

Abstract
This editorial refers to ‘Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype’†, by K. Yang et al., on page 2442.
Aortic aneurysm/dissections (AADs), prevalent in up to 5.1% of the population worldwide, is a life-threatening pathology affecting up to 9% of males over 65 years old and currently lacking effective pharmacotherapeutic management.1 The pathology of AAD derives from the inability of the vascular walls to withstand increased intraluminal pressure, culminating in the dilation of the aortic wall, disruption of the medial layer, and possible dissection followed by sudden death.2 Management of AAD is mainly achieved through surgical intervention, making it pertinent to identify a novel target for therapeutics. In this issue of the European Heart Journal, Yang et al. highlight the novel role of aldehyde dehydrogenase 2 (ALDH2) polymorphisms in the regulation of myocardin expression and speculate on ALDH2’s role as a critical denominator in the pathology of AAD.3 ALDH2, best known for its role in alcohol oxidation and recently described to inhibit myocardial infarction,4 is suggested to mediate a phenotypic switch within vascular smooth muscle cells (vSMCs), leading to AAD development. Specifically, this study investigates a G to A missense mutation at exon 12 within the ALDH2 gene resulting in the substitution of glutamate (Glu) by lysine (Lys) (Glu504Lys) and lower enzymatic activity.5 Whilst this mutation is only described in 8% of the world’s population, it is present in up to 50% of Asians,6 thus making it a relatively common polymorphism to investigate further.
Yang et al. highlight the correlation between the Glu504Lys polymorphism and AAD prevalence in two independent case–control studies (n = 406 and n = 300). Following adjustment for conventional risk factors, the GA/AA group displayed significantly lower AAD prevalence when compared with the wild-type GG allele group. Interestingly, the prevalence of coronary artery disease (CAD) was inversely higher in the GA/AA group compared with the wild-type GG group, thus creating a challenge for targeting ALDH2 to mitigate the progression of AAD as it may inadvertently make the patient more prone to CAD. The authors comment that a tissue-specific target may be a potential solution. Regardless, this finding does not negate the important biological role of ALDH2 in vascular pathogenesis described throughout the paper.
In order to investigate the biology behind this ALDH2 polymorphism, Yang et al. studied two mouse models of AAD treated with the ALDH2 inhibitor daidzin. The first is an angiotensin II infusion ApoE–/– model and the second is a toxin model using 3-aminopropionitrile fumarate (BAPN). Daidzin treatment attenuated AAD formation, stunted aneurysmal dilation, and led to a reduced rate of mortality within both models. Of note, ALDH2 inhibition caused a profound effect in both models which have unique facets: ApoE ApoE–/– angiotensin II infusion mostly caused aneurysm formation, while BAPN treatment mostly led to dissection formation. The authors chose to combine the aneurysm and dissection pathologies in their outcome. It might be interesting in the future to investigate this pathology in additional mice in order to separate these two outcomes, and also to examine thoracic and abdominal AAD independently since the degree of medial degeneration vs. atherosclerosis as drivers for AAD may be linked to its location. Furthermore, ALDH2 activation did not exacerbate disease severity, which may suggest a ceiling to the dose-responsive effects of ALDH2 enzyme activity and its ability to alter the course of AAD formation.
Differential gene expression between aortic tissue from both healthy and AAD patients demonstrated a decrease in expression of genes associated with structural development, extracellular matrix, and muscle contraction, and conversely an increase in expression of genes associated with the cell cycle, apoptosis, and inflammatory cell death. Most important was the discovery of a down-regulation of a cell differentiation cluster of genes indicating a phenotypic switch within the vSMCs in the AAD population. Notably, a reduction in myocardin, α-SMA, SM22-α, and calponin was shown. Since myocardin is known to play a significant role in the maintenance of a smooth muscle contractile phenotype, a potential link between myocardin and ALDH2 was further investigated. Specifically, the authors demonstrate that ALDH2 inhibition can reverse angiotensin II-induced myocardin repression in primary mouse aortic vSMCs in vitro. With no direct interaction between ALDH2 and myocardin identified, an miRNA microarray conducted on vSMCs from wild-type and ALDH2 knockout mice was performed and revealed a down-regulation of miR-31-5p in cells from mutant mice. miR-31-5p expression was also increased in vSMCs treated with angiotensin II, and miR-31-5p can bind to the 3'-untranslated region of myocardin in luciferase assays. More significantly, overexpression of miR-31-5p in vivo reversed the effects of daidzin on AAD incidence in angiotensin II-treated murine models, thus emphasizing the crucial downstream role of miR-31-5p in ALDH2-mediated vascular pathology. These experiments then raise the question of mechanistically how does ALDH2 enzymatic activity lead to changes in miR-31-5p expression? The authors further allude to the role of the master transcriptional regulator MAX binding within the promotor of miR-31-5p, suggesting its up-regulation in the absence of ALDH2; however, the precise underlying mechanism warrants further investigation.
The authors then thread their findings back to humans by extracting vSMCs from patients and demonstrating that those individuals with the GA genotype express significantly lower levels of miR-31-5p and have greater myocardin expression compared with individuals with the GG genotype. Undoubtedly, targeting miR-31-5p as opposed to directly inhibiting ALDH2 could be an attractive therapeutic for AAD. The benefit of a downstream target could curb the negative cardiovascular outcomes of ALDH2 knockdown such as CAD.7 , 8 Important considerations when choosing any miRNA as a therapeutic includes its potential to bind multiple targets, and its expression may vary widely in different tissue and during different disease states,8 thus possibly leading to new detrimental side effects. For example, miR-3105p has also been shown to play a role in cancer10 and endothelial dysfunction.11 That being said, the wealth of data presented here clearly highlights the tissue-specific role of ALDH2 in the vasculature and the potential of ALDH2 and its downstream mediator miR-31-5p in AAD management. These findings are concordant with previous associated studies between ALDH2 deficiency and other types of cardiovascular disease,12–14 such as heart failure and ischaemic heart disease.15
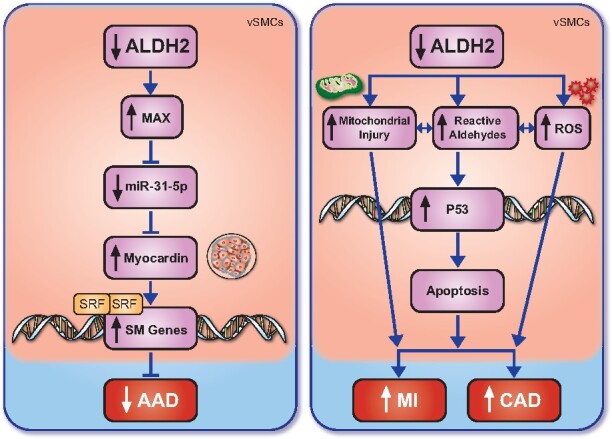
Overall, this comprehensive study provides compelling support for a role for ALDH2 deficiency in reducing the risk of AAD in both murine models and human patients through an miRNA-mediated phenotypic switch in aortic vSMCs. With the current management of AAD highly dependent on high-risk surgical interventions, the potential for a medical treatment strategy involving the ALDH2 pathway is long overdue. Care must be taken when using the ALDH2 pathway for therapeutic targets given the conflicting effects of ALDH2 enzyme activity on cardiovascular health (Take home figure).
Take home figure .
The role of ALDH2 in cardiovascular pathologies. ALDH2 mutation or inhibition reduces MAX levels and the expression of miR-31-5p, reducing myocardin production, thus inhibiting the production of contractile smooth muscle proteins and ultimately leading to the increased incidence of AAD. Conversely, a reduction in ALDH2 has been demonstrated to increase mitochondrial injury, reactive aldehyde production, and the presence of ROS, the latter two increasing p53 expression, leading to cellular apoptosis and ultimately an increase in myocardial infarction and coronary artery disease. AAD, aortic aneurysm/dissections; ALDH2, aldehyde dehydrogenase 2; CAD, coronary artery disease; MI, myocardial infarction; ROS, reactive oxygen species; SM Genes, smooth muscle cells; SRF, serum response factor .
Funding
E.A. is supported by NIH grants R01 HL 136431, R01 HL 141917, and R01 HL 147095.
Conflict of interest: none declared.
Footnotes
†doi:10.1093/eurheartj/ehaa352.
Contributor Information
Francesca Bartoli-Leonard, Center for Interdisciplinary Cardiovascular Sciences, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Louis Saddic, Department of Anaesthesiology and Perioperative Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA, USA.
Elena Aikawa, Center for Interdisciplinary Cardiovascular Sciences, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA; Center for Excellence in Vascular Biology, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA; Department of Human Pathology, Sechenov First Moscow State Medical University, Moscow, Russia.
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.
References
- 1. Thompson RW. Detection and management of small aortic aneurysms. N Engl J Med 2002;346:1484–1486. [DOI] [PubMed] [Google Scholar]
- 2. Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation 2005;111:816–828. [DOI] [PubMed] [Google Scholar]
- 3. Yang K, Ren J, Li X, Wang Z, Xue L, Cui S, Sang W, Xu T, Zhang J, Yu J, Liu Z, Shang H, Pang J, Huang X, Chen Y, Xu F. Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur Heart J 2020;41:2442–2452. [DOI] [PubMed] [Google Scholar]
- 4. Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med 2011;3:107ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ebert AD, Kodo K, Liang P, Wu H, Huber BC, Riegler K, Chukro J, Lee J, de Almeida P, Lan F, Diecke S, Burridge PW, Gold JD, Mochley-Rosen D, Wu JC. Characterisation of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci Transl Med 2014;6:255ra130–255ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo HR, Wu GS, Pakstis AJ, Tong L, Oota H, Kidd KK, Zhang YP. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487Lys. Gene 2009;435:96–103. [DOI] [PubMed] [Google Scholar]
- 7. Xu F, Chen YG, Xue L, Li RJ, Zhang H, Bian Y, Zhang C, Lv RJ, Feng JB, Zhang Y. Role of aldehyde dehydrogenase 2 Glu504Lys polymorphism in acute coronary syndrome. J Cell Mol Med 2011;15:1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan C, Zhao Y, Bian Y, Shang R, Wang JL, Xue L, Wei SJ, Zhang H, Chen YG, Xu F. Aldehyde dehydrogenase 2 Glu504Lys variant predicts a worse prognosis of acute coronary syndrome patients. J Cell Mol Med 2018;22:2518–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doxakis E. Principles of miRNA-target regulation in metazoan models. Int J Mol Sci 2013;14:16280–16302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu Z, He Q, Liang J, Li W, Su Q, Chen Z, Wan Q, Zhou X, Cao L, Sun J, Wu Y, Liu L, Wu X, Hou J, Lian K, Wang A. miR-31-5p is a potential circulating biomarker and therapeutic target for oral cancer. Mol Ther Nucleic Acids 2019;16:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim S, Lee KS, Choi S, Kim J, Lee DK, Park M, Park W, Kim TH, Hwang JY, Won MH, Lee H, Ryoo S, Ha KS, Kwon YG, Kim YM. NF-κB-responsive miRNA-31-5p elicits endothelial dysfunction associated with preeclampsia via down-regulation of endothelial nitric-oxide synthase. J Biol Chem. 2018;293:18989–19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma C, Yu B, Zhang W, Wang W, Zhang L, Zeng Q. Associations between aldehyde dehydrogenase 2 (ALDH2) rs671 genetic polymorphisms, lifestyles and hypertension risk in Chinese Han people. Sci Rep 2017;7:11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu Y, Ni J, Cai X, Lian F, Ma H, Xu L, Yang L. Positive association between ALDH2 rs671 polymorphism and essential hypertension: a case–control study and meta-analysis. PLoS One 2017;12:e0177023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li ZM, Kong CY, Sun KY, Wang LS. The ALDH2 gene rs671 polymorphism is not associated with essential hypertension. Clin Exp Hypertens 2017;39:691–695. [DOI] [PubMed] [Google Scholar]
- 15. Liu X, Sun A. Aldehyde dehydrogenase-2 roles in ischemic cardiovascular disease. Curr Drug Targets 2017;18:1817–1823. [DOI] [PubMed] [Google Scholar]