

Abstract
Diffuse midline glioma (DMG) is a highly malignant childhood tumor with an exceedingly poor prognosis and limited treatment options. The majority of these tumors harbor somatic mutations in genes encoding histone variants. These recurrent mutations correlate with treatment response and are forming the basis for molecularly guided clinical trials. The ability to detect these mutations, either in circulating tumor DNA (ctDNA) or cerebrospinal fluid tumor DNA (CSF-tDNA), may enable noninvasive molecular profiling and earlier prediction of treatment response. Here, the authors review ctDNA and CSF-tDNA detection methods, detail recent studies that have explored detection of ctDNA and CSF-tDNA in patients with DMG, and discuss the implications of liquid biopsies for patients with DMG.
Keywords: ctDNA, diffuse midline glioma, diffuse intrinsic pontine glioma, liquid biopsy
DIFFUSE midline gliomas (DMGs) are highly malignant childhood tumors with poor clinical outcomes. The anatomical location of these lesions, the majority of which are found in the brainstem, often precludes attempts at resection (Fig. 1). Thus, treatment often consists of fractionated radiation therapy (RT).7 Several chemotherapy regimens have been trialed with minimal benefit achieved.52 Given the paucity of treatment options for these patients, molecularly informed targeted therapies are being actively investigated.
FIG. 1.
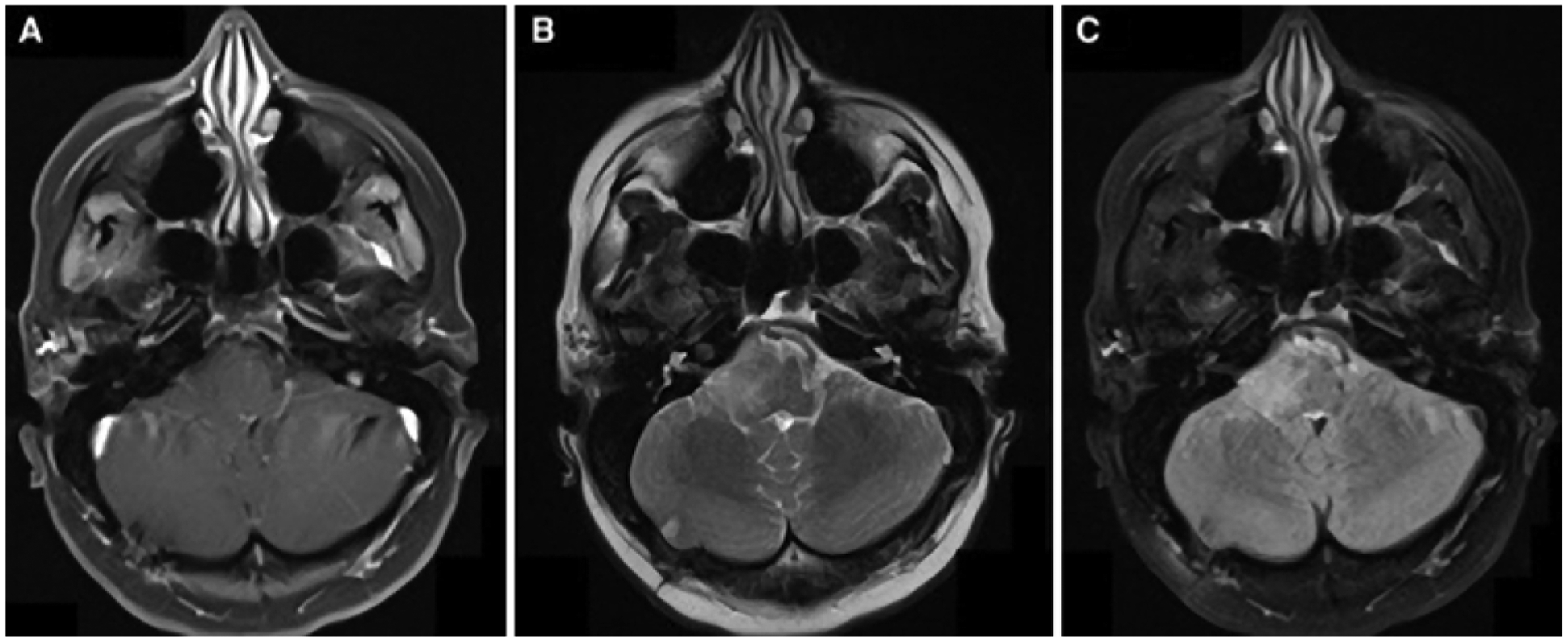
Axial MR images obtained in a patient with a newly diagnosed brainstem glioma: T1-weighted postcontrast (A), T2-weighted (B), and FLAIR (C).
DMGs are characterized by recurrent somatic mutations in the genes encoding histone H3 variants. Mutations in H3F3A, encoding H3.3, and in HIST1H3B, encoding H3.1, are found in the majority of pediatric DMG, with the substitution of lysine 27 to methionine (K27M) in H3.3 or H3.1 reported in up to 80% of DMG.47,56 Tumors with H3 mutations are more aggressive and demonstrate worse responses to treatment, allowing molecular profiling to inform treatment planning and patient counseling.3 Several recent studies of pediatric brain tumors have demonstrated the feasibility of molecular profiling of biopsy tissue in order to guide treatment decisions.8,23,24,42,44 Patients with DMG were more likely to have molecular features that changed management decisions than patients with other tumor types.30 In light of these studies, consensus guidelines were recently released, recommending tumor next-generation sequencing (NGS) of cancer-related genes for pediatric high-grade glioma patients in addition to immunohistochemistry (IHC) of H3K27M for DMG.30 Increasing the availability of tumor tissue for molecular profiling is a key step forward in enabling targeted therapies for DMG patients. Historically, offering surgical biopsies to these patients was controversial given an uncertain risk-benefit tradeoff. However, with possible targeted therapies and a prospective multicenter study demonstrating the safety of neurosurgical biopsy for DMG,16 the historical paradigm has begun to shift.
Alternatives to neurosurgical biopsy for DMG patients are beginning to emerge. Specifically, the sampling of cerebrospinal fluid (CSF) and peripheral blood to detect tumor mutations offers diagnostic and prognostic leverage without the risks associated with a neurosurgical procedure. We review these recent developments in “liquid biopsy” for DMG, providing an overview of circulating tumor DNA (ctDNA) and CSF tumor DNA (CSF-tDNA) and methods of detecting these molecules. We further detail the recent studies that have demonstrated the feasibility of detecting ctDNA and CSF-tDNA in patients with DMG and provide a framework for incorporating these assays into the clinical care of DMG.
Methods
We restricted the scope of this review to the detection of cell-free DNA (cfDNA) molecules in the blood or CSF. Other liquid biopsy approaches (e.g., cell-free RNA, circulating tumor cells, circulating proteins) are well described in a recent review by Bonner and Bornhorst et al.2 We searched the PubMed/MEDLINE electronic database and the references of relevant articles for English-language articles published up to August 1, 2019. To identify ctDNA publications related to DMG, the following search phrase was employed: “(DMG OR DIPG OR “diffuse glioma” OR “diffuse intrinsic pontine glioma” OR “diffuse midline glioma”) AND (cfDNA OR “cell free” OR “cell-free” OR ctDNA OR “circulating DNA” OR “liquid biopsy” OR “plasma DNA” OR “circulating tumor DNA” OR “circulating tumour DNA”).” To identify CSF-tDNA publications related to DMG, the above Boolean phrase was modified to “(DMG OR DIPG OR “diffuse glioma” OR “diffuse intrinsic pontine glioma” OR “diffuse midline glioma”) AND (“cerebrospinal fluid” OR CSF).” Articles describing results of ctDNA or CSF-tDNA assays in the blood or CSF of DMG patients were retained (Fig. 2).
FIG. 2.
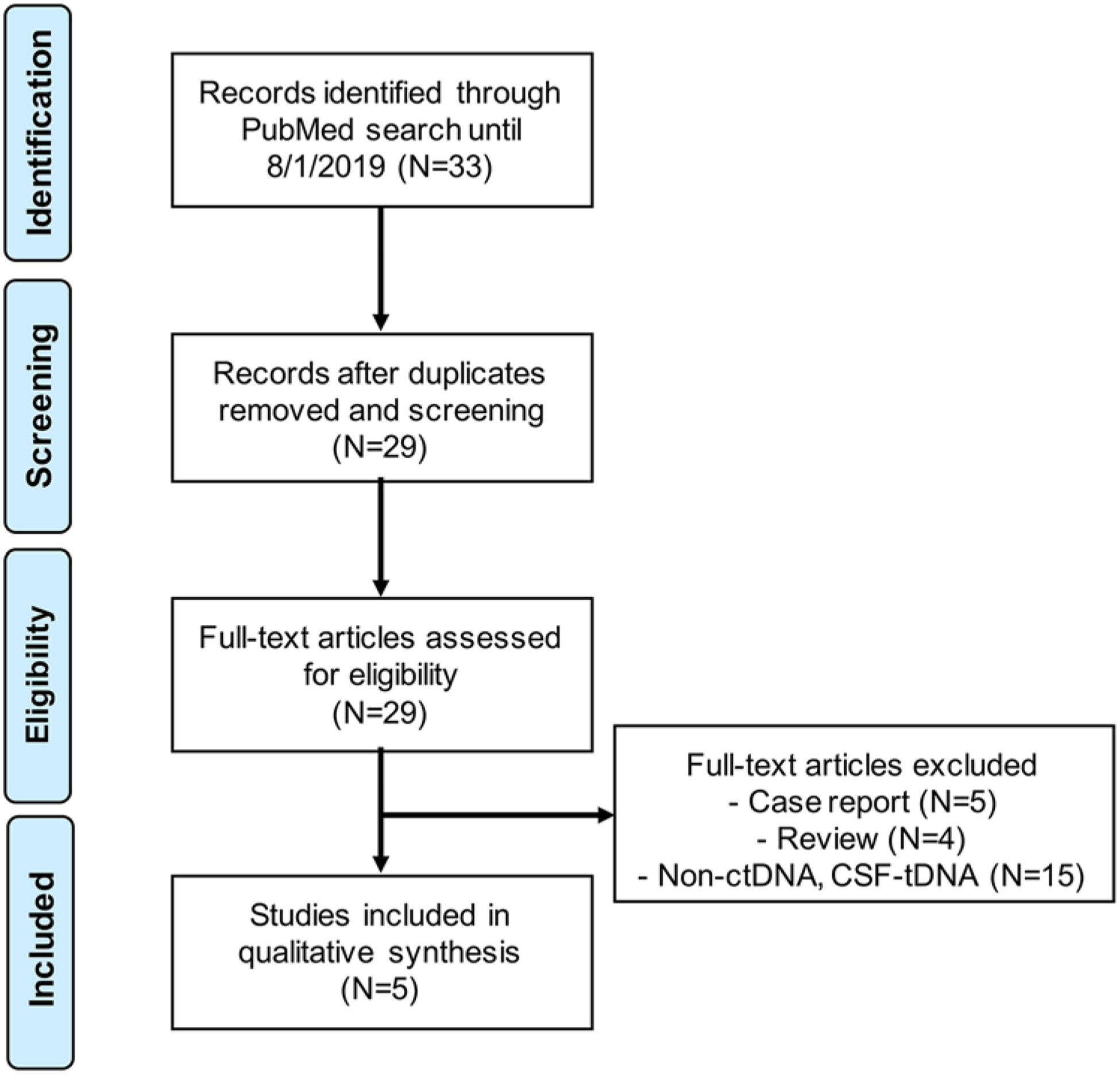
PRISMA flowchart illustrating literature review strategy, including included and excluded articles. Excluded full-length papers were review articles (n = 4) or case reports (n = 5) or did not describe results of ctDNA or CSF-tDNA assays in DMG (n = 15).
Results
Overview of ctDNA and CSF-tDNA
Cell-free DNA is composed of nuclear DNA released into the peripheral blood by dying cells and circulates as short fragments between 170 and 340 bp in length.17,51 Circulating tumor DNA, the fraction of cfDNA that originates from the tumor, holds promise as an emerging cancer biomarker for both the genotyping of tumors and the detection of disease during or after therapy.1,17,19,36 Particularly when assessing serially collected samples, the noninvasive nature of ctDNA acquisition offers considerable advantages over biopsies and allows for robust monitoring of tumor response to therapy.5,17,54 Furthermore, ctDNA summarizes the entirety of the tumor, reducing bias associated with spatial heterogeneity and metastatic disease.54 Applications of ctDNA have been explored in a number of cancers, notably lymphoma, lung cancer, and colorectal cancer; however, there have been relatively fewer studies exploring ctDNA in the context of central nervous system (CNS) tumors.
Detection of ctDNA from CNS malignancies remains difficult. The blood-brain barrier remains a formidable obstacle isolating the CNS from the peripheral circulation. Furthermore, as ctDNA quantitation relies on the accuracy of somatic variant identification to interrogate the circulating tumor burden, the relatively low mutational burden in CNS tumors complicates ctDNA assessment. This conundrum is even more pronounced in pediatric brain tumors, which have been shown to carry approximately 10-fold fewer coding mutations.15 In these patients, tumor-derived DNA fragments spanning a somatic mutation are exceedingly rare, likening the problem of tumor detection to “finding a needle in a haystack.”
The use of CSF-tDNA circumvents this dilemma by reducing the size of the proverbial haystack. The CNS, while not as immune-privileged as once believed, is home to far fewer myeloid, epithelial, and endothelial cells than the rest of the body. By reducing the mass of cfDNA originating from myeloid, epithelial, and endothelial derivatives, which constitute more than 95% of peripheral blood cfDNA in healthy individuals, the fraction of CSF-tDNA that is tumor derived can be greatly enriched.41 Early CSF-tDNA studies demonstrated improved sensitivity compared to peripheral blood ctDNA and recapitulated tumor response to treatment using quantitation of the CSF tumor burden.10,33,41,55 More recent investigations profiling WHO grade II–IV gliomas using CSF cfDNA obtained through lumbar puncture comprehensively characterized somatic alterations associated with the pretreatment sample and uncovered temporal evolution of the tumor genome during therapy in a subset of the cohort.31
Methods of ctDNA and CSF-tDNA Detection
As with the genotyping of nuclear DNA, ctDNA and CSF-tDNA can be profiled to assess the mutational landscape, structural alterations, and tumor burden. Multiple high-throughput sequencing approaches have evolved to allow for accurate and scalable interrogation of nucleotide content, including digital polymerase chain reaction (PCR),11–13,53 amplicon sequencing,14,22 hybrid capture sequencing,36,37 whole-exome sequencing (WES),35 and whole-genome sequencing (WGS).18,26 An overview, including advantages and disadvantages of these methods, is provided in Table 1.
TABLE 1.
Overview of ctDNA detection approaches
| Approach | Summary | Advantages | Disadvantages | DMG Study |
|---|---|---|---|---|
| Digital PCR | ||||
| Emulsion PCR | Based on end-amplification detection of fluorescence in partitioned samples of the input DNA | Simple quantification of DNA of interest; high sensitivity & specificity per mutation | Requires a predefined list of mutations of interest; not optimized for genotype discovery | Huang et al., 2017; Martinez-Ricarte et al., 2018; Stallard et al., 2018; Panditharatna et al., 2018 |
| BEAMing | Integration of superparamagnetic beads & flow cytometry to improve identification of mutant DNA templates | Additional bead capture & flow sorting improves ability to detect mutant DNA fragments compared to traditional PCR | Additional reagents required compared to traditional digital PCR; does not have the genotyping capabilities of WES/WGS | |
| NGS | ||||
| Hybrid capture | Enrichment of regions of interest through direct capture of DNA using probes hybridizing to predefined genomic regions | Improved coverage uniformity compared to amplicon-based sequencing | Library preparation can be lengthy; current technologies require multiplexing of samples to reduce unit costs | |
| Targeted | Deep sequencing of a predefined set of regions spanning alterations of interest; custom gene panels are required for isolation of DNA fragments of interest | Increased depth compared to approaches w/broader sequencing coverage; improved recovery of rare variants | Limited hypothesis space restricts genotype discovery; increased sequencing depth can result in over-sequencing of duplicate input molecules | Pan et al., 2019 |
| WES | Broad sequencing of the exome using a hybridization panel spanning coding regions of the genome | Spans the coding region of the genome, allowing for the genotyping of functional mutations w/o restriction; more affordable than WGS | Does not cover noncoding regions of the genome, limiting genotyping of broad alterations (e.g., whole arm copy number gains); more expensive than targeted sequencing | |
| Amplicon | Enrichment of regions of interest through PCR-based amplification using paired probes | Improved on-target rates compared to hybridization capture methods; sequencing library preparation may be faster than hybridization capture methods | Most effective for highly targeted sequencing (compared to WGS); limited ability to genotype genome-wide alterations | |
| WGS | Bulk sequencing of input DNA w/o restriction to prespecified genomic coordinates; conducted using shotgun sequencing principles | Broad hypothesis space allows for improved discovery of genome-wide aberrations | Deep sequencing can be costly compared to either WES or targeted sequencing; increased hypothesis space could increase detection of spurious variants not associated w/pathology of interest | |
BEAMing = beads, emulsion, amplification, magnetics.
Digital PCR harnesses in vitro PCR to quantitate target sequences within an input mass of DNA fragments by detecting the presence of fluorescence in individual partitions of the original sample after amplification.43,53 Methods that generate nanoliter-scale water-oil emulsions called “droplets” prior to amplification can be grouped under the category of “droplet digital PCR” (ddPCR), a process that has markedly increased the scalability of digital PCR.43 In contrast, “NGS” refers to a broad range of highly scalable approaches built off the principles of Sanger sequencing. Fundamentally, these can be broken down into hybridization-based, amplicon-based, and whole-genome approaches. Sequencing scope can range from a few hundred base pairs to the entirety of the genome.27 Hybridization-based approaches utilize hybridization probes aligning to a predefined set of genomic coordinates to isolate DNA fragments spanning regions of interest.20,38 An alternative approach, amplicon sequencing, interrogates amplicons generated from PCR using paired probes flanking regions of interest.46 These approaches are further summarized in Table 1.
More specifically, each of these approaches has advanced in numerous ways to create a diverse spectrum of options for ctDNA and CSF-tDNA profiling. The development of BEAMing (beads, emulsion, amplification, magnetics) in the early 21st century further enhanced emulsion-based ddPCR by capturing PCR products on superparamagnetic beads.13 In contrast to emulsion digital PCR, a downstream flow cytometry step of the fluorescently tagged bead-DNA output allows for the precise determination of mutant DNA from wild-type fragments.11–13 “NGS” is an umbrella term that spans a multitude of chemical approaches, including methods based on sequencing by synthesis and sequencing by ligation, the specifics of which are beyond the scope of this review.48 Notably, the scope and scale of NGS is highly flexible for both genotype discovery and recovery. Applications of NGS include deep sequencing of select regions, which can range from hundreds of base pairs to the entirety of the exome to shallow coverage of the full genome. This flexibility allows for a broad range of applications ranging from the recovery of rare mutations in samples with low tumor content to the comprehensive profiling of point mutations, structural alterations, and copy number alterations across the genome.48
CSF-tDNA for DMG
Our literature review returned 4 publications that reported the results of CSF-tDNA assays in diffuse gliomas.21,29, 39,49 We describe these studies in the order in which they were published.
Huang et al. analyzed archival CSF from 11 patients with pediatric brain tumors, including 6 patients with DMG (diffuse intrinsic pontine glioma [DIPG], n = 4; thalamic anaplastic astrocytoma, n = 2).21 CSF was collected (volume range 0.4–2 ml) either by placement of a CSF diversion device (ventricular shunt, external ventricular drain, or indwelling CSF reservoir, 2/11) or by accessing an existing CSF diversion device (9/11 [82%]). cfDNA concentrations isolated from the CSF of the DMG patients ranged from 10 to 280 ng/ml. Two independent techniques to detect H3 mutations in the CSF were employed: Sanger sequencing of H3F3A and HIST1H3B, and nested PCR with primers specific to H3F3A c.83A > T transversion (H3.3K27M). The authors reported sufficient DNA (accounting for quality and quantity) for analysis of 5/6 DMG samples by nested PCR and 3/6 samples by Sanger sequencing. The PCR strategy resulted in detection of the H3F3A mutation in 4/5 samples, while Sanger sequencing detected H3F3A mutations in 2/3 samples. Concordance of CSF-tDNA mutation status was confirmed by IHC or Sanger sequencing of matched tumor biopsy DNA in 3/3 DMG patients with available tissue.21
Martínez-Ricarte et al. investigated detection of CSF-tDNA in a series of 20 diffuse gliomas, including 3 cases with DMG (pediatric, n = 2).29 Two milliliters of CSF were collected from these patients by lumbar puncture. cfDNA concentrations isolated from the CSF of the DMG patients ranged from 4.8 to 37.5 ng/ml. For non-DMG diffuse gliomas, the authors performed amplicon sequencing of IDH1, IDH2, ATRX, and TP53. However, for DMG patients, ddPCR assays for H3F3A K27M and HIST1H3B K27M were designed and applied to tumor DNA, germline DNA from peripheral blood lymphocytes, and CSF-tDNA. H3F3A K27M was detected in the CSF of all 3 DMG patients, with variant allele fractions (VAFs) ranging from 7% to 17.3%.29 Stallard et al. also used a ddPCR approach to detect mutant H3F3A K27M in the CSF of 4 pediatric brain tumor patients (DIPG, n = 2; glioblastoma [GBM], n = 2).49 In the 2 DIPG patients, VAFs were markedly higher (43% and 65%) than those in the ddPCR-based study by Martínez-Ricarte et al. Notably, while Martínez-Ricarte et al. collected CSF via lumbar puncture, Stallard et al. collected CSF directly from the ventricles (Rickham reservoir, n = 1; endoscopic third ventricle, n = 1). This point is further reinforced by the finding of Stallard et al. via multifocal sampling of CSF from the autopsy of a DIPG patient—the quantity of K27M copies was twice as high in lateral ventricle CSF than in lumbar puncture CSF.49
Pan et al. took a different approach, using a 68-gene NGS panel covering specific brain tumor–related genes.39 They applied this panel to profile tumor DNA, germline DNA, and CSF-tDNA from 57 patients with brainstem tumors. Their cohort included 12 pediatric patients with grade IV tumors (DMG, n = 3; pontine GBM, n = 9). CSF was obtained by ventriculoperitoneal shunt, during resection, or by lumbar puncture. These authors reported detection of H3F3A mutations in the CSF-tDNA of 3/3 DMG and 8/9 pontine GBM cases. In the pontine GBM case without H3F3A mutations detected, a HIST1H3B mutation was detected in the CSF-tDNA. The median H3F3A CSF-tDNA VAF was 35.6% (IQR 19.2%–45.4%). The authors also attempted to detect mutations in the peripheral blood of a subset of their entire cohort (n = 8; DMG, n = 1). No ctDNA was detected in the DMG patient, and overall the sensitivity of their assay was markedly lower in peripheral blood than in CSF.39
ctDNA for DMG
We identified one recent publication reporting the results of plasma ctDNA assays in DMG.40 Panditharatna et al. applied an H3K27M ddPCR assay to CSF and plasma samples from 48 DMG patients (79% with H3.3K27M, 15% with H3.1K27M) through an ongoing clinical trial (NCT02274987). The authors detected CSF-tDNA in 3/4 patients at the initial collection time and in 19/21 patients postmortem. Histone mutations were detected in the upfront/diagnostic plasma sample of 16/20 patients with known H3K27M mutations. H3K27M ctDNA was detected in 2 additional patients at a post-RT point. As anticipated, VAF in the CSF was significantly higher than in matched plasma samples. The comparatively lower ctDNA levels in plasma, compared to CSF, underscore a key limitation to the use of peripheral blood instead of CSF to monitor the disease status of patients with CNS tumors.
Beyond demonstrating the feasibility of detecting H3K27M mutations in plasma, Panditharatna et al. provided initial data describing how serial ctDNA sampling can be used to monitor treatment response.40 They showed that ctDNA levels in DMG appear to decrease following RT and may correlate with decreased post-RT tumor volume on MRI. These authors present an additional compelling vignette of a different DMG patient with progression on panobinostat and mebendazole, suggested by an increasing H3K27M VAF. They then observed a decreasing H3K27M VAF after enrollment in a different clinical trial, potentially suggesting response to therapy.
Clinical Utility of ctDNA and CSF-tDNA for DMG
The utility of detecting tumor-associated cfDNA in the blood or CSF of DMG patients falls into two general categories: minimally invasive molecular profiling and longitudinal monitoring. In light of recent guidelines30 recommending tumor NGS of cancer-related genes for pediatric high-grade glioma patients, less invasive means of genotyping are warranted. While tumor tissue is likely to remain the gold standard, neurosurgical biopsy is never without risk and may not be available to all DMG patients. For DMG patients, there is clear benefit in ascertaining H3K27M status and, in the future, potentially additional genetic information that could be used to drive therapeutic choices. The ability to reliably determine these data through venipuncture or lumbar puncture would likely expedite the care of these patients. Moreover, it would facilitate enrollment in future clinical trials for DMG patients, as many recent or upcoming trials require knowledge of H3K27M status (NCT02960230, NCT02274987, NCT01182350). The feasibility of obtaining this information was demonstrated in a recent DIPG pilot trial, in which detection of H3F3A or HIST1H3B K27M mutations in peripheral blood using ddPCR was successful in 92% of H3K27M-mutant cases.34 A key limitation of using mutation-specific ctDNA assays such as ddPCR is that molecular information about the remainder of the exome or genome, which could potentially influence treatment decisions, is unavailable. Approaches to ctDNA that survey greater swathes of the genome, such as targeted sequencing panels,37 WES,28 low-pass WGS,32 or hybrid approaches,6,9 offer avenues to overcome this limitation.
In addition to minimally invasive molecular profiling, monitoring the response to therapy is a key application of ctDNA and CSF-tDNA for patients with DMG. Panditharatna et al. began to explore this in their study of CSF-tDNA and ctDNA,40 but further studies are required. The use of serial ctDNA measurements in diffuse large B-cell lymphoma (DLBCL)25 has recently been shown to enable superior prediction of patient outcome and could be used to guide treatment decisions longitudinally. While the treatment lines for DMG are markedly more limited than in DLBCL, a parallel paradigm does exist. Specifically, disease status could be monitored sequentially using CSF sampling or venipuncture. Serial CSF sampling has been a long-standing part of care for some pediatric malignancies, such as medulloblastoma, where cytology is utilized to assess disease status and response to therapy. In addition, CSF-tDNA or ctDNA levels may be able to demonstrate early disease progression that may not be radiographically detectable and would enable modifications of the treatment regimen or enrollment into a clinical trial. We illustrate this paradigm using a hypothetical DMG patient vignette in Fig. 3. The utility of ctDNA for this purpose has been demonstrated in other malignancies such as colon and lung cancer, where ctDNA changes precede clinical and radiographic progression by 5 or more months.4,50 In lung, breast, and other advanced cancers, ctDNA-guided trials are beginning to emerge (NCT02284633, NCT02743910).45
FIG. 3.
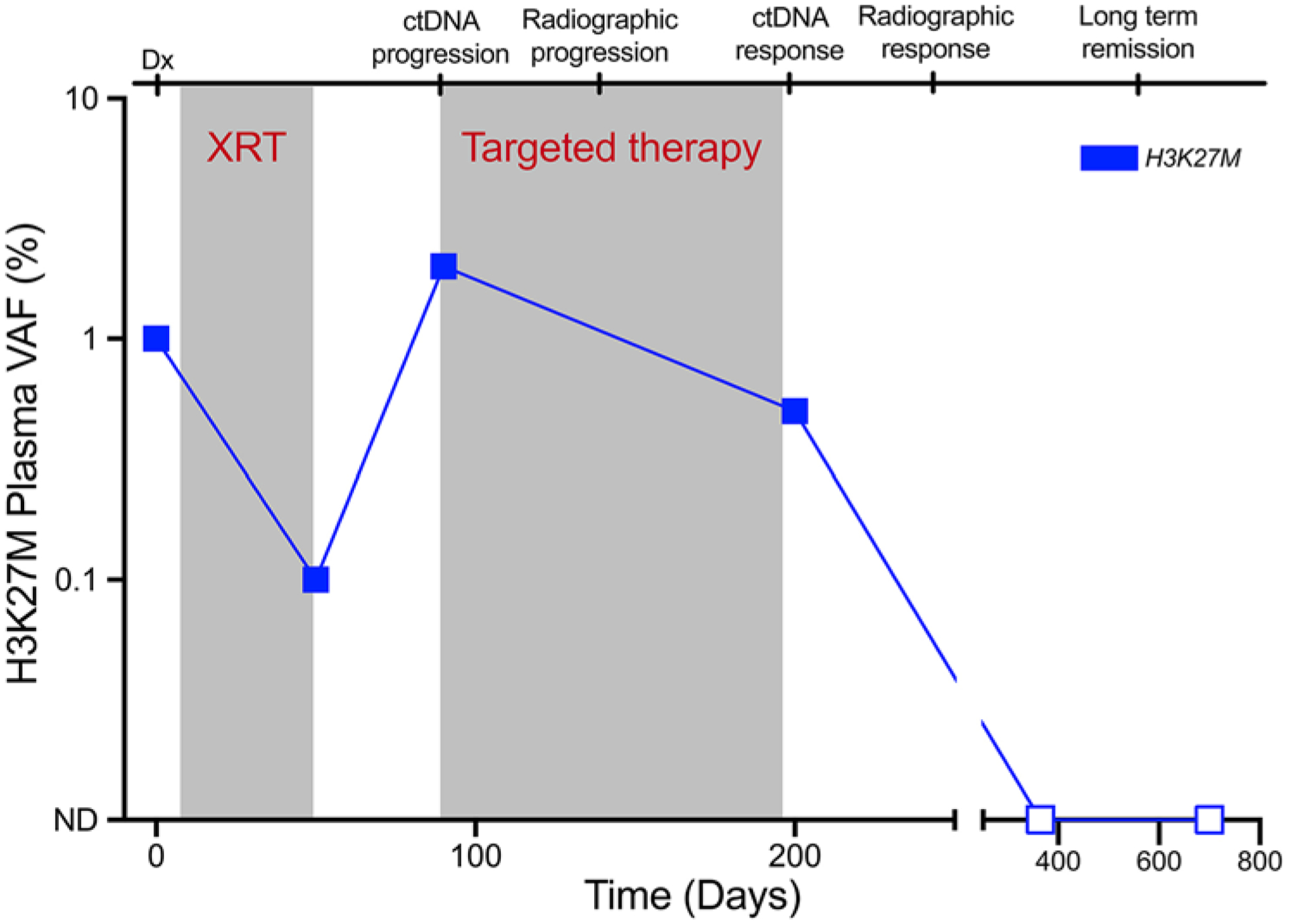
Hypothetical vignette illustrating the use of ctDNA in a DMG patient to guide therapeutic decision-making. Dx = diagnosis; ND = not detected; XRT = radiation therapy.
Conclusions
The treatment options for patients with DMG, unfortunately, remain limited. However, the advent of targeted and cell-based therapies provides reason for cautious optimism. The ability to less invasively obtain molecular information via ctDNA or CSF-tDNA to aid in prognostication and treatment decision-making may afford a significant step forward for patients and families. Improved monitoring of therapy response and early allocation to molecularly informed clinical trials represent important potential benefits of regularly using ctDNA or CSF-tDNA in the care of patients with DMG.
Acknowledgments
This work was supported by the Thomas M. Hohman Memorial Cancer Research Fund, a Burroughs Wellcome Career Award for Medical Scientists, the Chi-Li Pao Foundation, and National Institutes of Health grants R21-CA208723 and R37-CA230400.
ABBREVIATIONS
- cfDNA
cell-free DNA
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CSF-tDNA
CSF tumor DNA
- ctDNA
circulating tumor DNA
- ddPCR
droplet digital PCR
- DIPG
diffuse intrinsic pontine glioma
- DMG
diffuse midline glioma
- GBM
glioblastoma
- NGS
next-generation sequencing
- PCR
polymerase chain reaction
- RT
radiation therapy
- VAF
variant allele fraction
- WES
whole-exome sequencing
- WGS
whole-genome sequencing
Footnotes
Disclosures
Dr. Bettegowda is a consultant for DePuy-Synthes.
References
- 1.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. : Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224ra24, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonner ER, Bornhorst M, Packer RJ, Nazarian J: Liquid biopsy for pediatric central nervous system tumors. NPJ Precis Oncol 2:29, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. : Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. : Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 7:1394–1403, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin RI, Chen K, Usmani A, Chua C, Harris PK, Binkley MS, et al. : Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol Diagn Ther 23:311–331, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. : Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359:926–930, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen KJ, Jabado N, Grill J: Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol 19:1025–1034, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole BL, Lockwood CM, Stasi S, Stevens J, Lee A, Ojemann JG, et al. : Year 1 in the molecular era of pediatric brain tumor diagnosis: application of universal clinical targeted sequencing in an unselected cohort of children. JCO Precision Oncology, 2018 [DOI] [PubMed] [Google Scholar]
- 9.Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. : Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570:385–389, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. : Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 6:8839, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. : Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 102:16368–16373, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D: BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 3:551–559, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B: Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A 100:8817–8822, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. : Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4:136ra68, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. : The landscape of genomic alterations across childhood cancers. Nature 555:321–327, 2018 [DOI] [PubMed] [Google Scholar]
- 16.Gupta N, Goumnerova LC, Manley P, Chi SN, Neuberg D, Puligandla M, et al. : Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol 20:1547–1555, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heitzer E, Haque IS, Roberts CES, Speicher MR: Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 20:71–88, 2019 [DOI] [PubMed] [Google Scholar]
- 18.Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. : Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med 5:30, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heitzer E, Ulz P, Geigl JB: Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 61:112–123, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, et al. : Genome-wide in situ exon capture for selective resequencing. Nat Genet 39:1522–1527, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, et al. : Detection of histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun 5:28, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B: Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 108:9530–9535, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. : Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol 19:699–709, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koschmann C, Wu YM, Kumar-Sinha C, Lonigro R, Vats P, Kasaian K, et al. : Clinically integrated sequencing alters therapy in children and young adults with high-risk glial brain tumors. JCO Precision Oncology (2):1–34, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, et al. : Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell 178:699–713.e19, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. : Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2:20ra14, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, et al. : Target-enrichment strategies for next-generation sequencing. Nat Methods 7:111–118, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Manier S, Park J, Capelletti M, Bustoros M, Freeman SS, Ha G, et al. : Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun 9:1691, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martínez-Ricarte F, Mayor R, Martínez-Sáez E, Rubio-Pérez C, Pineda E, Cordero E, et al. : Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumor DNA from cerebrospinal fluid. Clin Cancer Res 24:2812–2819, 2018 [DOI] [PubMed] [Google Scholar]
- 30.Miklja Z, Pasternak A, Stallard S, Nicolaides T, Kline-Nunnally C, Cole B, et al. : Molecular profiling and targeted therapy in pediatric gliomas: review and consensus recommendations. Neuro Oncol 21:968–980, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. : Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565:654–658, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. : Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 10:10, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouliere F, Mair R, Chandrananda D, Marass F, Smith CG, Su J, et al. : Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol Med 10:10, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mueller S, Jain P, Liang WS, Kilburn L, Kline C, Gupta N, et al. : A pilot precision medicine trial for children with diffuse intrinsic pontine glioma-PNOC003: a report from the Pacific Pediatric Neuro-Oncology Consortium. Int J Cancer 145:1889–1901, 2019 [DOI] [PubMed] [Google Scholar]
- 35.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. : Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497:108–112, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. : An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20:548–554, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. : Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34:547–555, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME: Microarray-based genomic selection for high-throughput resequencing. Nat Methods 4:907–909, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Pan C, Diplas BH, Chen X, Wu Y, Xiao X, Jiang L, et al. : Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol 137:297–306, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panditharatna E, Kilburn LB, Aboian MS, Kambhampati M, Gordish-Dressman H, Magge SN, et al. : Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient-derived liquid biopsy. Clin Cancer Res 24:5850–5859, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, et al. : Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol 34:2404–2415, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfaff E, El Damaty A, Balasubramanian GP, Blattner-Johnson M, Worst BC, Stark S, et al. : Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. Eur J Cancer 114:27–35, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Quan PL, Sauzade M, Brouzes E: dPCR: a technology review. Sensors (Basel) 18:18, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramkissoon SH, Bandopadhayay P, Hwang J, Ramkissoon LA, Greenwald NF, Schumacher SE, et al. : Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol 19:986–996, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. : Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med 25:738–743, 2019 [DOI] [PubMed] [Google Scholar]
- 46.Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, et al. : Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum Mutat 36:903–914, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. : Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, et al. : DNA sequencing at 40: past, present and future. Nature 550:345–353, 2017 [DOI] [PubMed] [Google Scholar]
- 49.Stallard S, Savelieff MG, Wierzbicki K, Mullan B, Miklja Z, Bruzek A, et al. : CSF H3F3A K27M circulating tumor DNA copy number quantifies tumor growth and in vitro treatment response. Acta Neuropathol Commun 6:80, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. : Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 8:346ra92, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. : Fragment length of circulating tumor DNA. PLoS Genet 12:e1006162, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanan MI, Eisenstat DD: DIPG in children - what can we learn from the past? Front Oncol 5:237, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vogelstein B, Kinzler KW: Digital PCR. Proc Natl Acad Sci U S A 96:9236–9241, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. : Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17:223–238, 2017 [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. : Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A 112:9704–9709, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becks-fort J, et al. : Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]