

Abstract
T cell receptor (TCR) stimulation activates diverse kinase pathways, which include the mitogen-activated protein kinases (MAPKs) ERK and p38, the phosphoinositide 3-kinases (PI3Ks), and the kinase mTOR. Although TCR stimulation activates the p38 pathway through a “classical” MAPK cascade that is mediated by the adaptor protein LAT, it also stimulates an “alternative” pathway in which p38 is activated by the kinase ZAP70. Here, we used dual-parameter, phosphoflow cytometry and in silico computation to investigate how both classical and alternative p38 pathways contribute to T cell activation. We found that basal ZAP70 activation in resting T cell lines reduced the threshold (“primed”) TCR-stimulated activation of the classical p38 pathway. Classical p38 signals were reduced after T cell-specific deletion of the guanine nucleotide exchange factors Sos1 and Sos2, which are essential LAT signalosome components. As a consequence of Sos1/2 deficiency, production of the cytokine IL-2 was impaired, differentiation into regulatory T cells was reduced, and the autoimmune disease EAE was exacerbated in mice. These data suggest that the classical and alternative p38 activation pathways exist to generate immune balance.
INTRODUCTION
A member of the mitogen-activated protein kinase (MAPK) family, p38 (also known as MAPK14), is expressed in various mammalian cells including immune cells (1, 2). There are four isoforms of p38; p38α is ubiquitously expressed, whereas the expression patterns of p38β, p38γ, and p38δ are relatively tissue specific (3). Receptor-induced p38 activation typically occurs through a canonical/classical MAPK cascade initiated by either a kinase or activated guanosine triphosphatase (GTPase), which stimulates a MAPK kinase kinase (MAPKKK) to activate a MAPK kinase (MAPKK) that activates p38 MAPK. Thus, upstream signals stimulate dual phosphorylation of threonine180 (T180) and tyrosine182 (Y182) in the activation loop of p38 through sequential the activation of MKK3, MKK4, and MKK6 (4).
The dominant form expressed in T cells is p38α, which we will simply refer to p38 hereafter. Proper regulation of p38 activity is important for early thymocyte development, CD4+ T helper (TH) cell differentiation, and cytokine production (5, 6). The activity of p38 is greatest in CD4−CD8− double-negative thymocytes, a very early T cell developmental stage, and critical for proper transition to the next stage of thymic T cell development (7, 8). However, the requirement of p38 activity for double-positive thymocyte selection is controversial (9). In mature CD4+ T cells, pharmacological inhibition of p38 inhibits in vitro TH1 and induced regulatory T (iTreg) cells (10, 11) and in vitro or in vivo interleukin-17 (IL-17) production important for TH17 function (12).
Engagement of the T cell receptor (TCR) on peripheral T cells stimulates proximal signaling events that include activation of the ZAP70 (also 70-kDa zeta-associated protein) kinase. Proximal TCR signals are transduced not only through the adapter molecule LAT (also linker for activation of T cells) to downstream kinase pathways, which includes extracellular signal-regulated kinase (ERK) and p38 MAPKs, but also through the mechanistic target of rapamycin (mTOR) kinases (13). The activation of these kinases can depend on the incoming TCR signal strength (14, 15), and activity through specific kinase pathways can stimulate differentiation of CD4-positive (CD4+) T cells into distinct CD4+ TH cell subsets. For example, both mTOR complex 1 (mTORC1) and mTORC2 signals drive TH1 cell differentiation, whereas mTORC2 signals promote TH2 cells differentiation [reviewed in (16)].
How p38 is activated in T cells has remained poorly understood and somewhat controversial. Two separate p38 pathways have been proposed to exist downstream of TCR; a MAPKKK-MAPKK-MAPK “classical pathway” and an “alternative pathway” [reviewed in (6, 17)]. The classical pathway involves TCR signals through proximal Src and ZAP70 kinases that result in the assembly of a LAT “signalosome,” an intracellular signaling hub in T cells. Phosphorylation of LAT on multiple tyrosine residues by ZAP70 provides docking sites for the recruitment of SLP-76 [Src homology 2 (SH2) domain containing leukocyte protein of 76 kDa] and other adapter molecules, which bind guanine nucleotide exchange factors (GEFs) such as SOS (Son of sevenless), RasGRP1 (RAS guanyl-releasing protein 1), dedicator of cytokinesis 2 (DOCK2), and VAV [reviewed in (18)]. These GEFs activate the RAS and RAC (Ras-related C3 botulinum toxin substrate 1) family small GTPases, which can activate MAPK cascades such as ERK and p38 (19–23). Our work shows that LAT and SOS are required for optimal activation of p38 in both B and T cells (24). Although the requirement of SOS for p38 activation is independent of its enzymatic activity, which suggests that SOS functions as an adapter in the p38 pathway. In further support of a classical Rac-MAPKKK-MAPKK-P38 pathway, deficiency in Rac2 is associated with reduced TCR-induced p38 activation (25). Disruption of MAPKKK MAP3K2 (also Mekk2), a component in MAPK cascade, decreases p38 activation in response to TCR engagement (26). GADD45β (growth arrest and DNA damage-inducible β) promotes p38 activation through MAP3K4 (also MEKK4) (27), and TCR-induced p38 activation is greatly decreased in T cells lacking GADD45β (28). Conversely, enhanced activity of the MAPKKK MAP3K6 (also MKK6 or ASK2) is associated with increased p38 activity (10, 29).
In T cells, an alternative p38 pathway connects TCR-induced ZAP70 activity directly to auto-activation of p38. The C terminus of p38α and p38β contain a tyrosine, Tyr323, that can be phosphorylated by ZAP70 but not by the related kinase Syk, which is mostly expressed in B lymphocytes. When phosphorylated at Tyr323 (pTyr323), p38 auto-activation can induce monophosphorylation of pThr180 within the activation motif (30, 31). X-ray crystallography indicates that pTyr323 increases the accessibility of the activation motif, which allows for monophosphorylation of Thr180 (32). Genetic substitution of Y323F interferes with full activation of p38 as measured by double phosphorylation of Thr180 and Tyr182 in the Thr180-X-Tyr182 activation loop (pp38-Thr180Tyr182) in vitro and in vivo (31, 33, 34). Genetic perturbation of this alternative pathway by Y323F knock-in p38α and p38β (p38αβ Y323F) expression impairs interferon-γ (IFN-γ) synthesis in TH1 cells and IL-17 production in TH17 cells, indicating that the alternative p38 pathway is required for proinflammatory T helper functions (33–35). Along this line, the selective disruption of the alternative p38 pathway by p38αβ Y323F reduces the severity of experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (34). These studies revealed that T cells can bypass a classical, MAPKKK-MAPKK-MAPK p38 pathway and suggested that the adapter LAT is not required for T cell p38 activation. However, this immediately introduced a conundrum where conflicting evidence in support of both the classical and alternative p38 pathways exists [reviewed in (6) and (36)].
To address the p38 activation conundrum and explore potential contributions from alternative and classical pathways in T cells, we combined dual-parameter flow cytometry-based assays with traditional Western blot analysis and iterative computational modeling. Predictions of in silico models were experimentally tested in lymphocyte cell lines and primary CD4+ T cells. We found that basal TCR signals through the ZAP70-p38 alternative pathway lowered the threshold for full pp38-Thr180Tyr182 activation of the classical pathway. Complete p38 activation occurred with delayed kinetics, required LAT, SOS, and the catalytic activity of p38 through a classical pathway. CD4+ T cells require SOS-dependent p38 signals for IL-2 production and efficient differentiation into induced regulatory T cells. In addition, loss of Sos1 and Sos2 in CD4+ T cells results in aggravated autoimmune disease in the EAE mouse model. These data suggest that p38 alternative and classical activation pathways cooperate to balance appropriate CD4+ T cell responses.
RESULTS
Two distinct pathways connect TCR stimulation to p38 activation
To confirm that p38 activation is important for the development of distinct helper T cell populations (10–12), we in vitro-polarized cells into TH1, TH2, TH17, and iTreg subsets in parallel. Exposure of cells to the p38 inhibitor SB203580 (p38i) decreased the efficiency in TH1, TH17, and iTreg differentiation but increased the efficiency of cells to polarize to TH2 (Fig. 1A).
Fig. 1. p38 activity is essential for TH cell differentiation, and two distinct pathways connect TCR to p38 activation.
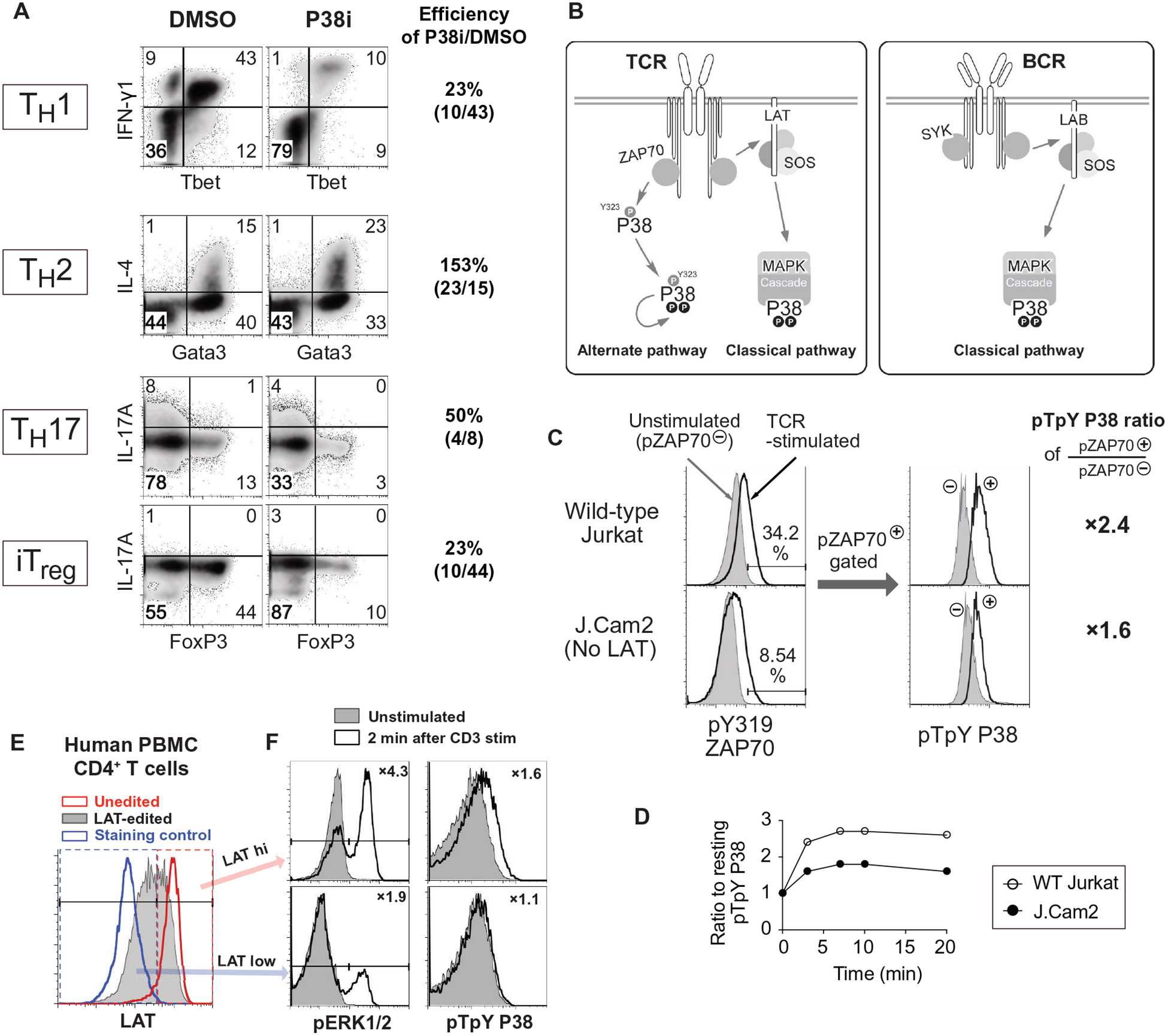
(A) Flow cytometry analysis of the indicated cytokine and transcription factors abundance in naïve CD4+ T cells cultured in vitro TH1 (IFN-γ+TBet+), TH2 (Gata3+IL-4+), TH17 (IL-17A+FoxP3−), and iTreg (IL-17A FoxP3+) polarizing conditions after control DMSO or p38 inhibitor SB203583 (p38i) treatment. Data are representative of two independent experiments. (B) Model of the distinct p38 pathways that exist in T cells and B cells, where a ZAP70-dependent alternative pathway that is unique to T cells. (C and D) Flow cytometry analysis of pZAP70 (Tyr319) and pp38 (Thr180Tyr182) in wild-type Jurkat cells and LAT-deficient J.Cam2 cells at rest (filled) or at 3 min after TCR activation (open). Activation of p38 after TCR stimulation was compared among cells that activated ZAP70 comparably (pZAP70+ gate), and the fold increase relative to unstimulated cells is indicated (right). Histograms (C) and quantified data (D) are representative of two independent experiments (see also fig. S1, A and B). (E and F) Flow cytometry analysis of LAT, pERK1/2, and pp38 in wild-type and CRISPR-Cas9-mediated LAT knockout human CD4+ T cells. Gates separating LAT-hi and LAT-low cells (E) were determined by LAT staining of unedited control cells (red histogram) and staining control (blue histogram). The activation of ERK1/2 and p38 (F) were determined in LAT-hi and LAT-low cells after TCR stimulation, and insets indicate the fold increase after activation (open histogram) relative to unstimulated cells (shaded histogram). Histograms are representative of two independent experiments (see also fig. S1E).
In T cells, the ZAP70 kinase can directly activate p38, whereas in B cells, the closely related Syk kinase cannot (Fig. 1B). T cells may solely use the alternative ZAP70-p38 pathway as they do not require signals that depend on the adapter molecule LAT to activate p38 (31). However, TCR-induced p38 activation is impaired in thymocytes that are deficient or haploinsufficient for the adapter Grb2, which binds to phosphorylated LAT (37, 38). In addition, our work shows that SOS is essential for optimal activation of p38 in both B and T cells (24). Because Grb2 and SOS are part of the LAT signalosome in T cells (18), we revisited the role of LAT in p38 activation.
In LAT-deficient Jurkat cells (J.Cam2), p38 activation is impaired after TCR cross-linking as measured by Western blot, which evaluates the population average activation (15). We noted that J.Cam2 cells express a reduced amount of surface TCR and exhibit reduced ZAP70 activation in response to TCR cross-linking (15, 39). For a fair comparison with single-cell resolution, we performed a dual-parameter phospho-flow (pFLOW) assay of p38 phosphorylation at Thr180 and Tyr182 (pp38-Thr180Tyr182) with simultaneous detection of ZAP70 phosphorylation at Tyr319 (pZAP70-Tyr319), which is indicative of ZAP70 activation. By gating on cells with similar amounts of pZAP70-Tyr319, we could analyze wild-type Jurkat and LAT-deficient J.Cam2 cells with equal ZAP70 activation (Fig. 1C and fig. S1A). In J.Cam2 cells with ZAP70 activation (pZAP70+) equal to wild-type Jurkat cells, we found reduced amounts of pp38-Thr180Tyr182 (Fig. 1C and fig. S1A). The total magnitude of pp38-Thr180Tyr182 induction was consistently lower in J.Cam2 cells when comparing distinct time points after TCR stimulation, indicating that full p38 activation also requires a LAT-dependent pathway (Fig. 1D and fig. S1B). Because CRISPR-Cas9-mediated editing in human peripheral blood CD4+ T cells (fig. S1C) did not delete LAT in primary CD4+ T cells with 100% efficiency (fig. S1D), we optimized pp38-Thr180Tyr182 analysis by pFLOW together with LAT staining. This approach allowed us to analyze cells at equal LAT abundance. In agreement with J.Cam2-based observations, CD3-stimulated activation of p38 was decreased in human CD4+ T cells with low amounts of LAT (LAT-low) when compared to higher amounts of LAT (LAT-hi; Fig. 1F and fig. S1E). When we analyzed ERK phosphorylation as a control, we found that ERK phosphorylation was reduced in LAT-low primary CD4+ T cells, as expected from LAT’s well-established role in Ras-ERK signaling (Fig. 1F and fig. S1E). Thus, our results from immortalized T cell line and primary CD4+ T cells indicated that two distinct pathways are necessary to connect TCR stimulation to full p38 activation.
Strong p38 activation is delayed and requires feedback from p38 kinase activity
We used pFLOW assays and traditional Western blot analysis to investigate the quantitative and qualitative differences between the alternative and classical p38 activation pathways. To this end, we tested and validated reagents to measure p38 phosphorylation (fig. S2). These tests proved to be critical quality controls. For example, we found that, although one can specifically interrogate the alternative pZAP70-p38 pathway through Tyr323 phosphorylation, the antibody that recognizes phosphorylation at this site does not perform reliably in the pFLOW assay (figs. S2 and S3).
We used Jurkat and DT40 cells, which have been widely used to the study of MAPK signaling (14, 15, 40, 41), as models of T and B cell activation and followed up with studies on primary murine CD4+ T cells. Because DT40 B cells do not express ZAP70, they therefore lack the alternative ZAP70-p38 pathway (Fig. 1B, right). TCR-stimulated Jurkat T cells rapidly achieved maximal pp38-Thr180Tyr182 amounts, which appeared in an anti-CD3 dose-dependent manner (Fig. 2A). In DT40 B cells, p38 activation had two distinct phases; an initial dose-dependent pp38-Thr180Tyr182 increase at 3 min after B cell receptor (BCR) stimulation that was followed by a subsequent phase of substantially stronger p38 activation (Fig. 2B and fig. S4). We postulated that the delayed strong activation of pp38-Thr180Tyr182 likely relied on a positive feedback loop. To test this, we examined BCR-induced pp38-Thr180Tyr182 in the presence of the p38 inhibitor SB203580 (P38i) and the MEK inhibitor U0126 (MEKi) (42–44). Inhibition of p38 activity abolished the late phase of strong p38 activation but left modest pp38-Thr180Tyr182 induction intact (Fig. 2C and fig. S5, A and B). By contrast, the MEKi did not affect p38 activation but completely blocked the activation of ERK in the RAF-MEK-ERK pathway (Fig. 2C and fig. S5, A and B). Of note, inhibition of p38 activity did not affect ERK activation (fig. S5A). These results suggest that the ERK and p38 MAPK pathways are fairly isolated from each other and put forth a multistep model for full p38 activation with early p38 activity-independent but late p38 activity-dependent signaling components.
Fig. 2. Strong activation of p38 is delayed and requires p38 kinase activity.
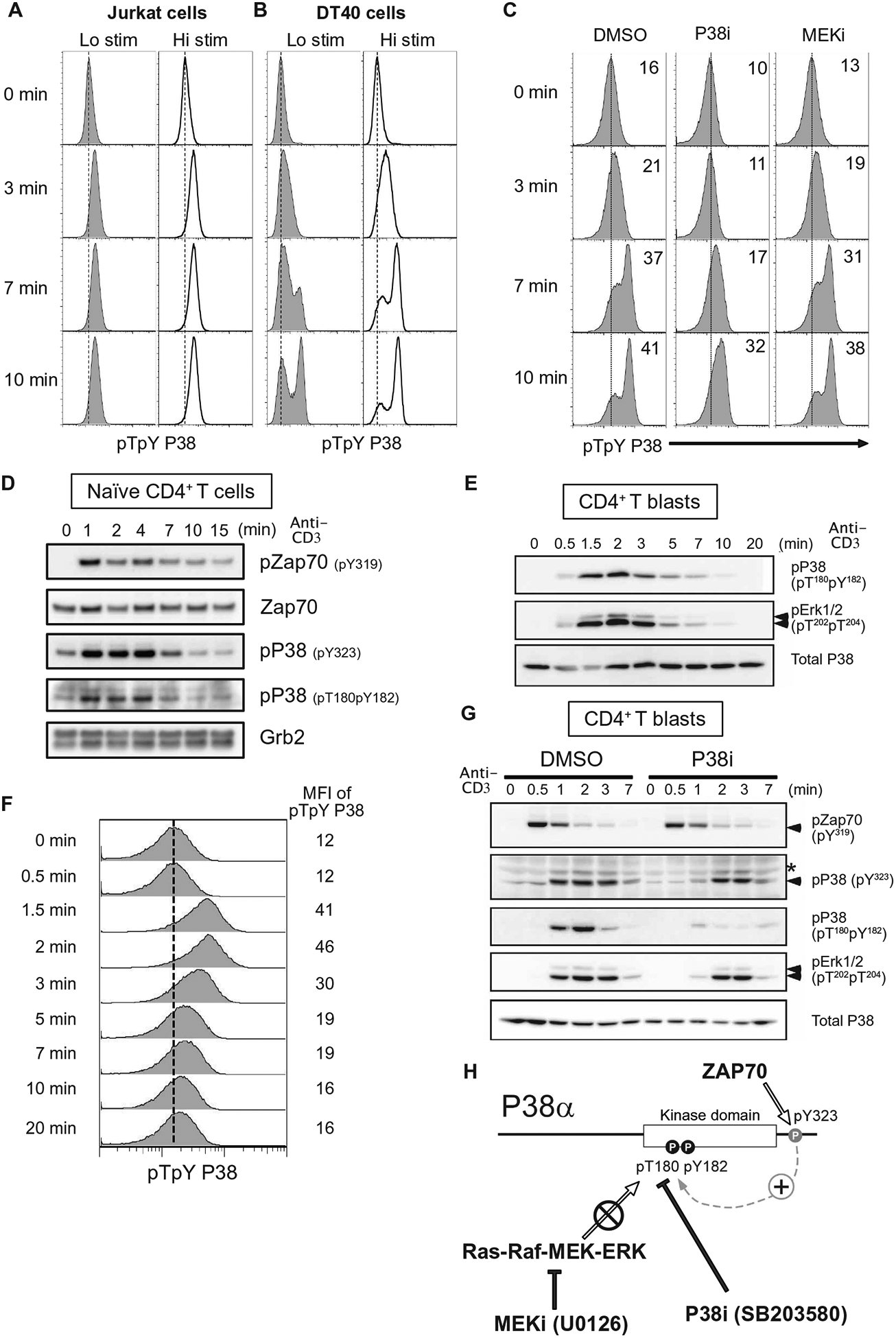
(A and B) Flow cytometry analysis of p38 activation in Jurkat T cells stimulated with two doses of antibody against mouse TCR (A) or DT40 B cells stimulated with two doses of antibody against mouse BCR (B). Histograms are representative of two independent experiments (see also fig. S4). (C) Flow cytometry analysis of p38 activation DT40 B cells treated with DMSO, P38i (SB203580), or MEKi (U0126) and stimulated with antibody against mouse BCR for the indicated times. Dashed lines indicate p38 activation in the resting cells, and insets indicate the mean fluorescence intensity (MFI) of pp38. Histograms are representative of two independent experiments (also, fig. S5). (D and E) Western blot analysis of pZAP70 and pp38 in lysates from primary naïve CD4+ T cells (D) and CD4+ T blast cells (E) stimulated with antibodies against CD3ε. Blots are representative of two independent experiments (see also fig. S6A). (F) Flow cytometry analysis of p38 activation of CD4+ T blast cells stimulated with antibodies against CD3ε. Histograms are representative of two independent experiments (see also fig. S6B). (G) Western blot analysis of phosphorylation of the indicated proteins in lysates from CD4+ T cell blasts treated with DMSO or P38i and stimulated with antibodies against CD3ε. Blots are representative of two independent experiments (see also fig. S6C). (H) In the alternative pathway, TCR-induced ZAP70 activity phosphorylates Tyr323 in the p38 C terminus. Phosphorylation at that site induces autophospho-rylation at Thr180 within the Thr180-X-Tyr182 activation loop. The p38 inhibitor SB203580 but not the MEK inhibitor U0126 blocks phosphorylation of p38 at Thr180 and Tyr182.
We examined p38 activation in primary naïve murine CD4+ T cell and T cell blasts in detail. Focusing on pp38-Thr180Tyr182 and pp38-Tyr323 abundance by Western blot analysis of naïve murine CD4+ T cells, we observed that both phosphorylation events were rapidly induced after TCR stimulation (Fig. 2D and fig. S6A). Furthermore, we observed substantial amounts of pp38-Thr180Tyr182 and pp38-Tyr323 in unstimulated naïve CD4+ T cells, which suggested that there is tonic activation of these pathways in these cells (Fig. 2D and fig. S6A). We analyzed the kinetics of p38 activation more extensively in primary CD4+ T cell blasts that were expanded in vitro, rested, and subsequently stimulated. By pFLOW assay and Western blot analysis, we observed that anti-CD3 TCR stimulation induced maximum pp38-Thr180Tyr182 and pp38-Tyr323 abundance roughly at 2 min and then decreased (Fig. 2, E to G, and fig. S6, B and C). Because the antibody for pp38-Tyr323 does not work in pFLOW, we could not use that assay to measure pp38-Tyr323 and pp38-Thr180Tyr182 simultaneously in the same cell. By Western blot, we found that CD3ε cross-linking stimulated pZAP70-Tyr319 acutely at 30 s, which was rapidly followed by induction of pp38-Tyr323 and ERK phosphorylation (Fig. 2G and fig. S6C). By contrast, pp38-Thr180Tyr182 amounts peaked with slower kinetics in these cells after TCR stimulation (Fig. 2G). In these primary CD4+ T cell blasts, full phosphorylation of pp38-Thr180Tyr182 was reduced by exposure to the p38 inhibitor SB203580 (Fig. 2G), in agreement with the Jurkat cell line data (Fig. 2C). However, p38 inhibitor exposure left a substantial portion of pp38-Tyr323 intact (Fig. 2G and fig. S6C), arguing that induction or stabilization of pp38-Tyr323 does not fully rely on p38 kinase activity. These results agree with x-ray crystallographic studies revealing that phosphorylation of Tyr323 increases the accessibility of p38’s activation motif, which leads to monophosphorylation of Thr180 (32). Together, these data suggest a model where an initial alternative ZAP70-p38 signals that hinge on phosphorylation of Tyr323 are followed by a subsequent amplification of p38 activation that is p38 activity dependent (Fig. 2H).
In silico modeling of the two pathways driving T cell p38 activation
To explore the consequences of the two distinct pathways to p38, we built minimal computational models connecting the TCR to p38 activation through the classical and the alternative pathways (Fig. 3A). The purpose of these models was to explore the qualitative consequences of different hypotheses not a quantitative recapitulation of the experimental data described above. For the alternative pathway, active ZAP70 activates p38 directly at Tyr323 (31). For the classical pathway, ZAP70 phosphorylates LAT. Mass spectrometry-based time-resolution studies indicate that LAT signalosome assembly occurs early, around 2 min after TCR stimulation of primary murine CD4+ T cells (45). The exact identity of all signal transducers upstream of the classical p38 pathway is unclear, although Grb2 (37, 38), SOS (15), Vav (46), Rac2 (25), and MKK6 (10) all play a role in T cell activation, and we confirmed a role for LAT here (Fig. 1). Together, this fits a canonical adapter exchange factor, small GTPase-MAPKKK-MAPKK-MAPK cascade. Because it was our goal to understand the two pathways leading to p38 activation and not to resolve the entire p38 signaling cascade, we classified the molecules connected in the LAT signalosome that signal to p38 as one hypothetical unit, called the LAT-GPS (generalized protein signaling) complex. Our results demonstrated that, in T cells, there is tonic pp38-Tyr323 (Figs. 1 and 2), which increases in two phases after TCR stimulation. Full pp38-Thr180Tyr182 activation requires the presence of both LAT and p38 kinase activity. To represent these characteristics in our coarse grain model, we considered two possible models.
Fig. 3. In silico modeling of p38 activation predicts distinct roles for alternative and classical pathways.
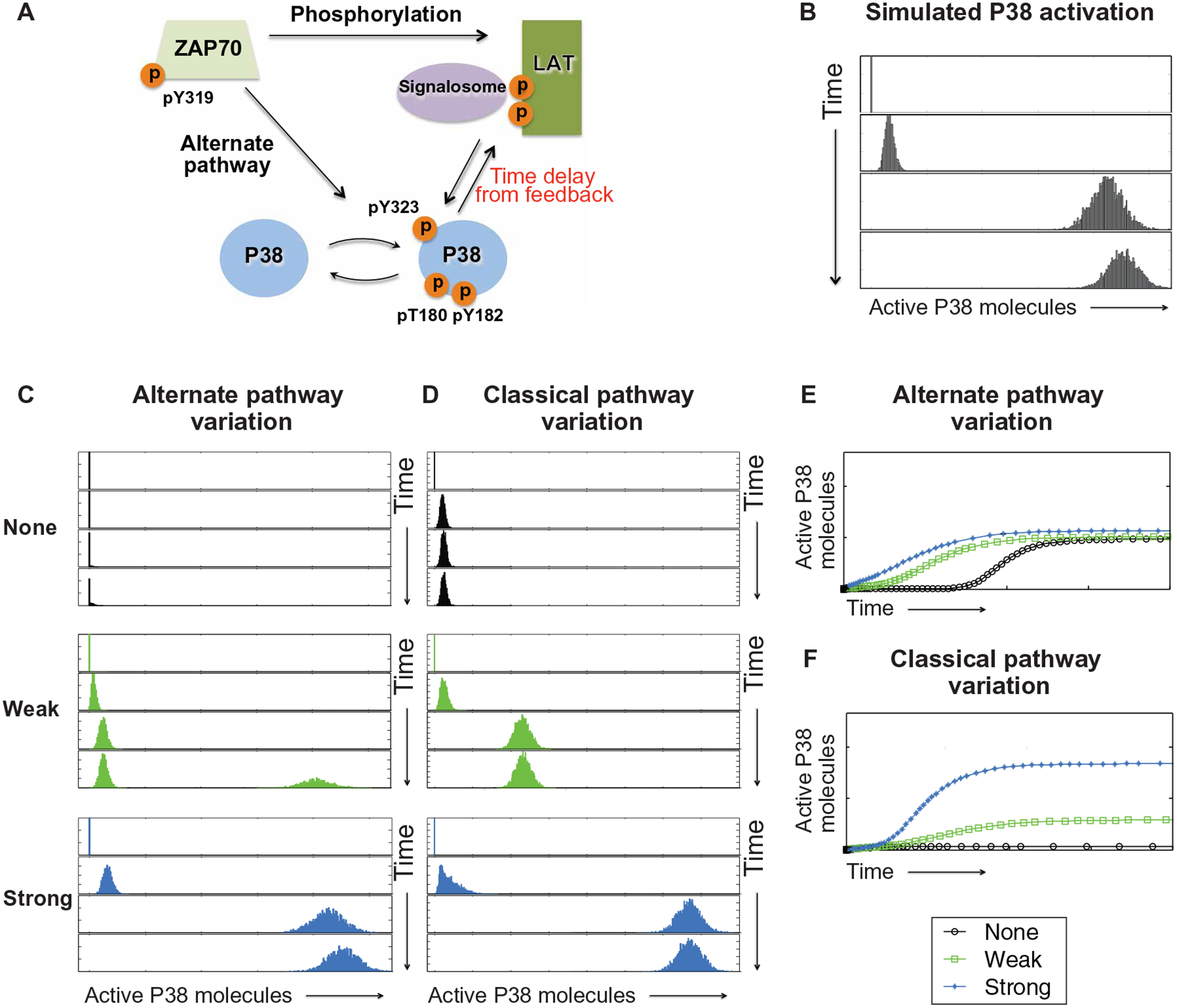
(A) A coarse grain model of p38 activation pathways in T cells where the alternative (alternate) pathway is directly connected to ZAP70 activation, and the classical pathway requires LAT signalosome formation and MAPK cascade activation. Temporal delay is introduced to reflect p38 activity-dependent full p38 activation. (B) In silico prediction of p38 activation with intermediate contribution from both the alternative and classical pathways. (C to F) Predicted p38 activation after varying the strength (none, weak, and strong) of alternative pathway (C) or classical pathway (D) activation. Projection in each model is graphically represented for alternative pathway (E) or classical pathway (F). The rate constant characterizing the phosphorylation of p38 by active ZAP70 was varied, from a value of zero (no contribution of alternative pathway) to stronger values (0.005 and 0.02).
In model I, we assumed that there was a delay in the assembly process of the LAT-GPS complex such that classical pathway p38 activation lagged behind the activation through the alternative pathway. To represent the time delay, we took the rate constant of the p38 activation by LAT-GPS as a step function. We assumed that, at early times, the activation rate of p38 by GPS-LAT was zero. After a certain threshold time (t), the classical pathway was activated with a nonzero activation rate of p38 by GPS-LAT. Mathematically, the rate constant of the phosphorylation process of p38 by GPS-LAT could then be described as a Heaviside step function, 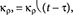 where τ is the critical tie point. When we carried out simulations of model I with different values of the critical time point (|), we found that model I does not recapitulate the qualitative characteristics of the experimental data, regardless of the choice of |. Specifically, full p38 activation could not occur, because when the classical pathway was activated (after the critical time point), the peak of pP38 moved directly to the final stable state in an analog fashion. In other words, the delay of the assembly of the LAT-GPS complex did not explain the experimental data.
where τ is the critical tie point. When we carried out simulations of model I with different values of the critical time point (|), we found that model I does not recapitulate the qualitative characteristics of the experimental data, regardless of the choice of |. Specifically, full p38 activation could not occur, because when the classical pathway was activated (after the critical time point), the peak of pP38 moved directly to the final stable state in an analog fashion. In other words, the delay of the assembly of the LAT-GPS complex did not explain the experimental data.
In model II, we assumed that a certain threshold amount of pp38 needs to be generated, before a positive feedback loop from p38 to the LAT-GPS complex could kick in. This feature reflected a p38 activity-dependent element (Fig. 3A). To represent the delay in feedback regulation of the LAT-GPS complex by p38, we took the rate constant of the feedback reaction as a step function. When the amount of phosphorylated p38 was small, the rate constant of the feedback loop from pP38 to GPS-LAT was zero. After pp38 accumulated to a certain threshold (〉P380), the feedback loop was triggered with a nonzero rate constant. Mathematically, this was modeled by the function 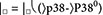 . Simulations using this minimal model were carried out using the Gillespie algorithm (47), and the rate of each reaction was modeled using standard mass action kinetics. Simulation of model II showed an initial modest increase in the number of active p38 molecules after ZAP70 activation, which was used as a proxy for TCR stimulation (Fig. 3B). As time progressed, a large increase in the number of active p38 molecules was predicted (Fig. 3B). The simulation also showed that the threshold of amount of pP380 and the strength of the classical pathway affected the behavior of p38 activation in a coordinated manner. To determine whether p38 activation is bistable in this model, we carried out a linear stability analysis of the steady-state equations corresponding to the minimal model and found that there were three predicted steady states. However, one of these steady states (the largest one) was not a biologically relevant solution, and another (close to zero) was unstable. Thus, p38 activation in this system was not bistable.
. Simulations using this minimal model were carried out using the Gillespie algorithm (47), and the rate of each reaction was modeled using standard mass action kinetics. Simulation of model II showed an initial modest increase in the number of active p38 molecules after ZAP70 activation, which was used as a proxy for TCR stimulation (Fig. 3B). As time progressed, a large increase in the number of active p38 molecules was predicted (Fig. 3B). The simulation also showed that the threshold of amount of pP380 and the strength of the classical pathway affected the behavior of p38 activation in a coordinated manner. To determine whether p38 activation is bistable in this model, we carried out a linear stability analysis of the steady-state equations corresponding to the minimal model and found that there were three predicted steady states. However, one of these steady states (the largest one) was not a biologically relevant solution, and another (close to zero) was unstable. Thus, p38 activation in this system was not bistable.
To determine how the kinetics or magnitude of p38 signaling differs when the contribution of each pathway is varied, we simulated three different strengths of the alternative (Fig. 3C) or classical (Fig. 3D) p38 pathway activation. In this minimal model, zero input from the alternative pathway predicted complete impairment of p38 activation (Fig. 3C, none). The outcome of our in silico modeling differed from our cellular experiments with B cells, and it was possible that the alternative pathway contribution to pp38-Tyr323 was never completely nil in lymphocytes. Increasing the contribution of the alternative pathway in our model predicted faster acquisition of full p38 activation (Fig. 3C). Eliminating all contribution of the classical pathway predicted blunted p38 activation (Fig. 3D, none), which is akin to LAT deletion or to the p38 inhibitor results in our cellular experiments. Increasing the contribution of the classical pathway predicted an increase in the magnitude of the maximal p38 activation (Fig. 3D). Collectively, the modeling results predicted convergence of the alternative and classical pathways in which the alternative pathway may lower the threshold for p38 activation and the classical pathway sets the magnitude of p38 activation after TCR stimulation (Fig. 3, E and F).
Basal ZAP70 activity lowers p38 activation threshold and facilitates rapid full activation of p38
Analysis of unstimulated primary thymocytes and peripheral T cells from lymphoid organs reveals that the TCR CD3ζ chain is constitutively phosphorylated and bound to ZAP70 (48–50). Constitutive ZAP70 activation may influence p38 activation, as direct comparisons of intracellular pFLOW stain for pp38-Thr180Tyr182 revealed that baseline abundance was consistently higher in Jurkat T cells when compared to DT40 B cells that do not express ZAP70 (Fig. 4A and fig. S7A). Given that Src family kinases phosphorylate immuno-receptor tyrosine-based activation motifs in the TCR CD3ζ chain, which is a prerequisite for ZAP70 activation (51), we used a Src family kinase inhibitor PP2 which reduces ZAP70 activity (52, 53). When we treated cells with PP2 for 30 min at 37°C, we found that PP2 treatment reduced the basal pp38-Thr180Tyr182 abundance (Fig. 4A and fig. S7A). On the basis of these findings, we modified our in silico p38 activation model and included a basal amount of active ZAP70 at the beginning of the simulation as an initial condition (Fig. 4B). Simulations with this iteration of our p38 activation model predicted that basal ZAP70 activity may lower the threshold for p38 activation (Fig. 4C). This computational prediction and the fact that ZAP70, but not SYK, can enhance activation of p38 in T cells (30, 31) suggested that ZAP70, but not SYK, may facilitate TCR-stimulated full p38 activation.
Fig. 4. Basal ZAP70-dependent alternative pathway lowers the activation threshold to facilitate strong activation of p38.
(A) Flow cytometry analysis of p38 activation Jurkat and DT40 B cells (top) or Jurkat cells treated with DMSO or the Src kinase inhibitor PP2. Histograms are representative of two independent experiments (see also fig. S7A). (B and C) Iteration of the p38 activation model that includes basal ZAP70 kinase activity (B) and in silico predictions of p38 activation with or without basal ZAP70 activity (C). (D) Flow cytometry analysis of pZAP70 and pp38 in DT40 B cells transfected with expression constructs for human ZAP70 or SYK kinases. Three subpopulations were arbitrarily defined as low (black), medium (blue), and high (red) for pZAP70 or pSYK (left), and histograms indicate pp38 of the indicated sub-population with the inset of MFI values. The black vertical divider marks the basal p38 activation in parental DT40 cells. Data are representative examples of two independent experiments (see also fig. S7B). (E) Western blot analysis of the indicated proteins in lysates from ZAP70-deficient Jurkat T cells (P116) transfected with ZAP70 or SYK expression constructs and stimulated with antibody against CD3ε for the indicated times. Band intensity values are given relative to the indicated control. Blots are representative examples of two independent experiments (see also fig. S7C). (F to H) Flow cytometry analysis of pZAP70 and pp38 in ZAP70-deficient Jurkat T cells (P116) transfected with ZAP70 or SYK expression constructs (F) and stimulated with antibody against CD3ε for the indicated times (G). Subpopulations with low (black), medium (blue), or high (red) pZAP70 or pSYK abundance were identified (left), and histograms (right) indicate p38 activation in cells with equivalent ZAP70 (filled histogram) or SYK (open histogram) activation. Flow cytometry plots and quantified p38 values after CD3ε stimulation (H) are representative of two independent experiments (see also fig. S7, C and D).
To test this prediction, we transiently transfected cell lines with constructs for either human ZAP70 or SYK and measured active ZAP70/SYK in tandem with pp38-Thr180Tyr182 by dual-parameter pFLOW. We found that the antibody against both pZAP70-Tyr319 and pSYK-Tyr352 does not detect phosphorylation of endogenously expressed avian SYK in chicken DT40 B cells. In unstimulated DT40 cells, we found that the amount of phosphorylation on ectopically expressed pZAP70/pSyk correlated with basal pp38-Thr180Tyr182 phosphorylation (Fig. 4D and fig. S7B). We only found pp38-Thr180Tyr182 in unstimulated DT40 cells that also had high amounts, and not medium amounts, of phosphorylated, ectopically expressed SYK (Fig. 4D and fig. S7B). We also confirmed these findings in a P116 Jurkat T cell line that is deficient for ZAP70 (54). When we quantitatively compared reconstitution of P116 cells with either ZAP70 or SYK kinase by immunoblot (Fig. 4E and fig. S7C), we found that the magnitude of TCR-stimulated pSYK was substantially lower than the pZAP70. Although SYK-expressing P116 cells efficiently activate phospholipase C-γ1 and ERK1/2 after TCR stimulation, p38 phosphorylation is reduced when compared to ZAP70-expressing P116 cells (Fig. 4E and fig. S7C). Similarly, in pFLOW-based assays, only cells that exhibited strong pZAP70 also showed basal pp38-Thr180Tyr182, but cells with similar amounts of phosphorylated ectopically expressed SYK did not (Fig. 4F). Furthermore, P116 cells transfected with ZAP70 displayed robust pp38-Thr180Tyr182 after TCR stimulation, which was less apparent in P116 cells expressing SYK (Fig. 4, G and H, and fig. S7D). Collectively, these data demonstrated that the alternative ZAP70-p38 pathway converges on the classical pathway even in resting cells to facilitate full p38 activation.
SOS1/2 deficiency attenuates TCR-induced p38 activation and induced regulatory T cell differentiation
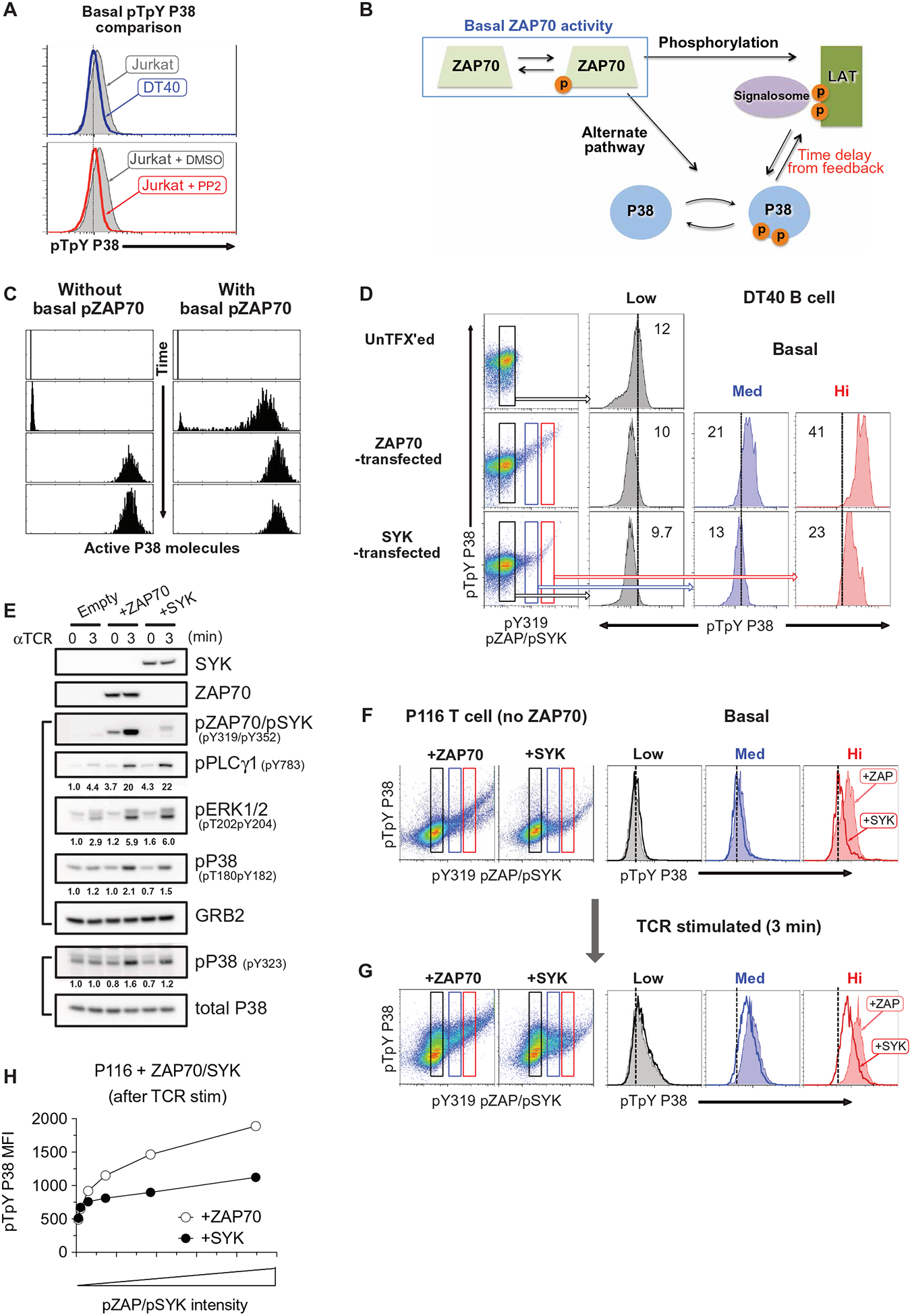
Although SOS GEFs are critical for optimal p38 pathway activation, the catalytic activity of SOS1 is not required (15). These data suggest that SOS1 likely acts as an adapter molecule to promote p38 pathway function, possibly within the LAT signalosome (Fig. 5A). By pFLOW, we confirmed that p38 activation was impaired in SOS1/2 double-deficient DT40 cells, especially the later phase strong p38 activation (Fig. 5B and fig. S8, A and B). In primary CD4+ T cells from Sos1fl/fl:Sos2−/−:CD4cre+ (Sos1/2 dKO) mice (55), when we compared p38 activation to wild-type CD4+ T cells, we found that Sos1/2 deletion reduced p38 activation in response to weak anti-CD3ε stimulation (Fig. 5C and fig. S8C). Loss of Sos1/2 was not associated with decreased TCR-stimulated ZAP70 activation, and the alternative p38 pathway activation as measured by pp38-Tyr323 was comparable to wild-type CD4+ T cells (Fig. 5D and fig. S8D).
Fig. 5. SOS1/2 deficiency reduces p38 activation and regulatory T cell differentiation.
(A) Iteration of the p38 activation model that includes SOS and Grb2 molecules added as a component of the LAT signalosome. (B) Flow cytometry analysis of pp38 in wild-type (filled) and SOS1/2−/− (open) DT40 B cells stimulated with antibody against the BCR for the indicated times. Histograms are representative of two independent experiments (see also fig. S8, A and B). (C) Flow cytometry analysis of pp38 in wild-type and SOS1/2 dKO naïve CD4+ T cells stimulated with antibody against CD3ε for the indicated times. Histograms are representative of two independent experiments (see also fig. S8C). (D) Western blot analysis of the indicated proteins in lysates from wild-type or Sos1/2 dKO naïve CD4+ T cells with antibody against CD3ε for the indicated times. Blots are representative of three independent experiments (see also fig. S8D). (E to H) Flow cytometry analysis of the indicated cytokine and transcription factor expression in wild-type (top) and Sos1/2 dKO (bottom) naïve T cells cultured under TH1 (E), TH2 (F), TH17 (G, left), or iTreg (G, right) polarizing conditions. Dot plots (E to G) are representative of two independent experiments (see also figs. S8, E and F, and S9). The frequency of iTreg induced by stimulation of wild-type (filled) and Sos1/2 dKO (open) CD4+ T cells with increasing anit-CD3ε doses (H) of at least five mice are from two independent experiments. *P < 0.05, **P < 0.01, and n.s., not significant by two-tailed Mann-Whitney test. WT, wild type.
Although SOS deficiency in T cells increases PI3K activity and augments T cell migration (56), the biological impact of Sos1/2 deletion in CD4+ T cells has remained largely unexplored (55). Because p38 activation is required for TH1, TH17, and iTreg differentiation (10–12), we examined whether in vitro TH cell differentiation was affected by Sos1/2 deletion. When TH1, TH2, and TH17 cell differentiation were assessed by intracellular staining of IL-4, IFN-γ, and IL-17A cytokines, Sos1/2 dKO CD4+ T cells were comparable to wild-type CD4+ T cells (Fig. 5, E and F, and fig. S8, E and F). However, iTreg differentiation was reduced in CD4+ T cells lacking Sos1/2 (Fig. 5G and fig. S8, E and F). The observed impairment in iTreg differentiation was consistent over a range of TCR stimulation strengths, but most notable when TCR stimulation was more modest (Fig. 5H and fig. S9). TH17 cell differentiation was affected by Sos1/2 deletion after weak TCR stimulation (Fig. 5H and fig. S9). Thus, SOS contributes to p38 activation and iTreg differentiation, especially when TCR stimulatory signals are more modest.
SOS1/2 deficiency leads to reduced IL-2 production and increased sensitivity to EAE
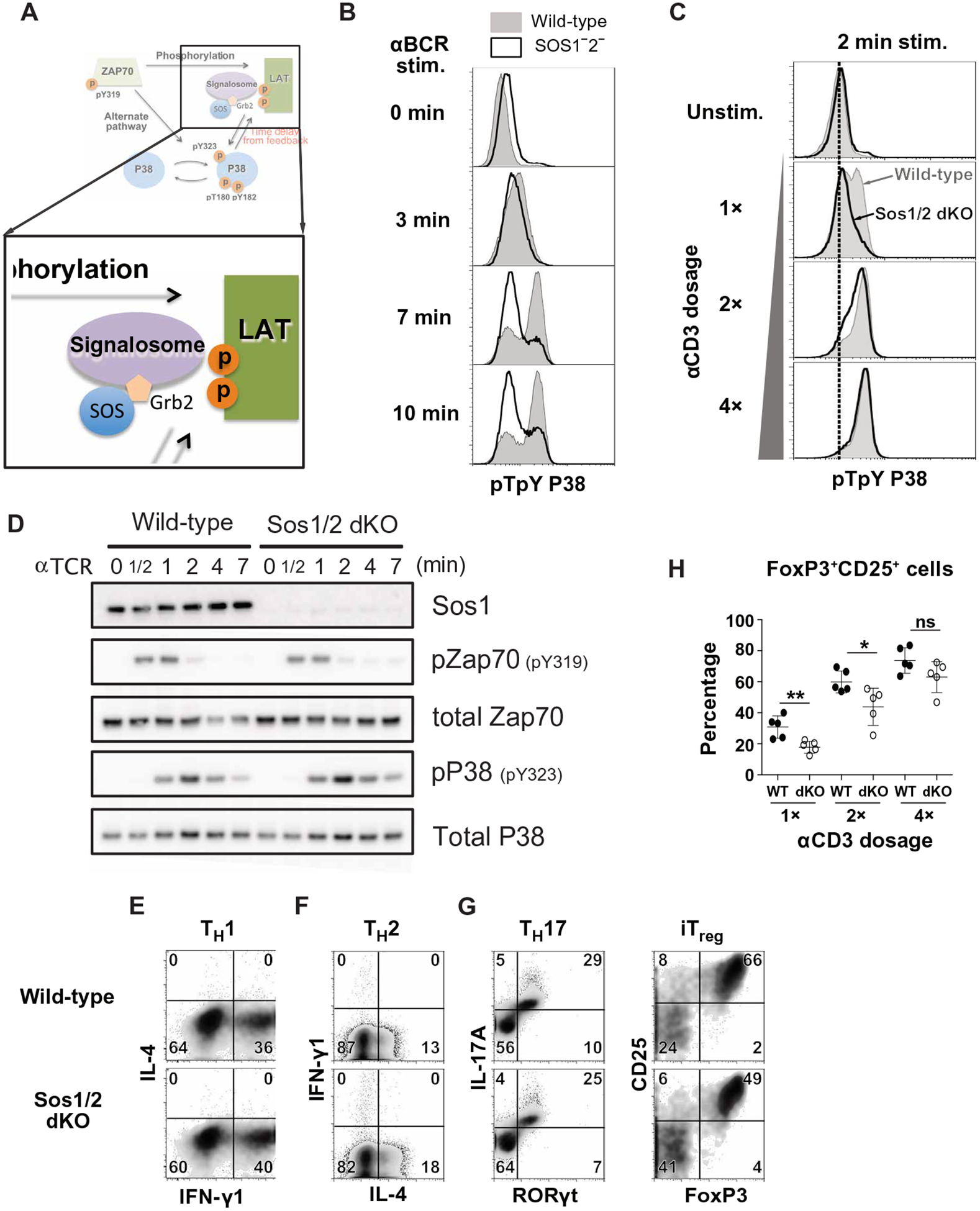
Because optimal p38 activation through TCR/CD28 is important for IL-2 production (57–59) and regulatory T cell differentiation and Treg homeostasis show strong dependence on IL-2 (60–63), we explored the possibility that Sos1/2-dependent p38 activation promotes IL-2 production. When we cultured sorted naïve CD4+ T cells from wild-type or Sos1/2 dKO animals and assayed IL-2 production, we found that loss of Sos1/2 reduced the production of IL-2 (Fig. 6A). Treatment with the p38 inhibitor SB203580 reduced the frequency of IL-2+ cells in cultures of wild-type but not Sos1/2 dKO CD4+ T cells (Fig. 6B).
Fig. 6. SOS1/2 deficiency leads to reduced IL-2 production and increased sensitivity to EAE.
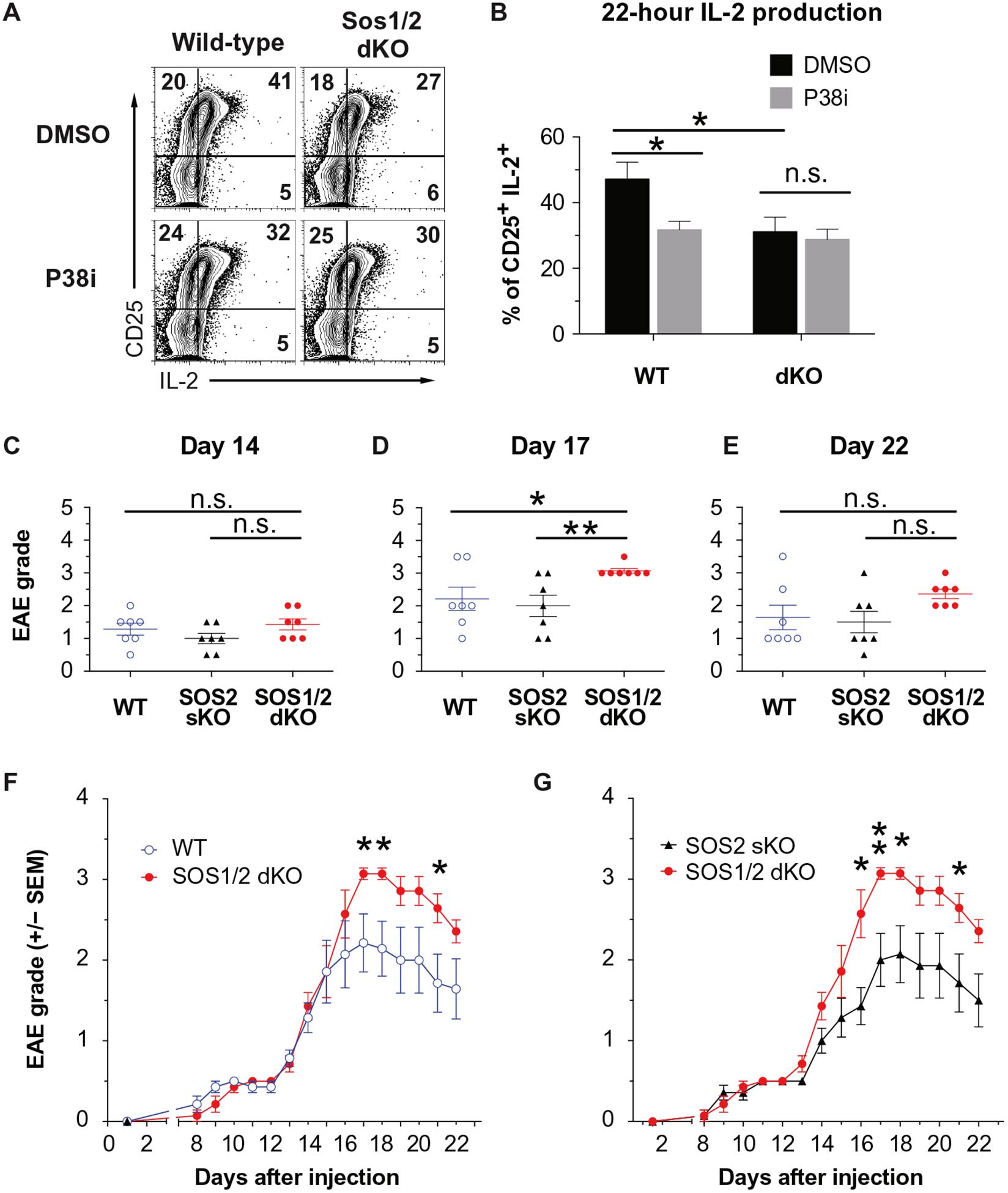
(A and B) Flow cytometry analysis of CD25 and IL-2 abundance in wild-type (left) and Sos1/2 dKO (right) naïve T cells treated with DMSO or p38i after TCR stimulation. The relative percentage of cells in each quadrant is indicated. Histograms (A) are representative of two independent experiments, and quantified frequency of CD25+IL-2+ (B) are means ± SEM from two experiments performed in duplicate. (C to G) EAE disease severity in wild-type, SOS2 sKO, and SOS1/2 dKO mice was clinically scored at the indicated times after immunization with MOG35–55 peptide and CFA, which correlated with the induction (C), peak severity (D), or recovery (E) phases. Data with means ± SEM (D to F) of seven mice are from two independent experiments. Cumulative clinical score over time (F and G) are means ± SEM from all experiments. *P < 0.05, **P < 0.01 by two-tailed Mann-Whitney test.
Young Sos1/2 dKO mice are healthy, have normal lymphoid compartments, and show no signs of autoimmune diseases (55). We used a common mouse model of TH1 cell-mediated human multiple sclerosis [reviewed in (64)] to assess the contribution of SOS1/2-p38 activation in vivo. In mice, experimental autoimmune encephalitis (EAE) is induced by injection of myelin oligodendrocyte glycoprotein 35 to 55 (MOG35–55) in complete Freund’s adjuvant (CFA) with pertussis toxin (PTX) (65–67). In this model, Treg cells are required to control inflammatory autoimmune reactions (68–71). The peak disease severity and rate of resolution are determined by balance between autoimmune pathogenic effector cells and protective Treg cells. To assess a potential role for SOS in T cells during induced autoimmune disease, we followed the onset, peak disease, and recovery phase of EAE in wild-type, Sos1/2 dKO, and Sos2-deficient mice (Sos2 sKO). In all three cohorts of mice, EAE disease onset occurred at day 14 (Fig. 6C); however, the peak of EAE disease was more severe in Sos1/2 dKO than in wild-type or Sos2 sKO mice (Fig. 6, D, F, and G). Although, eventually, EAE disease resolves in all groups, the kinetics are delayed in Sos1/2 dKO mice (Fig. 6, E to G). Thus, loss of Sos1 and Sos2 in CD4+ T cells exacerbated EAE in mice.
DISCUSSION
TCR-induced p38 activation has remained poorly understood over the past decade, and the conflicting reports on the alternative and classical pathways added to the confusion. Here, we capitalized on computational models and dual-parameter pFLOW to clarify how TCR stimulation activates p38 and unify the alternative and classical pathways. We found that a basal alternative ZAP70-dependent pp38-Tyr323 pathway lowers the threshold (“primes”) for the classical pathway, leading to full pp38-Thr180Tyr182 activation. The classical pathway requires the adapter LAT, SOS1, and catalytic activity of p38. Without the SOS-p38 axis, CD4+ T cells produce less IL-2, and differentiation into iTreg cells is impaired. Genetic perturbation of alternative pathway through p38-Y323F knock-in approaches reveals that the alternative p38 pathway is required for proinflammatory TH1 and TH17 functions (33–35). Together, all the data suggest that alternative and classical p38 activation pathways need to coexist to create balance between proinflammatory and anti-inflammatory responses (Fig. 7).
Fig. 7. The balance between distinct p38 pathways tunes pro- and anti-inflammatory T cell responses.
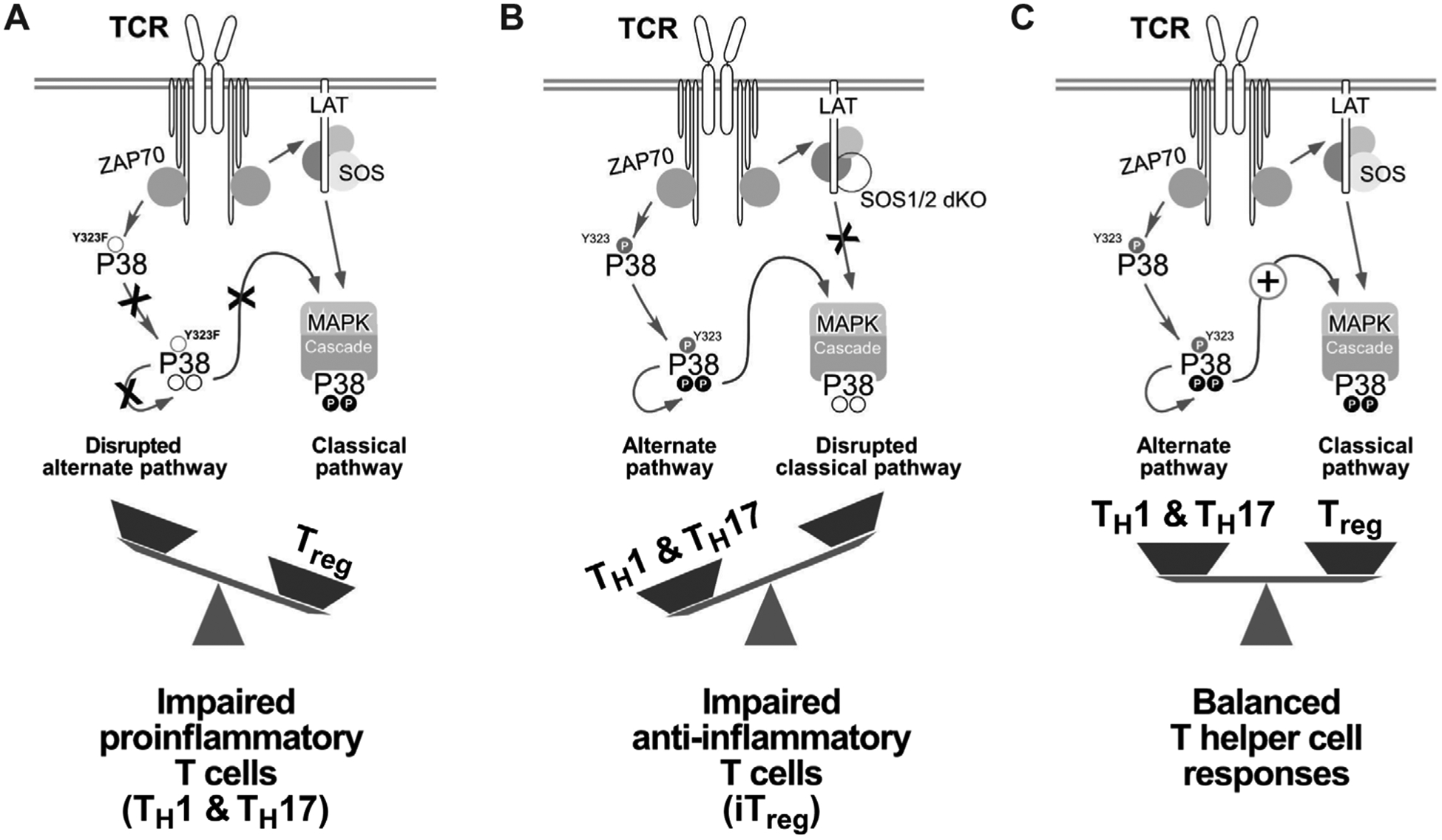
(A) Selective disruption of the alternative p38 pathway by Y323F substitution in P38α and P38β (P38αβY323F) impairs proinflammatory TH1 and TH17 cell IFN-γ and IL-17 production (35, 36). (B) Uncoupling of the classical p38 pathway by SOS1/2 deletion reduces iTreg cell differentiation, which counteracts the proinflammatory T cell functions of p38 activation. (C) The functional dichotomy between pro- and anti-inflammatory T cell responses is determined by the cooperation of two distinct p38 activation pathways.
Incoming TCR signal strength determines the activation of a number of downstream kinase pathways in peripheral T cells (13). SOS is required for digital ERK responses (14, 15, 72), which depends on active RasGTP allosterically activating SOS to produce more RasGTP (15, 73–75). In addition, binding of RasGTP to the allosteric, noncatalytic, pocket of SOS provides another membrane anchor for SOS and retains it longer at the membrane where it activates Ras (72). However, a different RasGEF, RasGRP1, activates Ras in an analog manner and that its produced RasGTP can prime this digital SOS-Ras-ERK pathway (14, 73).
In our studies, we found that SOS also amplified the p38 pathway, particularly under conditions of suboptimal TCR stimulation. Although the presence but not the catalytic activity of SOS is required to amplify p38 signals (15), the exact molecular connection between SOS and p38 activation is not known. Whereas SOS may act as an adaptor, other signaling molecules such as Vav and Rac are likely involved, which we defined as signalosome in our computational models. Our results suggested that priming of SOS-p38 signals occurs through a different mechanism than for Ras-ERK signaling and involves the alternative ZAP70-p38 pathway. Thus, SOS appears to be an amplifier for both the ERK and p38 kinase pathways. We speculate that this step-wise “priming and amplification” mechanism that we observed for p38 activation may be a mechanism to precisely control kinase pathway activation and T cell responses that occur when only a small number of TCR-pMHC (peptide-MHC) are engaged (76, 77).
Whereas the biochemical underpinnings of digital SOS-Ras-ERK signals are well established (14, 15), mice that lack Sos1 and Sos2 in T cells (Sos1/2 dKO) are grossly normal (55). Young Sos1/2 dKO mice are healthy, have normal lymphoid compartments, and show no signs of autoimmune diseases (55). In contrast, when Lck-Cre drives deletion of Sos1 and Sos2 in thymocytes, Sos1/2 dKO mice (with the CD4-Cre driver) do not develop peripheral T cells. Stimulating thymocytes with anti-CD3ε cross-linking does not rescue this developmental blockade, which suggests that Sos1 plays a critical role in transmitting pre-TCR signals (55). Here, we demonstrated that SOS-mediated p38 activation is critical for optimal IL-2 production and differentiation in iTreg cells, particularly when weaker TCR stimulation is applied. In addition, mice with Sos1/2 dKO T cells were more susceptible to experimentally induced EAE. Aged Sos1/2 dKO T cell animals also develop an unexpected chondrocyte dysplasia phenotype leading to joint deformation. SOS deficiency in a chondrocyte-like cell where the CD4-Cre must be active causes this phenotype (56). Relevant to our work here, the chondrocyte dysplasia phenotype appears more penetrant when Sos1/2 deficiency is crossed onto a Rag2-deficient background, raising the possibility that altered or impaired regulatory T cell differentiation affects the unexpected joint phenotype.
We anticipate that more peripheral T cell functions may depend on SOS function, particularly when these functions require full activation of ERK and p38 pathways that are amplified by SOS and cannot be generated by more modest RasGRP1-Ras-Erk or by ZAP70-p38 signals. A likely additional T cell subset where SOS could also play a critical role are TH17 cells, because we observed that SOS deletion and p38 inhibition partially impaired the efficiency of TH17 differentiation. Both Treg and TH17 cells are abundantly found in the intestinal mucosa of healthy mice (78, 79). TH17 cells are important for normal protective mucosal immunity against pathogens, and auto-inflammation in the intestine (80, 81) and normal intestinal health depends on homeostatic balance between Treg and TH17 cells (82). Our study enables a new model of balanced proinflammatory and anti-inflammatory responses as a function of distinct p38 pathway signals (Fig. 7). Future studies are needed on which signaling cues that can tip the balance between p38 pathways and specific effector molecules that may be downstream of the alternative versus classical p38 pathways.
MATERIALS AND METHODS
Cell line maintenance, plasmid transfection, inhibitor treatment, and stimulation
Both wild-type and SOS1−2− chicken DT40 B cells were gifts from T. Kurosaki (RIKEN). Wild-type human leukemic Jurkat, LAT-deficient J.Cam2, and ZAP70-deficient Jurkat variant P116 cells were obtained from A. Weiss lab [University of California, San Francisco (UCSF)]. Wild-type and mutant variant cultures of Jurkat T cells and chicken DT40 B cells were carried out as described before (15, 72). Plasmids encoding full-length human ZAP70 or SYK kinases were gifts from A. Weiss lab (UCSF). For transient transfection of human ZAP70 or SYK in cell lines, cells were harvested and re-suspended at 66 × 106cells/ml plain RPMI 1640. For each transfection, a total of 20 × 106 cells were transfected with 10 μg of plasmid DNA by Bio-Rad electroporator (Bio-Rad) at exponential decay, 250 V, and 950 F. For routine cell stimulation, healthy cells were harvested and rested for 30 min in plain RPMI 1640 at 37°C. Stimulation of Jurkat cells was carried out in plain RPMI 1640 at 37°C with either a 1:600 (high) or 1:6000 (low) dilution of immunoglobulin M (IgM) mouse ascites fluid from the C305 hybridoma against TCR (14). Stimulation of DT40 cells was carried out in plain RPMI 1640 at 37°C with either a 1:1000 (high) or 1:40,000 (low) dilution of IgM mouse ascites fluid from the M4 hybridoma against BCR (14, 73). Inhibition of Src kinase, MEK, and p38 activity was mediated by preincubating cells with 20 μM final PP2 [4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo [3,4-d]pyrimidine; Calbiochem], 10 μM final U0126 (MEKi; Promega), or 10 μM final SB203580 (p38i; Calbiochem) for 30 min during the cell resting stage before antigen receptor stimulation. All inhibitors were diluted in dimethyl sulfoxide (DMSO). DMSO was added to control samples at equivalent concentrations. For TH differentiation assay, p38i was applied during the entire duration of culture.
Human peripheral CD4+ T cell culture, CRISPR-Cas9 editing of LAT, and T cell stimulation
Human PBMC (peripheral blood mono-nuclear cells) CD4+ T cells were edited as previously described (83). For LAT gene deletion, purified CD4+ T cells were stimulated for 48 hours on anti-CD3ε-coated plates [coated with αCD3ε UCHT1 (10 μg/ml), Tonbo Biosciences] in the presence of soluble anti-CD28 [5 μg/ml; CD28.2, Tonbo Biosciences]. LAT-targeting CRISPR RNA (5′-GCAGATGGAGGAGGCCATCC-3′) and the matching tracer RNA were designed by using Benchling.com online tool and were chemically synthesized (Dharmacon). Recombinant Streptococcus pyogenes Cas9 protein containing a C-terminal hemagglutinin tag and two nuclear localization signal peptides was obtained from the QB3 Macrolab (University of California, Berkeley) (84). Cas9 ribonuclear protein (RNP) electroporation was performed using 96-well reaction cuvette with program EH-115 on the Amaxa 4D-Nucleofector (Lonza) as previously described (83). Electroporated cells were allowed to recover for 30 min at 37°C in prewarmed complete culture medium and then transferred to containing complete T cell medium. After culturing for 36 hours at 37°C, cells were washed twice with plain RPMI. For each condition, 1 × 106 cells were rested in 100 μl of plain RPMI in a 96-well plate for 2 hours before stimulation. Cells were stimulated with final concentration of anti-CD3ε IgM (20 μg/ml; MEM92; Envigo Bioproducts Inc., USA).
Immunoblot analysis
Immunoblot analysis was performed as previously described (16). The following antibodies were purchased from Cell Signaling Technology: pZAP70 (pY319; #2701), phospho-p38 pT180/pY182 (clone 3D7; #9215), phospho-p44/42 MAPK ERK1/2 pT204/pY204 (#9101), and total p38 (#9212). The antibody for phospho-p38 (pY323) was purchased from Thermo Fisher Scientific (#PA5–12868). Horseradish peroxidase-conjugated goat anti-rabbit (Thermo Fisher Scientific) and sheep anti-mouse (VWR) were used as secondary antibodies. Immunoblot signal was visualized using enhanced chemiluminescence Western blot substrate (Pierce) and the LAS-4000 image system that allows for pixel densitometry measurements (Fujifilm Life Science).
Mice, primary CD4+ T cell preparation, stimulation, and blast preparation
Sos1/2 dKO animals were a gift from L. Samelson lab (National Institutes of Health/National Cancer Institute, MD) and were previously described (55, 85). All mice were bred and maintained under specific pathogen-free conditions at the laboratory animal resource center of UCSF. All mice were housed and treated in accordance with the guidelines of the Institutional Animal Care and Use Committee guidelines of the UCSF (AN098375–03B). All experimental mice except for EAE study were used at the age of 8 to 12 weeks in an age- and sex-matched manner. Naïve CD4+ T cells were purified from peripheral lymph nodes and splenic lymphocytes by magnetic negative selection (95 to 98% purity CD4+CD25− CD62LhighCD44low) (#130-104-453, Miltenyi Biotec) according to the manufacturer’s protocol. CD4+ T cell blasts were prepared by culturing purified naïve CD4+ T cells with plate-bound anti-CD3ε (clone 2C11, UCSF antibody core; clone 500A2, BD Pharmingen #553238; both used at 1 μg/ml) and soluble anti-CD28 [clone 37.51 (2 μg/ml); UCSF antibody core) for 2 days. Stimulated cells were harvested and then expanded with IL-2 (100 U/ml; PeproTech) for three more days. For acute TCR stimulation, purified CD4+ T cells or blasts were rested in full culture medium for 16 to 18 hours at 37°C, followed by resting in serum-free plain RPMI for 60 min. For stimulation, a gradient of anti-CD3ε antibody was applied at 0.5 to 5 μg/ml, subsequently super-crosslinked by adding 10-fold higher concentration (5 to 50 μg/ml) of goat anti-Armenian or Syrian hamster secondary antibody (Jackson ImmunoResearch Laboratories) 30 s later.
pFLOW analysis
Flow cytometry analysis of ZAP70 and p38 activation was performed according to established procedures (15, 72). Briefly, cells were stimulated with either TCR or BCR cross-linking mouse IgM (clone M4, 8300–01, Southern Biotech) for the desired time period. Stimulation was then stopped by addition of 4% paraformaldehyde in phosphate-buffered saline (PBS), and cells were fixed for 20 min at room temperature. Fixed cells were washed three times with fluorescence-activated cell sorting (FACS) wash buffer (PBS, 1% bovine serum albumin, and 10 mM EDTA) and subsequently permeabilized overnight with pre-chilled 90% methanol. Permeabilized cells were washed and stained for LAT (clone LAT1111, #623902, BioLegend), pZAP70 Tyr319 (#2701, Cell Signaling Technology), pERK Thr202/Tyr204 (#9101, Cell Signaling Technology), Alexa Fluor 647-conjugated pERK Thr202/Tyr204 (#13148, Cell Signaling Technology), or pp38 Thr180/Tyr182 (clone 3D7, #4092, Cell Signaling Technology). Probed primary antibodies were visualized by secondary staining with phycoerythrin (PE)- or allophycocyanin (APC)-conjugated strepavidin (S866, Invitrogen) or AffiniPure F(ab′)2 fragment donkey anti-rabbit IgG (#711-136-152, Jackson ImmunoResearch Laboratories). For dual pFLOW assays, pp38 was probed with mouse anti-sera (clone 36, #612288, BD Transduction) and visualized with Alexa Fluor 488-conjugated donkey anti-mouse IgG antibody (Invitrogen). Stained cells were acquired with a FACS Calibur machine (BD) and analyzed with FlowJo software.
In vitro TH differentiation assay
For differentiation of TH1 or TH2 CD4+ T cells, CD4+ T cells were isolated by MACS (Magnetic Activated Cell Sorting) negative isolation (Miltenyi) or by FACS (staining for CD4, CD25, CD44, and CD5) in the UCSF Flow Cytometry Core. For differentiation of TH1 or TH2 CD4+ T cells, CD4+ T cells were stimulated for 2 days with plate-bound anti-CD3ε (1 μg/ml) and soluble CD28 antibodies (2 μg/ml; BD, Heidelberg, Germany) in the presence of recombinant IL-12 (10 ng/ml; PeproTech) and anti-IL-4 antibody (10 μg/ml; clone 11B11, Tonbo) or recombinant IL-4 (50 ng/ml; PeproTech) with anti-IFN-γ blocking antibody (10 μg/ml; clone XMG1.2, UCSF antibody core), respectively. On day 3, activated cells were blasted in the presence of rIL-2 (100 U/ml; PeproTech) with TH1- or TH2-polarizing cytokine and blocking antibodies. Differentiated cells are harvested on day 4 or 5 as indicated. For the conversion into TH17 and iTreg cells, sorted CD4+ T cells were activated as above in the presence of hTGFβ1 (1 ng/ml; R&D Systems) with or without rIL-6 (40 ng/ml; PeproTech) and anti-IFN-γ, respectively. Differentiated cells are harvested on day 4. For TH1, TH2, and TH17 cells, before culture harvest, cells are restimulated with PMA (phorbol 12-myristate 13-acetate) (50 ng/ml; EMD Millipore) and ionomycin (1 μg/ml; EMD Millipore) for 4 hours in the presence of GolgiStop (BD). Differentiated cells were assessed by staining with antibodies against T-Bet (#12-5825-80, eBioscience), IFN-γ (#17-7311-81, eBioscience), Gata3 (#50-9966-42, eBioscience), IL-4 (#554435, BD Pharmingen), IL-17A (#45-7177-82, eBioscience), and RORγt (#12-6988-80, eBioscience). For iTreg cell assay, the expression of Foxp3 was assessed by a FoxP3 staining kit (eBioscience) with APC-conjugated anti-FoxP3 antibody (#17-5775-80, eBioscience).
Intracellular IL-2 production assay
Purified naïve CD4+ T cells (2 × 106 cells/ml) from wild-type and Sos1/2 dKO mice were stimulated with plate-bound anti-CD3ε (1 μg/ml; clone 500A2) and soluble anti-CD28 (2 μg/ml; clone 37.51) antibodies overnight. After 18 hours after stimulation, GolgiStop was added to each sample and cultured for 4 more hours. After a total of 22 hours of culture, cells were harvested and treated for intracellular IL-2 staining using a BD cytofix/cytoperm staining kit (#554714, BD). PE-conjugated rat anti-IL-2 antibody was purchased from BD (#554435).
EAE induction and scoring
EAE disease was induced in wild-type or Sos2 sKO and Sos1/2 dKO female littermate mice that were 13 to 15 weeks old by immunization with MOG35–55 peptide and CFA emulsion with PTX (catalog no. EK-2110, Hooke Laboratories). Briefly, 100 μl of MOG/CFA emulsion was subcutaneously injected at two sites per mouse on day 0. PTX (100 μl) was administered intraperitoneally on one side. PTX injection was repeated on the opposite side on day 2. For 22 days after EAE induction, mice were monitored and assigned disease scores daily according to manufacturer’s protocol. Once EAE was noticed, symptoms were clinically scored as follows: 0, no sign of disease; 0.5, limp tail tip; 1, complete limp tail; 1.5, one hind leg inhibition; 2, two hind leg inhibition; 2.5, two hind leg paralysis with some movement; 3, total hind leg paralysis/dragging; 3.5, unable to self-right or both legs dragged on the same side; 4, partial front leg paralysis and reduced mobility; 5, moribund.
In silico simulation method and parameters
The various models tested are shown in the schematic diagrams (Figs. 3A, 4B, and 5A). The rate of each reaction was modeled using standard mass action kinetics. We solved the stochastic version of these equations using the Gillespie algorithm (48) in manners analogous to our previous work (14, 15). In all stochastic simulations, we used a spatially homogeneous simulation box of size V = area (4 mm2) × height (0.02 mm2). This choice of the system size ensures that the system is well mixed. The initial numbers of species and the rate constants are listed in Tables 1 and 2.
Table 1.
Initial numbers of species.
| Species | Initial number of molecules |
|---|---|
| ZAP70 | 100 |
| GPS | 100 |
| LAT | 200 |
| GPS | 1000 |
Table 2.
Rate constants.
| Reactions | Rate constant k (1/s) |
|---|---|
| Activated ZAP70 phosphorylates LAT, kzl | 0.05 |
| Activated ZAP70 phosphorylates p38, kzp | 0.005 |
| pLAT self-decays to the inactive form, kdl | 1 |
| GPS binds to pLAT, kbg | 0.0001 |
| GPS unbinds to pLAT, kug | 5 |
| GPS-LAT complex phosphorylates p38, ka | 0.1 |
| pp38 self-decays to the inactive form, kdp | 40 |
| The positive feedback of pp38 to GPS-LAT complex, kf | 0.01 |
Supplementary Material
Fig. S1. The role of LAT in p38 activation in Jurkat T cells and human PBMC CD4+ T cells.
Fig. S2. Experimental validation of anti-phospho p38 antisera for pTpY p38 and pY323 p38 detection.
Fig. S3. Testing anti-pY323 p38 antibody for pFLOW assay.
Fig. S4. Full activation of p38 through the classical pathway is thresholded and time delayed regardless of stimulus strength.
Fig. S5. Impact of p38 or MEK1 inhibition on BCR-induced ERK activation.
Fig. S6. p38 activation kinetics in primary T cells.
Fig. S7. Zap70 expression facilitates p38 activation.
Fig. S8. The role of SOS in p38 activation and iTreg formation.
Fig. S9. Sos1/2 double deficiency has more substantial impact on iTreg than TH17 cell differentiation.
Acknowledgments:
We thank members of the Roose and Chakraborty teams and also the lab of A. Weiss for helpful discussions. We thank R. Kortum and L. Samelson for sharing the Sos-deficient mice. We appreciate W.-L. Lo in A. Weiss lab (UCSF) for provision of LAT-targeting guide RNA sequence. V. Tobin and D. Simeonov in Marson lab (UCSF) provided human PBMC CD4+ T cells and assisted in establishing CRISPR-Cas9-editing protocol.
Funding: These studies were supported by grants from the NIH-NIAID (R01-AI104789 to J.P.R. and P01-AI091580 to J.P.R. and A.C.).
Footnotes
Competing interests: J.P.R. is a co-founder and scientific advisor of Seal Biosciences Inc. and on the scientific advisory committee for the Mark Foundation for Cancer Research. All other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Dong C, Davis RJ, Flavell RA, MAP kinases in the immune response. Annu. Rev. Immunol 20, 55–72 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Rincón M, Davis RJ, Regulation of the immune response by stress-activated protein kinases. Immunol. Rev 228, 212–224 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Hale KK, Trollinger D, Rihanek M, Manthey CL, Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J. Immunol 162, 4246–4252 (1999). [PubMed] [Google Scholar]
- 4.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ, Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem 270, 7420–7426 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Dodeller F, Skapenko A, Kalden JR, Lipsky PE, Schulze-Koops H, The p38 mitogen-activated protein kinase regulates effector functions of primary human CD4 T cells. Eur. J. Immunol 35, 3631–3642 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Ashwell JD, The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol 6, 532–540 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Sen J, Kapeller R, Fragoso R, Sen R, Zon LI, Burakoff SJ, Intrathymic signals in thymocytes are mediated by p38 mitogen-activated protein kinase. J. Immunol 156, 4535–4538 (1996). [PubMed] [Google Scholar]
- 8.Diehl NL, Enslen H, Fortner KA, Merritt C, Stetson N, Charland C, Flavell RA, Davis RJ, Rincón M, Activation of the p38 mitogen-activated protein kinase pathway arrests cell cycle progression and differentiation of immature thymocytes in vivo. J. Exp. Med 191, 321–334 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rincón M, Flavell RA, Davis RJ, Signal transduction by MAP kinases in T lymphocytes. Oncogene 20, 2490–2497 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Rincón M, Enslen H, Raingeaud J, Recht M, Zapton T, Su MS, Penix LA, Davis RJ, Flavell RA, Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 17, 2817–2829 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber S, Schrader J, Fritz G, Presser K, Schmitt S, Waisman A, Lüth S, Blessing M, Herkel J, Schramm C, p38 MAP kinase signaling is required for the conversion of CD4+CD25-T cells into iTreg. PLOS ONE 3, e3302 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noubade R, Krementsov DN, del Rio R, Thornton T, Nagaleekar V, Saligrama N, Spitzack A, Spach K, Sabio G, Davis RJ, Rincon M, Teuscher C, Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 118, 3290–3300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myers D, Roose JP, Kinase and phosphatase effector pathways in T cells. Encyclopedia Immunobiol. 3, 25–37 (2016). [Google Scholar]
- 14.Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, Chakraborty AK, Roose JP, Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell 136, 337–351 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jun JE, Yang M, Chen H, Chakraborty AK, Roose JP, Activation of extracellular signal-regulated kinase but not of p38 mitogen-activated protein kinase pathways in lymphocytes requires allosteric activation of SOS. Mol. Cell. Biol 33, 2470–2484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi H, Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol 12, 325–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodeller F, Schulze-Koops H, The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res. Ther 8, 205 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malissen B, Grégoire C, Malissen M, Roncagalli R, Integrative biology of T cell activation. Nat. Immunol 15, 790–797 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Kortum RL, Rouquette-Jazdanian AK, Samelson LE, Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. 34, 259–268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minden A, Lin A, Claret F-X, Abo A, Karin M, Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 81, 1147–1157 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Bubeck Wardenburg J, Pappu R, Bu J-Y, Mayer B, Chernoff J, Straus D, Chan AC, Chan AC, Chan AC, Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity 9, 607–616 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Salojin KV, Zhang J, Delovitch TL, TCR and CD28 are coupled via ZAP-70 to the activation of the Vav/Rac-1-/PAK-1/p38 MAPK signaling pathway. 163, 844–853 (1999). [PubMed] [Google Scholar]
- 23.Sanui T, Inayoshi A, Noda M, Iwata E, Oike M, Sasazuki T, Fukui Y, DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-θ and LFA-1, in T cells. Immunity 19, 119–129 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Jun JE, Rubio I, Roose JP, Regulation of ras exchange factors and cellular localization of ras activation by lipid messengers in T cells. Front. Immunol 4, 239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Leitenberg D, Li B, Flavell RA, Deficiency of small GTPase Rac2 affects T cell activation. J. Exp. Med 194, 915–926 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Zhang F, Chen F, Liu D, Zheng Y, Zhang Y, Dong C, Su B, MEKK3 regulates IFN-gamma production in T cells through the Rac1/2-dependent MAPK cascades. J. Immunol 186, 5791–5800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takekawa M, Saito H, A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95, 521–530 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Lu B, Ferrandino AF, Flavell RA, Gadd45beta is important for perpetuating cognate and inflammatory signals in T cells. Nat. Immunol 5, 38–44 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Perchonock CE, Fernando MC, Quinn WJ III, Nguyen CT, Sun J, Shapiro MJ, Shapiro VS, Negative regulation of interleukin-2 and p38 mitogen-activated protein kinase during T-cell activation by the adaptor ALX. Mol. Cell. Biol 26, 6005–6015 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mittelstadt PR, Yamaguchi H, Appella E, Ashwell JD, T cell receptor-mediated activation of p38α by mono-phosphorylation of the activation loop results in altered substrate specificity. J. Biol. Chem 284, 15469–15474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, Fornace AJ Jr., Ashwell JD, Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol 6, 390–395 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Tzarum N, Diskin R, Engelberg D, Livnah O, Active mutants of the TCR-mediated p38α alternative activation site show changes in the phosphorylation lip and DEF site formation. J. Mol. Biol 405, 1154–1169 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Jirmanova L, Sarma DN, Jankovic D, Mittelstadt PR, Ashwell JD, Genetic disruption of p38alpha Tyr323 phosphorylation prevents T-cell receptor-mediated p38alpha activation and impairs interferon-gamma production. Blood 113, 2229–2237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD, Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood 118, 3280–3289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alam MS, Gaida MM, Ogawa Y, Kolios AGA, Lasitschka F, Ashwell JD, Counter-regulation of T cell effector function by differentially activated p38. J. Exp. Med 211, 1257–1270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudd CE, MAPK p38: Alternative and nonstressful in T cells. Nat. Immunol 6, 368–370 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Gong Q, Cheng AM, Akk AM, Alberola-Ila J, Gong G, Pawson T, Chan AC, Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nat. Immunol 2, 29–36 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Jang IK, Zhang J, Chiang YJ, Kole HK, Cronshaw DG, Zou Y, Gu H, Grb2 functions at the top of the T-cell antigen receptor-induced tyrosine kinase cascade to control thymic selection. Proc. Natl. Acad. Sci. U.S.A 107, 10620–10625 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markegard E, Trager E, Yang C.-w. O., Zhang W, Weiss A, Roose JP, Basal LAT-diacylglycerol-RasGRP1 signals in T cells maintain TCRα gene expression. PLOS ONE 6, e25540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, Kurosaki T, Involvement of guanosine triphosphatases and phospholipase C-gamma2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J. Exp. Med 188, 1287–1295 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh-hora M, Johmura S, Hashimoto A, Hikida M, Kurosaki T, Requirement for Ras guanine nucleotide releasing protein 3 in coupling phospholipase C-γ2 to Ras in B cell receptor signaling. J. Exp. Med 198, 1841–1851 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young PR, McLaughlin MM, Kumar S, Kassis S, Doyle ML, McNulty D, Gallagher TF, Fisher S, McDonnell PC, Carr SA, Huddleston MJ, Seibel G, Porter TG, Livi GP, Adams JL, Lee JC, Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J. Biol. Chem 272, 12116–12121 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL, Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 47, 185–201 (2000). [DOI] [PubMed] [Google Scholar]
- 44.DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, Scherle PA, Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J. Immunol 160, 4175–4181 (1998). [PubMed] [Google Scholar]
- 45.Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, Kanduri K, Joly R, Malzac A, Lähdesmäki H, Lahesmaa R, Yamasaki S, Saito T, Malissen M, Aebersold R, Gstaiger M, Malissen B, Quantitative proteomics analysis of signalosome dynamics in primary T cells identifies the surface receptor CD6 as a Lat adaptor-independent TCR signaling hub. Nat. Immunol 15, 384–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salojin KV, Zhang J, Meagher C, Delovitch TL, ZAP-70 is essential for the T cell antigen receptor-induced plasma membrane targeting of SOS and Vav in T cells. J. Biol. Chem 275, 5966–5975 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Gillespie DT, Approximate accelerated stochastic simulation of chemically reacting systems. J. Chem. Phys 115, 1716–1733 (2001). [Google Scholar]
- 48.van Oers NS, Tao W, Watts JD, Johnson P, Aebersold R, Teh HS, Constitutive tyrosine phosphorylation of the T-cell receptor (TCR) zeta subunit: Regulation of TCR-associated protein tyrosine kinase activity by TCR zeta. Mol. Cell. Biol 13, 5771–5780 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stefanová I, Dorfman JR, Germain RN, Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature 420, 429–434 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Goodfellow HS, Frushicheva MP, Ji Q, Cheng DA, Kadlecek TA, Cantor AJ, Kuriyan J, Chakraborty AK, Salomon AR, Weiss A, The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci. Signal 8, ra49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Au-yeung BB, Deindl S, Hsu LY, Palacios EH, Levin SE, Kuriyan J, Weiss A, The structure, regulation, and function of ZAP-70. Immunol. Rev 228, 41–57 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA, Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem 271, 695–701 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Karni R, Mizrachi S, Reiss-Sklan E, Gazit A, Livnah O, Levitzki A, The pp60c-Src inhibitor PP1 is non-competitive against ATP. FEBS Lett. 537, 47–52 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Williams BL, Schreiber KL, Zhang W, Wange RL, Samelson LE, Leibson PJ, Abraham RT, Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: Reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell. Biol 18, 1388–1399 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kortum RL, Sommers CL, Alexander CP, Pinski JM, Li W, Grinberg A, Lee J, Love PE, Samelson LE, Targeted Sos1 deletion reveals its critical role in early T-cell development. Proc. Natl. Acad. Sci. U.S.A 108, 12407–12412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guittard G, Gallardo DL, Li W, Melis N, Lui JC, Kortum RL, Shakarishvili NG, Huh S, Baron J, Weigert R, Kramer JA, Samelson LE, Sommers CL, Unexpected cartilage phenotype in CD4-Cre-conditional SOS-deficient mice. Front. Immunol 8, 343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward SG, Parry RV, Matthews J, O’Neill L, A p38 MAP kinase inhibitor SB203580 inhibits CD28-dependent T cell proliferation and IL-2 production. Biochem. Soc. Trans 25, 304S (1997). [DOI] [PubMed] [Google Scholar]
- 58.Xu W, Yan M, Lu L, Sun L, Theze J, Zheng Z, Liu X, The p38 MAPK pathway is involved in the IL-2 induction of TNF-β gene via the EBS element. Biochem. Biophys. Res. Commun 289, 979–986 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Smith JL, Collins I, Chandramouli GV, Butscher WG, Zaitseva E, Freebern WJ, Haggerty CM, Doseeva V, Gardner K, Targeting combinatorial transcriptional complex assembly at specific modules within the interleukin-2 promoter by the immunosuppressant SB203580. J. Biol. Chem 278, 41034–41046 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY, An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol 17, 1322–1333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY, A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol 6, 1142–1151 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Bouguermouh S, Fortin G, Baba N, Rubio M, Sarfati M, CD28 co-stimulation down regulates Th17 development. PLOS ONE 4, e5087–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ, Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26, 371–381 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KHG, T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 162, 1–11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendel I, Kerlero de Rosbo N, Ben-Nun A, A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor Vβ expression of encephalitogenic T cells. Eur. J. Immunol 25, 1951–1959 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Zamvil SS, Steinman L, The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol 8, 579–621 (1990). [DOI] [PubMed] [Google Scholar]
- 67.Krementsov DN, Thornton TM, Teuscher C, Rincón M, The emerging role of p38 mitogen-activated protein kinase in multiple sclerosis and its models. Mol. Cell. Biol 33, 3728–3734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kohm AP, Carpentier PA, Anger HA, Miller SD, Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol 169, 4712–4716 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G, Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol. Commun 2, 163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stephens LA, Gray D, Anderton SM, CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc. Natl. Acad. Sci. U.S.A 102, 17418–17423 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK, Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A 101, 15434–15439 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christensen SM, Tu H-L, Jun JE, Alvarez S, Triplet MG, Iwig JS, Yadav KK, Bar-Sagi D, Roose JP, Groves JT, One-way membrane trafficking of SOS in receptor-triggered Ras activation. Nat. Struct. Mol. Biol 23, 838–846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roose JP, Mollenauer M, Ho M, Kurosaki T, Weiss A, Unusual interplay of two types of Ras activators, RasGRP and SOS, establishes sensitive and robust Ras activation in lymphocytes. Mol. Cell. Biol 27, 2732–2745 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sondermann H, Soisson SM, Boykevisch S, Yang S-S, Bar-Sagi D, Kuriyan J, Structural analysis of autoinhibition in the Ras activator son of sevenless. Cell 119, 393–405 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Boykevisch S, Zhao C, Sondermann H, Philippidou P, Halegoua S, Kuriyan J, Bar-Sagi D, Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr. Biol 16, 2173–2179 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Kimachi K, Croft M, Grey HM, The minimal number of antigen-major histocompatibility complex class II complexes required for activation of naive and primed T cells. Eur. J. Immunol 27, 3310–3317 (1997). [DOI] [PubMed] [Google Scholar]
- 77.Huang J, Brameshuber M, Zeng X, Xie J, Li Q.-j., Chien Y.-h., Valitutti S, Davis MM, A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT, Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3-precursor cells in the absence of interleukin 10. Nat. Immunol 8, 931–941 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Khader SA, Gaffen SL, Kolls JK, Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2, 403–411 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Curtis MM, Way SS, Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126, 177–185 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Omenetti S, Pizarro TT, The Treg/Th17 Axis: A dynamic balance regulated by the gut microbiome. Front. Immunol 6, 639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hultquist JF, Schumann K, Woo JM, Manganaro L, McGregor MJ, Doudna J, Simon V, Krogan NJ, Marson A, A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep 17, 1438–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anders C, Jinek M, In vitro enzymology of Cas9. Methods Enzymol. 546, 1–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guittard G, Kortum RL, Balagopalan L, Çuburu N, Nguyen P, Sommers CL, Samelson LE, Absence of both Sos-1 and Sos-2 in peripheral CD4 +T cells leads to PI3K pathway activation and defects in migration. Eur. J. Immunol 45, 2389–2395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The role of LAT in p38 activation in Jurkat T cells and human PBMC CD4+ T cells.
Fig. S2. Experimental validation of anti-phospho p38 antisera for pTpY p38 and pY323 p38 detection.
Fig. S3. Testing anti-pY323 p38 antibody for pFLOW assay.
Fig. S4. Full activation of p38 through the classical pathway is thresholded and time delayed regardless of stimulus strength.
Fig. S5. Impact of p38 or MEK1 inhibition on BCR-induced ERK activation.
Fig. S6. p38 activation kinetics in primary T cells.
Fig. S7. Zap70 expression facilitates p38 activation.
Fig. S8. The role of SOS in p38 activation and iTreg formation.
Fig. S9. Sos1/2 double deficiency has more substantial impact on iTreg than TH17 cell differentiation.