ClysΔ/Δ) or a complemented strain (ece1Δ/Δ+ECE1, herein called Rev) were described (61).
Flow cytometry
Flow cytometry of tongue homogenates was performed as described (44). Tongues were digested with DNaseI (1 mg/ml, Roche), and collagenase IV (0.7 mg/ml) in HBSS at 37°C. Filtered cell suspensions were stained directly or separated by Percoll gradient centrifugation. Abs were from eBioscience, BD Biosciences, BioLegend or Abcam. Proliferation was assessed using the Foxp3/Transcription factor Buffer kit (eBioscience) with Ki67-APC/PerCP (Cell Signaling #9129). To assess apoptosis, CD45−EpCAM+ cells were stained with caspase-3 apoptosis kit (BD Biosciences). For cell cycle analysis, mice were injected i.p. with 1 mg bromodeoxyuridine (Abcam, BrdU flow kit, BD Biosciences, # 552598) on day 1 p.i. and tongues harvested 24 h later. Data were acquired on an LSR Fortessa and analyzed with FlowJo (BD Biosciences).
qPCR, RNASeq and GSEA
Total tongue RNA was extracted using RNeasy kits (Qiagen) after homogenization in a Gentle MACS Dissociator (Miltenyi Biotec). RNA from TR146 cells was extracted in RLT buffer. Real time PCR (qPCR) was performed as described and normalized to Gapdh (29). Primers were from QuantiTect Primer Assays (Qiagen). For RNASeq, cDNA libraries were prepared from tongue RNA harvested day 1 p.i. (Nextera XT Kit) and RNASeq was performed on the Illumina NextSeq 500 platform by the Health Sciences Sequencing Core at the University of Pittsburgh. Sequencing reads were annotated and aligned to the UCSC mouse reference genome (mm10, GRCm38.75) using HISAT (111). HISAT alignment files were used to generate read counts for each gene, and determination of differential gene expression was performed using the DE-seq package from Bioconductor (112). Unbiased hierarchical clustering of differentially expressed genes with P<0.05 was calculated using CLC Genomics Workbench and Partek software. Relative expression values in heat maps are TPM (transcripts per kilobase million) values per sample that have been divided by the average expression across all samples. Partekflow and GSEA from the Broad Institute were used to calculate enrichment of genes in each set. Additional bioinformatics assistance was from the University of Pittsburgh Center for Research Computing and Health Sciences Library.
Histology, immunofluorescence and immunocytochemistry
Cryosections were stained per the Cell Signaling immunofluorescence protocol (https://www.cellsignal.com/contents/resources-protocols/immunofluorescence-general-protocol/if). Abs: IL-22RA1 (R&D Systems, clone MAB42941), Keratin 13 and Keratin 14 (Abcam, EPR3671 and EPR17350; Invitrogen LL002), Ki67 and anti-p-STAT3 (Tyr705) (Cell Signaling, #9145), IL-17RA (Amgen, clone M751). TUNEL staining was performed with an Apoptosis Detection kit (Millipore). Images were acquired on an EVOS FL microscope (Life Technologies). TUNEL+ cells were enumerated from 10 random fields per slide.
Cell culture, cytokine stimulations and immunoblotting
TR146 cells were cultured in DMEM-F12 with 15% FBS and antibiotics as described (31). For immunoblotting, 3–5 × 105 cells were seeded in serum-free DMEM-F12 overnight prior to cytokine stimulation. Recombinant human IL-22 (Peprotech) was used at 100 ng/ml. Abs to STAT3 were from Cell Signaling (#12640) and Abs to actin were from Millipore (clone C4-EMD).
Statistics
Data were analyzed on Prism (GraphPad). PCR data was analyzed by one-way ANOVA, Student’s t-test and post-hoc tests were used as indicated in figure legends. Fungal load data are presented as geometric mean ± SD and analyzed by ANOVA and Mann-Whitney U test. Each symbol represents one mouse unless indicated. *P<0.05, **<0.01, ***<0.001, ****<0.0001.
Supplementary Material
ACKNOWLEDGMENTS
Bernhard Hube (University of Jena) kindly provided C. albicans mutant strains. We thank U. Siebenlist (NIH) for Act1−/− mice, Amgen for Il17ra−/− mice and anti-IL-17RA Abs, Genentech for Il22−/− mice and anti-IL-22 Abs, Janssen for anti-IL-17A antibodies, X. Lin (MD Anderson) for Card9−/− mice, and F. De Beer (U. Kentucky) for Saa1/2−/− mice. We thank Scott Filler, Heather Conti, Paula Sundstrom, Mandy McGeachy, Harry Mullin, Christina Morse, Nate Weathington, Tim Hand, John Alcorn, Robert Binder and Akash Verma for helpful input and critical reading.
FUNDING
The following NIH grants supported this work: SLG, DE022550, DE023815, AI107825; PSB, DK104680; JRN, DE022550; GTN, HL135476; DHK, AR071720, AR060744; JKK, HL139930; VMB, AI110820. Equipment grant 1S1OD011925 supported flow cytometry. JRN was supported by the Wellcome Trust (214229_Z_18_Z) and the NIH Research at Guys and St. Thomas’s NHS Foundation Trust and the King’s College London Biomedical Research Centre (IS-BRC-1215-20006). This work was partially supported by the Division of Intramural Research of the NIAID.
Abbreviations:
- AhR
aryl hydrocarbon receptor
- AMP
antimicrobial peptide
- BD
β-defensin
- BEL
basal epithelial layer
- BM
bone marrow
- BrdU
bromodeoxyuridine
- CFU
colony forming units
- Clys
candidalysin
- CMC
chronic mucocutaneous candidiasis
- GSEA
gene set enrichment analysis
- HIES
hyper-IgE syndrome (Job’s)
- OEC
oral epithelial cell
- nTh17
natural Th17
- NES
normalized enrichment score
- OPC
oropharyngeal candidiasis
- SAA
serum amyloid A
- SEL
superficial epithelial layer
- TUNEL
terminal deoxynucleotidyl transferase nick-end labeling
Footnotes
COMPETING INTERESTS
SLG previously received a grant from Janssen to evaluate anti-IL-17A antibody function in OPC (110). Candidalysin has been patented by King’s College London, UK and Hans-Knöll Institut, Jena, DE (US Patent No.: 9,969,796; EU Patent No.: 2984103). The other authors declare no competing interests.
DATA AND MATERIALS AVAILABILITY
Act1−/− mice are available by MTA from NIH. Il17ra−/− mice and anti-IL-17RA antibodies (clone M751 are available under an MTA with Amgen, Corp. Anti-IL-17A antibodies are available by MTA with Janssen Research & Development LLC.). IL-22TdTomato mice are available by MTA from the National Cancer Institute. Anti-IL-22 Abs and Il22−/− mice are available by MTA from Genentech. Card9−/− mice are available by MTA from the MD Anderson Cancer Center. Saa1/2−/− mice are available by MTA from the University of Kentucky. All raw sequencing reads from the RNASeq experiments were submitted to the NCBI sequence read archive (SRA) under BioProject accession number PRJNA599123.All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Brown GD et al. , Hidden killers: human fungal infections. Sci Transl Med 4, 165rv113 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Cassone A, Development of vaccines for Candida albicans: Fighting a skilled transformer. Nat Rev Microbiol 11, 884–891 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Naglik JR, Gaffen SL, Hube B, Candidalysin: discovery and function in Candida albicans infections. Curr Opin Microbiol 52, 100–109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fidel PL Jr., Candida-Host Interactions in HIV Disease: Implications for Oropharyngeal Candidiasis. Adv Dent Res 23, 45–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conti HR, Gaffen SL, IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J Immunol 195, 780–788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glocker E, Grimbacher B, Chronic mucocutaneous candidiasis and congenital susceptibility to Candida. Curr Opin Allergy Clin Immunol 10, 542–550 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Li J, Vinh DC, Casanova JL, Puel A, Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr Opin Microbiol 40, 46–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyerich K et al. , Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 128, 2640–2645 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Milner J, Holland S, The cup runneth over: lessons from the ever-expanding pool of primary immunodeficiency diseases. Nat Rev Immunol 13, 635–648 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Farah C, Hu Y, Riminton S, Ashman R, Distinct roles for interleukin-12p40 and tumour necrosis factor in resistance to oral candidiasis defined by gene targeting. Oral Microbiol Immunol 21, 252–255 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Kamai Y et al. , New model of oropharyngeal candidiasis in mice. Anti-microb. Agents Chemo 45, 3195–3197 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti H et al. , Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206, 299–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho A et al. , IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J. Immunol 185, 1063–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira MC et al. , Interleukin-17-induced protein lipocalin 2 is dispensable for immunity to oral candidiasis. Infect Immun 82, 1030–1035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutz S, Eidenschenk C, Ouyang W, IL-22, not simply a Th17 cytokine. Immunol Rev 252, 116–132 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Puel A et al. , Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207, 291–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisand K et al. , Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 207, 299–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browne SK, Holland SM, Anti-cytokine autoantibodies explain some chronic mucocutaneous candidiasis. Immunol Cell Biol 88, 614–615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husebye ES, Anderson MS, Autoimmune polyendocrine syndromes: clues to type 1 diabetes pathogenesis. Immunity 32, 479–487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bichele R et al. , IL-22 neutralizing autoantibodies impair fungal clearance in murine oropharyngeal candidiasis model. Eur J Immunol 48, 464–470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goupil M et al. , Defective IL-17- and IL-22-dependent mucosal host response to Candida albicans determines susceptibility to oral candidiasis in mice expressing the HIV-1 transgene. BMC Immunol 15, 49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naglik JR, Konig A, Hube B, Gaffen SL, Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr Opin Microbiol 40, 104–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moyes DL et al. , A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8, 225–235 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moyes DL et al. , Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J Infect Dis 209, 1816–1826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu W et al. , EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci U S A 109, 14194–14199 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swidergall M, Solis NV, Lionakis MS, Filler SG, EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat Microbiol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho J et al. , Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun 10, 2297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altmeier S et al. , IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa. PLoS Pathog 12, e1005882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma A et al. , Oral epithelial cells orchestrate innate Type 17 responses to Candida albicans through the virulence factor Candidalysin. Sci Immunol 2, eeam8834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma AH et al. , IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J Immunol 201, 627–634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conti H et al. , IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606–617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond RA, Gaffen SL, Hise AG, Brown GD, Innate Defense against Fungal Pathogens. Cold Spring Harb Perspect Med 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomalka J et al. , beta-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J Immunol 194, 1788–1795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaffen SL, Moutsopoulos N, Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci Immunol in press, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Presland R, Dale B, Epithelial structural proteins of the skin and oral cavity: Function in health and disease. Crit Rev Oral Biol Med 11, 383–408 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Alcolea MP, Jones PH, Lineage analysis of epidermal stem cells. Cold Spring Harb Perspect Med 4, a015206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moutsopoulos NM, Konkel JE, Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwakura Y, Ishigame H, Saijo S, Nakae S, Functional specialization of interleukin-17 family members. Immunity 34, 149–162 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Monin L, Gaffen SL, Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb Perspect Biol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zelante T, Iannitti R, De Luca A, Romani L, IL-22 in antifungal immunity. Eur J Immunol 41, 270–275 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Solis NV, Filler SG, Mouse model of oropharyngeal candidiasis. Nat Protoc 7, 637–642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conti HR, Huppler AR, Whibley N, Gaffen SL, Animal models for candidiasis. Curr Protoc Immunol 105, 19 16 11–19 16 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gladiator A, Wangler N, Trautwein-Weidner K, Leibundgut-Landmann S, Cutting Edge: IL-17-Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J Immunol 190, 521–525 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Conti H et al. , Oral-resident ‘natural’ Th17 cells and γδ-T cells control opportunistic Candida albicans infections. J Exp Med 211, 2075–2084 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandiyan P et al. , CD4+CD25+Foxp3+ regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 infection model. Immunity 34, 422–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernández-Santos N et al. , Th17 cells confer long term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol 6, 900–910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bär E et al. , A novel Th cell epitope of Candida albicans mediates protection from fungal infection. J Immunol 188, 5636–5643 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Marks BR et al. , Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol 10, 1125–1132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huppler AR, AH V, HR C, Gaffen SL, Neutrophils do not express IL-17A in the context of acute oropharyngeal candidiasis. Pathogens 4, 559–572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C, IL-17 and IL-22: siblings, not twins. Trends Immunol 31, 354–361 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Whibley N et al. , Antibody blockade of IL-17-family cytokines in immunity to acute murine oral mucosal candidiasis. J Leukoc Biol 99, 1153–1164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kashem SW et al. , Candida albicans Morphology and Dendritic Cell Subsets Determine T Helper Cell Differentiation. Immunity 42, 356–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leibundgut-Landmann S et al. , Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8, 630–638 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Trautwein-Weidner K et al. , Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis. PLoS Pathog 11, e1005164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drummond RA, Brown GD, The role of Dectin-1 in the host defence against fungal infections. Curr Opin Microbiol 14, 392–399 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Dennehy KM, Willment JA, Williams DL, Brown GD, Reciprocal regulation of IL-23 and IL-12 following co-activation of Dectin-1 and TLR signaling pathways. Eur J Immunol 39, 1379–1386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dennehy KM et al. , Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 38, 500–506 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu W, Hsu YM, Bi L, Songyang Z, Lin X, CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat Immunol 10, 1208–1214 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Sano T et al. , An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veldhoen M et al. , The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Moyes D et al. , Candidalysin: A fungal peptide toxin critical for mucosal infection. Nature 532, 64–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen W, Hixon JA, McLean MH, Li WQ, Durum SK, IL-22-Expressing Murine Lymphocytes Display Plasticity and Pathogenicity in Reporter Mice. Front Immunol 6, 662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hirota K et al. , Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolk K, Witte E, Witte K, Warszawska K, Sabat R, Biology of IL-22. Semin Immunol 32, 17–31 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Vasioukhin V, Degenstein L, Wise B, Fuchs E, The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A 96, 8551–8556 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trevejo-Nunez G, Elsegeiny W, Conboy P, Chen K, Kolls JK, Critical Role of IL-22/IL22-RA1 Signaling in Pneumococcal Pneumonia. J Immunol, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Belle AB et al. , IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol 188, 462–469 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Borghi M et al. , Pathogenic NLRP3 Inflammasome Activity during Candida Infection Is Negatively Regulated by IL-22 via Activation of NLRC4 and IL-1Ra. Cell Host Microbe 18, 198–209 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Yano J et al. , The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS One 7, e46311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A, IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol 185, 5453–5462 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peters BM et al. , The IL-17R/IL-22R signaling axis is dispensable for vulvovaginal candidiasis regardless of estrogen status. J Infect Dis, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levy R et al. , Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A 113, E8277–E8285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puel A et al. , Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tengvall S, Che KF, Linden A, Interleukin-26: An Emerging Player in Host Defense and Inflammation. J Innate Immun 8, 15–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S, IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol 8, 221–231 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Pandiyan P, Bhaskaran N, Zhang Y, Weinberg A, Isolation of T cells from mouse oral tissues. Biol Proced Online 16, 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bishu S et al. , CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun 82, 1173–1180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bacher P et al. , Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176, 1340–1355 e1315 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Aggor FEY, Way SS, Gaffen SL, Fungus Among Us: The Frenemies Within. Trends Immunol 40, 469–471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shao TY et al. , Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417 e406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Acosta-Rodriguez EV et al. , Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8, 639–646 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Solis NV, Swidergall M, Bruno VM, Gaffen SL, Filler SG, The Aryl Hydrocarbon Receptor Governs Epithelial Cell Invasion during Oropharyngeal Candidiasis. MBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Swidergall M, Filler SG, Oropharyngeal Candidiasis: Fungal Invasion and Epithelial Cell Responses. PLoS Pathog 13, e1006056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aujla SJ et al. , IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14, 275–281 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Presland R, Jurevic R, Making sense of the epithelial barrier: What molecular biology and genetics tell us about the functions of the oral mucosal and epidermal tissues. J Dent Educ 66, 564–574 (2002). [PubMed] [Google Scholar]
- 86.Sparber F et al. , Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog 14, e1007069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yano J, Lilly E, Barousse M, Fidel PL Jr., Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun 78, 5126–5137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verma A, Gaffen SL, Swidergall M, Innate Immunity to Mucosal Candida Infections. J Fungi 3, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blanpain C, Fuchs E, Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 344, 1242281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Naik S et al. , Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gronke K et al. , Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566, 249–253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Luca A et al. , IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3, 361–373 (2010). [DOI] [PubMed] [Google Scholar]
- 93.Zelante T et al. , IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 37, 2695–2706 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Qian Y et al. , The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8, 247–256 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Lee J et al. , Interleukin-23-dependent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maxwell J et al. , Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 43, 739–750 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Song X et al. , Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 43, 488–501 (2015). [DOI] [PubMed] [Google Scholar]
- 98.Whibley N, Gaffen SL, Gut-Busters: IL-17 Ain’t Afraid of No IL-23. Immunity 43, 620–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hueber W et al. , Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saunte DM, Mrowietz U, Puig L, Zachariae C, Candida infections in psoriasis and psoriatic arthritis patients treated with IL-17 inhibitors and their practical management. Br J Dermatol, (2016). [DOI] [PubMed] [Google Scholar]
- 101.Guttman-Yassky E et al. , Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J Am Acad Dermatol 78, 872–881 e876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Conti H et al. , New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol 4, 448–455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Okuma A et al. , Enhanced apoptosis by disruption of the STAT3-IkappaB-zeta signaling pathway in epithelial cells induces Sjogren’s syndrome-like autoimmune disease. Immunity 38, 450–460 (2013). [DOI] [PubMed] [Google Scholar]
- 104.Wang T et al. , JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab 27, 136–150 e135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee M et al. , Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol, 101073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Genini D et al. , Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc Natl Acad Sci U S A 114, E4924–E4933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Richardson JP, Moyes DL, Adaptive immune responses to Candida albicans infection. Virulence, 0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hsu YM et al. , The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 8, 198–205 (2007). [DOI] [PubMed] [Google Scholar]
- 109.Eckhardt ER et al. , Intestinal epithelial serum amyloid A modulates bacterial growth in vitro and pro-inflammatory responses in mouse experimental colitis. BMC Gastroenterol 10, 133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shen F et al. , Combined Blockade of TNF-alpha and IL-17A Alleviates Progression of Collagen-Induced Arthritis without Causing Serious Infections in Mice. J Immunol 202, 2017–2026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim D, Langmead B, Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anders S, Huber W, Differential expression analysis for sequence count data. Genome Biol 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.