

SUMMARY
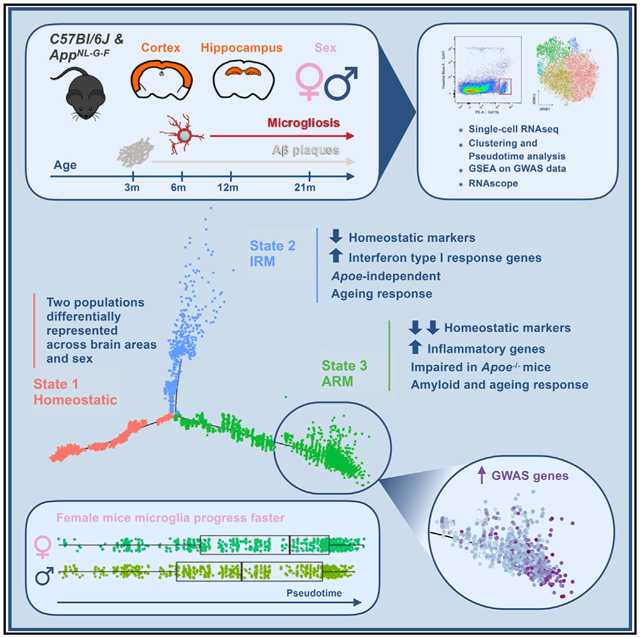
Gene expression profiles of more than 10,000 individual microglial cells isolated from cortex and hippocampus of male and female AppNL-G-F mice over time demonstrate that progressive amyloid-b accumulation accelerates two main activated microglia states that are also present during normal aging. Activated response microglia (ARMs) are composed of specialized subgroups overexpressing MHC type II and putative tissue repair genes (Dkk2, Gpnmb, and Spp1) and are strongly enriched with Alzheimer’s disease (AD) risk genes. Microglia from female mice progress faster in this activation trajectory. Similar activated states are also found in a second AD model and in human brain. Apoe, the major genetic risk factor for AD, regulates the ARMs but not the interferon response microglia (IRMs). Thus, the ARMs response is the converging point for aging, sex, and genetic AD risk factors.
Graphical Abstract
In Brief
Sala Frigerio et al. show how microglia respond to amyloid-b, the Alzheimer’s disease (AD)-causing factor. Their major response, the ARMs response, is enriched for AD risk genes, is abolished by Apoe deletion, develops faster in female mice, and is also part of normal aging. Thus, major AD risk factors converge on microglia.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by typical biochemical lesions (β-amyloid peptide [Ab] plaques and tau tangles) accompanied by extensive cellular changes (neuronal dystrophic alterations, neuronal cell loss, astrogliosis, and microgliosis) (De Strooper and Karran, 2016; Serrano-Pozo et al., 2011). Rare mutations in amyloid precursor protein (APP), presenilin (PSEN) 1 and 2 (Karch et al., 2014), ADAM10 (Suh et al., 2013), and ADAM17 (Hartl et al., 2018) trigger Aβ plaque accumulation and are sufficient to induce the full biochemical and morphological signature of AD. While this clearly indicates a major role for Aβ in AD pathology (Jack et al., 2013; Selkoe and Hardy, 2016), even in these genetic forms, a decades-long asymptomatic phase is present (Bateman et al., 2012). Thus, in addition to Aβ plaques, other pathological processes, either in response to or in parallel to Aβ accumulation, need activation to cause neurodegenerative disease (De Strooper and Karran, 2016).
The search for the genetic risk determinants in sporadic AD has highlighted the central role of non-neuronal genes in pathways that do not appear directly related to Aβ metabolism (Cruchaga et al., 2014; Guerreiro et al., 2013; Lambert et al., 2013; Marioni et al., 2018; Salih et al., 2018; Sims et al., 2017; Villegas-Llerena et al., 2016). Most of the genes associated with the ~40 loci identified by genome-wide association (GWA) analysis or by rare variant sequencing studies are expressed in glial cells (e.g., APOE, TREM2, BIN1, CD33, INPP5D, CTSB, CTSD, and PICALM). Moreover, analysis of available single-cell transcriptome datasets for human brain cells reported an association between AD GWA signals and microglia as well as astrocytes (Calderon et al., 2017). Analysis of regulatory networks of genes differentially expressed in AD patients indicates that immune- and microglia-specific gene modules are key contributors to AD pathology (Zhang et al., 2013). Thus, genetic and molecular evidence suggest that Aβ accumulation is the trigger of a series of pathogenic processes in which microglia play a central role. No consistent hypothesis, however, links the causality implied by the mutations in the amyloid pathway genes to the genetic risk linking sporadic AD to inflammatory pathways. This has caused severe criticism on the amyloid cascade hypothesis for AD, raising questions about its validity in sporadic AD (Behl and Ziegler, 2017; Harrison and Owen, 2016; Herrup, 2015; Karran and Hardy, 2014; Makin, 2018; Selkoe and Hardy, 2016). One possible resolution is that amyloid pathology acts only as a trigger in sporadic AD (Karran et al., 2011); i.e., Aβ accumulation is necessary but insufficient to cause full-blown disease. The cellular response, determined by the genetic makeup of the patients, tilts the table from a rather benign Aβ proteopathy to the severe neurodegeneration with inflammation and Tau pathology that characterizes AD (De Strooper and Karran, 2016). In this regard, further understanding of the microglia response to amyloid pathology and the role of risk factors for AD in this response is key.
Several groups have applied high-throughput single-cell genomics and proteomics methods to characterize the shift in microglial cell states after different kinds of insults (Friedman et al., 2018; Hammond et al., 2019; Keren-Shaul et al., 2017; Mathys et al., 2017; Mrdjen et al., 2018). In neurodegenerative settings, microglia develop disease-associated phenotypes, such as the microglial neurodegenerative phenotype (MGnD) (Krasemann et al., 2017) or the disease-associated microglia (DAMs) (Keren-Shaul et al., 2017), but it is still an open question whether different subsets of such phenotypes exist, whether there are brain-area- or sex-specific differences in the microglial responses to Aβ, and which are the roles of AD risk genes expressed in microglia.
Here, we set out to address in a systematic way the question of how microglia respond over time, in cortex and hippocampus, to progressive Aβ deposition and whether this is affected by the three major risk factors for AD, i.e., age, sex, and genetics. We use an App knockin mouse model (AppNL-G-F), which displays progressive amyloidosis and microgliosis while avoiding overexpression of APP via artificial promotors (Masuda et al., 2016; Saito et al., 2014; Sasaguri et al., 2017) (Figure 1A). We show that the microglial responses to Aβ pathology are complex but, surprisingly, largely reproducible cell states that are also appearing during normal aging, albeit slower and quantitatively more limited. Moreover, we show that microglia in female mice tend to react earlier and in a more pronounced way than microglia in male mice, particularly in older mice. Interestingly, the major response of microglia to amyloid pathology is enriched for AD risk genes, with Apoe expression, in particular, becoming highly upregulated. This is partially confirmed in human tissue. Analysis of microglia from an Apoenull AD mouse model showed that the main Aβ response is severely impaired in the absence of Apoe.
Figure 1. Microglia Mount a Heterogeneous Response to β-amyloid, Marked by Apoe Overexpression.
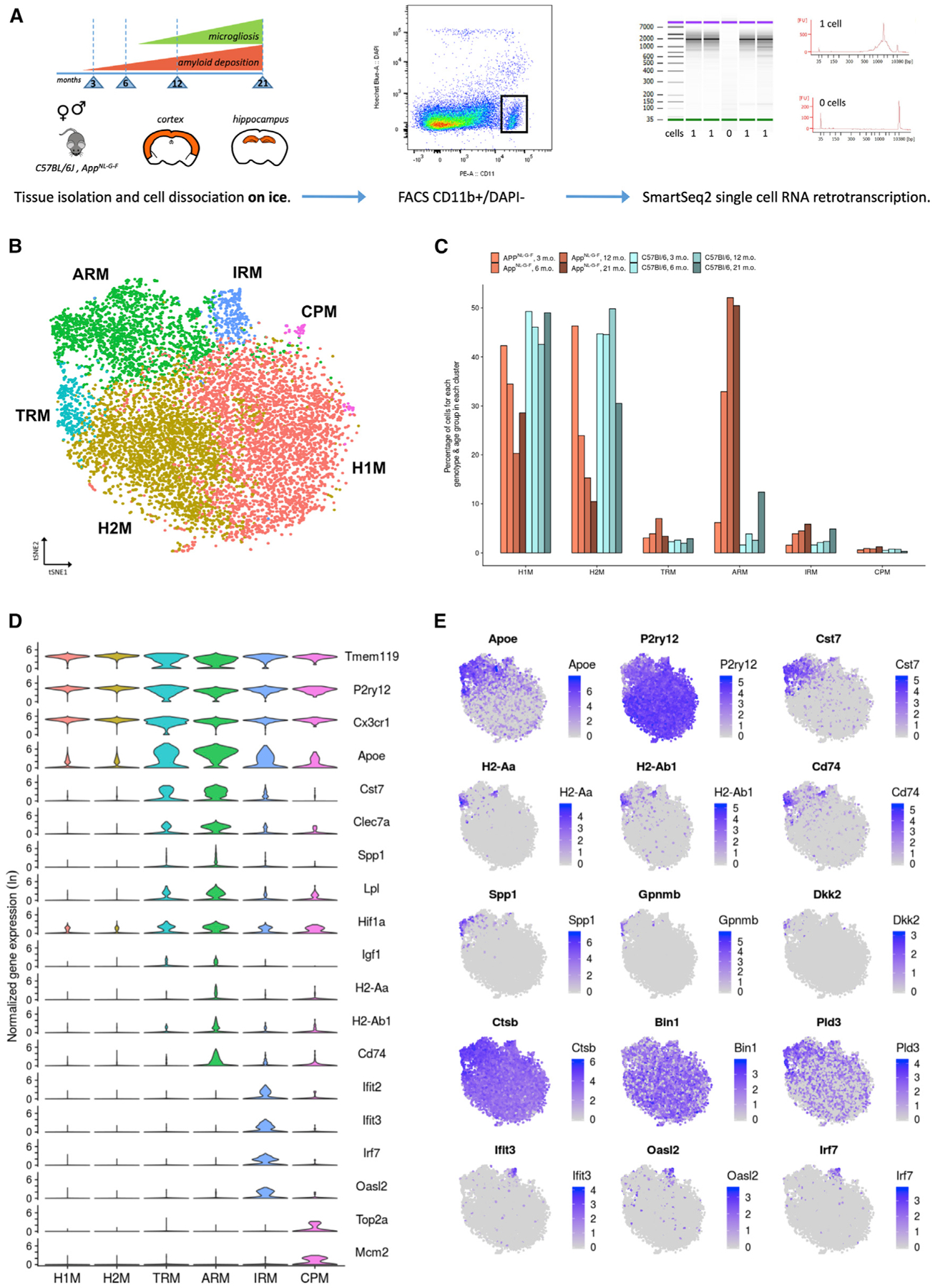
(A) Dataset presentation. We used male and female AppNL-G-F and wild-type C57BL/6J mice from four time points over the course of amyloid pathology and microgliosis as indicated. We dissected separately cortex and hippocampal tissues. The tissue from two animals for each experimental condition (age, sex, tissue, genotype) was pooled before microglia isolation. All procedures were performed on ice. Single live microglial cells were isolated by FACS (CD11b+, DAPI), and single-cell RNA-sequencing (RNA-seq) libraries were prepared according to the SmartSeq2 and Nextera methods.
(B) t-distributed stochastic neighbor embedding (t-SNE) plot visualizing the 10,801 single microglial cells passing quality control after removal of peripheral cells. Cells are colored according to clusters identified with Seurat’s k-nearest neighbors (kNN) approach (H1M and H2M, homeostatic microglia; TRM, transiting response microglia; ARM, activated response microglia; IRM, interferon response microglia; CPM, cycling and proliferating microglia).
(C) Percentage of cells from each genotype-age group for each cluster identified. AppNL-G-F cells are indicated in shades of red, while wild-type cells are indicated in shades of blue.
(D) Violin plots of selected marker genes for each identified cluster. The y axis indicates normalized gene expression (ln scale).
(E) t-SNE plots as in (B), colored by the level of ln normalized expression of selected genes. Clusters of TRMs and ARMs display increased expression of Apoe and inflammation markers (Cst7) and concurrently display a reduction of homeostatic markers (P2ry12). Two distinct regions of the ARMs cluster display increased expression of MHC class II genes (H2-Aa, H2-Ab1, and Cd74), suggesting the existence of microglial subpopulations. Further, a small subset of the ARMs cluster displays an enrichment for tissue repair genes (Spp1, Gpnmb, and Dkk2). The ARMs cluster also displays differential expression of several AD-related genes (e.g., Ctsb, Bin1, and Pld3) compared to clusters of H1Ms and H2Ms. The cluster of IRMs is enriched for interferon genes (Ifit3, Oasl2, and Irf7).
See also Figure S5.
RESULTS
We analyzed cortical and hippocampal microglia in female and male C57BL/6J wild-type and in AppNL-G-F mice at four different time points: at the beginning of Aβ deposition (3 months old [m.o.]), at the beginning of overt histologically detectable microgliosis (6 m.o.), when both processes are well underway (12 m.o.), and at a late stage (21 m.o.) (Masuda et al., 2016) (Figure 1A). All together, we analyzed 32 different experimental conditions, taking into consideration the combinations of genotype, age, sex, and tissue (Figure 1A).
Tissue and cell suspensions were kept at <4°C during all steps of the isolation of microglia, to minimize artifactual activation (see STAR Methods). We isolated single live microglial cells (Cd11b+/DAPI−) by fluorescence-activated cell sorting (FACS) (Figure S1) and prepared single-cell full-length mRNA-sequencing libraries, using a modified SmartSeq2 method (Picelli et al., 2013, 2014; Trombetta et al., 2014) (see Figure 1A and STAR Methods). We sequenced a total of 12,024 single cells across the different experimental conditions. After quality control and removal of peripheral neutrophils (see Figure S2A and STAR Methods), we retained 10,801 microglial cells for further analysis.
Aging and Aβ Deposition Induce Similar Responses in Microglia
We next performed clustering analysis (see STAR Methods), which resulted in the identification of 6 major subpopulations of microglia (Figure 1B), displaying different abundances between genotypes and age groups (Figure 1C).
Two clusters expressing high levels of known homeostatic microglia markers (Tmem119, P2ry12, and Cx3cr1; Figure 1D) (Butovsky et al., 2014) dominated the whole microglial population (homeostatic 1 microglia and homeostatic 2 microglia; H1Ms and H2Ms, respectively; Figure 1B). In wild-type mice, H1Ms and H2Ms together accounted for 80%–90% of the total microglial population. They were roughly equally large at every time point, although H2Ms decreased by ~10% in 21-m.o. mice (Figure 1C). In AppNL-G-F mice, both H1Ms and H2Ms were equally (>40% of all microglia each) represented at 3 months but then showed a drastic decrease with age over the course of pathology, particularly H2Ms (Figures 1C and S3A). Thus, H2Ms seem to be more sensitive to aging and amyloid pathology than H1Ms. H1Ms and H2Ms display subtle differences in expression of more than 700 genes, with statistically significant fold changes of 1.1–1.5 in either direction. While no clear gene expression signature differentiates the two subsets, genes such as C1qa, C1qb, C1qc, Ctsb, Ctsd, Fth1, and Lyz2 were significantly upregulated in H2Ms. Therefore, the cluster of H2Ms, although displaying a canonical homeostatic gene expression profile, seems more primed toward synapse pruning and heightened lysosomal activity and might therefore engage faster into more activated states. Interestingly, the clusters of H1Ms and H2Ms display a differential distribution relative to sex and tissue groups (Figures S3A–S3D). Therefore, our data uncover previously unrecognized heterogeneity in homeostatic microglia.
The other clusters were quite divergent from the homeostatic ones (Figure 1D). One cluster (ARMs; Figure 1B) increased strongly in the AppNL-G-F mice. It is characterized by the expression of gene sets involved in inflammatory processes (Cst7, Clec7a, and Itgax), in major histocompatibility complex (MHC) class II presentation (Cd74, H2-Ab1, H2-Aa, Ctsb, and Ctsd), and possibly involved in tissue regeneration (Spp1, Gpnmb, and Dkk2) (Figures 1D and 1E). A second cluster (TRMs; transiting response microglia; Figure 1B) has an overall transcriptomic profile similar to that of the ARMs cluster but had lower expression levels of Apoe and other inflammatory genes, particularly of MHC class II genes, while it did not express tissue regeneration genes (Figures 1D and 1E). A third cluster (IRMs [interferon response microglia]; Figure 1B) displayed a high expression of several genes involved in innate immune response and interferon response type I pathways (e.g., Ifit2, Ifit3, Ifitm3, Irf7, and Oasl2), which were otherwise not expressed in other clusters (Figures 1D and 1E). Finally, we identified a very small cluster of cells corresponding to 0.3%–1.2% of the total microglial pool (CPMs, cycling/proliferating microglia) (Figures 1B and 1C) that was enriched in genes involved in DNA replication, chromatin rearrangement, and cell cycle (e.g., Top2a, Mcm2, Tubb5, Mki67, and Cdk1) (Figure 1D). The cluster of CPMs did not display any selective enrichment for specific phenotypic groups (Figure 1C and S3A–S3D) and may represent a small pool of cycling microglia (Askew et al., 2017).
The proportion of cells in the clusters of reactive microglia (ARMs, TRMs, and IRMs) increased over age in both AppNL-G-F and wild-type mice (Figures 1C and S3A–S3D). Importantly, each cluster is present even in young wild-type mice (all together constituting 5.5% of the total wild-type microglia at 3 m.o.; Figure 1C), demonstrating that these clusters are physiological states of microglia. Although amyloid pathology does not induce a unique pathological microglial state in this animal model, there is, however, a very outspoken quantitative effect on the ARMs cluster, with 87% of the microglia in this cluster coming from AppNL-G-F mice. The number of ARMs increased 5-fold (6% to 33%) between 3 and 6 months, and by 12 months, they were the major type of microglia (52%) in the AppNL-G-F mice, outnumbering the two homeostatic clusters. ARMs are, nevertheless, also increasing with aging in wild-type mice, reaching 12% of the total number of microglia at 21 months of age (Figure 1C). IRMs were present at a low percentage in both wild-type and AppNL-G-F mice, but their number increased over aging, particularly in AppNL-G-F mice (Figures 1C and S3A). Thus, aging alone induces transcriptome changes leading to two subtypes of activated microglia (ARMs and IRMs), and Aβ deposition enhances both cell states significantly.
Microglia Differentiate into Mutually Exclusive Response States
We wondered whether the different responses represented separate differentiation fates or consecutive steps on a continuous differentiation trajectory. Semi-supervised pseudotime analysis with Monocle 2 (Qiu et al., 2017a, 2017b; Trapnell et al., 2014) aligned microglial cells along a two-branched trajectory (Figure 2A). By identifying the distribution of cells from each cluster along these three branches, it becomes evident that homeostatic microglial cells (state 1) progressed either toward a state corresponding to the interferon-based IRMs response (state 2) or to a state corresponding to the heterogeneous ARMs response (state 3) (Figure 2A). This is quantitatively represented in Figure 2B, showing that 86% of homeostatic microglia (H1Ms and H2Ms) were retrieved in state 1, while 90% of the interferon response cells from the cluster of IRMs were in state 2, and 93% of the main Aβ-responsive ARMs cluster cells were in state 3 (Figure 2B; Figure S4). Cells from the cluster of TRMs were distributed between state 1 (36%) and state 3 (63%) (Figure 2B; Figure S4), further suggesting that this cluster represents cells transitioning between the two stages. The few cells in the cluster of CPMs were mostly found in state 1 (54%; 43 cells) but were represented also in state 2 (18%; 14 cells) and state 3 (28%; 22 cells) (Figure 2B; Figure S4).
Figure 2. Microglia Diversify into Two Cell State Branches during Response to β-amyloid Plaques.
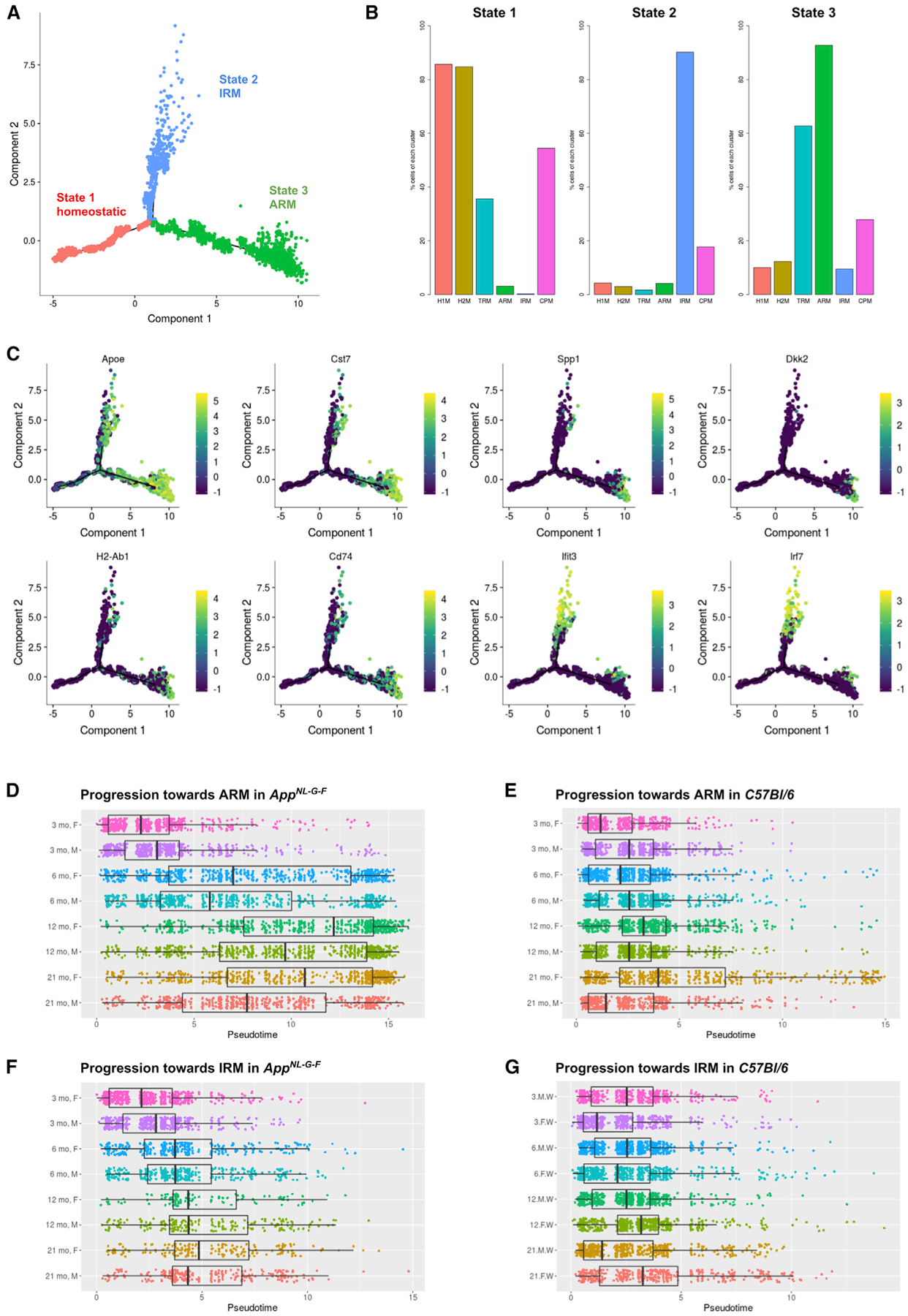
(A) Plot of cell trajectories for all microglial cells, obtained by a semi-supervised pseudotime ordering with Monocle 2. Microglia are grouped into three stages (red: stage 1; blue: stage 2; green: stage 3). Homeostatic microglia (red) progress toward two separate fates: either the multifunctional ARMs response (green) or the IRMs response (blue).
(B) Percentage of cells from each cluster (Figure 1B) per state. The majority (>80%) of homeostatic microglia (clusters of H1Ms and H2Ms) are in state 1, the majority of interferon response cells (cluster of IRMs) are in state 2, and the majority of the activated response cells (cluster of ARMs) are in state 3.
(C) Expression levels of selected marker genes are plotted over a plot of cell trajectories, as in (A).
(D) Pseudotime progression plot of homeostatic AppNL-G-F microglial cells to the ARMs branches, presented separately for each age-gender experimental condition. Box represents the interquantile range, and the thick bar represents the group median.
(E) Pseudotime progression plot from homeostatic to ARMs for each wild-type microglial cell.
(F) Pseudotime progression plot from homeostatic to IRMs response for each AppNL-G-F microglial cell.
(G) Pseudotime progression plot from homeostatic to IRMs response for each wild-type microglial cell.
Remarkably, Apoe expression continually increases along pseudotime in the ARMs (state 3) branch, together with several inflammatory markers (e.g., Cst7) (Figure 2C). MHC class II genes (e.g., H2-Ab1 and Cd74) were predominantly expressed by cells toward mid-to-late pseudotime in the ARMs (state 3) branch (Figure 2C). Genes involved with tissue repair (e.g., Spp1 and Dkk2) were expressed only by cells at the farthest end of the ARMs (state 3) branch (Figure 2C), which also expressed the highest Apoe levels, indicating that this subpopulation represents an advanced evolution of the microglial response. Most (94%) of these cells are derived from the AppNL-G-F mice. Although Apoe is also expressed in some interferon response (state 2) branch cells, its expression levels were lower compared to those in state 3 cells (Figure 2C), in agreement with differential expression analysis conducted at the cluster level (discussed earlier).
Differential expression analysis did not yield tissue- or sex-specific responses (Figures S3E and S3F), indicating that microglial responses to Aβ deposition is actuated stereotypically in hippocampus and cortex. Interestingly, female microglia displayed a faster progression in the deployment of the ARMs response in 6 m.o. AppNL-G-F and onward (Figures 2D–2G), which is reflected by the higher proportion of female ARMs cells compared to that of males (Figure S3B). Thus, although the response types and the genes involved are conserved between the two sexes, microglia in female mice appear to react earlier to Aβ.
ARMs Display Distinct Subpopulations Enriched for GWAS AD Risk Genes
Strikingly, Apoe, the main genetic risk factor for AD (Corder et al., 1993; Lambert et al., 2013), is strongly upregulated in ARMs, representing the major Aβ-response population (Figures 1D and 1E). Therefore, we wondered whether other AD risk genes are enriched among the genes that are differentially expressed (both up and down) in each of the reactive microglial clusters, compared to homeostatic microglia, using gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005). We used a list of genes associated with family history of AD from a recent extensive GWA study (GWAS) (Marioni et al., 2018). Taking into account that recent studies on polygenic risk scores show that genes with no genome-wide significance in GWASs still carry information with regard to risk of AD (Escott-Price et al., 2017), we decided to test for enrichment of GWAS genes at a number of different p value cutoffs (Figure 3A). For all tested p value cutoffs (ranging from p < 1e−6 to p < 0.01), only the ARMs cluster shows a strong enrichment for GWAS-associated AD genes (between p < 1e−6 and p <0.005), suggesting that polygenic risk associates with the ARMs cell state. The GSEA algorithm yields a set of GWAS genes most responsible for the observed enrichment (which we refer to as the GWAS core enrichment genes). In our case, using the set with the strongest enrichment (p < 1e−5), these genes are the well-established AD risk genes Apoe, H2-Eb1, Inpp5d, Bin1, and Ms4a6b but also Siglech (or CD33) and H2-Ab1, which have been suggested to be associated with AD (Lambert et al., 2013). Interestingly, Siglech, Inpp5d, Bin1, and Ms4a6b were significantly downregulated in the ARMs cluster, while the others were significantly upregulated. Further work is needed to determine whether these changes act in a protective or harmful way, but it is known that a SNP in the Siglech locus suppressing CD33 expression is protective in AD (Griciuc et al., 2013), while a SNP increasing its expression also increases AD risk (Bradshaw et al., 2013). We further looked specifically at the expression level (Figure S5) of a list of AD-related genes (Table S2) compiled from an extensive literature review. The expression levels of cathepsins Ctsb and Ctsd, Trem2 and Tyrobp, H2-Eb1, Pld3, and Aplp2 were all significantly upregulated in the ARMs cluster, while Adam10, Bin1, Cass4, Cd33, Ctsf, Inpp5d, Ms4a6d, and Picalm showed decreased expression in the same cluster (Figure S5).
Figure 3. Enrichment Analysis of AD Genes Highlights Substructures of ARMs.
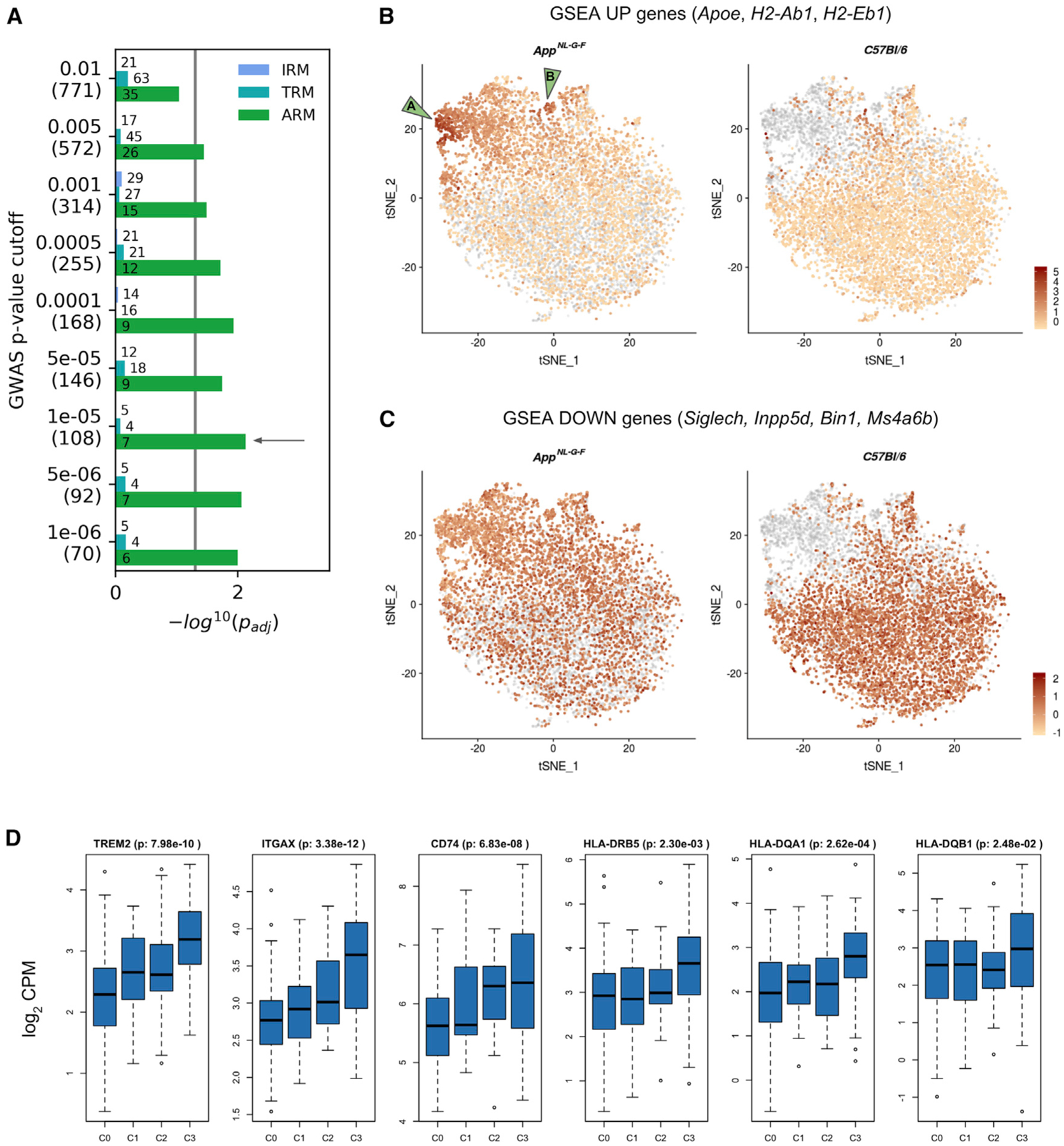
(A) Bar plot showing the significance (−log10 p value adjusted for false discovery rate; padj) of enrichment of AD GWAS genes among the genes differentially expressed in each of the three reactive microglial clusters (IRMs, TRMs, and ARMs) compared to the homeostatic microglia (H1Ms + H2Ms), calculated using GSEA. A significant enrichment indicates that more AD GWAS genes than expected are found among genes most strongly affected in the differential expression analysis for each comparison. We tested a number of different AD GWAS sets using different p value cutoffs: the numbers in the parentheses indicate the number of genes for that specific cutoff. The numbers in the bars indicate the size of the GSEA-predicted leading edge (core enrichment genes), which can be interpreted as the genes responsible for the observed enrichment. The enrichment for the p < 1e−5 cutoff yielded the lowest padj.
(B and C) Of the 7 core enrichment genes for the p < 1e−5 cutoff, 3 (Apoe, H2-Ab1, and H2-Eb1) were upregulated in ARMs compared to homeostatic microglia, while 4 genes (Siglech, Inpp5d, Bin1, and Ms4a6b) were downregulated. For each set of up- and downregulated core enrichment genes, we calculated a signature score (i.e., a composite expression score of a set of genes) using Seurat’s AddModuleScore function. Each cell’s score for either the upregulated (B) or downregulated (C) gene set is visualized on a t-SNE plot (as in Figure 1B). In both cases, cells of the two genotypes are plotted separately, as indicated in the titles, with cells of the other genotype plotted in gray. In (B), the AppNL-G-F cells clearly display a strong signature score for AD GWAS genes showing in the ARMs cluster. The two green arrowheads on the left in (B) indicate two areas with particularly strong expression of these genes.
(D) Boxplots of gene expression across CERAD stages in the parahippocampal brain region from the MSBB cohort. The adjusted p values of CERAD score C3 (Alzheimer’s disease) versus C0 comparisons are displayed next to the gene names. Similar results were found in the ROSMAP dataset (data not shown).
We next investigated the genes upregulated in the ARMs cluster relative to the homeostatic cluster in bulk tissue from human brains obtained from the Accelerating Medicines Partnership-AD (AMP-AD) portal (Hodes and Buckholtz, 2016). 62 of the top 100 genes upregulated in the ARMs cluster were also significantly (adjusted p < 0.05) upregulated in subjects with high plaque burden (Consortium to Establish a Registry for AD [CERAD] stage C3) compared to the stage-C0 subjects. The parahippocampal region from the Mount Sinai Brain Bank (MSBB) cohort region had the highest number of differentially expressed genes, with TREM2, ITGAX, and CD74 displaying the highest effect sizes (Figure 3D). Some of the GSEA core enrichment genes were also significantly upregulated in C3 subjects: HLA-DRB5, HLA-DQA1, and HLA-DQB1 (orthologs of H2-Eb1 and H2-Ab1) (Figure 3D). We then sought to determine whether these expression changes due to plaque increase were independent of the presence of tangles. To test this hypothesis, we analyzed gene expression between subjects with different plaque load and without tangles (Braak stages 0, I, and II), the human conditions most analogous to AppNL-G-F mice. The top genes that increased with plaque mean score were TREM2, TYROBP, and CD68 in the MSBB parahippocampal dataset, with TREM2 displaying a 2.7-fold increase (p = 0.2 · 10−4).
As indicated earlier, the ARMs from AppNL-G-F mice do not appear as one homogeneous cluster. Therefore, we assessed whether we could further subdivide the ARMs cells. In Figure 3B, we visualized the signature score (see STAR Methods) for the upregulated GWAS core enrichment genes, showing two sub-clusters within the ARMs cluster (labeled “A” and “B” in Figure 3B). Both subclusters are highly enriched (96%) for AppNL-G-F cells, suggesting that these particular subsets of ARMs might constitute specific responses to Aβ accumulation. The ARMs A sub-cluster expresses typical ARMs genes, along with genes possibly involved in tissue repair and remyelination, e.g., Spp1 (osteopontin, a chemoattractant and an adhesion protein involved in wound healing), Gpnmb (osteoactivin, involved in extracellular matrix remodelling), and Dkk2 (a secreted Wnt pathway antagonist) (Figure 1E). The ARMs B subcluster expresses Tmem119 but no genes overexpressed by other ARMs (e.g., Spp1, Lpl, Gpnmb, Apoe, Clec7a, and Cst7) (Figure 1E). Thus, in AppNL-G-F mice, Aβ boosts the heterogeneous ARMs response, with a gene signature that is enriched with AD risk genes, and elements of which can be retrieved in human subjects displaying a high plaque load.
Apoe-Expressing Microglia Cluster around β-amyloid Plaques
We were particularly intrigued by the high expression of Apoe in ARMs. As Apoe is mainly expressed by astrocytes in non-pathological conditions, we wondered whether astrocytes would also increase Apoe expression upon exposure to Aβ plaques. We performed single-molecule fluorescence in situ hybridization (smFISH) with RNAscope probes against Apoe and Itgam or Slc1a3 to mark microglia and astrocytes, respectively. smFISH was coupled with immunofluorescent detection of Aβ plaques using 6E10 antibody (Figures 4A–4F). Itgam and Apoe signals strongly colocalized in cells associated with plaques, while this was rare in cells located further away from plaques (Figures 4A, 4B, and 4G). Staining with the astrocyte marker Slc1a3 (Figures 4D and 4E) confirmed that astrocytes, but no microglia, were expressing Apoe in wild-type mice (Figures 4C and 4F).
Figure 4. Microglia Are the Major Contribu tors of Apoe Expression in the Vicinity of β-amyloid Plaques.
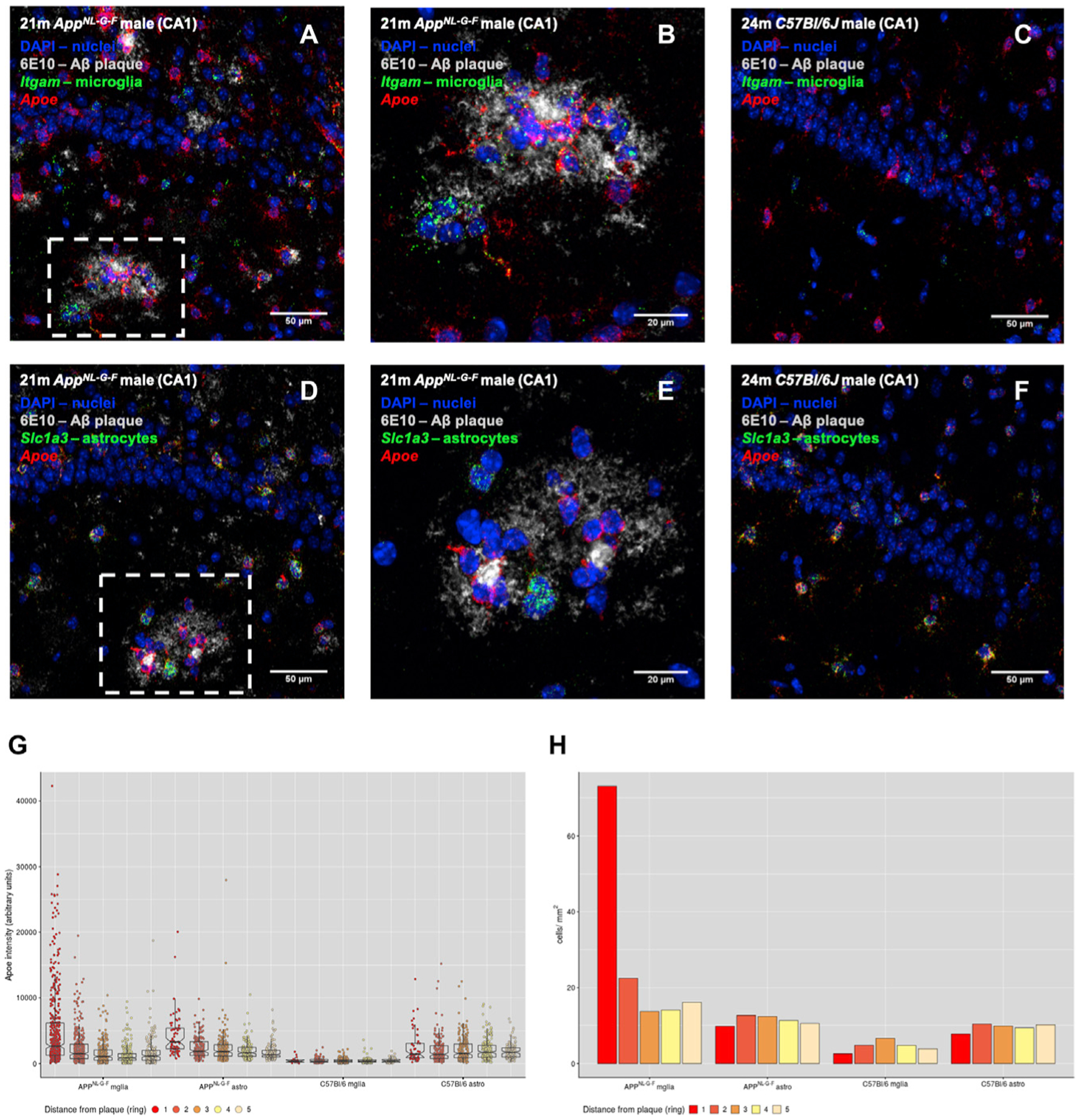
(A–F) Combined RNAscope and immunofluorescent analyses of Apoe expression by microglia and astrocytes in the vicinity of β-amyloid plaques. Expressions of Apoe, the microglia marker Itgam (A–C), and the astrocyte marker Slc1a3 (D and E) were visualized using RNAscope probes, while plaques were visualized by staining with the anti-Aβ antibody 6E10. Nuclei were visualized with DAPI. Photos are representative of three mice per genotype. (A and D) are representative images of AppNL-G-F CA1 stained for microglia (Itgam) and astrocytes (Slc1a3), respectively. (B) Zoom-in of the boxed area in (A), taken as a separate image with a higher magnification lens. Similarly, (E) is a zoom-in of the boxed area in (D). (C) and (D) are representative images of male wild type C57Bl/6J CA1, stained for microglia (Itgam) and astrocytes (Slc1a3), respectively. Scale bars in (A), (C), (D), and (F) represent 50 μm, while in (B) and (E) represent 20 μm.
(G) Quantification of Apoe staining intensity per cell, classified based on the genotype (AppNL-G-F or C57BL/6J), cell type (microglia [mglia] or astrocyte [astro]), and distance from a plaque (ring). For wild-type mice, measurements were made by selecting random regions of interest (ROIs) in the same brain areas as in AppNL-G-F. Measurements were made from at least 25 plaques or ROIs for each condition (AppNL-G-F mglia, C57BL/6J mglia, AppNL-G-F astro, and C57BL/6J astro), collected from 3 mice per genotype.
(H) Number of microglia and astrocytes next to plaques. As in (G), cells were classified based on genotype and distance from plaques (AppNL-G-F) or random ROIs (C57BL/6J).
We quantified the intensity of Apoe staining in microglia and astrocytes at different distances from plaques. The intensity of Apoe staining gradually increased in both cell types when closer to plaques, but the increase was much higher in microglia than in astrocytes (Figure 3G). Microglia, furthermore, were clustered around Aβ plaques, while astrocytes were evenly distributed across the tissue (Figure 4H).
These results indicate that microglia constitute the main cellular response close to plaques and that ApoE production at plaques is mainly of microglial origin. We did not see an increase of cycling cells (cluster of CPMs) in AppNL-G-F mice, suggesting that the increased number of microglia next to plaques is not due to increased proliferation and represents likely the evolution of microglia from homeostatic H1 and H2 to reactive ARMs cell states in AppNL-G-F.
Deletion of Apoe Suppresses the Microglial Responseto β-amyloid
We wondered whether Apoe was directly involved in the microglial responses to Aβ pathology. An Apoenull mouse strain is not available on an AppNL-G-F background at this moment; therefore, we used a second AD mouse model (APP/PS1) for which an Apoenull strain has been recently developed (APP/PS1-Apoenull; [E.H., J. Klickstein, C. Cannavo, R. Jackson, A. Muzikansky, S. Gandhi, D. Urick, T. Sargent, L. Wrobleski, A.D. Roe,S., Hou, K.V. Kuchibhotla, R.A. Betensky, T. Spires-Jones, B.T. Hyman, unpublished data]). We sequenced 2,304 single cells from 18-m.o. male APP/PS1 and APP/PS1-Apoenull mice and from the respective control strains, C57BL/6J and C57BL/6J-Apoenull. After quality control and removal of peripheral neutrophils (Figure S6), we retained 1,880 microglial cells for further analysis. Clustering analysis of cells from all four genotypes identified 4 major microglial populations (Figure 5A, middle) and was consistent with a similar analysis performed after removal of Apoe from the original gene count matrix (data not shown).
Figure 5. Apoe Deletion Prevents the Estab lishment of a Main Inflammatory Response to β-amyloid Plaques.
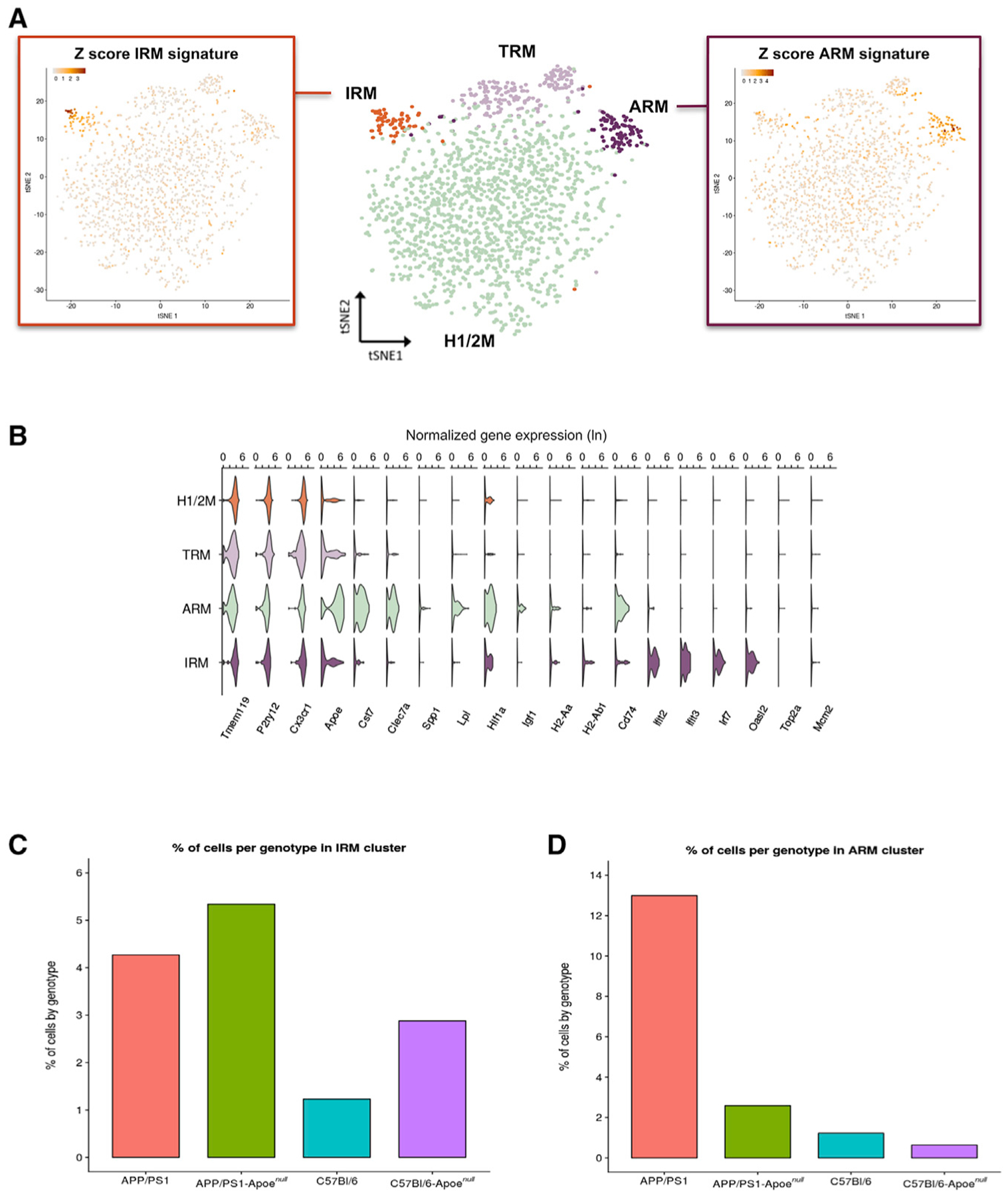
Analysis by single-cell RNA-seq of single live microglial (CD11b+/DAPI−) cells, prepared as described in Figure 1A, from APP/PS1 and APP/PS1-Apoenull mice and their respective wild-type control strains C57BL/6J and C57BL/6J-Apoenull.
(A) Middle: t-SNE plot visualizing the 1,880 single microglial cells passing quality control after removal of peripheral cells. Cells are indicated according to clusters identified with Seurat’s kNN approach (H1/2M, homeostatic microglia; TRM, transiting response microglia; ARM, activated response microglia; IRM, interferon response microglia). Left: t-SNE plot as in the middle, colored by the Z-score of gene signatures for the interferon response (IRM). Right: t-SNE plot as in the middle, colored by the Z score of gene signatures for the activated response (ARM).
(B) Violin plots of selected marker genes for each identified cluster. The x axis indicates normalized gene expression (ln scale).
(C) Percentages of cells from each mouse genotype in the IRMs cluster.
(D) Percentages of cells from each mouse genotype in the ARMs cluster.
We confirmed that the overall microglial responses in APP/PS1 mice were similar to those seen in AppNL-G-F mice. The major cluster displayed high expression of homeostatic genes (Figure 5B) (H1/2M [homeostatic microglia 1 and 2]; Figure 5A). A second cluster displayed a high score for the ARMs signature (ARMs cluster; Figure 5A, right), while a third cluster displayed high scores for the interferon response signature (IRMs cluster; Figure 5A, left). A fourth cluster displayed high levels of homeostatic genes but also a higher ARMs score compared to that of cluster H1/2M, thus representing a transiting state between H1/2M and ARMs (TRMs cluster; Figure 5A). Notably, the ARMs cluster was strongly enriched for APP/PS1 microglia (15% of all APP/PS1 microglia; p = 0.08e–10) but contained only very few (2%) APP/PS1-Apoenull cells (Figure 5D). Conversely, APP/PS1-Apoenull mice showed 6% cells in the IRMs cluster versus 3% of microglia in APP/PS1 mice (Figure 5C). Thus, deletion of Apoe impairs the normal deployment of the ARMs response but does not block the IRMs response. Moreover, lack of Apoe boosted the interferon-type response independently of Aβ deposition, as C57BL/6J-Apoenull showed a tendency for a higher percentage of IRMs compared to C57BL/6J mice (2.9% versus 1.2%, respectively) (Figure 5C). This is in agreement with gene expression data from AppNL-G-F mice, which displayed high levels of Apoe in ARMs cells, while IRMs cells showed only a moderate increase in Apoe expression (Figure 2C).
Plaques of APP/PS1 mice appear bigger and more amorphous compared to plaques in AppNL-G-F mice (Figures 6A and 6B), as evidenced elsewhere (E.H.,J. Klickstein, C. Cannavo, R. Jackson,A. Muzikansky, S. Gandhi, D. Urick,T. Sargent, L. Wrobleski, A.D. Roe,S., Hou, K.V. Kuchibhotla, R.A. Betensky,T. Spires-Jones, B.T. Hyman, unpublished data). In both AppNL-G-F and APP/PS1 mice, plaques are stained heavily for ApoE, and microglia appear to develop ramifications and to invade plaques (Figures 6A and 6B). Lack of Apoe expression did not alter the global amyloid burden (Figures 6B, 6D, and 6E); however, it caused a significant decrease in the amounts of Metoxy-XO4-stained amyloid (Figure 6F), as previously reported for other Apoenull AD mouse models (Holtzman et al., 2000; Irizarry et al., 2000; Krasemann et al., 2017; Ulrich et al., 2018), with a consequent shift of the total versus dense core amyloid ratio (Figure 6G). Moreover, lack of ApoE causes a significant decreased density of microglial cells around amyloid deposits (p < 0.001; Figure 6H). Thus, ApoE, next to be involved in amyloid aggregation, also appears required to mount a full ARMs transcriptional response against Aβ plaques.
Figure 6. Lack of Apoe Prevents Migration of Microglia toward Plaques.
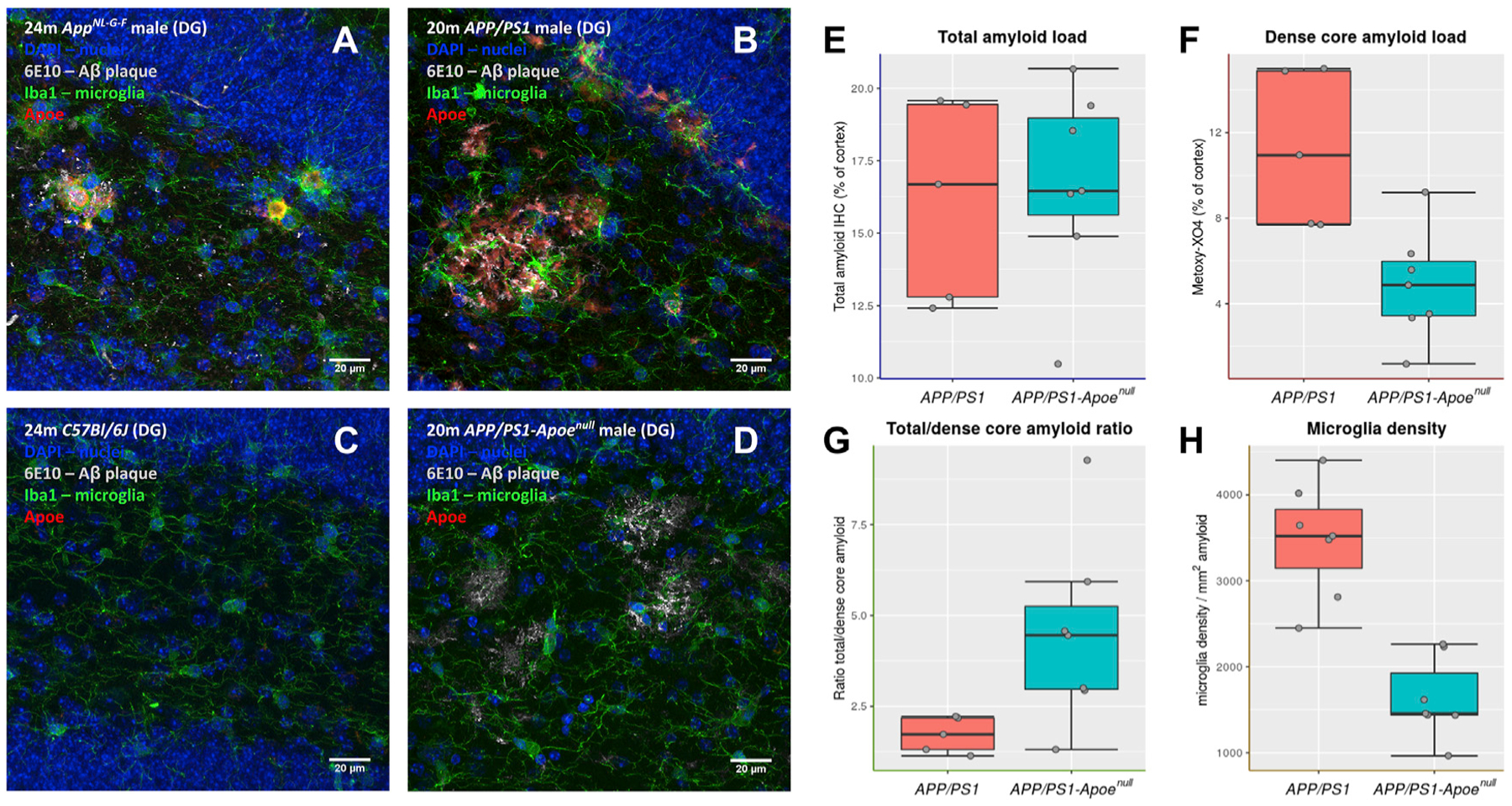
(A–D) Immunofluorescent staining of microglia (Iba1, green), ApoE (red), and Aβ plaques (6E10, white) in sagittal sections of AppNL-G-F (A), APP/PS1 (B), C57BL/6J (C), and APP/PS1-Apoenull (D). Images are zoomed on the dentate gyrus (DG) region. Scale bars represent 20 μm.
(E–G) Analysis of amyloid burden in 18- to 20-m.o. APP/PS1 and APP/PS1-Apoenull mice. Total amyloid burden as detected by anti-Aβ immunostaining (E), dense core plaque burden identified by Metoxy-XO4 staining (F), and the ratio between total amyloid burden and dense core plaques (G) are presented as boxplots (boxes represent the 25%–75% quartile range; whiskers represent the ±1.5 interquantile range; each experimental point is represented by a gray dot; n = 5–7 mice per group; *p < 0.05, Mann-Whitney test).
(H) Stereological evaluation of the density of Iba-1 reactive microglia around amyloid deposits in APP/PS1 and APP/PS1-Apoenull mice; boxplot is as in (E), n = 5–7 mice per group. **p < 0.001, Mann-Whitney test.
DISCUSSION
Our data show previously unrecognized heterogeneity in both homeostatic and reactive microglia. We identified two populations of homeostatic microglia (H1Ms and H2Ms), which display subtle but significant gene expression differences and are differentially enriched in different brain areas and in female compared to male mice. The separation in these two clusters is relevant, as H2Ms cells also decline more rapidly than H1Ms cells with aging. We identified also two major populations of reactive microglia. One displays a multifunctional gene response that we call here the ARMs response. Although the ARMs response overlaps with the DAMs response described by Keren-Shaul et al. (2017), we provide evidence that these cells are part of the normal evolution of microglia in healthy aging, and we find the term “disease associated” misleading. In fact, ARMs cells are already detected in the brains of wild-type mice at young ages, further underlining that they are not necessarily disease associated. The presence of amyloid plaques in AppNL-G-F mice boosts the redistribution of homeostatic microglia to ARMs cells, which eventually become the predominant population. At odds with the original DAMs response description is also the heterogeneity in the ARMs group uncovered here: several subpopulations can be discerned by the expression of specific subsets of genes, such as genes involved in MHC class II presentation (H2-Ab1, H2-Aa, and Cd74) and genes potentially involved in tissue repair (Spp1, Gpnmb, and Dkk2). These subsets are highly enriched for AppNL-G-F microglia, suggesting that they could represent an exaggerated or modified response to the plaques and that these sub-responses might provide the real pathological signature of microglia in AD.
The second population of reactive microglia is enriched for interferon response genes and therefore called the IRMs response. IRMs are again present in wild-type and AppNL-G-F mice in all age groups and, therefore, again part of the normal physiological heterogeneity of microglia in the healthy mouse brain. The IRMs population is much less affected by the presence of amyloid plaques than the ARMs population. Interestingly, gene enrichment analysis indicates that ARMs, but not IRMs, are enriched for known AD risk genes such as Apoe, Trem2, Tyrobp (DAP12), Ctsb, Ctsd, H2-Eb1, and Pld3 (Cruchaga and Goate, 2015a, 2015b; Cruchaga et al., 2014; Fazzari et al., 2017; Heilmann et al., 2015; Hooli et al., 2015; Lambert et al., 2015; van der Lee et al., 2015). This strongly argues that the role of these genetic risk factors in AD is in the activation and function of this specific microglia subpopulation.
Microglia expressing interferon response genes have been described as a late response to neurodegeneration in a CK-p25 model (Mathys et al., 2017); however, these look quite different from the IRMs described here. For example, IRMs do not co-express MHC genes and, as stated earlier, are present also in healthy, normal brain, even at a young age. It should be noted that CK-p25 induces a rapid and severe neurodegeneration that might be different mechanistically from the very slow and mild disease caused in AppNL-G-F mice by Aβ deposition. AppNL-G-F mice are regarded as models of early AD pathology.
Pseudotime analysis is a good way to investigate how different microglial expression profiles relate to each other (Trapnell et al., 2014). This analysis suggests that the different cell states we describe here are, in fact, part of a continuous spectrum, with homeostatic microglia (H1Ms and H2Ms) adapting two major response branches, IRMs and ARMs, which increase during aging and especially upon exposure to Aβ. These branching trajectories broadly match the old classification of microglia as resting (M0), pro-inflammatory (M1), and pro-resolution (M2) microglia, but they also indicate that microglial phenotypes are highly dynamic (Butovsky and Weiner, 2018; Friedman et al., 2018; Keren-Shaul et al., 2017; Mrdjen et al., 2018; Ransohoff, 2016).
Heterogeneity of microglia between different brain areas (Grabert et al., 2016) and between different sexes (Villa et al., 2018) is increasingly recognized. We show here, however, that the response to Aβ deposition evolves stereotypically across brain areas (cortex and hippocampus) in animals of both sexes. Remarkably, we show that microglia in female mice progress faster over the ARMs trajectory compared to microglia in male mice (Figure 2D). This is in agreement with previous histological analysis (Masuda et al., 2016). Such sex-dependent differences in microglial responses are interesting to note, as there is a higher incidence of AD in women (Ferretti et al., 2018; van der Flier and Scheltens, 2005; Laws et al., 2018; Mazure and Swendsen, 2016).
We demonstrate here, in agreement with previous findings (Keren-Shaul et al., 2017; Krasemann et al., 2017), that ARMs induce Apoe expression up to 27-fold, compared to homeostatic microglia. In particular, in proximity to plaques, microglia express higher levels of Apoe than astrocytes (~2.3-fold higher median Apoe intensity per cell; Figure 3G). We show also that Apoe is not merely a marker for this cell state but is a key component of the ARMs response, as its deletion severely reduces the number of microglia displaying an ARMs signature (Figure 4) and affects the interaction of microglia with Aβ plaques (Figure 5). Given this spectacular influence of Apoe expression on the phenotype of microglia, we speculate that a large part of the genetic risk associated with the APOE4 genotype (Corder et al., 1993; Rebeck et al., 1993) is likely executed via modification of the microglial function rather than other mechanisms. While we cannot exclude that the abnormal plaques that are generated in the absence of ApoE affect the microglia response, it seems more likely that ApoE directly modulates the response of the microglia to the plaques and that ApoE is responsible for the induction of the ARMs response to amyloid, likely by interacting with Trem2 (Yeh et al., 2016). Thus, the major genetic risk factor for AD, together with most other identified risk genes in GWASs, modulates the neuroinflammatory response of the microglia to amyloid plaques.
Our observation that many AD GWAS-associated genes appear to change their expression in the ARMs indicates an overlap between the pathogenesis of AD in humans and the microglial response to the accumulation of amyloid in the brain of the mouse models. Thus, these GWAS variants, when present in a patient, likely affect the way that microglia cope with the accumulation of amyloid over age. In that capacity, they might either exacerbate or act protectively with regard to the onset of the disease. Such a view predicts that patients with a high load of amyloid but with a protective combined polygenic risk score (which, in the case of AD, covers mostly genes expressed in microglia) will respond in a beneficial way to amyloid and will not develop full-blown AD.
In conclusion, our data refine the concepts of homeostatic and reactive microglia and indicate that major AD risk factors (age, sex, and APOE) link to a multifunctional and complex microglial response to amyloid plaques that evolves over different branches of a continuous spectrum of molecular phenotypes. The complex response of the microglia, even when described as a unique cell state, “ARMs,” is probably not so surprising, as the responses of individual microglia will likely profoundly depend on factors in the microenvironment of the plaques requiring differential functions of the microglial toolkit to be engaged. A particular challenge for the future is to dissect the distribution of different ARMs subpopulations over the brain and to add functional significance to the different gene expression patterns. It is likely that such functional dissection will lead to a whole set of novel drug targets that each will be useful in modulating a microglial response that is most beneficial for AD.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Bart De Strooper (bart.destrooper@kuleuven.vib.be).
EXPERIMENTAL MODELS DETAILS
Mice
For the 3, 6 and 12 m.o. time points we used homozygous AppNL-G-F (Saito et al., 2014) mice, backcrossed for at least 2 generations with C57BL/6J mice in the De Strooper lab, and wild-type C57BL/6J mice themselves. For the 21 m.o. time point, both AppNL-G-F and C57BL/6J mice were transferred from the Saido lab to the De Strooper lab, and housed at least 1 month before being used for experiments. Both male and female AppNL-G-F and C57BL/6J mice were used for experiments. The novel APP/PS1-Apoenull strain is described in another manuscript [Hudry et al., submitted]. Briefly, APPswe/PS1dE9 (APP/PS1) were crossed with a C57BL/6J-Apoenull breeder to obtain homozygous APP/PS1-Apoenull mice. APP/PS1-Apoenull mice, along with parental APP/PS1 mice and the respective control strains C57BL/6J and C57BL/6J-Apoenull, were transferred from the Hyman lab to the De Strooper lab, and housed for at least 1 month before being used for experiments. Only male APP/PS1, APP/PS1-Apoenull, C57BL/6J and C57BL/6J-Apoenull were used. In every case, mice were housed according to the appropriate institution’s ethical requirements, and in compliance to the country’s laws for animal research.
METHOD DETAILS
Single cell suspension preparation
Mice were euthanized with CO2 and then rapidly perfused with ice cold PBS for 10–15 minutes using a pumping rate of 100 mL/h. The dissection (Srinivasan et al., 2016) and microglia isolation steps were carried out on ice or at +4°C, to minimize microglia activation due to technical artifacts. We removed the brain and placed it on a chilled glass plate sitting on wet ice. We quickly dissected the whole cortex and the whole hippocampus separately, using instruments chilled on ice. The tissue was finely chopped using a chilled razorblade and then transferred to a tube containing ice-cold Hibernate A medium without phenol red (BrainBits, Springfield, IL). For each experimental condition we pooled tissue from two mice. We prepared a single cell suspension from the minced tissue using the Adult Brain Dissociation kit from Miltenyi Biotec (Bergisch Gladbach, Germany) adapting manufacturer’s recommendations for manual tissue processing. First, we pelleted the tissue chunks by centrifugation (300 g for 2 minutes at 4°C), removed the Hibernate A medium and resuspended the tissue in a mix of buffer Z with enzymes P, A and Y prepared according to the manufacturer’s instructions. We next placed the tubes horizontally on ice for 30 minutes, with mechanical dissociation steps performed at 10 minutes intervals (first with 5 mL pipettes, then with fire-polished glass Pasteur pipettes, and lastly with P1000 tips). The resulting cell suspension was filtered on a 70 μm cell strainer, and the strainer was washed with 10 mL of D-PBS containing magnesium, calcium, glucose, and pyruvate (Life Technologies, Carlsbad, CA). Cells were centrifuged, resuspended in D-PBS, gently mixed with Debris Removal Solution, and overlaid with D-PBS according to manufacturer’s instructions. After centrifugation (3000 g for 10 minutes at 4°C), cells were washed once with D-PBS, resuspended in 1X Red Blood Cell Removal Solution, incubated for 10 minutes at 4°C, diluted 10 times with D-PBS containing 0.5% BSA (Miltenyi Biotec), and pelleted by centrifugation. Cells were finally resuspended in D-PBS containing 0.5% BSA.
Single microglia isolation by FACS
The single cell suspension was stained by incubation for 20 minutes on ice with anti-CD11b-PE conjugated primary antibody (Miltenyi Biotec) at a 1:10 dilution. Just before loading on the sorter, cells were stained with DAPI (1:5000 final dilution). Cell sorting was performed with a BD FACSAria III (BD Biosciences, Franklin Lakes, NJ). Gating was calibrated by running non-stained and single stained (anti-CD11b-PE only and DAPI-only) samples. Single live microglia cells (CD11b+/DAPI−) were sorted into independent wells of a 96-well plate, preloaded with 4 mL of 0.2% Triton in Ultrapure water (GIBCO), containing 1 U/μL of RNasinPlus RNase inhibitor (Promega, Madison, WI). After sorting, plates were sealed, briefly centrifuged, snap frozen on dry ice, and stored at −80°C until further processing.
Single cell mRNA libraries preparation and sequencing
Single cell retrotranscription and preparation of sequencing libraries was performed using a modified SmartSeq2 protocol (Picelli et al., 2013, 2014; Trombetta et al., 2014) to improve the yield of cDNA from single microglia cells. We used 5′-biotinylated primers throughout the protocol, oligo-dT and IS-PCR oligos were obtained from Integrated DNA Technologies (Skokie, IL), while template-switching oligo (TSO) was from Exiqon (Vedbaek, Denmark). First, 96-well plates with single sorted cells were thawed on ice, and we added to each well 1 μL of 10 mM dNTPs, 0.5 μL of 5 μM oligo-dT, and 0.5 μL of a 1:2660000 dilution of ERCC RNA Spike-In mix (ThermoFisher, Waltham, MA). After incubating the plate at 72°C for 3 minutes, we added to each well a retrotranscription mix (containing 1.5X First Strand buffer, 6.1 mM DTT, 1.7 M betaine, 10 mM MgCl2, 0.6 U/μL SUPERase IN, 11.1 U/μL SuperScript II polymerase, and 1.3 μM TSO oligo). All retrotranscription mix components were from the SuperScript II Reverse Transcriptase kit (ThermoFisher), except betaine (Sigma, St. Louis, MO), SUPERase IN (ThermoFisher), and the TSO oligo. Retrotranscription reaction was carried out for 90 minutes at 42°C, followed by 10 cycles of 2 minutes at 50°C and 2 minutes at 42°C, and completed by a 15 minutes incubation at 70°C. Next, we added to each well a preamplification mix containing 1X Kapa HiFi HS readymix (Kapa Bio-systems, Wilmington, MA) and 0.2 μM of IS-PCR primer. Preamplification was carried out for 23 cycles (98°C for 20 s, 67°C for 15 s, 72°C for 6 minutes). PCR products were purified using AMPure XP beads (Beckman Coulter, Brea, CA) at 1:0.6 ratio and according to manufacturer’s recommendations and eluted in 12 μL of EB buffer (QIAGEN, Hilden, Germany). cDNA preparation was checked by analyzing several random wells for each plate on an Agilent BioAnalyser DNA High Sensitivity chip (Agilent, Santa Clara, CA). We used the Nextera XT kit (Illumina, San Diego, CA) to tagment 1.25 μL of 1:15 diluted cDNA per reaction. Tagmentation reactions were assembled at room temperature (RT) using 2.5 μL of TD buffer and 1.25 μL of ATM mix per sample, incubated at 55°C for 8 minutes, and then immediately placed on hold at 10°C. We next added 1.25 μL of NT buffer (Illumina) and further incubated for 5 minutes at RT. To amplify tagmentation products, we added 1.25 μL of appropriate indexed forward and reverse primers (Nextera XT Index kit v2, sets A, B, C, or D, all from Illumina), and 3.75 μL of NPM mix (Illumina). Amplification reaction was carried out for 12 cycles (95°C for 10 s, 55°C for 30 s, 72°C for 30 s). Amplified products from each 96 well plate were pooled and purified using AMPure XP beads at 1:0.7 ratio. Purified pools were checked on an Agilent BioAnalyser DNA High Sensitivity chip, and quantified by quantitative PCR using the Kapa Library Quantification kit for Illumina libraries. Four 96-well plate pools with compatible indexes were further pooled to yield 384-samples libraries. Each library was sequenced on an Illumina NextSeq550 system using single end 75 base pair sequencing kits (Illumina).
Sequencing data analysis
Demultiplexed FASTQ files were aligned to the mouse genome (mm10 build) using STAR version 2.5.2 (Dobin et al., 2013) with default options. Reads aligned to each gene were counted using featureCounts version 1.5.1 (Liao et al., 2014) with options -g gene_name -Q10. The count table was imported in R (version 3.4.4) for analysis. Data generated from AppNL-G-F and C57BL/6J mice, and data generated from APP/PS1, APP/PS1-Apoenull, C57BL/6J and C57BL/6J-Apoenull mice are referred here as two separate datasets.
Quality control of cells - step 1
For each dataset, to exclude poorly sequenced cells, damaged cells and dying cells, we filtered out cells outside the limits of median ± 4 * median absolute deviations for both reads number and genes detected; moreover, we excluded cells with more than 10% of reads aligning to mitochondrial genes (Figure S2A and Figure S6A). Data was analyzed by principal component analysis (PCA) to identify obvious batch effects, in which case we would discard the entire 96-well plate (we excluded two plates following this procedure). For the first dataset, the median depth of sequencing was 572,294 reads/cell, with an interquantile range of 253,445 reads/cell, while the median number of genes detected per cell was 2,250 (interquantile range: 572). After quality control (Figure S2A) we retained 11,038 cells for further analysis, with at least 270 cells for each experimental condition (Table S1).
Quality control of cells - step 2
For each dataset, we applied a workflow based on the R package Seurat (version 2.3.1 (Butler et al., 2018)) to identify and remove non-microglia cells before proceeding with analysis. For the first dataset, after data normalization and scaling, we performed principal component analysis (PCA) on the 4,687 most variable genes detected. Based on a scree plot (i.e., a plot of the PC eigenvalues in decreasing order) of standard deviations of the principal components (PCs), we selected the first 17 PCs as input for the calculations to identify cell clusters (Seurat FindClusters function). We then performed non-linear dimensionality reduction and we visualized cell clusters on a t-SNE plot (Figure S3A). We identified 10 major cellular populations, most of them showing a tight distribution on the t-SNE plot, with only two clusters (7 and 9) clearly separating (Figure S2B). Based on a panel of marker genes (Figure S2C), we had no enrichment for markers of brain cells other than microglia, but two clusters (7 and 9, figure S2B–C) showed high expression levels of genes markers of perivascular macrophages (Mrc1) and neutrophils (Ccr2 and Pldb1). Indeed, clusters 7 and 9 scored low for a microglial genes signature (Figure S2D) and high for a neutrophil genes signature (Figure S2E). We did not find cells expressing gene signatures of other brain cells (Figure S2C: neurons (Gria2), astrocytes (Aqp4), oligodendrocytes (Olig1)), confirming that 97.9% of cells (10,801/11,038) in our post-QC dataset were microglia. Only the 10,801 microglia cells were retained for further analysis. For the second dataset, we considered the top 4,711 most variable genes, and we used the first 12 PCs for initial clustering. We identified several clusters, with one displaying high score for a neutrophil marker genes signature (cluster 12, Figure S6E), which was removed prior to subsequent analysis. The other cells scored high for a microglia marker genes signatures (Figure S6D), while expression of markers of other brain cell types were low or null (Figure S6C).
Clustering
Cells passing QC were analyzed using functions provided with the Seurat package. Data was log normalized and we regressed out the variables of read count and percentage of reads aligning on mitochondrial genes. Next, we identified the genes with highest variability and performed PCA on such gene set. We identified the most informative principal components based on a scree plot and we used these to perform cell clustering. Identification of differential expressed genes was performed using the Wilcox test implemented by Seurat’s FindMarker. t-SNE plots were prepared using Seurat’s t-SNE implementation. For the first dataset, we considered 4,777 highly variable genes for PCA and the first 14 PCs for clustering. The second dataset was analyzed similarly as described above, by performing PCA on the 4,967 most variable genes and by using the first 10 PCs to perform cluster analysis.
Pseudotime analysis
To infer the pseudotime of microglia progression toward response development we used functions provided with the Monocle 2 package (version 2.6.4 (Qiu et al., 2017a, 2017b; Trapnell et al., 2014)). We performed a semi-supervised identification of cell trajectories and states, based on marker genes identified during clustering. Briefly, we defined a series of genes (Apoe, Ifit3, H2-Aa, Dkk2), which are selectively expressed in each response type (main response and interferon-based) and in the subpopulations identified, which we used to build a classifier (CellTypeHierarchy) to assign cell identities in terms of microglia states of activation. We then calculated a covariance matrix for these marker genes, and we used the top 1000 genes (ranked by p value adjusted) to calculate single cell trajectories.
Differential Expression
Differential expression was performed using functions provided with the Seurat package; p values were calculated using the Wilcoxon rank-sum test, with all Seurat parameters set to default. Genes with adjusted p values (using a Bonferroni correction) <0.05, and ln fold changes > |0.2| were considered significantly differentially expressed.
Z scores of microglia responses signatures
For Figure 3, signatures were calculated using Seurat’s AddModuleScore function using a list of relevant genes identified from GSEA as input. For Figure 5, Z scores were calculated by considering the top genes showing differential expression between ARM and homeostatic cells in the AppNL-G-F dataset (n genes = 18) or between the IRM and homeostatic cells in the AppNL-G-F dataset (n genes = 28), respectively. Top genes were defined as being statistically significant after multiple testing correction and displaying at least 1 ln fold change. Apoe was excluded from the gene signature to avoid skewing the calculations in Apoenull samples.
Human RNaseq data analysis
We analyzed RNaseq data obtained from the AMP-AD (Accelerating Medicines Partnership – Alzheimer’s disease) portal (Hodes and Buckholtz, 2016) for the ROSMAP (Religious Orders Study and Memory and Aging Project) (Mostafavi et al., 2018) and MSBB (Mount Sinai Brain Bank) cohorts (Wang et al., 2018). The identifiers for these studies on the AMP-AD portal (www.synapse.org) are syn3157743 for MSBB and syn3388564 for ROSMAP. The reads were trimmed, aligned and mapped to the reference genome and the transcript expression levels quantified by their automated pipelines. The ROSMAP study provides RNaseq data from the Dorsolateral Prefrontal Cortex and the MSBB study has samples from the Prefrontal Cortex (PFC), Inferior Frontal Gyrus (IFG), Superior Temporal Gyrus (STG) and Parahippocampal Gyrus (PHG). Genes were considered to be microglial of their expression was at least two times the average expression in other cell types, according to cell type specific data provided by the “brain RNA seq” web portal (https://web.stanford.edu/group/barres_lab/brain_rnaseq.html). Sequencing data was analyzed as described in Bihlmeyer et al. (2019), and differential expression analysis was conducted using CERAD staging (Mirra et al., 1991) between C3 and C0 subjects.
Multiplex RNAscope and immunofluorescence staining
Mice were euthanized with CO2 and then rapidly perfused with ice cold PBS (10 minutes at 100 mL/h). Brain was removed and separated into two hemispheres. One hemisphere was then embedded in molds containing Tissue-Tek OCT (VWR, Radnor, PA), snap frozen in a bath of ethanol and dry ice, and stored at −80°C to perform RNAscope experiments, while the other hemisphere was processed for immunostaining as described below.
OCT-embedded hemispheres were cut into 14 mm sagittal sections using a CryoStar NX70 cryostat (ThermoFisher), layered on SuperFrost Plus glass slides (ThermoFisher) and further stored at −80°C before proceeding with experiments.
RNAscope experiments were performed using the Manual Fluorescent Multiplex kit v1 (Advanced Cell Diagnostics, Newark, CA) following manufacturer’s recommendations with minor adjustments. After fixation in 4% paraformaldehyde (PFA) for 30 minutes, sections were dehydrated using a series of ethanol dilution steps; protease digestion was performed for 20 min at RT using Protease IV for fresh frozen tissue provided in the kit, and probes hybridization was carried out for 2 hours at 40°C. We used the following probe sets (all from Advanced Cell Diagnostics): Mm-Slc1a3 (430781), Mm-Itgam-C2 (311491-C2), Mm-Apoe-C3 (313271-C3). Immediately after the RNAscope amplification steps, the sections were rinsed briefly with PBS and blocked for 1 hour at RT in PBS containing 0.3% Triton X-100 and 5% normal goat serum. Sections were then immunostained with the anti-β-amyloid 1–16 6E10 primary antibody (BioLegend, San Diego, CA) at 4°C overnight, washed 3 times for 5 minutes in PBS, and further stained for 1 hour at RT with an Atto-488-conjugated goat anti-mouse secondary antibody (Sigma-Aldrich, Saint Louis, MO). Sections were then incubated for 30 s in a 1X TrueBlack (Biotium, Fremont, CA) solution to reduce lipofuscin autofluorescence, washed briefly and stained with DAPI (Sigma-Aldrich). Sections were mounted with FluorSave Reagent (Merck Millipore, Burlington, MA). Imaging was carried out on a Leica TCS SP8 X confocal microscope (Leica Microsystems, Wetzlar, Germany) using a 40X objective, images were analyzed using Fiji ImageJ (Rueden et al., 2017; Schindelin et al., 2012).
RNAscope image analysis and quantification
Three 21–25 months old mice for each C57BL/6J and AppNL-G-F strain were used for the quantifications. For each mouse, four images of the hippocampus (two of CA1, one of CA2 and one of CA3) and five images throughout the isocortex were acquired and stacked in Fiji ImageJ. NIS-elements software (Nikon, Amsterdam, Netherlands) was used to detect nuclei, microglia, astrocytes and plaques using a custom-made protocol. Nuclei and cell body perimeter were established using the DAPI signal; microglia and astrocytes were identified by RNAscope puncta from the Itgam and Slc3a1 probes. All parameters were kept constant between images to allow unbiased detection. Plaque staining was judged individually in each image, as plaques vary greatly in size and intensity of staining. Arbitrary plaques were drawn in the images of C57BL/6J mice. Images from C57BL/6J and AppNL-G-F mice were analyzed in the same manner, for the latter mice we considered only the dense plaques. Around each plaque, five concentric circles of 18.2 mm were drawn, for each circle we counted microglia and astrocytes cells, and for each cell we measured the intensity of the signal from the Apoe probeset.
Immunofluorescence staining
Mice were sacrificed with CO2 and perfused with ice cold PBS (10 minutes at 100 mL/h), and the brain divided into two hemispheres as described above. One of the hemispheres was fixed in 4% PFA overnight and then stored in PBS containing 0.01% sodium azide at +4°C until sectioning on a vibratome. For sectioning, the hemispheres were embedded in UltraPure agarose (Invitrogen, Carlsbad, CA) and cut into 35 mm sections on a Vibratome Leica VT1000S. Antigen retrieval was performed using boiling citrate buffer (Sigma-Aldrich) at pH 6.0. The sections were then blocked in PBS with 0.3% Triton X-100 and 5% normal donkey serum for 1h at RT and incubated in primary antibody at 4°C overnight. The following day sections were washed with PBS and incubated with secondary antibodies for 1 hour at RT. Nuclei were stained with DAPI and sections were mounted with Fluorosave (Merck Millipore). The following primary antibodies were used: mouse anti-β-amyloid 1–16 6E10 (BioLegend), rabbit anti-Iba1 (Wako-Chemicals, Neuss, Germany), goat anti-ApoE (Sigma-Aldrich). Secondary antibodies were: donkey anti-mouse Alexa 488 (Invitrogen A21202), donkey anti-rabbit Alexa 594 (Invitrogen A211207), donkey anti-goat Alexa 647 (Invitrogen A21447).
Amyloid load analysis
Tissue collection and processing
Mice were euthanized by CO2 asphyxiation and the brain tissue collected for immunohistochemical analysis. After fixation in 4% paraformaldehyde and cryoprotection in 30% glycerol, 40 mm-thick floating sections were cut on a freezing microtome. Floating sections were permeabilized in 0.5% triton in TBS, blocked in 5% normal goat serum in TBS, and incubated with primary antibody (mouse anti-amyloid b Bam10, Sigma-Aldrich; rabbit anti-Iba-1, Wako) overnight at 4°C. Sections were then incubated with appropriate Alexa Fluor 488 or Alexa Fluor 568 conjugated secondary antibodies. Sections were mounted onto slides and coverslipped with Vectashield mounting medium with DAPI (Vector Labs, Burlingame, CA). For the counterstaining of dense-core amyloid plaques, the floating sections were incubated for 15 minutes in a solution of 1 μg/ml of Methoxy-XO4 (diluted in TBS) before mounting the slices with Fluoromount-G (No DAPI, SouthernBiotech, Birmingham, AL).
Stereology-based quantitative analyses
For the quantification of amyloid load, Alexa-568-anti-Amyloid immunolabeled and Methoxy-XO4 positive plaques were imaged using a NanoZoomer-XR Digital slide scanner (Hamamatsu Photonics, Shizuoka, Japan) under a 20X objective. The total surface of amyloid was determined using a custom-written script based on the “Analyze particle” function of Fiji (National Institutes of Health: http://fiji.sc/), after defining the cortex as region of interest. The total surface occupied by amyloid was then reported to the cortical area of each section considered. Stereology-based study of amyloid-associated microglia was performed on immunolabeled sections using an Olympus BX52 epifluorescent microscope equipped with motorized stage, DP70 digital CCD camera, and CAST stereology software (Olympus, Tokyo, Japan). The cortex was outlined and microglia counts were made using 20X high numerical aperture (1.2) objective. Using a meander sampling of 70% of cortical area, images were captured each time an amyloid deposit was encountered. Those images were then analyzed using Fiji, counting the number of Iba1-positive microglial cells close to a plaque (< 50 μm) and reporting this number to the surface of the plaque considered. All pathology quantification was carried out blinded until the last statistical analyses.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the respective figure legends and method sections. We provide here a brief summary.
Differential gene expression
Differential expression was performed using functions provided with the Seurat R package. To calculate p values, we used the Wilcoxon rank-sum test with default parameters, and we used a Bonferroni correction to calculate adjusted p values for multiple testing. Genes with adjusted p values < 0.05, and with a fold change (ln scale) > |0.2| were considered significantly differentially expressed.
RNAscope signal quantification and cell count
Around each plaque, or an arbitrary point in C57BL/6J mice, we drew five concentric circles of 18.2 μm. We counted microglia (Itgam+) and astrocytes (Slc1a3+) in each ring, and for each cell we measured the intensity of the signal from the Apoe probeset using the Fiji software.
Stereological quantification of amyloid burden and microglia density
Sections were imaged with a 20X objective and an epifluorescent microscope, and data were analyzed using a custom script based on the “Analyze particle” function of Fiji software. 5 to 7 mice per group werer analyzed, p values were calculated using the Mann-Whitney test.
DATA AND SOFTWARE AVAILABILITY
Raw and normalized gene expression data have been deposited in GEO (GEO: GSE127893). Data can be browsed interactively at: scope.bdslab.org
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD11b-PE, clone: M1/70.15.11.5 | Miltenyi Biotec | 130-113-797; RRID: AB_2726320 |
| anti-β-amyloid 1–16 6E10, mouse | BioLegend | 803004; RRID: AB_2715854 |
| anti-IBA1, rabbit | Wako-Chemicals | 019–19741; RRID: AB_839504 |
| anti-ApoE, goat | Sigma-Aldrich | AB947; RRID: AB_2258475 |
| atto-488-conjugated goat anti-mouse | Sigma-Aldrich | 62197–1ML-F; RRID: AB_1137649 |
| Alexa 488 donkey anti-mouse | Invitrogen | A21202; RRID: AB_1137649 |
| Alexa-594 donkey anti-rabbit | Invitrogen | A211207 |
| Alexa-647 donkey anti-goat | Invitrogen | A21447 |
| anti-β-amyloid BAM10, mouse | Sigma-Aldrich | A5213-.2ML; RRID: AB_476742 |
| Alexa-568 goat anti-mouse | Thermo Fisher | A-11004 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hibernate A without phenol red | Brain Bits | HAPR |
| D-PBS | Life Technologies | 14287072 |
| MACS BSA stock solution | Miltenyi Biotec | 130-091-376 |
| RNasin Plus RNase Inhibitor | Promega | N2615 |
| TrueBlack | Biotium | 23007 |
| Critical Commercial Assays | ||
| Adult Brain Dissociation kit | Miltenyi Biotec | 130-107-677 |
| Nextera XT kit | Illumina | FC-131–1096 |
| Nextera XT Index Kit v2 Set A (96 indexes, 384 samples) | Illumina | FC-131–2001 |
| Nextera XT Index Kit v2 Set B (96 indexes, 384 samples) | Illumina | FC-131–2002 |
| Nextera XT Index Kit v2 Set C (96 indexes, 384 samples) | Illumina | FC-131–2003 |
| Nextera XT Index Kit v2 Set D (96 indexes, 384 samples) | Illumina | FC-131–2004 |
| RNAscope | ACD | 320850 |
| Mm-Slc1a3 RNAscope probe | ACD | 430781 |
| Mm-Itgam-C2 RNAscope probe | ACD | 311491-C2 |
| Mm-Apoe-C3 RNAscope probe | ACD | 313271-C3 |
| Deposited Data | ||
| Raw data | This paper | GEO: GSE127893 |
| Analyzed data | This paper | scope.bdslab.org |
| Human RNaseq data (AMP-AD) | Allen et al., 2016 | MSBB synapse ID: syn3157743 ROSMAP synapse ID:syn3388564 |
| Experimental Models: Organisms/Strains | ||
| AppNL-G-F | Saito et al., 2014 | Available from the Saido lab. |
| C57BL/6J (control for AppNL-G-F) | Janvier | C57BL/6J@Rj |
| APPswe/PS1dE9 (APP/PS1) | Borchelt et al., 1997 | Available from The Jackson Laboratory, stock 34829-JAX |
| APP/PS1-Apoenull | Hudry et al., in preparation | Available from the Hyman lab. |
| C57BL/6J | The Jackson Laboratory | 000664 |
| C57BL/6J-Apoenull | The Jackson Laboratory | 002052 |
| Oligonucleotides | ||
| 5′-biotinylated template switching oligo: /5Biosg/AGCAGTGGTATCAACGCAGAGTACATrGrG+G | this paper | Exiqon/QIAGEN |
| 5′-biotinylated oligo-dT: /5Biosg/AAGCAGTGGTATCAACGCAGAGTACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN | Picelli et al., 2013 | IDT |
| 5′-biotinylated IS-PCR oligo: /5Biosg/AAGCAGTGGTATCAACGCAGAGT | Picelli et al., 2013 | IDT |
| Software and Algorithms | ||
| STAR RNA-seq aligner, v. 2.5.2 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Subread/Featurecounts, v. 1.5.1 | Liao et al., 2014 | http://subread.sourceforge.net/ |
| R, v. 3.4.4 | R core team | https://www.R-project.org/ |
| Seurat, v. 2.3.1 | Butler et al., 2018 | https://github.com/satijalab/seurat |
| Monocle, v. 2.6.4 | Qiu et al., 2017a | https://github.com/cole-trapnell-lab/monocle-release |
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
Highlights.
Exposure to Aβ in AD potentiates a microglia response present during normal aging
This microglia response is heterogeneous with potential synaptotoxic subtypes
Microglia in female mice develop this response faster than in male mice
Apoe deletion blocks the main response of microglia to Aβ
ACKNOWLEDGMENTS
Work in the De Strooper lab is supported by the Opening the Future campaign of the Leuven Universitair Fonds (LUF), the Alzheimer Research Foundation (SAO-FRA; P#16017), the Fonds voor Wetenschappelijk Onderzoek (FWO), VIB-KU Leuven, a Methusalem grant from KU Leuven and the Flemish Government, Vlaams Initiatief voor Netwerken voor Dementie Onderzoek (VIND; Strategic Basic Research Grant 135043), and the Alzheimer’s Association. B.D.S. is supported by the Geneeskundige Stichting Koningin Elisabeth and the Bax-Vanluffelen Chair for Alzheimer’s Disease. We were also supported by an anonymous foundation. Work in the Hyman lab is supported by a Massachusetts Alzheimer’s Disease Research Center grant (AG05134) and by the JPB Foundation. The ROSMAP project was supported by funding from the National Institute on Aging (AG034504 and AG041232). The MSBB data were generated from postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from the Mount Sinai School of Medicine. The authors thank Prof. Sarah Teichmann (Wellcome Sanger Institute) and members of her laboratory for helpful and useful discussions and Prof. Thierry Voet (KU Leuven) for help in the initial setup of the SmartSeq2 protocol. We thank Yannick Fourne (VIB-KU Leuven) and Kristofer Davie (VIB-KU Leuven) for help in the setup of the online data browser. Cell sorting was performed at the KU Leuven FACS core facility, and sequencing was carried out by the VIB Nucleomics Core.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.099.
DECLARATION OF INTERESTS
M.E.W., G.S., T.M., and E.K. are employed by AbbVie, Inc. AbbVie, Inc., has subsidized part of the study. B.D.S. is a consultant for several companies. All the other authors declare no competing interests.
REFERENCES
- Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, Burgess JD, Chai H-S, Crook J, Eddy JA, et al. (2016). Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, et al. (2017). Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. ; Dominantly Inherited Alzheimer Network (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C, and Ziegler C (2017). Beyond amyloid—widening the view on Alzheimer’s disease. J. Neurochem 143, 394–395. [DOI] [PubMed] [Google Scholar]
- Bihlmeyer NA, Merrill EM, Lambert Y, Srivastava GP, Clark TW, Hyman BT, and Das S (2019). Novel Methods for Integration and Visualization of Genomics and Genetics Data in Alzheimer’s Disease. Alzheimers Dement. Published online March 29, 2019. 10.1016/j.jalz.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, and Sisodia SS (1997). Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19, 939–945. [DOI] [PubMed] [Google Scholar]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, et al. ; Alzheimer Disease Neuroimaging Initiative (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci 16, 848–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, and Weiner HL (2018). Microglial signatures and their role in health and disease. Nat. Rev. Neurosci 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-b-dependent molecular and functional signature in microglia. Nat. Neurosci 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu AQ, and Pritchard JK (2017). Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet 101, 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, and Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, and Goate AM (2015a). Cruchaga & Goate reply. Nature 520, E5–E6. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, and Goate AM (2015b). Cruchaga & Goate reply. Nature 520, E10. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, et al. ; Alzheimer’s Research UK (ARUK) Consortium (2014). Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Shoai M, Pither R, Williams J, and Hardy J (2017). Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol. Aging 49, 214, e7–214.e11. [DOI] [PubMed] [Google Scholar]
- Fazzari P, Horre K, Arranz AM, Frigerio CS, Saito T, Saido TC, and De Strooper B (2017). PLD3 gene and processing of APP. Nature 541, E1–E2. [DOI] [PubMed] [Google Scholar]
- Ferretti MT, Iulita MF, Cavedo E, Chiesa PA, Schumacher Dimech A, Santuccione Chadha A, Baracchi F, Girouard H, Misoch S, Giacobini E, et al. ; Women’s Brain Project and the Alzheimer Precision Medicine Initiative (2018). Sex differences in Alzheimer disease—the gateway to precision medicine. Nat. Rev. Neurol 14, 457–469. [DOI] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee S-H, Haddick PCG, Ngu H, et al. (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847. [DOI] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, and McColl BW (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci 19, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, and Tanzi RE (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, et al. ; Alzheimer Genetic Analysis Group (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JR, and Owen MJ (2016). Alzheimer’s disease: the amyloid hypothesis on trial. Br. J. Psychiatry 208, 1–3. [DOI] [PubMed] [Google Scholar]
- Hartl D, May P, Gu W, Mayhaus M, Pichler S, Spaniol C, Glaab E, Bobbili DR, Antony P, Koegelsberger S, et al. ; AESG (2018). A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry, Published online July 9, 2018. 10.1038/s41380-018-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann S, Drichel D, Clarimon J, Fernández V, Lacour A, Wagner H, Thelen M, Hernández I, Fortea J, Alegret M, et al. (2015). PLD3 in non-familial Alzheimer’s disease. Nature 520, E3–E5. [DOI] [PubMed] [Google Scholar]
- Herrup K (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci 18, 794–799. [DOI] [PubMed] [Google Scholar]
- Hodes RJ, and Buckholtz N (2016). Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) knowledge portal aids Alzheimer’s drug discovery through open data sharing. Expert Opin. Ther. Targets 20, 389–391. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, and Paul SM (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 97, 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooli BV, Lill CM, Mullin K, Qiao D, Lange C, Bertram L, and Tanzi RE (2015). PLD3 gene variants and Alzheimer’s disease. Nature 520, E7–E8. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, and Hyman BT (2000). Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid beta-peptide deposition in homozygous APP(V717F) transgenic mice. Acta Neuropathol. 100, 451–458. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Cruchaga C, and Goate AM (2014). Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, and Hardy J (2014). A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol 76, 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, and De Strooper B (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. ; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-C, Grenier-Boley B, Bellenguez C, Pasquier F, Campion D, Dartigues J-F, Berr C, Tzourio C, and Amouyel P (2015). PLD3 and sporadic Alzheimer’s disease risk. Nature 520, E1. [DOI] [PubMed] [Google Scholar]
- Laws KR, Irvine K, and Gale TM (2018). Sex differences in Alzheimer’s disease. Curr. Opin. Psychiatry 31, 133–139. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Makin S (2018). The amyloid hypothesis on trial. Nature 559, S4–S7. [DOI] [PubMed] [Google Scholar]
- Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, Davies G, Ritchie CW, Gale CR, Starr JM, et al. (2018). GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Kobayashi Y, Kogo N, Saito T, Saido TC, and Itohara S (2016). Cognitive deficits in single App knock-in mouse models. Neurobiol. Learn. Mem 135, 73–82. [DOI] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, and Tsai L-H (2017). Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure CM, and Swendsen J (2016). Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 15, 451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, and Berg L (1991). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, Lelios I, Heppner FL, Kipnis J, Merkler D, et al. (2018). High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48, 380–395.e6. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017a). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma Y-A, and Trapnell C (2017b). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci 19, 987–991. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Reiter JS, Strickland DK, and Hyman BT (1993). Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11, 575–580. [DOI] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, and Saido TC (2014). Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci 17, 661–663. [DOI] [PubMed] [Google Scholar]
- Salih DA, Bayram S, Guelfi MS, Reynolds RH, Shoai M, Ryten M, Brenton J, Zhang D, Matarin M, Botia J, et al. (2018). Genetic variability in response to Aβ deposition influences Alzheimer’s risk. bioRxiv. 10.1101/437657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, and Saido TC (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36, 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, and Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, and Hyman BT (2011). Neuro-pathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. ; ARUK Consortium; GERAD/PERADES, CHARGE, ADGC, EADI (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, Borneo J, Poon C, Ho T, Cai F, et al. (2016). Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun 7, 11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, and Tanzi RE (2013). ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron 80, 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, and Regev A (2014). Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr. Protoc. Mol. Biol 107, 4–22, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nyström S, Nilsson KP, Song WM, Zhou Y, Reinartz M, Choi S, Jiang H, et al. (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med 215, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, and Scheltens P (2005). Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 76 (Suppl 5), v2–v7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee SJ, Holstege H, Wong TH, Jakobsdottir J, Bis JC, Chouraki V, van Rooij JGJ, Grove ML, Smith AV, Amin N, et al. (2015). PLD3 variants in population studies. Nature 520, E2–E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E, and Maggi A (2018). Sex-specific features of microglia from adult mice. Cell Rep. 23, 3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, and Pocock JM (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol 36, 74–81. [DOI] [PubMed] [Google Scholar]
- Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q, Ming C, Neff R, Ma W, Fullard JF, et al. (2018). The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data 5, 180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, and Sheng M (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and normalized gene expression data have been deposited in GEO (GEO: GSE127893). Data can be browsed interactively at: scope.bdslab.org