

Abstract
Histone methylation and acetylation participate in the modulation of gene expression. Here, chromatin immunoprecipitation sequencing (ChIP-Seq) was used to determine genome-wide patterns of three histone modifications, H3K4me3, H3K36me3, and H3K9ac (associated with actively expressed genes) and their associations with gene expression in Paulownia fortunei following phytoplasma infection and recovery from Paulownia witches’ broom (PaWB) disease after methyl methane sulfonate treatment. The three histone marks were preferentially deposited in genic regions, especially downstream of transcription start sites, and were highly concurrent with gene expression. Genes with all three histone marks exhibited the highest expression levels. Based on the comparison scheme, we detected 365, 2244, and 752 PaWB-associated genes with H3K4me3, H3K36me3, and H3K9ac marks, separately. KEGG pathway analysis showed that these genes were involved in plant-pathogen interaction, plant hormone signal transduction, and starch and sucrose metabolism. A small proportion of differentially modified genes showed changes in expression in response to phytoplasma infection, including genes involved in calcium ion signal transduction, abscisic acid signal transduction, and ethylene biosynthesis. This comprehensive analysis of genome-wide histone modifications and gene expression in Paulownia following phytoplasma infection provides new insights into the epigenetic responses to phytoplasma infection and will be useful for further studies on epigenetic regulation mechanisms in plants under biotic stress.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02331-0) contains supplementary material, which is available to authorized users.
Keywords: Chromatin immunoprecipitation sequencing, Epigenetic regulation, Histone marks, Gene expression
Introduction
In eukaryotes, gene expression is modulated by both genetic and epigenetic regulatory mechanisms. Histone modification is a well-known epigenetic modification. Eukaryotic DNA is organized in the form of chromatin, whose basic structural and functional unit is the nucleosome. Each nucleosome comprises a histone octamer (two subunits of each of H2A, H2B, H3, and H4) wrapped by 147 bp DNA and a linker histone H1 (H1). The amino-terminal tails of each histone are usually post-transcriptionally modified by various additions or reactions. These epigenetic modifications, which are known as histone modifications, include acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deimination, and proline isomerization. Histone modifications are an important epigenetic mechanism that modulate gene expression through changing the chromatin state, altering the interaction between histones and DNA, or recruiting enzyme complexes to chromatin (Kouzarides 2007). Moreover, function-specific histone modification profiles can be used to predict gene expression levels (Jung and Kim 2012). It has also been proposed that different histone modification types may function as a “histone code”; that is, they work together in a sequential, cooperative, antagonistic or redundant manner to maintain a chromatin conformation that is compatible with gene expression (Lennartsson and Ekwall 2009). Histone methylation and acetylation are two major forms of histone modifications that are implicated in the modulation of gene expression. In general, histone acetylation is associated with gene activation, while histone methylation can activate or repress gene expression depending on the modification site and extent (Berger 2007; Kouzarides 2007). The histone marks H3K9ac, H3K4me3, and H3K36me3 are positively correlated with active gene transcription. Many recent studies have provided evidence that histone methylation and acetylation are involved in modulating gene expression during plants development and in response to biotic and abiotic stresses (Ayyappan et al. 2015; Brusslan et al. 2015; Zong et al. 2013; Zhu et al. 2017). Histone methylation and acetylation is dynamically regulated by a variety of histone methyltransferases (HMTs) and demethylases (HDMs), and acetyltransferases (HATs) and deacetylases (HDACs), respectively. The functions of many histone modifiers have been investigated, and some have been shown to participate in plants’ responses to pathogen attack (Choi et al. 2012; Ding et al. 2012; Hou et al. 2015; Li et al. 2013).
Paulownia witches’ broom (PaWB) disease is a serious infectious disease for Paulownia fortunei in China. This disease greatly reduces the growth and biomass of Paulownia, resulting in enormous economic losses to the production industry. Paulownia witches’ broom phytoplasma, belonging to the aster yellows group (16SrI-D subgroup), is the causal agent of PaWB disease, and it is mainly transmitted via phloem-sucking insects, by parasitic plants, and by vegetative propagation (Bertaccini and Duduk 2009). Phytoplasma infection causes obvious morphological changes to Paulownia, including the classic witches’ broom appearance, leaf yellowing, phyllody, and shortened internodes (Hogenhout et al. 2008). Although PaWB disease has been studied for decades, its pathogenesis is still unclear, mainly because it is difficult to isolate the phytoplasma. Previously, we discovered that methyl methane sulfonate (MMS) is an effective reagent to minimize or eliminate the symptoms of PaWB disease (Cao et al. 2012). In our trials, phytoplasma-infected Paulownia plantlets treated with a low concentration of MMS exhibited reduced symptoms of PaWB disease, even though the phytoplasma was still present. Plantlets treated with a higher concentration of MMS (60 mg L−1) showed normal morphology, and the phytoplasma was eliminated and could not be detected by sensitive nested PCR. Therefore, MMS treatment makes it possible for Paulownia to recover from PaWB disease by eliminating the phytoplasma. At present, it is unknown how histone modifications contribute to the responses of Paulownia to phytoplasma infection. The recent development of highly efficient technologies has made it possible to detect alterations at the transcriptional (high-throughput mRNA sequencing) and epigenetic (chromatin immunoprecipitation sequencing) levels. Furthermore, the availability of P. fortunei genome provides an opportunity to profile the patterns of histone modifications in Paulownia and monitor their dynamics during phytoplasma infection and recovery processes.
In the current study, we explored the roles of H3K4me3, H3K36me3, and H3K9ac modifications in the epigenomic regulation of gene expression in Paulownia during infection by the PaWB phytoplasma and recovery from the disease. Profiles of three histone modifications, H3K4me3, H3K36me3, and H3K9ac in healthy P. fortunei (PF), phytoplasma-infected P. fortunei (PFI), phytoplasma-infected P. fortunei treated with 60 mg L−1 MMS (PFI-60), and healthy P. fortunei treated with 60 mg L−1 MMS (PF-60) were generated. Comparative analyses of these profiles allowed us to monitor the dynamics of histone marks during PaWB disease infection and recovery processes, and to identify genes closely associated with PaWB disease. An integrated genome-wide comparative analysis of histone modifications and mRNA transcriptional activity was also conducted.
Materials and methods
Plant materials
We first obtained 30-day-old tissue-cultured plantlets of healthy P. fortunei (PF) and phytoplasma-infected P. fortunei (PFI) according to the methods of Fan et al. (2002) and Yao et al. (2009). Subsequently, the PF and PFI plantlets were treated with 60 mg L−1 MMS as described by Fan et al. (2018). All materials were grown in the tissue culture chamber under the following conditions: 16-h-light/8-h-dark photoperiod, relative humidity of 70%, illumination intensity of 130 μmol m−2 s−1, and 25 ± 2 °C. The terminal buds of PF-60 and PFI-60 plantlets were harvested after treatment for 30 days. Three independent biological replicates were prepared for each sample to perform chromatin immunoprecipitation (ChIP)-Seq analyses.
Chromatin immunoprecipitation
The ChIP was conducted mainly as described previously (Zong et al. 2013) with some modifications. Briefly, samples were cross-linked with formaldehyde (final concentration, 1%). After stopping the crosslinking reaction with glycine (final concentration, 0.125 M), the chromatin was sheared into 100–300 bp fragments by sonication. The antibodies used for immunoprecipitating chromatin with the three histone marks were anti-H3K4me3 (Abcam), H3K36me3 (Abcam) and H3K9ac (Abcam). Accordingly, the input control was similarly processed without any antibody. The antibody-chromatin complexes were washed, eluted and reversely crosslinked. The ChIP DNA and input DNA were recovered using the phenol/chloroform extraction method and resuspended in distilled water for ChIP-Seq or ChIP-qPCR. For ChIP-Seq, the recovered ChIP DNA and input DNA were used to construct Illumina sequencing libraries, which were sequenced on the Illumina HiSeq 4000 platform with the PE 150 method, which were carried out by Wuhan IGENEBOOK Biotechnology Co., Ltd (www.igenebook.com) with three biological replicates for each sample.
ChIP-Seq data analysis
The H3K4me3, H3K36me3, and H3K9ac ChIP-Seq data for PF and PFI were obtained from our previous study with the accession number PRJNA488988 in NCBI database. Raw reads of ChIP-Seq were processed with Trimmomatic version 0.30 (Bolger et al. 2014), and clean reads were mapped to the P. fortunei reference genome (https://paulownia.genomics.cn/page/species/index.jsp) using BWA software (Li and Durbin 2010) with default parameters. Reproducibility between biological replicates was assessed using Pearson’s correlation analyses for genome-wide read distribution between the two members of each pair of biological replicates. Genomic regions marked with H3K4me3, H3K36me3, or H3K9ac were identified by comparing the ChIP library with each respective input DNA library using the Model-based analysis of ChIP-Seq (MACS) with default parameters (Zhang et al. 2008). The H3K4me3, H3K36me3 and H3K9ac modification level of each gene was quantified by the FPKM (fragments per kilobase of transcript per million mapped reads) method according to He et al. (2010) and Guo et al. (2015). Genes with fold change ≥ 1.2 and p value ≤ 0.05 were considered as significantly differentially histone-modified genes (DMGs). To screen histone-modified genes closely associated with PaWB disease, we performed comparison among the four samples according to the schemes as described by Wang et al. (2017b). Kyoto Encyclopedia of Genes and Genomes (KEGG) functional analysis was performed using OmicShare tools, a free online platform for data analysis (https://www.omicshare.com/tools).
ChIP-qPCR
For ChIP-qPCR, six genes from ChIP-Seq analysis were used to validate the reliability of ChIP antibodies to check whether the fold enrichment was at least greater than two. The fold enrichment was calculated as described by Mukhopadhyay et al. (2008). The primer pairs used for ChIP-qPCR are listed in Table S1. Each reaction was performed using three biological replicates.
RNA-Seq data analysis
The RNA-seq data for PF, PFI, PF-60 and PFI-60 were obtained from NCBI database with the accession number SRX1479063, SRX1479064, SRX7011163, and SRX1479066. The materials used for RNA-Seq were same as those used for ChIP-Seq. After filtering raw data, the clean reads were mapped to the P. fortunei reference genome (https://paulownia.genomics.cn/page/species/index.jsp) using Bowtie 2 (Langmead and Salzberg 2012). The expression level of each gene was normalized by the FPKM method (Mortazavi et al. 2008). Genes with fold change ≥ 2 and FDR ≤ 0.001 were defined as significantly differentially expressed genes (DEGs).
Results
Genome-wide analysis of H3K4me3, H3K36me3, and H3K9ac marks in P. fortunei
To explore the function of histone methylation and acetylation in PaWB, we analyzed ChIP-Seq data for PF, PFI, PF-60 and PFI-60 samples using antibodies that recognize H3K4me3, H3K36me3 and H3K9ac. Statistical details of the ChIP and input sequencing data for each sample are listed in Table S2. Repeatability analyses among the three biological replicates of each sample confirmed the high quality of the ChIP-Seq results (Table S3). More than 510 million H3K4me3, 499 million H3K36me3, and 465 million H3K9ac ChIP-Seq and 501 million input paired-end reads of the four samples were mapped to the P. fortunei reference genome for peak-calling by Model-based Analysis of ChIP-Seq (MACS). Totally, 17,202 H3K4me3, 11,652 H3K36me3, and 19,255 H3K9ac peaks were identified in PF by MACS software (Fig. S1), indicating that the three histone marks are widely distributed in the P. fortunei genome. The peaks of H3K4me3, H3K36me3, and H3K9ac in PF varied from 257 bp to 7831 bp, from 250 bp to 39,905 bp, and from 326 bp to 15,785 bp in length, respectively, with average lengths of 1679 bp, 2637 bp, and 2106 bp, respectively (for details, see Fig. S1). The majority of H3K4me3, H3K36me3 and H3K9ac marks in PF located in genic regions in the P. fortunei genome, including coding sequences (CDS), introns, promoters, 5′ untranslated regions (UTRs), and 3′ UTRs (Fig. 1a). The H3K4me3, H3K36me3, and H3K9ac marks in PF were enriched downstream of transcription start sites (TSSs) (Fig. 1b). The ChIP-Seq data were validated by independent ChIP-qPCR of six randomly selected genes. All the regions identified by ChIP-Seq and assayed by ChIP-qPCR showed clear immunoprecipitation enrichment in the H3K4me3, H3K36me3 and H3K9ac, and the fold enrichment values for most of them were greater than ten (Table S4).
Fig. 1.

The distribution of H3K4me3, H3K36me3 and H3K9ac marks within different genomic regions (a) and around transcription start site (TSS) ± 2 kb surrounding regions in protein-coding genes (b) in PF
From the ChIP-Seq results, 15,734 H3K4me3-, 9441 H3K36me3-, and 16,778 H3K9ac-modified genes were identified in PF. Of them, 14,257 (72.3%) were co-modified by at least two histone modifications, indicating a high concurrence of histone marks (Fig. 2a). Of note, the majority (13,230) of those genes were co-marked by H3K4me3 and H3K9ac, showing a highest concurrence frequency between them. Interestingly, more than 95.4% (9013 out of 9441) of the H3K36me3-modified genes in PF were modified by H3K4me3 and H3K9ac as well, whereas over 5000 genes were modified by H3K4me3 or H3K9ac, but not by H3K36me3. The combinations of histone marks did not differ drastically between PF and the other three samples (PFI, PF-60, and PFI-60). The histone modification patterns were similar among PFI, PF-60, and PFI-60.
Fig. 2.

Different combinations of H3K4me3, H3K36me3 and H3K9ac marks affect gene expression. (a) Venn diagram of genes
modified by H3K4me3, H3K36me3 and H3K9ac marks in PF; (b) Expression levels of genes with different modification patterns. X-axis stands for different combinations of histone marks; Y-axis stands for gene expression levels calculated by log10 (FPKM + 0.001)
To investigate the functional relationships between H3K4me3, H3K36me3, and H3K9ac marks and gene expression, we analyzed RNA-seq data obtained for the same samples and inspected the expression of H3K4me3/H3K36me3/H3K9ac-enriched genes. We found that over 95% of histone- modified genes were expressed in PF, confirming that these three histone marks are associated with gene expression. We also examined the expression levels of genes modified by different combinations of the three marks. As shown in Fig. 2b, genes marked by H3K9ac only exhibited higher expression than those marked by H3K4me3 or H3K36me3 only, and genes modified by a combination of any two histone marks showed higher expression than those modified by a single histone mark. What is more, genes modified by all three histone marks exhibited the highest expression levels. These results indicated that H3K4me3, H3K36me3, and H3K9ac are associated with active gene expression, and may work in a collaborative manner.
We observed similar histone modification distribution patterns among the other three samples (PFI, PF-60, and PFI-60). More detailed information about the peaks identified in the other three samples and their characteristics is provided in Fig. S1–S3. Taken together, our results illustrate the genome-wide profiles of gene expression and three histone modifications (H3K4me3, H3K36me3 and H3K9ac) of protein-coding genes in P. fortunei.
Changes in H3K4me3, H3K36me3, and H3K9ac in P. fortunei genome during phytoplasma infection and recovery processes
There is increasing evidence that histone methylation and acetylation play important roles in the regulation of gene expression in response to biotic and abiotic stresses (Ayyappan et al. 2015; Luo et al. 2012). To study changes in H3K4me3, H3K36me3, and H3K9ac during the phytoplasma infection and recovery processes, comparative analysis was conducted. Genes with fold change ≥ 1.2 and p value ≤ 0.05 were considered as DMGs. The results showed that the majority of genes modified by the three histone marks had similar histone modification levels during phytoplasma infection and recovery processes. In the phytoplasma infection process (PF vs. PFI comparison), 1169 genes showed increases in H3K4me3 levels, while 1265 genes showed decreases in H3K4me3 levels. The H3K36me3 and H3K9ac levels were induced in 5301 and 2688 genes, but reduced in 887 and 1228 genes, respectively. We noticed that the modification levels of most DMGs increased in response to phytoplasma infection. While relatively fewer genes displayed altered histone modifications levels during the recovery process (PFI vs. PFI-60 comparison) with 615 DMGs (282 up-regulated and 333 down-regulated) for H3K4me3, 594 DMGs (87 up-regulated and 507 down-regulated) for H3K36me3 and 701 DMGs (165 up-regulated and 436 down-regulated) for H3K9ac. The majority of genes in PFI showed decreased modification levels after treatment with MMS reagent (Fig. 3). The results of the KEGG analysis indicated that the DMGs affected by the phytoplasma and MMS reagent were mainly involved in plant-pathogen interaction (ko04626), plant hormone signal transduction (ko04075), starch and sucrose metabolism (ko00500), and RNA transport (ko03013) (Table S5; S6). These results indicated that, compared with MMS treatment, phytoplasma infection resulted in greater changes in the three histone marks on genes. Thus, phytoplasma infection may have a greater impact than MMS on histone modifications in Paulownia.
Fig. 3.
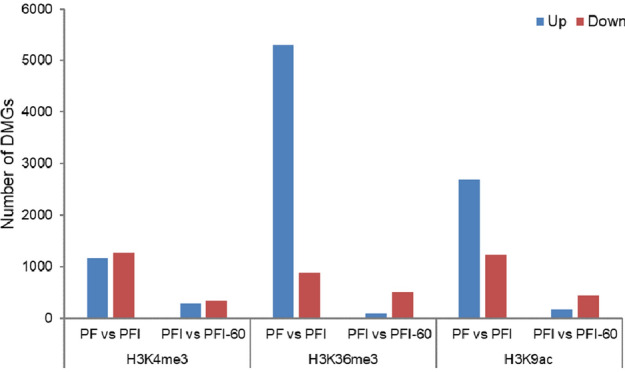
Genes differentially
modified by H3K4me3, H3K36me3 and H3K9ac marks during phytoplasma infection (PF vs. PFI) and recovery processes (PFI vs. PFI-60)
To study the potential roles of histone methylation and acetylation in regulating gene expression in Paulownia, we further analyzed the RNA-seq data obtained from the same plant materials. With the criteria of fold change ≥ 2 and FDR < 0.001, we found that the phytoplasma and MMS resulted in differential expression of 3563 genes (2066 increased, 1497 decreased) (PF vs. PFI comparison) and 2589 genes (1099 increased, 1490 decreased) (PFI vs. PFI-60 comparison), respectively (Fig. S4). The results of the KEGG pathway analysis indicated that the DEGs induced by the phytoplasma during the phytoplasma infection process were involved in 130 pathways in (Table S7a), while those affected by MMS treatment participated in 129 pathways (Table S7b), including plant-pathogen interaction (ko04626), starch and sucrose metabolism (ko00500), plant hormone signal transduction (ko04075) and phenylpropanoid biosynthesis (ko00940). We then determined the expression levels of all the histone-modified genes. During the phytoplasma infection process, 446 (18.3%) DMGs with H3K4me3, 818 (13.2%) DMGs with H3K36me3, 756 (19.3%) DMGs with H3K9ac showed changes in their expression levels (PF vs. PFI comparison). These results showed that histone modifications regulated only a small proportion of phytoplasma-responsive genes. Similarly, during the recovery process, it was also discovered that the DMGs having altered expression levels only accounted for a small part (PFI vs. PFI-60 comparison).
To further investigate the relationship between changes in histone modifications and gene expression levels, we analyzed the alterations in H3K4me3, H3K36me3 and H3K9ac levels of DEGs induced by stress. The scatter plot of these genes revealed significant positive correlations between DMGs with H3K4me3, H3K36me3, and H3K9ac and their gene expression levels during the Phytoplasma infection process (Pearson’s correlation coefficients of 0.54 for H3K4me3, 0.545 for H3K36me3, and 0.634 for H3K9ac) (Fig. 4). Surprisingly, the correlations between the DMGs for H3K4me3, H3K36me3, and H3K9ac and gene expression levels were weak during the recovery process (Fig. S5). Altogether, these results suggested that differential H3K4me3, H3K36me3, and H3K9ac modifications only affect the expression levels of a small number of genes, and that the three histone marks affect gene expression more strongly during the phytoplasma infection process than during recovery process.
Fig. 4.

Correlation analysis of differentially expressed and differentially (a) H3K4me3-, (b) H3K36me3- and (c) H3K9ac- modified genes in Paulownia during phytoplasma infection process (PF vs. PFI)
Genes with altered histone modification levels related to PaWB disease
To identify the DMGs closely associated with PaWB disease, we conducted comparative analyses based on the scheme described by Wang et al. (2017b). First, to exclude DMGs affected by MMS reagent, we retrieved the different DMGs between the comparisons of PFI vs. PFI-60 and PF vs. PF-60 (1113 genes with H3K4me3, 2658 genes with H3K36me3, and 1285 genes with H3K9ac) (Fig. 5). Next, we compared these genes with those identified as DMGs in the PF vs. PFI comparison to retrieve the common DMGs between them, which were regarded to be closely related to PaWB disease. In final, 365, 2244 and 752 DMGs with H3K4me3, H3K36me3, and H3K9ac marks, respectively, were found to be associated with PaWB disease (Table S8). KEGG analysis results indicated that the most represented pathways in which these DMGs involved were plant-pathogen interaction (ko04626), plant hormone signal transduction (ko04075), starch and sucrose metabolism (ko00500), RNA transport (ko03013), carbon metabolism (ko01200) and circadian rhythm-plant (ko04712) (Table S9). Among these genes, we found genes involved in calcium ion (Ca2+) signal transduction, abscisic acid (ABA) signal transduction and ethylene biosynthesis were induced by phytoplasma along with corresponding alterations in expression.
Fig. 5.

Venn diagrams based on the comparison scheme for (a) H3K4me3, (b) H3K36me3 and (c) H3K9ac
Discussion
In our previous studies on Paulownia infected by PaWB disease, we focused on changes at the transcriptional, post-transcriptional, and translational levels, and discovered numerous mRNAs, miRNAs, lncRNAs, and proteins potentially related to PaWB disease (Fan et al. 2014, 2016; Wang et al. 2017a, b). However, it was still unclear whether and how histone modifications affect gene expression in Paulownia. Histone methylation and acetylation, two main types of histone modifications, are known to be involved in the regulation of gene expression under biotic stress (Ayyappan et al. 2015; Crespo-Salvador et al. 2018). In the current study, we generated a global view of histone modification profiles of H3K4me3, H3K36me3 and H3K9ac in Paulownia and investigated their dynamic changes during phytoplasma infection and recovery processes using ChIP-Seq. Compared with ChIP-PCR and ChIP-chip, ChIP-Seq can provide genome-scale profiles of histone modifications with higher resolution and greater sensitivity, which is a powerful method to investigate the dynamic changes in histone marks associated with PaWB disease, and has been widely used in other species (Brusslan et al. 2015; Du et al. 2013; Hussey et al. 2017).
We found that the PFI, PF-60, and PFI-60 samples had genome-wide patterns of histone modifications consistent with the patterns in PF. The majority of H3K4me3, H3K36me3 and H3K9ac histone modifications were deposited in the genic regions, especially downstream of TSSs, in the genome of P. fortunei, which were in agreement with their distribution patterns in the genomes of Oryza sativa (Du et al. 2013), Eucalyptus grandis (Hussey et al. 2017), and Arabidopsis thaliana (Pajoro et al. 2017). However, the proportions of the histone marks in different genomic regions varied between the phytoplasma infection and recovery processes. The H3K4me3, H3K36me3 and H3K9ac histone marks targeted a large number of genes in Paulownia, and the number of modified genes increased after phytoplasma infection and MMS reagent treatment. This result indicated that both the phytoplasma and MMS reagent have influences on histone modifications of Paulownia. Studies have shown that distinct histone amino-terminal modifications can form synergistic or antagonistic combinations that represent a “histone code”, which is read by other proteins or protein modules to amplify the readout of upstream signaling pathways and alter the chromatin structure of target genes, which considerably extends the information potential of the genetic (DNA) code (Jenuwein and Allis 2001; Strahl and Allis 2000). In rice, different histone modifications frequently co-occur, and gene expression levels are positively correlated with histone marks (Du et al. 2013; He et al. 2010). In agreement with this, we observed that the genes co-occupied by at least two histone marks accounted for a large proportion of expressed genes in Paulownia. As reported for rice, Paulownia also showed a high concurrence between histone marks and gene expression. Genes with all three histone marks displayed the highest expression levels, indicating that these three histone marks have a combinatorial effect on gene expression.
Despite the conserved profiles of H3K4me3, H3K36me3, and H3K9ac marks in plants, differential histone modifications can modulate the expression of genes in response to abiotic and biotic stresses, such as drought, cold, and pathogen attack, allowing plants to respond rapidly to these stresses. As shown in previous studies, H3K4me3, H3K36me3, and H3K9ac are positively related to gene expression (Du et al. 2013; Kouzarides 2007). Surprisingly, our results showed that only a small proportion of DEGs also had significant changes in histone modifications (H3K4me3, H3K36me3, and H3K9ac). This can be partially explained by the stringent criteria used to identify the DEGs and DMGs in this study. Further analyses revealed a significant positive correlation between DEGs and DMGs (with H3K4me3, H3K36me3, and H3K9ac) during phytoplasma infection, but a weaker correlation between DEGs and DMGs (with H3K4me3, H3K36me3, and H3K9ac) after MMS treatment. This result highlights the regulatory roles of histone marks to promote an active chromatin configuration for gene expression during phytoplasma infection in Paulownia. The weak correlation between DMGs and DEGs after MMS treatment suggested that gene expression in Paulownia during recovery process from PaWB disease may not due to alterations of these three histone modifications for majority of genes in the genome, instead, it is likely that other histone marks and/or regulatory mechanisms might be more important in determining the transcriptional state of genes involved in recovery process that we are unable to capture here. Further analyses are required to explore such regulatory mechanisms. As well as histone modifications, many other complex mechanisms can regulate gene expression, including miRNA-mediated posttranscriptional regulation and DNA methylation-mediated epigenetic regulation. Posttranscriptional regulation mediated by small RNAs has been detected in Paulownia in our previous studies (Fan et al. 2016), and may explain inconsistencies between histone marks and gene expression. Two models have been proposed for the function of histone modifications in transcriptional regulation (Kouzarides 2002, 2007). In one model, histone modifications directly affect the interactions/contact of histones with DNA or interconnections between nucleosomes to alter the higher-order structure of chromatin, making it accessible or inaccessible to the transcriptional machinery. For instance, the acetylation of histones neutralizes the net positive charge of lysine residues, resulting in a less condensed chromatin architecture and increased accessibility of regulatory factors to DNA recognition sequences. In the second model, histone modifications recruit a series of ordered protein complexes with chromatin-remodeling activity and interact with each other via specific domains. For instance, methylation is recognized by chromo-like domains of the Royal family (chromo, tudor, MBT) and non-related PHD domains, and acetylation is recognized by bromodomains (BRD). Recently, a number of proteins recruited to specific modifications have been identified. However, further research is required to determine how these three histone marks regulate gene expression during phytoplasma infection.
Signaling pathways in plants have great regulatory potential to adapt to biotic stress rapidly and to utilize limited resources efficiently for growth and survival. Calcium ion (Ca2+) signal transduction has important functions in plant-pathogen interactions, and Ca2+ accumulation in the cytosol is an early signal that initiates defense responses to pathogens in plants. The perception of pathogens leads to the production of cyclic nucleotides and the activation of cyclic nucleotide-gated ion channels (CNGCs), which are non-selective cation channels. These channels are gated by the direct binding of cyclic nucleotides, and they control ion flux across the plasma-membrane and tonoplast. The genomes of Arabidopsis and rice contain 20 and 16 genes, respectively, encoding CNGCs (Mäser et al. 2001; Nawaz et al. 2014). There is growing evidence that CNGCs can facilitate Ca2+ uptake into the plant cell cytosol and participate in defense against pathogens (Ma 2011). In Arabidopsis, AtCNGC2, 4, 11, and 12 were reported to be involved in plant disease and defense signaling pathways (Jha et al. 2016). Calmodulins (CaMs) and CaM-like proteins (CMLs) are Ca2+ sensors with an EF-hand Ca2+-binding motif. These proteins can translate Ca2+ signals into cellular responses that lead to disease resistance. Gain-of-function or loss-of-function genetic experiments have confirmed the roles of several CMLs in plant immunity, including the Arabidopsis calcium sensor CML8 (Aldon et al. 2018; Zhu et al. 2016). Additionally, a plasmodesma-localized Ca2+-binding protein, CML41, was reported to mediate callose-dependent plasmodesmatal closure to reduce symplastic connectivity, serving as a critical defense against bacterial pathogens (Xu et al. 2017). We found that a gene encoding a CNGC (PAU019072.1) showed significantly increased H3K9ac levels during phytoplasma infection, along with increased transcript levels. Two genes encoding calcium-binding protein CML, PAU019652.1 and PAU002214.1, displayed significant increases in H3K9ac and H3K36me3 levels, accompanied by increased transcript levels, respectively. On the basis of these results, we inferred that CNGC and CML may play important roles in the Paulownia-phytoplasma interaction.
Phytohormones are small signaling molecules with vital functions in the regulation of plant development and the mediation of adaptive responses to a range of abiotic and biotic stresses (Bari and Jones 2009). ABA is a crucial phytohormone that regulates seed germination, stomatal movement, plant development, and biotic and abiotic stress responses. Several studies have demonstrated that ABA enhances disease resistance through inducing stomatal closure and promoting callose deposition, thus forming a physical barrier to prevent pathogen invasion (Ton and Mauch-Mani 2004). ABA has also been found to inhibit plant stem elongation and shoot growth (Arney and Mitchell 1969; Kaufman and Jones 1974). Mou et al. (2013) reported that the typical PaWB symptoms of phytoplasma-infected Paulownia, such as smaller leaves and shorter internodes, were associated with high ABA levels. Fan et al. (2015) found that the expression levels of genes associated with the ABA signaling pathway altered in response to phytoplasma infection. Pyrabactin resistance (PYR)-like (PYL)/regulatory component of ABA receptor (RCAR) family, protein phosphatase 2Cs (PP2Cs), and snf1-related kinase 2 (SnRK2) protein kinases are the core signaling components involved in ABA perception and signal transduction. Members of the PP2C family function as negative regulators of ABA signal transduction through interacting with SnRK2 (a positive regulator), resulting in inactivation of SnRK2 and suppression of signaling transduction. After ABA is perceived by PYR/PYL/RCAR receptors, the ABA-bound PYR/PYL/RCAR receptors interact with the phosphatase domain of PP2Cs and form an ABA-PYR/PYL/RCARs-PP2Cs complex. This breaks the physical interaction and inhibition of SnRK2s by PP2Cs and allows the activation of SnRK2s to target downstream components, such as ion channels, transcription factors, and NADPH oxidases (Lee and Luan 2012). The ABA signaling pathway is thought to mediate the majority of ABA-triggered plant responses, such as stomatal closure, inhibition of root growth, and seed germination (Cutler et al. 2010). Hayashi et al. (2014) reported that ABA suppressed hypocotyl elongation via dephosphorylation of the plasma membrane H+-ATPase in Arabidopsis thaliana, while ABA did not affect hypocotyl elongation and phosphorylation levels of H+-ATPase in the abi1-1 mutant. Those findings suggested that the ABA signaling pathway may be involved in mediating the inhibition of hypocotyl elongation. In this study, we found that genes involved in the ABA signaling pathway showed consistent tendency between alterations of histone modifications and gene expression during phytoplasma infection. For instance, two genes encoding PYR/PYL family receptors (PAU019318.1, PAU008762.1) showed increased H3K9ac levels, in accordance with their increased expression levels in response to phytoplasma infection. The gene encoding the negative regulator of ABA signal transduction, PP2C (PAU014089.1), showed decreased H3K9ac levels in response to phytoplasma infection, and significantly decreased transcript levels. The gene encoding ABA responsive element binding factor (ABF) (PAU003579.2) displayed significant increases in H3K36me3 mark and transcriptional levels in response to phytoplasma infection. These results suggested that changes in the state of chromatin near these genes during phytoplasma infection prepares them for future transcriptional changes, and the shorter internodes of PFI may be related to ABA signal transduction in Paulownia. Ethylene is one of the main phytohormones regulating various developmental processes and responses to pathogen attack (Broekaert et al. 2006). Pathogen challenge is known to enhance ethylene production in plants (Penninckx et al. 1998). We found that aminocyclopropanecarboxylate oxidase (ACO, PAU000476.1), a key enzyme in ethylene biosynthesis catalyzing the final step of ethylene synthesis using 1-aminocyclopropane-1-carboxylic acid (ACC) as substrate, was up-regulated after phytoplasma infection, accompanied by increased H3K4me3 levels, suggesting that the H3K4me3 mark is associated with changes in gene expression. Our results suggested that ethylene may be induced in response to phytoplasma infection, and that H3K4me3 might play a regulatory role in this process.
Conclusions
Taken together, we identified a large number of genes marked by H3K4me3, H3K36me3, and H3K9ac in PF, PFI, PF-60, and PFI-60 samples. All three histone marks were mainly distributed in genic regions, and were enriched downstream of the TSSs of genes in Paulownia. The three histone marks frequently co-occurred, and were associated with gene expression. The genes with all three histone marks showed the highest expression levels. The combined ChIP-Seq data and RNA-Seq data revealed that differential histone modifications only affect a small proportion of stress-responsive genes. We detected positive correlations between the H3K4me3, H3K36me3 and H3K9ac modification levels and gene expression levels for a subset of genes showing alterations both in modification and expression following phytoplasma infection. Furthermore, we obtained 365, 2244, and 752 PaWB-related DMGs with H3K4me3, H3K36me3, and H3K9ac marks, separately. These results provide new insights into the regulatory roles of histone modifications in modulating gene expression in Paulownia during phytoplasma infection process. Further work should focus on how these histone marks regulate gene expression in Paulownia during phytoplasma infection process, and on the functional roles of histone-modifying enzymes involved in this process.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study was funded by the National Key Research and Development Program (Grant No. 2016YFD0600106, 2017YFD060050604) and the Forestry Science and Technology Demonstration Project of Central Finance [Grant No. GTH (2017)15]. We thank Jennifer Smith, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author contributions
GF conceived and designed the experiments; XZ and ZZ performed the experiments; LY and ZZ analyzed the data; XZ contributed reagents/materials/analysis tools; LY wrote the manuscript; LY and GF revised the manuscript. All authors read and approved the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of authors.
Footnotes
Lijun Yan and Xiaoqiao Zhai authors contributed equally to this work.
References
- Aldon D, Mbengue M, Mazars C, Galaud JP. Calcium signalling in plant biotic interactions. Int J Mol Sci. 2018;19:E665. doi: 10.3390/ijms19030665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arney SE, Mitchell DL. The effect of abscisic acid on stem elongation and correlative inhibition. New Phytol. 1969;68:1001–1015. doi: 10.1111/j.1469-8137.1969.tb06500.x. [DOI] [Google Scholar]
- Ayyappan V, Kalavacharla V, Thimmapuram J, Bhide KP, Sripathi VR, Smolinski TG, Manoharan M, Thurston Y, Todd A, Kingham B. Genome-wide profiling of histone modifications (H3K9me2 and H4K12ac) and gene expression in rust (Uromyces appendiculatus) inoculated common bean (Phaseolus vulgaris L) PLoS ONE. 2015;10:e0132176. doi: 10.1371/journal.pone.0132176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari R, Jones JD. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69:473–488. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bertaccini A, Duduk B. Phytoplasma and phytoplasma diseases: a review of recent research. Phytopathol Mediterr. 2009;48:355–378. doi: 10.14601/Phytopathol_Mediterr-3300. [DOI] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekaert WF, Delauré SL, De Bolle MF, Cammue BP. The role of ethylene in host-pathogen interactions. Annu Rev Phytopathol. 2006;44:393–416. doi: 10.1146/annurev.phyto.44.070505.143440. [DOI] [PubMed] [Google Scholar]
- Brusslan JA, Bonora G, Rus-Canterbury AM, Tariq F, Jaroszewicz A, Pellegrini M. A genome-wide chronological study of gene expression and two histone modifications, H3K4me3 and H3K9ac, during developmental leaf senescence. Plant Physiol. 2015;168:1246–1261. doi: 10.1104/pp.114.252999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XB, Fan GQ, Zhai XQ. Morphological changes of the witches' broom seedlings of Paulownia tomentosa treated with methyl methanesulphonate and SSR analysis. Acta Phytopathologica Sinica. 2012;42:214–218. doi: 10.13926/j.cnki.apps.2012.02.014. [DOI] [Google Scholar]
- Choi SM, Song HR, Han SK, Han M, Kim CY, Park J, Lee YH, Jeon JS, Noh YS, Noh B. HDA19 is required for the repression of salicylic acid biosynthesis and salicylic acid-mediated defense responses in Arabidopsis. Plant J. 2012;71:135–146. doi: 10.1111/j.1365-313X.2012.04977.x. [DOI] [PubMed] [Google Scholar]
- Crespo-Salvador Ó, Escamilla-Aguilar M, López-Cruz J, López-Rodas G, González-Bosch C. Determination of histone epigenetic marks in Arabidopsis and tomato genes in the early response to Botrytis cinerea. Plant Cell Rep. 2018;37:153–166. doi: 10.1007/s00299-017-2218-9. [DOI] [PubMed] [Google Scholar]
- Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol. 2010;61:651–679. doi: 10.1146/annurev-arplant-042809-112122. [DOI] [PubMed] [Google Scholar]
- Ding B, Bellizzi Mdel R, Ning Y, Meyers BC, Wang GL. HDT701, a histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell. 2012;24:3783–3794. doi: 10.1105/tpc.112.101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Li H, Wei Q, Zhao X, Wang C, Zhu Q, Yi X, Xu W, Liu XS, Jin W, Su Z. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica Mol Plant. 2013;6:1463–1472. doi: 10.1093/mp/sst018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Zhai X, Jiang J, Liu X. Callus induction from Paulownia plant leaves and their plantlet regenerations. Scientia Silvae Sinicae. 2002;38:29–35. [Google Scholar]
- Fan G, Dong Y, Deng M, Zhao Z, Niu S, Xu E. Plant-pathogen interaction, circadian rhythm, and hormone-related gene expression provide indicators of phytoplasma infection in Paulownia fortunei. Int J Mol Sci. 2014;15:23141–23162. doi: 10.3390/ijms151223141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Cao X, Zhao Z, Deng M. Transcriptome analysis of the genes related to the morphological changes of Paulownia tomentosa plantlets infected with phytoplasma. Acta Physiol Plant. 2015;37:202. doi: 10.1007/s11738-015-1948-y. [DOI] [Google Scholar]
- Fan G, Niu S, Zhao Z, Deng M, Xu E, Wang Y, Yang L. Identification of microRNAs and their targets in Paulownia fortunei plants free from phytoplasma pathogen after methyl methane sulfonate treatment. Biochimie. 2016;127:271–280. doi: 10.1016/j.biochi.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Fan G, Cao Y, Wang Z. Regulation of long noncoding RNAs responsive to phytoplasma infection in Paulownia tomentosa. Int J Genomics. 2018;3:174–352. doi: 10.1155/2018/3174352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Song G, Liu Z, Qu X, Chen R, Jiang D, Sun Y, Liu C, Zhu Y, Yang D. Global epigenomic analysis indicates that epialleles contribute to Allele-specific expression via Allele-specific histone modifications in hybrid rice. BMC Genomics. 2015;16:232. doi: 10.1186/s12864-015-1454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Takahashi K, Inoue S, Kinoshita T. Abscisic acid suppresses hypocotyl elongation by dephosphorylating plasma membrane H+-ATPase in Arabidopsis thaliana. Plant Cell Physiol. 2014;55:845–853. doi: 10.1093/pcp/pcu028. [DOI] [PubMed] [Google Scholar]
- He G, Zhu X, Elling AA, Chen L, Wang X, Guo L, Liang M, He H, Zhang H, Chen F, Qi Y, Chen R, Deng XW. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenhout SA, Oshima K, Ammar D, Kakizawa S, Kingdom HN, Namba S. Phytoplasmas: bacteria that manipulate plants and insects. Mol Plant Pathol. 2008;9:403–423. doi: 10.1111/j.1364-3703.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Wang L, Wang L, Liu L, Li L, Sun L, Rao Q, Zhang J, Huang S. JMJ704 positively regulates rice defense response against Xanthomonas oryzae pv oryzae infection via reducing H3K4me2/3 associated with negative disease resistance regulators. BMC Plant Biol. 2015;15:286. doi: 10.1186/s12870-015-0674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey SG, Loots MT, van der Merwe K, Mizrachi E, Myburg AA. Integrated analysis and transcript abundance modelling of H3K4me3 and H3K27me3 in developing secondary xylem. Sci Rep. 2017;7:3370. doi: 10.1038/s41598-017-03665-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jha SK, Sharma M, Pandey GK. Role of cyclic nucleotide gated channels in stress management in plants. Curr Genomics. 2016;17:315–329. doi: 10.2174/1389202917666160331202125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung I, Kim D. Histone modification profiles characterize function-specific gene regulation. J Theor Biol. 2012;310:132–142. doi: 10.1016/j.jtbi.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Kaufman PB, Jones RA. Regulation of growth in Avena (Oat) stem segments by gibberellic acid and abscisic acid. Physiol Plantarum. 1974;31:39–43. doi: 10.1111/j.1399-3054.1974.tb03674.x. [DOI] [Google Scholar]
- Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Luan S. ABA signal transduction at the crossroad of biotic and abiotic stress responses. Plant Cell Environ. 2012;35:53–60. doi: 10.1111/j.1365-3040.2011.02426.x. [DOI] [PubMed] [Google Scholar]
- Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta. 2009;1790:863–868. doi: 10.1016/j.bbagen.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen X, Zhong X, Zhao Y, Liu X, Zhou S, Cheng S, Zhou DX. Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell. 2013;25:4725–4736. doi: 10.1105/tpc.113.118802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Liu X, Singh P, Cui Y, Zimmerli L, Wu K. Chromatin modifications and remodeling in plant abiotic stress responses. Biochim Biophys Acta. 2012;1819:129–136. doi: 10.1016/j.bbagrm.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Ma W. Roles of Ca2+ and cyclic nucleotide gated channel in plant innate immunity. Plant Sci. 2011;181:342–346. doi: 10.1016/j.plantsci.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Mäser P, Thomine S, Schroeder JI, Ward JM, Hirschi K, Sze H, Talke IN, Amtmann A, Maathuis FJ, Sanders D, Harper JF, Tchieu J, Gribskov M, Persans MW, Salt DE, Kim SA, Guerinot ML. Phylogenetic relationships within cation transporter families of Arabidopsis. Plant Physiol. 2001;126:1646–1667. doi: 10.1104/pp.126.4.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Mou HQ, Lu J, Zhu SF, Lin CL, Tian GZ, Xia X, Zhao WJ. Transcriptomic analysis of Paulownia infected by Paulownia witches’-broom phytoplasma. PLoS ONE. 2013;8:e77217. doi: 10.1371/journal.pone.0077217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3:698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Kakar KU, Saand MA, Shu QY. Cyclic nucleotide-gated ion channel gene family in rice, identification, characterization and experimental analysis of expression response to plant hormones, biotic and abiotic stresses. BMC Genomics. 2014;15:853. doi: 10.1186/1471-2164-15-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajoro A, Severing E, Angenent GC, Immink RGH. Histone H3 lysine 36 methylation affects temperature-induced alternative splicing and flowering in plants. Genome Biol. 2017;18:102. doi: 10.1186/s13059-017-1235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninckx IA, Thomma BP, Buchala A, Métraux JP, Broekaert WF. Concomitant activation of jasmonate and ethylene response pathways is required for induction of a plant defensin gene in Arabidopsis. Plant Cell. 1998;10:2103–2113. doi: 10.1105/tpc.10.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Ton J, Mauch-Mani B. Beta-amino-butyric acid-induced resistance against necrotrophic pathogens is based on ABA-dependent priming for callose. Plant J. 2004;38:119–130. doi: 10.1111/j.1365-313X.2004.02028.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu W, Fan G, Zhai X, Zhao Z, Dong Y, Deng M, Cao Y. Quantitative proteome-level analysis of paulownia witches' broom disease with methyl methane sulfonate assistance reveals diverse metabolic changes during the infection and recovery processes. Peer J. 2017;5:e3495. doi: 10.7717/peerj.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhai X, Cao Y, Dong Y, Fan G. Long non-coding RNAs responsive to witches’ broom disease in Paulownia tomentosa. Forests. 2017;8:348. doi: 10.3390/f8090348. [DOI] [Google Scholar]
- Xu B, Cheval C, Laohavisit A, Hocking B, Chiasson D, Olsson TSG, Shirasu K, Faulkner C, Gilliham M. A calmodulin-like protein regulates plasmodesmal closure during bacterial immune responses. New Phytol. 2017;215:77–84. doi: 10.1111/nph.14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Cao X, Zhai X, Fan G. Establishment of in vitro plantlet regeneration system by witches’ broom organ of two Paulownia species. J Henan Agr Univ. 2009;43:145–150. doi: 10.16445/j.cnki.1000-2340.2009.02.016. [DOI] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Robe E, Jomat L, Aldon D, Mazars C, Galaud JP. CML8, an Arabidopsis calmodulin-like protein plays a role in Pseudomonas syringae plant immunity. Plant Cell Physiol. 2016;58:307–319. doi: 10.1093/pcp/pcw189. [DOI] [PubMed] [Google Scholar]
- Zhu A, Greaves IK, Dennis ES, Peacock WJ. Genome-wide analyses of four major histone modifications in Arabidopsis hybrids at the germinating seed stage. BMC Genomics. 2017;18:137. doi: 10.1186/s12864-017-3542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W, Zhong X, You J, Xiong L. Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol Biol. 2013;81:175–188. doi: 10.1007/s11103-012-9990-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.