

Abstract
Fibrinogen levels and extent of fibrin polymerization have been associated with such pathological conditions as cardiovascular disease, arteriosclerosis, and coagulation disorders. Activated Factor XIII (FXIIIa) introduces γ-glutamyl-ε-lysinyl isopeptide bonds between reactive glutamines and lysines in the fibrin network to form a blood clot resistant to fibrinolysis. FXIIIA crosslinks the γ-chains and at multiple sites in the αC region of fibrinogen. Fibrinogen αC (Fbg αC) contains a FXIII binding site involving αC (389–402) that is located near three glutamines whose reactivities rank Q237 >> Q366 ≈ Q328. Mass spectrometry and 2D HSQC Nuclear Magnetic Resonance assays were used to probe the anchoring role that αC E396 may play in controlling FXIII function and characterize the effects of Q237 on the reactivities of Q328 and Q366. Studies with αC (233–425) revealed that the E396A mutation does not prevent the transglutaminase function of FXIII A2 or A2B2. Other residues must play a compensatory role in targeting FXIII to αC. Unlike full fibrinogen, Fbg αC (233–425) did not promote thrombin cleavage of FXIII, an event contributing to activation. With the αC (233–425) E396A mutant, Q237 exhibited slower reactivities compared to αC WT consistent with difficulties in directing this N-terminal segment toward an anchored FXIII interacting at a weaker binding region. Q328 and Q366 became less reactive when Q237 was replaced with inactive N237. Q237 crosslinking is proposed to promote targeting of Q328 and Q366 to the FXIII active site. FXIII thus uses Fbg αC anchoring sites and distinct Q environments to regulate substrate specificity.
Keywords: Fibrinogen αC, Factor XIII, coagulation, mass spectrometry, NMR
Introduction
Fibrinogen (Fbg), Factor XIII (FXIII), and thrombin play important roles at the end of the blood coagulation cascade helping to form a stable clot that is resistant to fibrinolysis.1,2 The most abundant of these three clotting factors is fibrinogen (AαBβγ).3,4 The N-termini of the fibrinogen chains form the central E region. The fibrinogen chains then extend via a coil-coiled region to the two distal D regions. The highly flexible αC region extends further from the D region and becomes tethered to the E region.1,5,6
Fibrin polymerization is initiated by thrombin-catalyzed cleavage of fibrinopeptides A and B from the fibrinogen Aα and Bβ chains.7 The αC region becomes exposed following cleavage of fibrinopeptide B. Thrombin-activated Factor XIII (FXIIIa) is later responsible for introducing γ-glutamyl-ε-lysyl isopeptide bonds between specific glutamines (Q) and lysines (K) on the fibrin α and γ chains.8–11 FXIIIa first crosslinks fibrin to form γ-γ dimers followed by α-α dimers, γ-α hybrids, and higher order α polymers.12,13 Fibrinogen also provides a suitable surface for binding several components required for clot stability including platelets, red blood cells, and antifibrinolytic agents.14–16
Recent studies have emphasized the role of the αC region in promoting several functions of fibrinogen. FXIIIa-catalyzed crosslinking of the fibrin α-chain resulted in increased fiber thickness and tautness and decreased fibrin clot lysis rate.17 Moreover, the αC crosslinking was found to increase clot stiffness to a greater extent than γ crosslinking. Microscopy studies revealed that removal of the αC regions slows down fibrin clot formation leading to a denser network composed of longer protofibrils and thinner fibers.18,19 Interestingly, the absence of the αC region causes red blood cell retention in the fibrin clot to decrease.14,20,21 Furthermore, FXIIIa-catalyzed crosslinking within the αC region is an important contributor to this red blood cell retention.14,20,21
Previous studies demonstrated that activation of FXIII A2B2 was enhanced by a segment on the αC region, αC (242–424).22 In the presence of this αC region, the optimal calcium concentration for FXIII activation reduced to 1.5 mM. In this environment, more effective thrombin-catalyzed hydrolysis of the FXIII activation peptide was reported, later followed by dissociation of FXIII B2 from thrombin-cleaved A2’. A FXIII A2 binding site on fibrinogen was localized to αC (389–402) using antibodies specific for Fbg Aα, Bβ, and γ chains.23 The anti-Aα 389–402 antibody decreased the ability of FXIII to bind to fibrinogen further supporting the proposal that the FXIII binding site was positioned in this region.23
Using Surface Plasmon Resonance, Smith et al. demonstrated that FXIII A2B2 and thrombin-activated FXIII both bound to recombinant Fibrinogen αC (233–425).24 However, higher affinity was observed for FXIII A2B2. The short Fbg αC segment (389PDWGTFEEVSGNVS402) is highly conserved across species.24 The presence of Pep1, a synthetic version of this conserved αC (389–402) region, substantially inhibited FXIII’s binding interaction with αC and delayed crosslinking activity. Results revealed that the αC (389–402) E396A mutation caused the greatest loss of this inhibitory effect.25 Together these findings suggested that E396 and possibly neighboring residues promote FXIII’s ability to bind to fibrinogen. However, the data remained unclear whether the E396A substitution also alters FXIII’s ability to crosslink each reactive glutamine.
Like other transglutaminases, FXIIIa exhibits a higher selectivity and specificity for the glutamine substrate and a relatively low selectivity for the amine substrate.8,10 The source of the glutamine substrate specificity however remains unclear. The lysine mimic glycine ethyl ester (GEE) has been routinely used in FXIIIA experiments to identify reactive glutamines within fibrinogen.26–28 In prior studies, crosslinking of individual reactive glutamines Q237, Q328, and Q366 in Fbg αC (233–425) was examined using a combination of mass spectrometry and NMR assays.29–31 Our mass spectrometry assays revealed reactive glutamine ranking to GEE of Q237 >> Q366 ≈ Q328.29 This ranking aligns with the number of glutamine–lysine crosslinking pairs identified by Wang et al. in full-length fibrinogen.32 Q237 was involved in more lysine crosslinking pairs than either Q366 or Q328, correlating with our reactivity ranking for Q237.
The aims of the current project were thus two-fold: 1) to probe the anchoring role that αC E396 may play in controlling FXIIIa enzymatic function and 2) to characterize the effects of Q237 on the reactivities of Q328 and Q366. Our MALDI-TOF mass spectrometry assays and 15N HSQC NMR studies revealed that FXIIIa crosslinks GEE to Q237, Q328, and Q366 to similar extents in Fbg αC (233–425) and the αC E396A mutant. Both the active site subunit FXIII A2 and circulatory FXIII A2B2 were examined. Although Fbg αC E396 is a key anchoring residue for FXIII binding, our results suggest that this residue is not required for controlling FXIII catalytic function. Fbg αC Q237 remains the most reactive glutamine within αC (233–425), irrespective of the E396A mutation. Intriguingly, Q237 reactivity is somewhat hindered suggesting difficulties targeting this Q residue to FXIII anchored at a weaker αC binding site. To further explore Q237 influences, an LC mass spectrometry-based assay was used to assess the reactivities of Q328 and Q366 within αC (233–425) Q237N. Results revealed that Q328 and Q366 become less reactive when Q237 is replaced with inactive N237. The ability of Q237 to crosslink with a lysine partner is, therefore, hypothesized to promote a conformation that directs Fbg αC residues Q328 and Q366 toward the FXIII active site.
Materials and Methods
Proteins and Materials
FXIII A2 expressed in S. cerevisiae was generously provided by the late Dr. Paul Bishop (ZymoGenetics). Plasma FXIII A2B2 was purchased from Enzymes Research Laboratories (South Bend, IN). Molar concentration in this work refers to the A subunit protomers of FXIII. Bovine thrombin, glycine ethyl ester (GEE), and other assay components were obtained from Sigma Aldrich (St. Louis, MO). 15N-labeled GEE was from Cambridge Isotopes (Tweksbury, MA).
Fibrinogen αC (233–425) WT (Fbg αC WT) was expressed and purified as described previously.29 To introduce the E396A mutation into this fibrinogen segment, primer sets were designed with codon GAG (for E396) mutated to codon GCG (for A396) (Supplementary Table S1). The QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) was then employed with the WT cDNA serving as the template. The E396A mutation was confirmed by DNA sequencing. The DNA was subsequently transformed into BL21-Gold(DE3) E. coli cells. The Fbg αC (233–425) E396A protein was then expressed and purified using the same methods as Fbg αC (233–425) WT.29 A similar mutagenesis strategy was employed to generate Fbg αC (233–425) Q237N.
All variants of GST-Fbg αC (233–425) were purified by affinity chromatography with on-column cleavage of the GST-tag achieved using the human rhinovirus 3C protease.29 Protein concentrations were determined at 280 nm employing an extinction coefficient of 41480 M−1cm−1 (expasy.org). Fbg αC (233–425) mutations were further verified using MALDI-TOF mass spectrometry (Voyager DE-PRO, Applied Biosystems). GluC and chymotrypsin digests of Fbg αC (233–425) were employed to confirm the E396A and the Q237N mutations. In addition, the presence of peptides containing Q237, Q328, and/or Q366 were also verified (Supplementary Table S2).
2D 1H-15N HSQC NMR Assay
To test the ability of FXIIIa to crosslink all three reactive glutamines in Fbg αC E396A, a 2D 1H-15NHSQC NMR experiment was performed.29 Briefly, FXIII A2 was activated by incubating 800 nM FXIII A-subunits with thrombin (21 NIH U/mL) and CaCl2 (5 mM) for 10 min at 37°C. Thrombin was inhibited with 200 nM PPACK. The crosslinking reaction was initiated by adding [15N]-GEE (10mM) and the glutamine-containing substrate Fbg αC E396A (40 μM) to activated FXIIIA. The total assay volume was 400 μL in 20 mM borate buffer (pH 8).29 2D HSQC NMR experiments were performed on a Varian Inova 700 MHz NMR spectrometer as described previously.29
MALDI-TOF mass spectrometry kinetic assay
A modified version of our mass spectrometry kinetic assay was used to monitor the FXIIIa -catalyzed crosslinking reaction between reactive glutamines in Fbg αC and the lysine mimic GEE.29 Physiologically, the FXIII A2B2 concentration in plasma is 14–28 mg/L corresponding to 86–173 nM of FXIII A-subunits.33 Our assays were carried out with 500 and 2000 nM FXIIIA. Previously published mass spectrometry studies also used higher concentrations in the 360–600 nM FXIIIA range to monitor crosslinking.30,32
The crosslinking ability of individual glutamines in Fbg αC (233–425) WT versus the E396A mutant were compared in the presence of activated FXIII A2 and FXIII A2B2. FXIII A2 or FXIII A2B2 (500 nM of the protomer FXIIIA) was first incubated with GEE (17 mM) and (Fbg αC WT or E396A) (13.6 μM final concentration) for 5 min at 37°C in MALDI Kinetic buffer (100 mM Tris acetate, 150 mM NaCl and 0.1% PEG8000 pH 7.4). This step provided the zymogen FXIII with an opportunity to interact with the Fbg αC surface. Thrombin (8.4 U/ml) and CaCl2 (4 mM) were added to the reaction mixture to activate the FXIII A2 or FXIII A2B2. In the continued presence of calcium, the transglutaminase reaction was initiated. At different time points, 25 μL aliquots were removed from this reaction and quenched with 1.6 μL of 160 mM EDTA (10 mM). Samples from this reaction were digested separately with Chymotrypsin or GluC and then analyzed by mass spectrometry.29 The peak height ratio method was used to compare crosslinking within Fbg αC WT versus E396A.29 For some studies, the FXIIIA concentration was raised to 2 μM, and the amount of thrombin for FXIII activation was increased accordingly. The different assays were carried out in triplicate. Averages for the individual Q-consumption time points were determined and their standard deviations calculated. The data were analyzed by Student T-test and by p-values (GraphPad InStat3).
Liquid chromatography-mass spectrometry (LC-MS) kinetic assay
A LC-MS assay was employed to quantitatively rank the reactivities of Q328 and Q366 when the most reactive glutamine residue Q237 was mutated to an inactive asparagine (N). FXIIIa-catalyzed Q-GEE crosslinking reactions with Fbg αC (233–425) WT were compared to Q237N. Kinetic reactions were performed in the same manner as for the MALDI-TOF MS assay.29 Final concentrations of activated FXIII A2 and Fbg αC (233–425) species were maintained at 500 nM FXIIIA and 13.6 μM αC. Quenched time point aliquots were digested separately with Chymotrypsin or GluC, and then analyzed using the LC-MS approach.
The digested samples were then prepared for analysis as follows. Individual samples were subjected to a ZipTip cleanup procedure and frozen. SpeedVac dried samples were dissolved in 30 μL 2% v/v acetonitrile / 0.1% v/v formic acid and 4 μL were analyzed. A 250 μm ID × 15 cm Radel R tube (Idex Health & Science LLC, Oak Harbor, WA, USA) was packed in-house with Aeris Peptide 3.6 μm XB-C18 material (Phenomenex, Torrance, CA, USA) and run on a Acquity M-Class UPLC® system (Waters Corporation, Milford, MA, USA). Sample separation was accomplished with a 40 min linear gradient from 2% B to 40% B (water to acetonitrile in 0.1% v/v formic acid), followed by a 5 min linear gradient from 40% B to 85% B, and then a 5-minute wash with 85% B. A Low Flow Electron Spray Ionization (ESI) probe was employed to introduce sample into a ZSpray LockSpray source (Waters).
A Synapt G2-Si mass spectrometer was used to collect data from the LC eluate. An MSe Continuum method was created in MassLynx v4.1 SCN924 (Waters). The spectrometer was operated in positive resolution mode. Leucine enkephalin (556.2771 Da/e) at 300 pg/μL in 1:1 acetonitrile : water was used as the lock mass. Skyline data analysis software (https://skyline.ms/, v3.6) and FASTA files containing the Fbg αC amino acids 233–425 (± the Q237N variant sequence) were employed in this project. This combination was utilized to identify chymotryptic- and GluC- precursor ions and fragment ion series needed for peptide validation and for precursor quantification. Post-translational modifications were constrained to the addition of H6C4O2 (monoisotopic mass 86.036779) from glycine ethyl ester (GEE; www.unimod.org) to glutamine residues.34 The peak height ratio method was later used to follow the glutamine reactions over time.35
Influence of αC (233–425) on thrombin-catalyzed activation of FXIII A2 and A2B2
An SDS-PAGE assay was used to monitor the possible influence of Fbg αC (233–425) on thrombin’s ability to cleave the FXIII activation peptide. FXIII A2 or A2B2 (1 μM of FXIIIA) was incubated with 5 μM αC (233–425) and 4 mM CaCl2 in Tris-Acetate buffer for 10 minutes at 37° C. During this time period, the FXIII species could interact with the Fbg αC. A final concentration of 30 nM human recombinant thrombin was added to the mixture. Control experiments were also performed in the absence of αC (233–425). Aliquots were removed at timed intervals and quenched with the thrombin inhibitor PPACK. Samples were run under reducing conditions on 8% SDS-PAGE gels and stained with Commassie Blue. ImageJ was used for densitometric analysis. Thrombin-cleaved FXIIIA appeared at a lower molecular weight consistent with removal of the 4kDa activation peptide. Plots were generated for % cleavage as a function of time.
Results
FXIIIa crosslinks all three reactive glutamines in Fbg αC (233–425) E396A
A 2D 1H −15N HSQC NMR assay was used to monitor the ability of FXIIIa to crosslink all three reactive glutamines in Fbg αC (233–425) E396A to 15N-labeled GEE. This NMR approach provided a direct method to quickly assess whether the loss of the acidic E396 residue in Fbg αC (233–425) would markedly hinder the reactivity of FXIIIa. With this heteronuclear NMR method, a single NMR peak is expected for each reactive glutamine that becomes crosslinked to [15N]-GEE.29 The 2D 15N-HSQC approach effectively tests for reactivity in a 40 μM αC (233–425) sample after a single 30 min incubation time point followed by a two hour NMR experiment. The presence or absence of all possible crosslinking reactions with GEE can be visualized in a single spectrum. The NMR results on Fbg αC E396A revealed three distinct peaks with each 2D cross peak appearing at a similar 1H and 15N chemical shift position as observed previously with Fbg αC WT.29 (Fig 1) These results suggest that the E396A mutation does not detrimentally affect the ability of FXIIIa to crosslink Q237, Q328, and Q366 in Fbg αC (233–425).
Figure 1: 2D 1H-15N HSQC NMR spectrum demonstrating that FXIIIA can crosslink 15N-labeled GEE to the reactive glutamines Q237, Q328, and Q366 of Fbg αC (233–425) E396A.
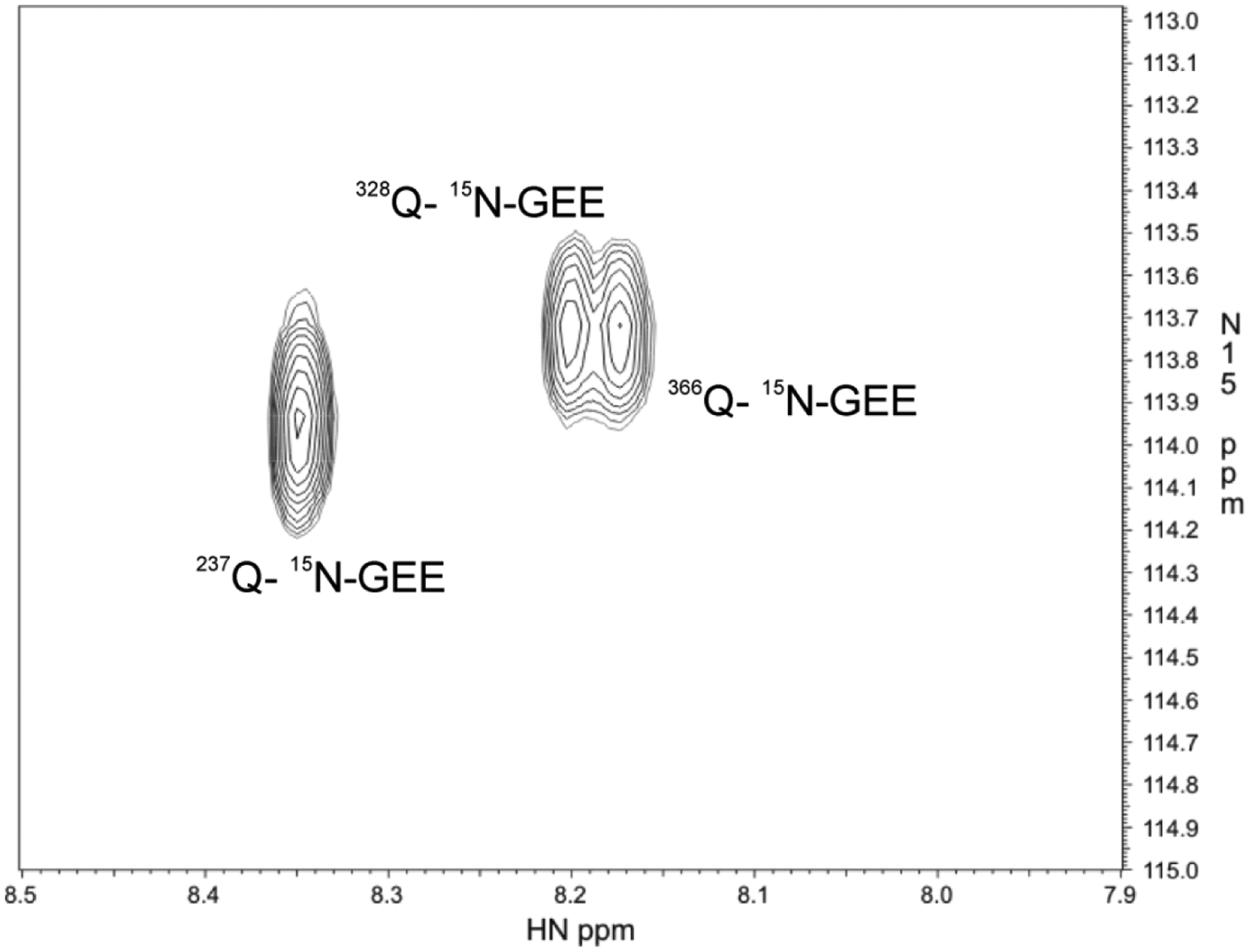
The glutamine side chains of Fbg αC (233–425) E396A can each be crosslinked with a 15N labeled version of the lysine mimic glycine ethyl ester. The individual crosspeaks then correspond to a proton (1H) that is covalently attached to a specific 15N label. A series of Q to inactive N mutants was used previously to match each crosspeak to a particular αC glutamine.
MALDI –TOF mass spectrometry kinetic assays comparing FXIII A2 catalyzed crosslinking of reactive glutamines Q237, Q328, and Q366 in Fbg αC (233–425) WT versus E396A.
Our mass spectrometry-based kinetic assay has the advantage of better monitoring over time the crosslinking of each reactive Q to GEE. Moreover, subtle differences in reactivities of the glutamines can thus be probed with mass spectrometry. Under our kinetic assay conditions, Q237 becomes completely crosslinked to GEE within 10 minutes of the reaction.29 To allow for quantitative comparisons between the reactive Q237 in Fbg αC WT versus Fbg αC E396A, the MS assay was modified to include a series of shorter time points. In earlier studies, FXIII A2 was pre-activated with thrombin and calcium, and the reaction was initiated by adding Fbg αC (233–425), the glutamine substrate.29 For the current project, zymogen FXIII was first incubated with Fbg αC (WT or E396A), GEE, and MALDI kinetic buffer to allow FXIII binding and interaction with the Fbg αC substrate. The transglutaminase reaction was then initiated by adding thrombin and calcium to activate FXIII (an in-assay activation approach).
The MALDI-TOF MS assay results revealed that activated FXIII A2 crosslinked the Q237 of Fbg αC E396A to GEE at a somewhat slower rate than with Fbg αC WT (Fig 2A). The 0 min time point corresponds to a reaction quench that occurs right after all components have been added and the mixture vortexed. For the first six minutes, the reactivity of Q237 with Fbg αC E396A was slower than that with Fbg αC WT. A statistical difference was confirmed between the rate of Q-reactant consumption of WT and E396A Fbg αC for such early time points. For the first 2–5 minutes, the p-value range was 0.01–0.013 and at 6 minutes the p-value was 0.059. Overall, the E396A substitution did not drastically hinder the reactivity of Q237; however, the rate was affected. The putative binding region within αC (233–425) may still contribute a modest effect in assuring that FXIII is positioned at or near αC and thus ready to target Q residues.
Figure 2: Comparing FXIII A2-catalyzed crosslinking of (Q237, Q328, and Q366) in WT Fbg αC (233–425) and the mutant αC E396A.
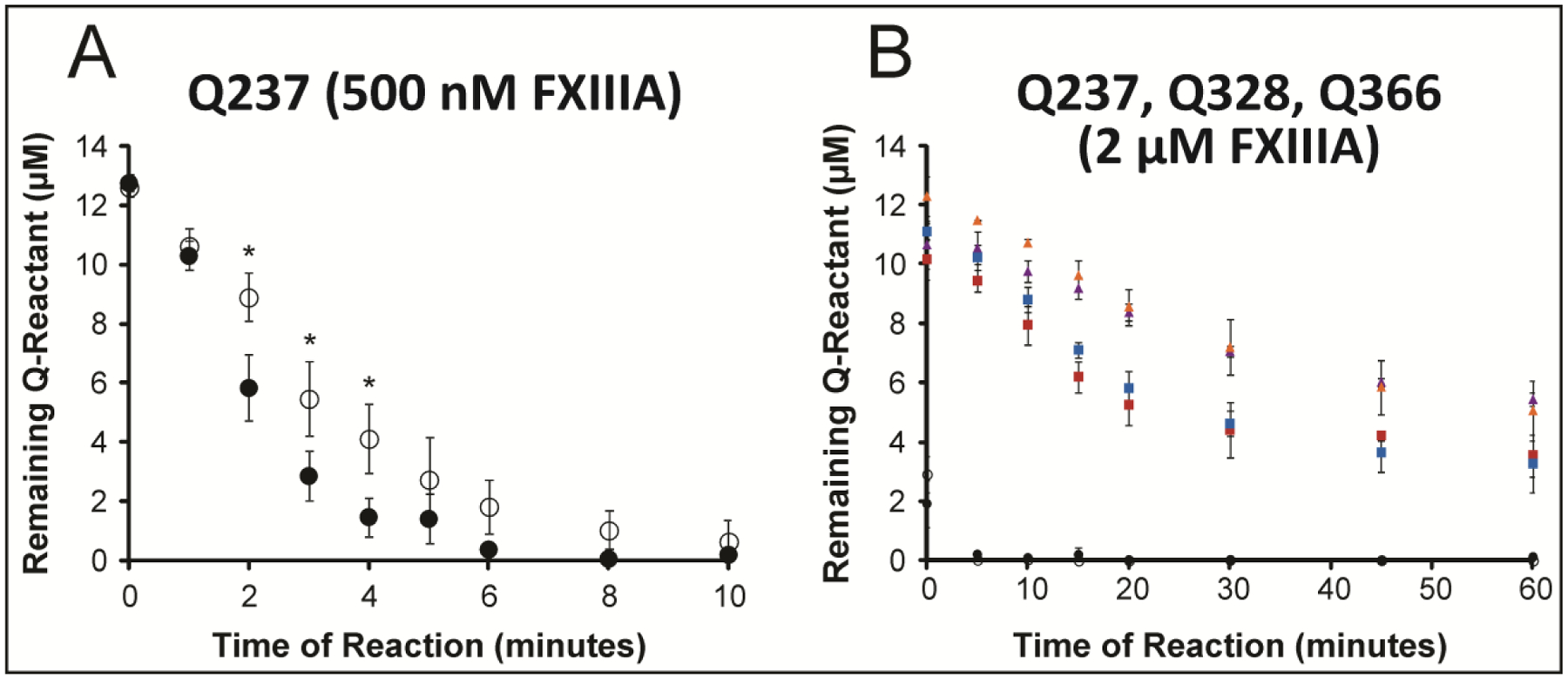
Two FXIIIa concentrations were employed in these trials. A) Reactivity of Q237 in WT Fbg αC (233–425) (black filled circles) versus the E396A mutant (black open circles) was monitored for 10 minutes in the presence of FXIII A2 (500 nM FXIIIA). For 2–4 minutes, the p-values were (0.008, 0.039, 0.128) and at 6 minutes the p-value was 0.059. B) Glutamine reactivities in WT Fbg αC (233–425) versus the E396A mutant were monitored for 60 minutes in the presence of 2 μM FXIIIA. The individual Q residues include Q328 WT (purple triangle), Q328 E396A (golden triangle), Q366 WT (red square), Q366 E396A (blue square), Q237 WT (black filled circle), Q237 E396A (black open circle). For A) and B), the peak-height ratio method was employed to calculate the amount of Q reactant left following reactions with GEE. Experiments were performed in triplicate, and the results reported as mean ± SD. Asterisk (*) for p-values less than 0.05.
To examine the effect of Fbg αC E396A on the other two reactive glutamines Q328 and Q366, a higher concentration of FXIII A2 (2 μM FXIIIA instead of 0.5 μM) was used, and reactivities were monitored over an hour. Q-rankings would not change while the assay would benefit from more extensive Q-substrate consumption over that time period. The data collected reveal that Q328 and Q366 are crosslinked in Fbg αC E396A with a similar ranking as in the WT Fbg αC. During the first few minutes of the reaction, the amount of remaining Q328 reactant from Fbg αC E396A is somewhat greater compared to the Fbg αC WT (Fig 2B) suggesting lower reactivity. With αC E396A, FXIII may not be as well directed to the αC (389–402) binding site as observed with WT αC. Nonetheless, the ability for FXIIIa to crosslink all reactive glutamines, with a similar ranking to the WT, is still maintained.
In previous studies, the FXIIIA concentration was 500 nM and the time points were from 0 to 35 minutes.29 The glutamine rankings were reported as Q237 >> Q366 ≈ Q328. When the FXIIIA concentration was increased 4-fold to 2 μM and the time points extended to 60 minutes, the Q366 was found to be more reactive than Q328. The glutamine rankings can thus be updated to Q237 >> Q366 ≥ Q328.
MALDI –TOF mass spectrometry kinetic assay comparing crosslinking with FXIII A2B2 in Fbg αC (233–425) WT and E396A.
To investigate the role of the carrier FXIII B-subunits in controlling FXIII’s activation and subsequent crosslinking ability, the MALDI-TOF mass spectrometry kinetic assay was performed with FXIII A2B2 in the presence of Fbg αC WT versus E396A. The FXIII B-subunits have been proposed to contribute to the higher affinity of FXIII A2B2 for αC (233–425).24 During plasma FXIII activation, regulatory FXIII B2 subunits are released after FXIII A2 is thrombin-cleaved.8 FXIII A2B2 (500 nM FXIIIA final) was incubated with Fbg αC (233–425) WT or E396A in the presence of MALDI kinetic buffer and GEE. The reaction was initiated by adding calcium and thrombin. Quenched time points were analyzed by MALDI-TOF mass spectrometry.
To examine the most reactive glutamine Q237, time points were first collected with one-minute intervals (Fig 3A). Results suggest that Q237 can be crosslinked to GEE in the presence of FXIII A2B2 for both substrates. The extent of crosslinking was modestly reduced for the Q237 of Fbg αC E396A when compared to the WT (Figure 3A). For the time period of 2–5 minutes, the p-value range (0.01–0.02) was statistically significant. Interestingly, the rate of Q237 consumption was slower for reactions with FXIII A2B2 than for FXIII A2 (Fig 2A, 3A). Reactive Q328 and Q366 were also monitored at longer time points and the extent of crosslinking compared for Fbg αC WT and E396A. Results indicate that all three reactive glutamines can still be crosslinked with a similar ranking when FXIII A2B2 is used (Fig 3B). As observed with FXIII A2, crosslinking is slightly reduced in the presence of Fbg αC E396A.
Figure 3: Comparing FXIII A2B2-catalyzed crosslinking of (Q237, Q328, and Q366) in WT Fbg αC (233–425) and the mutant αC E396A during two different assay time frame.
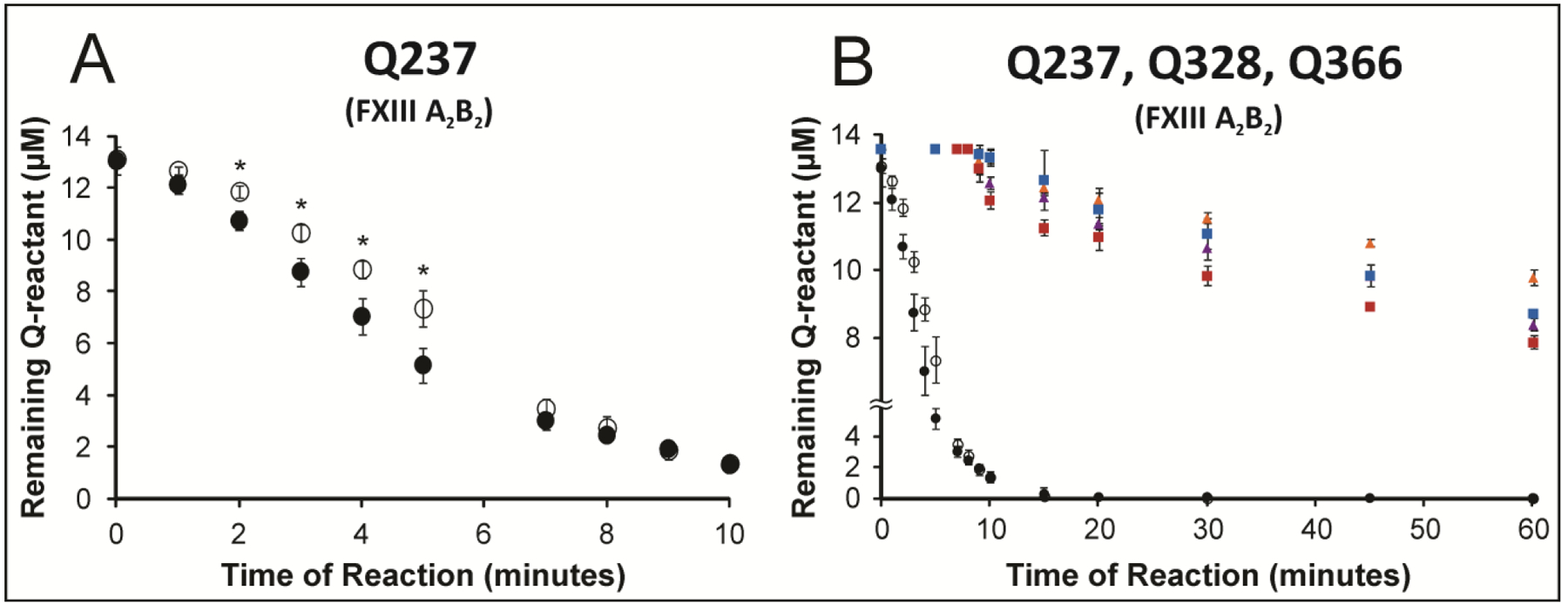
Two different assay times were used for the same FXIIIa concentration. A) Reactivity of Q237 in WT Fbg αC (233–425) (black filled circle) versus the E396A mutant (black open circle) was monitored for 10 minutes in the presence of FXIII A2B2 (500 nM FXIIIA). For 2–5 minutes, the p-values were (0.011, 0.014, 0.016, 0.017), respectively. B) Glutamine reactivities in WT Fbg αC (233–425) versus the E396A mutant were monitored for 60 minutes in the presence FXIII A2B2 (500 nM FXIIIA). The individual Q residues include Q328 WT (purple triangle), Q328 E396A (golden triangle), Q366 WT (red square), Q366 E396A (blue square), Q237 WT (black filled circle), Q237 E396A (black open circle). For A) and B), the peak-height ratio method was used to calculate the amount of Q reactant left following reactions with GEE. Experiments were performed in triplicate and the results reported as mean ± SD. Asterisk (*) for p-values less than 0.05.
Influence of αC (233–425) on thrombin-catalyzed activation of FXIII A2 and A2B2
Thrombin was used to proteolytically activate both FXIII A2 and FXIII A2B2. Fbg αC (233–425) has been reported to enhance FXIII activation, but the actual cleavage of the FXIII activation peptide segment at the R37-G38 peptide bond has not been studied extensively.22 For cleavage of FXIII A2, a new lower molecular weight band appears (Fig 4A) for the thrombin-cleaved FXIIIA (83 kDa decreased to 79 kDa). More than 50% of the FXIII A2 units are cleaved by 5 minutes (Fig 4B). The presence of Fbg αC (233–425) WT (20 kDa) did not accelerate the cleavage of the FXIII A2 as shown by the gel results. The lack of an effect was also observed for FXIII A2B2 in the absence and presence of αC (233–425) even though FXIII A2B2 exhibits a higher affinity for αC (233–425) than activated FXIII A2.24 (Fig 4C, 4D)
Figure 4: Evaluating the influence of WT Fbg αC (233–435) on thrombin-catalyzed cleavage of the FXIII A2 and FXIII A2B2 activation peptide.
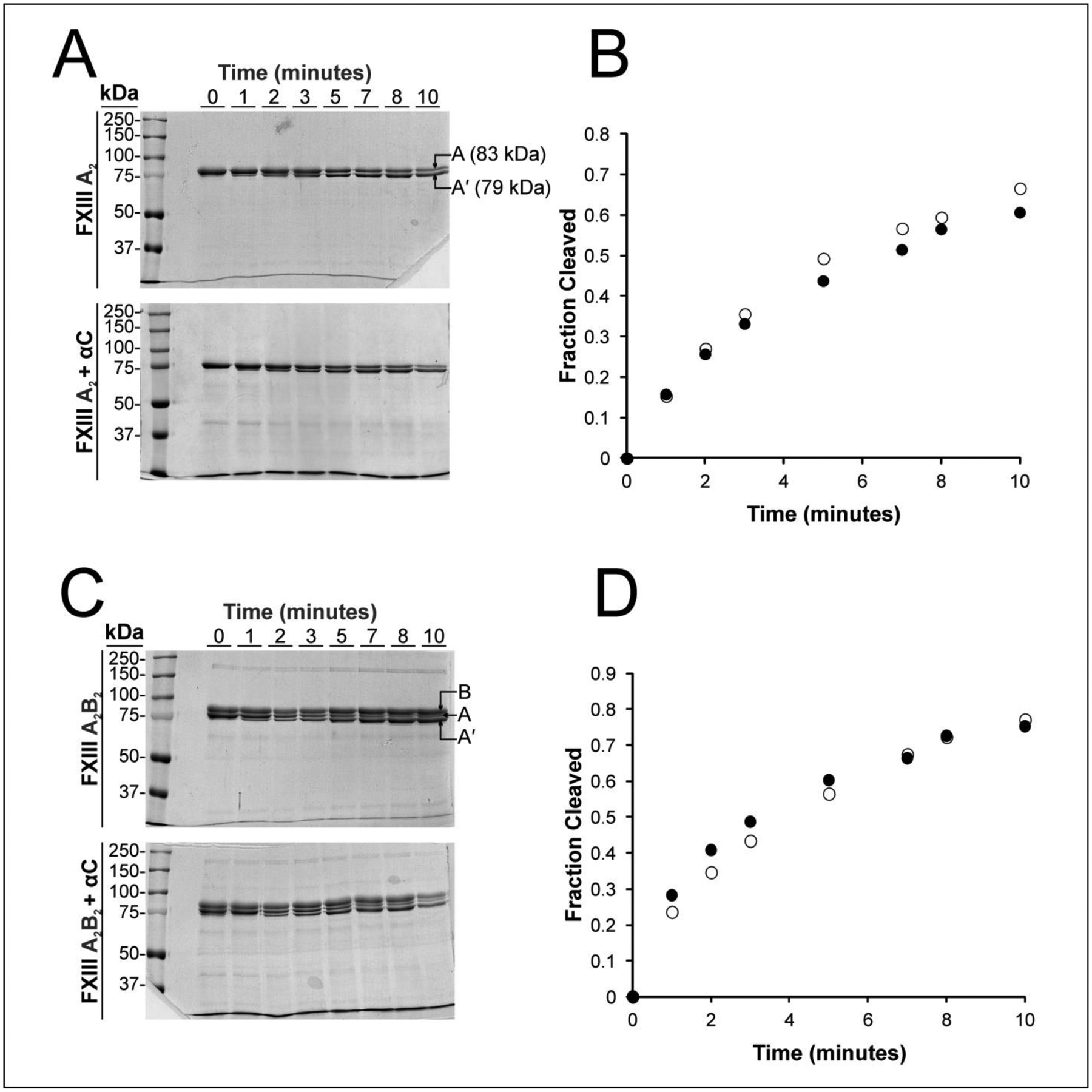
Cleavage reactions contained 5 μM αC (233–425), 1μM FXIIIA (from FXIII A2 or FXIII A2B2), and 4mM CaCl2 in Tris-acetate buffer. Incubations were maintained at 37°C and quenched at defined time points using the thrombin inhibitor PPACK. Samples were run under reducing conditions on 8% SDS-PAGE gels and stained with Coomassie Blue. A) Gels showing reactions with FXIII A2 (top) versus FXIII A2 + αC (233–425) bottom. Following cleavage of the FXIII activation peptide segment at the R37-G38 peptide bond, the MW for the A subunit (83 kDa) decreases to A’ (79 kDa). In the lower figure, the dark band found at the bottom of the gel corresponds to αC (233–425). B) The gels were dried and the fractions of FXIII A-chains cleaved over time were calculated using Image J. The filled circles in the plot correspond to free FXIII A2 and the open circle to FXIII A2 in the presence of αC (233–425). The αC region does not seem to promote thrombin cleavage of FXIII A2. C) Gels showing reactions with FXIII A2B2 (top) versus FXIII A2B2 + αC (233–425) bottom. Following cleavage of the FXIII activation peptide, the MW for the A subunit (83 kDa) decreases to A’ (79 kDa). D) The filled circles in the plot correspond to free FXIII A2B2 and the open circle to FXIII A2B2 in the presence of αC (233–425). The αC region does not seem to promote thrombin cleavage of FXIII A2B2. For both FXIII forms, representative data from a set of independent triplicates are displayed.
Examining the influences of Q237 on the reactivities of Q328 and Q366.
Our 2D 1H-15N HSQC NMR assay demonstrated that, in the absence of one or more reactive glutamines, FXIIIa is still able to crosslink the remaining reactive glutamines.29 There was thus interest in characterizing the crosslinking abilities of Fbg αC (233–425) Q328 and Q366 in the absence of the most reactive glutamine Q237 (Q237N mutant). With a mass spectrometry assay, short time points could be run and detailed information on Q reactivities obtained.
All studies were carried out with FXIII A2 (500 nM FXIIIA) pre-activated by thrombin and calcium before introducing the Fbg αC (233–425) WT. Using this strategy, data could be directly compared with previously published studies.29 Moreover, our current results suggest that preincubation with Fbg αC does not have a critical influence on promoting FXIII activation cleavage or FXIIIa reactivity. The quenched transglutaminase reactions were examined using an LC-MS based method. Retention times and m/z values for each peptide fragment containing a reactive glutamine are summarized in Supplementary Table S3.
In the absence of Q237 (i.e Q237N), the remaining glutamines Q328 and Q366 were both crosslinked to a lesser extent than in Fbg αC WT (Fig 5). These observations suggest that the fast crosslinking of Q237 to GEE has a positive influence on the ability of FXIIIa to crosslink Q328 and Q366. This effect occurs even though Q237 is located rather distant in sequence from Q328 and Q366 and also the putative FXIII binding site (αC 389–402).
Figure 5: Plot comparing FXIII A2-catalyzed crosslinking of three reactive Qs in WT Fbg αC (233–425) versus αC Q237N.
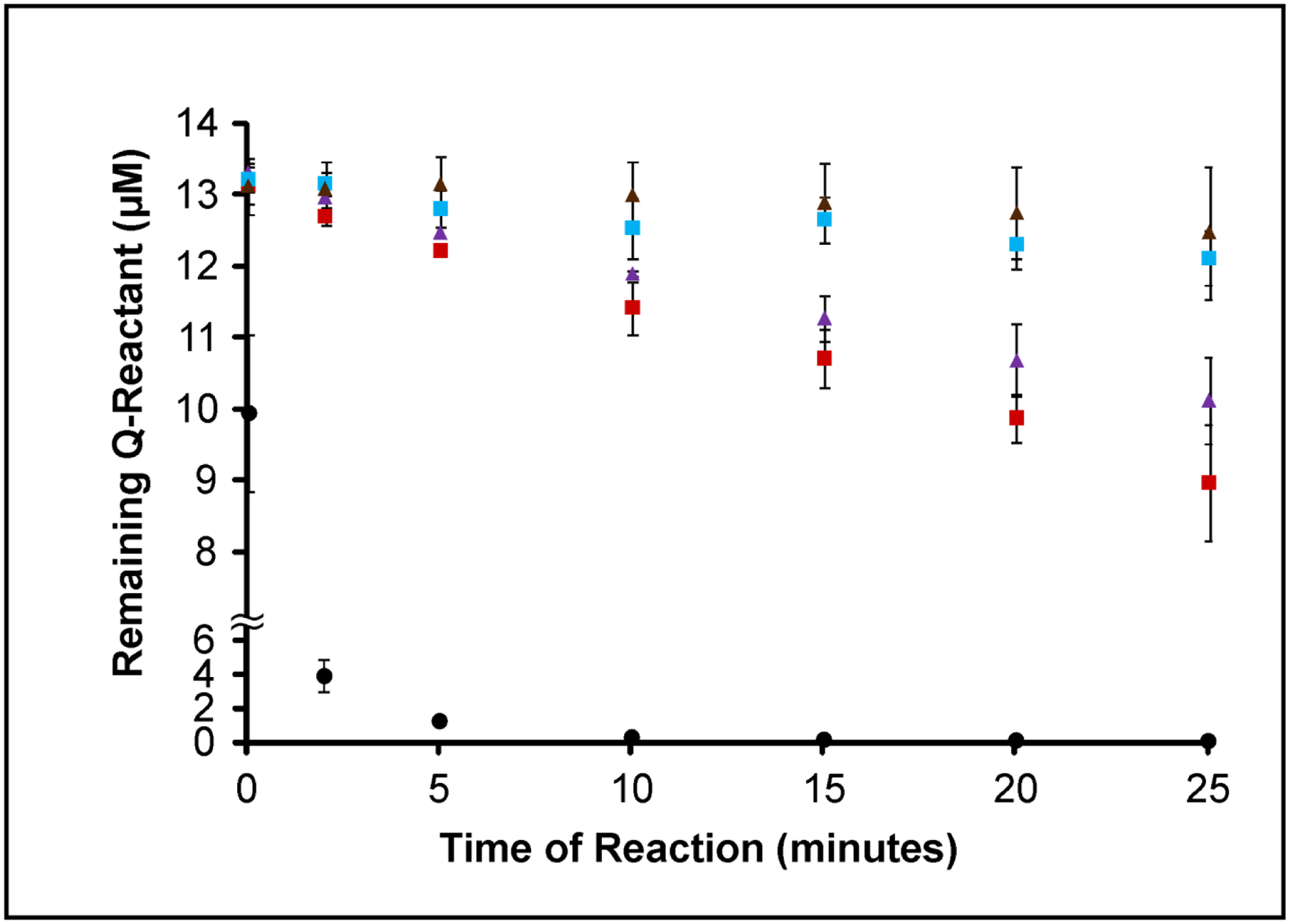
Glutamines reactivities in WT Fbg αC (233–425) versus the Q237N mutant were monitored for 25 minutes in the presence of 500 nM FXIIIA (pre-activated FXIII A2). The individual Q residues include Q328 WT (purple triangle), Q328 within Q237N (brown triangle), Q366 WT (red square), Q366 within Q237N (blue square), and Q237 WT (black circle). The peak-height ratio method was used to calculate the amount of Q reactant left following reactions with GEE. Experiments were performed in triplicate and the results reported as mean ± SD
Discussion
In blood coagulation, the most highly abundant protein in the cascade is fibrinogen. Much research has focused on its γ chain and the important role of fibrin γ-γ crosslinking in maintaining fibrin clot architecture.1–3 Recent research suggests that the αC region of fibrinogen plays important roles in clot stability and disease leading to renewed interest in this region.17–19 The αC region also plays a critical role in red blood cell retention.14,20,21 From a genetic perspective, mutations of Q residues within the αC region have been associated with fibrinogen deficiencies.36,37 FXIIIa catalyzed crosslinking reactions involving this αC region confers biophysical properties that help decrease fibrin clot lysis rate.17 In this study, we highlight new knowledge on the contributions of individual reactive glutamines within Fbg αC (233–425) and the role of the putative FXIII binding site αC (389–402).17,38–41 As a result, a better understanding of blood clotting physiology is achieved.
FXIII’s reactivity towards individual fibrinogen glutamines is selective and specific. Our previous study led to a ranking of Q237 >> Q328 ≈ Q366 in Fbg αC(233–425), which correlates closely with crosslinking trends seen in full length fibrinogen.29,32 In the current study, the abilities of activated FXIII A2 and A2B2 to crosslink GEE to the three reactive glutamines (Q327, Q328, Q366) of both WT and E396A Fbg αC were investigated. The acidic αC E396 residue is an important anchoring component within Fbg αC (389–402), a putative binding site for FXIII.24 Our results revealed that thrombin-activated FXIII A2 and A2B2 could still crosslink all three reactive glutamines located within the αC (233–425) E396A mutant. This characteristic was confirmed by both NMR 2D 1H-15N HSQC and mass spectrometry based crosslinking assays. No major differences were observed in the ranking of reactive glutamines for WT αC versus mutant E396A. Although Fbg E396 is critical in the binding site for FXIII, this fibrinogen residue is not required to ensure FXIII transglutaminase activity.
The Fbg αC region is highly flexible and its structure cannot be documented by X-ray crystallography.42,43 FXIII reactive glutamines are frequently found in highly flexible substrate regions.8 In this study, the most reactive Q237 is positioned near the αC N-terminus and located rather distant in sequence from the anchoring residue E396. Intriguingly, Q237 was crosslinked to a significantly lesser extent in the presence of αC E396A compared to WT during early reaction time points. It is possible that Q237 and surrounding residues become oriented toward the FXIII binding site αC (389–402) during crosslinking activity. Activated FXIII A2 anchored to weaker binding Fbg αC E396A may have some hindered ability to interact with Q237.
Unlike Q237, Fbg αC Q328 and Q366 are located just N-terminal of the putative FXIII binding region. As a result, Q328 and Q366 may be expected to experience a greater hindrance in crosslinking in the presence of Fbg αC (233–425) E396A. Using chemical crosslinking studies and molecular modeling efforts, Smith et al. demonstrated that αC residues 389–402 (PDWGTFEEVSGNVS) would bind to the FXIII β-sandwich domain near where the FXIII activation peptide binds.25 In particular, αC E396 would make a salt bridge with FXIII R158 within this β-sandwich region. Furthermore, Q366 would be positioned closer to the FXIII active site than Q328. With higher FXIII concentrations, we showed that Fbg αC Q366 could be crosslinked by thrombin-activated FXIII A2 to a greater extent than Q328. Surprisingly, the presence of the αC E396A mutation did not substantially affect reactivity ranking for Q328 or Q366.
FXIII A2B2 interacts with αC (233–425) with a higher affinity than FXIII A2.24 In our additional mass spectrometry assays, thrombin-cleaved FXIII A2B2 could still crosslink all three reactive glutamines to a similar extent and ranking for both WT and Fbg αC E396A (233–425). This feature suggests again that the transglutaminase function of FXIIII A2B2 does not have a strong dependence on αC E396. Other residues surrounding E396 may play a compensatory role and could contribute to other FXIII-related functions in binding and activation.
Intriguingly, the Q237 crosslinking reaction was slower with FXIII A2B2 than for FXIII A2. The presence and/or release of FXIII B2 likely plays a role in this delay. In circulation, FXIII B2 is the carrier subunit and has been shown to bind to Fibrinogen’s γ and αC surfaces helping anchor the enzymatic subunit FXIII A2 for activation and subsequent crosslinking.14,20,24,44 The delayed release of the large biomolecule FXIII B from the αC surface may slightly hinder the availability of the Q-reactive substrate regions.
Zymogen FXIII A2 does not exhibit much affinity for αC (233–425) whereas FXIII A2B2 has a KD of 7 nM.24 We used SDS-PAGE experiments to monitor thrombin-catalyzed activation of FXIII A2 and A2B2 in the presence and absence of αC (233–425). As to be expected, this stretch of Fbg αC WT does not promote cleavage of the FXIII A2 activation peptide segment. Interestingly, activation cleavage of FXIII A2B2 was also not enhanced in the presence of αC (233–425) indicating unique features of this Fbg αC segment.
Unlike Fbg αC (233–425), full length fibrinogen does promote cleavage of FXIII A2B2.14,45,46 FXIII A2B interactions occur within the D-E-D structure of polymerized fibrin.1 Studies have shown that FXIII A2B2 binds to the D domain and then thrombin to the E domain. Binding of an N-terminal segment of the fibrin α chain to thrombin’s anion binding exosite I (ABE I) helps to promote thrombin-catalyzed activation of FXIII A2B2.46 PAR1 and thrombomodulin do utilize the ABE I exosite to enhance cleavage events at the thrombin active site.47,48 FXIII A, itself, does not have a segment that can target this thrombin exosite. By contrast, this binding property is observed with the N-terminal fibrin α chain.
Recent studies have further defined interactions between FXIII A2B2, FXIII A2, and the fibrinogen D-domain. Souri et al. examined the binding properties of plasma FXIII A2B2 and recombinant versions of FXIII A2B2, FXIII A2, and FXIII B2 to fibrinogen.44 Moreover, FXIII activation rates were investigated. Their studies effectively demonstrated that the FXIII B subunits are responsible for helping to improve FXIII binding to fibrinogen.44 In addition, they provided further evidence that proper positioning of FXIII on the fibrinogen surface assists in accelerating thrombin-dependent FXIII activation. Byrnes and coworkers then elucidated that FXIII A2B2 targets the Fbg γ (390–396) segment via the binding of FXIII B2.14,45 Acceleration of FXIII activation was lost with a Fbg γ390−396A mutant. Plasma FXIII B2 was proposed to circulate bound to Fbg γ (390–396).14,45 After activation, FXIIIa is well positioned to perform γ-γ crosslinking (Q398/Q399 -K406) followed by crosslinking events involving the αC region.
In our previous study, we characterized and ranked, for the first time, the three reactive glutamines involved in Fbg αC crosslinking.29 Our current studies suggest that FXIII and Fibrinogen αC use unique features to control anchoring, activation, and substrate specificity for crosslinking. FXIII A2B2 and activated FXIII A2 may be directed toward αC (389–402) so that the FXIII is poised to later target specific glutamines and/or lysines within fibrinogen. Reactive Qs 237, 328, and 366 are positioned both close and distant from this anchoring site. Although αC E396 has been reported to be a critical residue for FXIII binding, its absence does not drastically hinder the transglutaminase function of activated FXIII A2 or A2B2. Furthermore, Fbg αC (233–425) does not promote thrombin-catalyzed cleavage of FXIIIA. Interestingly, the reactivity of distant Q237 is more affected by the E396A substitution within αC (233–425) than Q328 and Q366. We previously demonstrated that activated FXIII A2 was able to crosslink αC Q237, Q328, and Q366 independently following Q to N mutations.29 For the current study, a Q237N substitution to WT αC hinders reactivity at Q328 and Q366. The Q237 crosslink is thus hypothesized to play a role in promoting an αC conformation that enhances the ability of Q328 and Q366 to interact with the FXIII active site.
In summary, molecular details on fibrin substrate specificity have been readily elucidated by working with the fibrinogen segment αC (233–425). This segment contains reactive glutamines whose crosslinking rankings correlate with physiological, full-length fibrinogen. If fibrin α-α crosslinking could be selectively hindered, the resultant clot would be less stiff and may have longer protofibrils and thinner fibers.17–19 Moreover, there would be some loosening of red blood cell retention.20,21 The current studies predict that novel strategies to regulate FXIII’s ability to crosslink Fbg αC may result from altering individual reactive Qs within αC (233–425), controlling the distinct local environments surroundings these Qs, and/or further manipulating the FXIII anchoring segment αC (389–402).
Supplementary Material
Summary Table.
What is known on this topic?
Fibrinogen αC (389–402) is a binding site for Factor XIII A2B2 and thrombin-activated FXIII A2 with αC E396 reported to be a key anchoring residue.
Within the highly flexible Fbg αC (233–425) region, Factor XIIIa catalyzes crosslinking of reactive glutamines with rankings of Q237 >> Q366 ≈ Q328, similar to full length fibrinogen.
Crosslinking of the fibrin α chain by Factor XIIIa is important for increasing fibrin thickness and decreasing clot lysis rate.
What does this paper add?
Activated FXIII (A2 and A2B2) crosslinks Q237, Q328, and Q366 to similar extents with Fbg αC (233–425) and the mutant E396A. These studies reveal that the Fbg αC E396 residue is not critical for maintaining FXIII transglutaminase function.
The orientation and crosslinking of Q237 are proposed to play roles in helping direct Q328 and Q366 to the FXIII active site.
Altering Fbg αC anchoring sites and the distinct Q-substrate local environments may be strategies to regulate FXIII’s ability to crosslink the αC.
Acknowledgements:
The authors appreciate helpful discussions related to this project with B. Anokhin, R. Billur, and F.D. Ablan. The authors also thank O. Ahmed for helping with some preliminary experiments related to the FXIII activation studies.
Funding
This research project was funded by National Institutes of Health grant: NIH R15 HL120068
Footnotes
Conflict of Interest
None
REFERENCES
- 1.Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121(10):1712–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ariens RA. Fibrin(ogen) and thrombotic disease. Journal of thrombosis and haemostasis : JTH. 2013;11 Suppl 1:294–305 [DOI] [PubMed] [Google Scholar]
- 3.Litvinov RI, Weisel JW. What Is the Biological and Clinical Relevance of Fibrin? Seminars in thrombosis and hemostasis. 2016;42(4):333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosesson MW. Fibrinogen and fibrin structure and functions. Journal of thrombosis and haemostasis : JTH. 2005;3(8):1894–1904 [DOI] [PubMed] [Google Scholar]
- 5.Weisel JW. Fibrinogen and fibrin. Advances in protein chemistry. 2005;70:247–299. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z, Mochalkin I, Veerapandian L, Riley M, Doolittle RF. Crystal structure of native chicken fibrinogen at 5.5-A resolution. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(8):3907–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Standeven KF, Ariens RA, Grant PJ. The molecular physiology and pathology of fibrin structure/function. Blood reviews. 2005;19(5):275–288 [DOI] [PubMed] [Google Scholar]
- 8.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiological reviews. 2011;91(3):931–972 [DOI] [PubMed] [Google Scholar]
- 9.Bagoly Z, Koncz Z, Harsfalvi J, Muszbek L. Factor XIII, clot structure, thrombosis. Thrombosis research. 2012;129(3):382–387 [DOI] [PubMed] [Google Scholar]
- 10.Richardson VR, Cordell P, Standeven KF, Carter AM. Substrates of Factor XIII-A: roles in thrombosis and wound healing. Clinical science. 2013;124(3):123–137 [DOI] [PubMed] [Google Scholar]
- 11.Standeven KF, Carter AM, Grant PJ, et al. Functional analysis of fibrin {gamma}-chain cross-linking by activated factor XIII: determination of a cross-linking pattern that maximizes clot stiffness. Blood. 2007;110(3):902–907 [DOI] [PubMed] [Google Scholar]
- 12.Cottrell BA, Strong DD, Watt KW, Doolittle RF. Amino acid sequence studies on the alpha chain of human fibrinogen. Exact location of cross-linking acceptor sites. Biochemistry. 1979;18(24):5405–5410 [DOI] [PubMed] [Google Scholar]
- 13.Chen R, Doolittle RF. - cross-linking sites in human and bovine fibrin. Biochemistry. 1971;10(24):4487–4491 [DOI] [PubMed] [Google Scholar]
- 14.Byrnes JR, Wilson C, Boutelle AM, et al. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues gamma390–396 and the FXIIIB subunits. Blood. 2016;128(15):1969–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KN, Lee CS, Tae WC, Jackson KW, Christiansen VJ, McKee PA. Cross-linking of wild-type and mutant alpha 2-antiplasmins to fibrin by activated factor XIII and by a tissue transglutaminase. The Journal of biological chemistry. 2000;275(48):37382–37389 [DOI] [PubMed] [Google Scholar]
- 16.Tsurupa G, Yakovlev S, McKee P, Medved L. Noncovalent interaction of alpha(2)-antiplasmin with fibrin(ogen): localization of alpha(2)-antiplasmin-binding sites. Biochemistry. 2010;49(35):7643–7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duval C, Allan P, Connell SD, Ridger VC, Philippou H, Ariens RA. Roles of fibrin alpha- and gamma-chain specific cross-linking by FXIIIa in fibrin structure and function. Thrombosis and haemostasis. 2014;111(5):842–850 [DOI] [PubMed] [Google Scholar]
- 18.Protopopova AD, Barinov NA, Zavyalova EG, Kopylov AM, Sergienko VI, Klinov DV. Visualization of fibrinogen alphaC regions and their arrangement during fibrin network formation by high-resolution AFM. Journal of thrombosis and haemostasis : JTH. 2015;13(4):570–579 [DOI] [PubMed] [Google Scholar]
- 19.Protopopova AD, Litvinov RI, Galanakis DK, et al. Morphometric characterization of fibrinogen’s alphaC regions and their role in fibrin self-assembly and molecular organization. Nanoscale. 2017;9(36):13707–13716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrnes JR, Duval C, Wang Y, et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015;126(16):1940–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aleman MM, Byrnes JR, Wang JG, et al. Factor XIII activity mediates red blood cell retention in venous thrombi. The Journal of clinical investigation. 2014;124(8):3590–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Credo RB, Curtis CG, Lorand L. Alpha-chain domain of fibrinogen controls generation of fibrinoligase (coagulation factor XIIIa). Calcium ion regulatory aspects. Biochemistry. 1981;20(13):3770–3778 [DOI] [PubMed] [Google Scholar]
- 23.Procyk R, Bishop PD, Kudryk B. Fibrin--recombinant human factor XIII a-subunit association. Thrombosis research. 1993;71(2):127–138 [DOI] [PubMed] [Google Scholar]
- 24.Smith KA, Adamson PJ, Pease RJ, et al. Interactions between factor XIII and the alphaC region of fibrinogen. Blood. 2011;117(12):3460–3468 [DOI] [PubMed] [Google Scholar]
- 25.Smith KA, Pease RJ, Avery CA, et al. The activation peptide cleft exposed by thrombin cleavage of FXIII-A(2) contains a recognition site for the fibrinogen alpha chain. Blood. 2013;121(11):2117–2126 [DOI] [PubMed] [Google Scholar]
- 26.Doolittle RF, Cassman KG, Cottrell BA, Friezner SJ. Amino acid sequence studies on the alpha chain of human fibrinogen. Isolation and characterization of two linked alpha-chained cyanogen bromide fragments from fully cross-linked fibrin. Biochemistry. 1977;16(8):1715–1719 [DOI] [PubMed] [Google Scholar]
- 27.Karpati L, Penke B, Katona E, Balogh I, Vamosi G, Muszbek L. A modified, optimized kinetic photometric assay for the determination of blood coagulation factor XIII activity in plasma. Clinical chemistry. 2000;46(12):1946–1955 [PubMed] [Google Scholar]
- 28.Cleary DB, Maurer MC. Characterizing the specificity of activated Factor XIII for glutamine-containing substrate peptides. Biochimica et biophysica acta. 2006;1764(7):1207–1217 [DOI] [PubMed] [Google Scholar]
- 29.Mouapi KN, Bell JD, Smith KA, Ariens RA, Philippou H, Maurer MC. Ranking reactive glutamines in the fibrinogen alphaC region that are targeted by blood coagulant factor XIII. Blood. 2016;127(18):2241–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuka YV, Medved LV, Migliorini MM, Ingham KC. Factor XIIIa-catalyzed cross-linking of recombinant alpha C fragments of human fibrinogen. Biochemistry. 1996;35(18):5810–5816 [DOI] [PubMed] [Google Scholar]
- 31.Schwartz ML, Pizzo SV, Hill RL, McKee PA. Human Factor XIII from plasma and platelets. Molecular weights, subunit structures, proteolytic activation, and cross-linking of fibrinogen and fibrin. The Journal of biological chemistry. 1973;248(4):1395–1407 [PubMed] [Google Scholar]
- 32.Wang W Identification of respective lysine donor and glutamine acceptor sites involved in factor XIIIa-catalyzed fibrin alpha chain cross-linking. The Journal of biological chemistry. 2011;286(52):44952–44964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katona E, Haramura G, Karpati L, Fachet J, Muszbek L. A simple, quick one-step ELISA assay for the determination of complex plasma factor XIII (A2B2). Thrombosis and haemostasis. 2000;83(2):268–273 [PubMed] [Google Scholar]
- 34.Schilling B, Rardin MJ, MacLean BX, et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol Cell Proteomics. 2012;11(5):202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doiphode PG, Malovichko MV, Mouapi KN, Maurer MC. Evaluating factor XIII specificity for glutamine-containing substrates using a matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay. Analytical biochemistry. 2014;457:74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridgway HJ, Brennan SO, Faed JM, George PM. Fibrinogen Otago: a major alpha chain truncation associated with severe hypofibrinogenaemia and recurrent miscarriage. British journal of haematology. 1997;98(3):632–639 [DOI] [PubMed] [Google Scholar]
- 37.Park R, Doh HJ, An SS, Choi JR, Chung KH, Song KS. A novel fibrinogen variant (fibrinogen Seoul II; AalphaGln328Pro) characterized by impaired fibrin alpha-chain cross-linking. Blood. 2006;108(6):1919–1924 [DOI] [PubMed] [Google Scholar]
- 38.Hethershaw EL, Cilia La Corte AL, Duval C, et al. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. Journal of thrombosis and haemostasis : JTH. 2014;12(2):197–205 [DOI] [PubMed] [Google Scholar]
- 39.Kurniawan NA, Grimbergen J, Koopman J, Koenderink GH. Factor XIII stiffens fibrin clots by causing fiber compaction. Journal of thrombosis and haemostasis : JTH. 2014;12(10):1687–1696 [DOI] [PubMed] [Google Scholar]
- 40.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophysical journal. 1999;77(5):2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Helms CC, Ariens RA, Uitte de Willige S, Standeven KF, Guthold M. alpha-alpha Cross-links increase fibrin fiber elasticity and stiffness. Biophysical journal. 2012;102(1):168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kollman JM, Pandi L, Sawaya MR, Riley M, Doolittle RF. Crystal structure of human fibrinogen. Biochemistry. 2009;48(18):3877–3886 [DOI] [PubMed] [Google Scholar]
- 43.Tsurupa G, Tsonev L, Medved L. Structural organization of the fibrin(ogen) alpha C-domain. Biochemistry. 2002;41(20):6449–6459 [DOI] [PubMed] [Google Scholar]
- 44.Souri M, Osaki T, Ichinose A. The Non-catalytic B Subunit of Coagulation Factor XIII Accelerates Fibrin Cross-linking. The Journal of biological chemistry. 2015;290(19):12027–12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrnes JR, Wolberg AS. Newly-Recognized Roles of Factor XIII in Thrombosis. Seminars in thrombosis and hemostasis. 2016;42(4):445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naski MC, Lorand L, Shafer JA. Characterization of the kinetic pathway for fibrin promotion of alpha-thrombin-catalyzed activation of plasma factor XIII. Biochemistry. 1991;30(4):934–941 [DOI] [PubMed] [Google Scholar]
- 47.Jacques SL, LeMasurier M, Sheridan PJ, Seeley SK, Kuliopulos A. Substrate-assisted catalysis of the PAR1 thrombin receptor. Enhancement of macromolecular association and cleavage. The Journal of biological chemistry. 2000;275(52):40671–40678 [DOI] [PubMed] [Google Scholar]
- 48.Ye J, Liu LW, Esmon CT, Johnson AE. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion-binding exosite of thrombin and alter its specificity. The Journal of biological chemistry. 1992;267(16):11023–11028 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.