

Abstract
Society’s demand for metals is ever increasing while stocks of high-grade minerals are being depleted. Biomining, for example of chalcopyrite for copper recovery, is a more sustainable biotechnological process that exploits the capacity of acidophilic microbes to catalyze solid metal sulfide dissolution to soluble metal sulfates. A key early stage in biomining is cell attachment and biofilm formation on the mineral surface that results in elevated mineral oxidation rates. Industrial biomining of chalcopyrite is typically carried out in large scale heaps that suffer from the downsides of slow and poor metal recoveries. In an effort to mitigate these drawbacks, this study investigated planktonic and biofilm cells of acidophilic (optimal growth pH < 3) biomining bacteria. RNA and proteins were extracted, and high throughput “omics” performed from a total of 80 biomining experiments. In addition, micrographs of biofilm formation on the chalcopyrite mineral surface over time were generated from eight separate experiments. The dataset generated in this project will be of great use to microbiologists, biotechnologists, and industrial researchers.
Subject terms: Soil microbiology, Environmental impact
| Measurement(s) | biofilm formation • Proteome • protein expression data • RNA • transcriptome |
| Technology Type(s) | fluorescence microscopy • mass spectrometry • RNA sequencing |
| Sample Characteristic - Organism | Leptospirillum ferriphilum • Sulfobacillus thermosulfidooxidans • Acidithiobacillus caldus |
| Sample Characteristic - Environment | mine |
Machine-accessible metadata file describing the reported data: 10.6084/m9.figshare.12173637
Background & Summary
Biomining is an industrial biotechnology involving sulfide mineral dissolution for the recovery of metals such as copper, nickel, and gold1. For low-grade ores, biomining is often carried out in very large heaps whereby the ore is stacked on an impermeable membrane, acid and in some cases microbes are added to the surface, and solubilized metals are collected at the base of the heap that are subsequently recovered2. During biomining, the sulfide mineral is attacked by ferric ions that results in soluble metal ions and the resulting ferrous iron is re-oxidized by acidophilic microorganisms3 to complete the abiotic-biotic catalytic cycle4. Further products of the ferric iron attack on the metal sulfide are inorganic sulfur compounds that are oxidized to generate the acidity required by the acidophiles5.
One of the major challenges for further exploitation of industrial biomining is for copper recovery from the refractory mineral chalcopyrite (CuFeS2), the largest reserve of copper containing mineral in the world. The problems associated with chalcopyrite bioleaching (i.e., when the target metal forms part of the mineral matrix) are slow rates and poor total recoveries often attributed to passivation of the mineral surface that has recently been suggested to be by iron-oxyhydroxides6. One method to avoid chalcopyrite passivation is to carry out the bioleaching at low redox potentials and high temperatures7,8 and several methods have been suggested to maintain the redox potential in the desired range including controlling the oxygen concentration9 or utilizing a microbial community that maintains the potential in the desired range10,11. An additional critical factor for chalcopyrite bioleaching, especially in early stages of bioheap inoculation, is the attachment and biofilm formation on the mineral surface12. However, how to control the redox potential to achieve high copper dissolution rates and recoveries has not been solved in several square kilometer large industrial bioheaps.
In this Data Descriptor, we present data for (meta)-transcriptomes, (meta)-proteomes, microscope images, and the accompanying metadata of axenic and defined consortia of bioleaching bacteria growing in continuous cultures and in the presence of chalcopyrite mineral (Fig. 1). The data set contains twelve continuous culture samples (Table 1), omics data from 80 bioleaching experiments (Table 1), and microscopy images tracking biofilm formation on chalcopyrite grains for eight distinct conditions over time (Table 1). The complete data in this descriptor have not been previously reported although parts have been included in published articles. These include RNA transcripts and protein concentrations of Leptospirillum ferriphilumT in axenic culture13, RNA transcripts and protein concentrations of simple defined mixed cultures11, microscopy images14,15, and on reverse engineering of omics data to generate gene regulatory networks16.
Fig. 1.
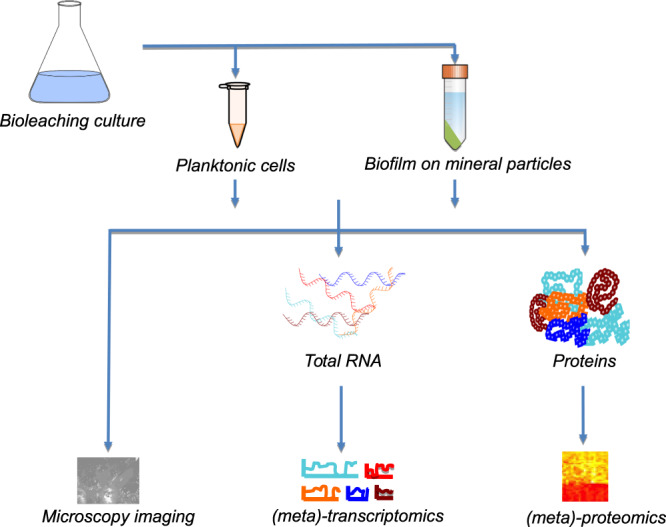
Workflow depicting the cultivation of (mixed) bioleaching bacteria cultures, proteins, and transcripts extraction and generation of microscopy, (meta)-transcriptomics, and (meta)-proteomics datasets.
Table 1.
Number of samples for which RNAseq and proteomics data are provided.
| Condition | Combination of strains* | RNA samples | Protein samples | Both | Imaging |
|---|---|---|---|---|---|
| Cn | AXX | 6 | 3 | 3 | |
| Cn | LXX | 3 | 0 | 0 | |
| Cn | SXX | 3 | 0 | 0 | |
| M | AXX | 2 | 0 | 0 | yes |
| M | LXX | 2 | 4 | 2 | yes |
| M | S–AL** | 2 | 4 | 1 | yes |
| M | S–LX** | 2 | 2 | 0 | yes |
| P | ALX | 3 | 0 | 0 | |
| P | ASL | 3 | 0 | 0 | |
| P | ASX | 11 | 20 | 11 | yes |
| P | AXX | 4 | 2 | 2 | |
| P | LSX | 3 | 4 | 3 | yes |
| P | LXX | 2 | 5 | 2 | yes |
| P | S–AL** | 3 | 5 | 3 | yes |
| P | S–AX** | 4 | 11 | 3 | yes |
| P | SLX | 4 | 0 | 0 | yes |
| P | SXX | 4 | 4 | 3 | yes |
Condition or cellular fraction indicates if the samples were derived from continuous culture (Cn) or bioleaching cultures and here either from the biofilm fraction (M) or the planktonic fraction (P). The number of samples for which RNA and proteins derived data could be generated are given in column 4 (Both).
*The combination of strains were composed of A. caldus (A), L. ferriphilum (L) and S. thermosulfidooxidans (S) and used as pure or mixed cultures, resulting in the following categories: A, L, S, AS, LS, and ASL (X stands for no species). A description corresponding to the strain composition and sample naming is found in the FAIRDOMHub repository (“SysMetEx – Dataset collection”55, “omics samples”).
**The dash sign (“–”) in the combination of strain names indicates sequential inoculation for pre-colonization by the species corresponding to the letter before the dash sign.
Methods
These methods are expanded versions of descriptions in our related work11–16.
Transcriptomics and proteomics
Mineral preparation for transcriptomics and proteomics
Chalcopyrite mineral concentrate from the Aitik copper mine (N 67°4′ 24″, E 20°57′ 51″) was provided by Boliden AB (Sweden). It was of high purity (>98%) and high copper content (29.5%) as revealed by aqua regia digestion and elemental analysis. The concentrate was sieved to a size fraction of 50–100 μm and washed in three volumes of 0.1 M EDTA in 0.4 M NaOH for 10 min while stirring to remove iron and copper compounds resulting from mineral weathering17,18. After elemental sulfur was removed from the mineral surfaces by three washing iterations with one volume of acetone, the mineral was dried at 60 °C overnight, and subsequently autoclaved at 120 °C for 10 h under a nitrogen atmosphere to prevent changes in its structure. This procedure likely prevented the growth of contaminating microbes as flotation chemicals are toxic to the acidophile, Sulfolobus metallicus19.
Microbial species cultivation for transcriptomics and proteomics
Three bacterial acidophile species were used: Leptospirillum ferriphilumT DSM 1464720, Sulfobacillus thermosulfidooxidansT DSM 929321, and Acidithiobacillus caldusT DSM 858422. Cells were maintained in the exponential growth phase at 38 °C in three separate axenic continuous cultures11 until inoculation for further experiments. Continuous cultures (1 L working volume) containing Mackintosh basal salt (MAC) medium23 and ferrous sulfate (100 mM) as electron donor were adjusted to pH 1.4 for L. ferriphilum, or with 5 mM potassium tetrathionate adjusted to pH 2.3 and pH 2.0 for S. thermosulfidooxidans and A. caldus, respectively. The continuous culture vessels containing MAC medium plus tubing, connectors etc. were autoclaved while the ferrous sulfate and potassium tetrathionate were sterile filtered (0.2 μm pore size, cellulose acetate filter, PALL).
Bioleaching experiments were carried out and analyzed as previously reported11. Quadruplets of 100 mL MAC medium were adjusted to pH 1.8 by addition of sulfuric acid and supplemented with 2% (wt/vol) chalcopyrite concentrate. Different combinations of the three bacteria were inoculated in aliquots of 107 cells per mL per species that were captured by 20 min centrifugation from the continuous cultures at 12,500 g. Sample inoculation occurred with all species simultaneously according to the three-letter sample names, in which the initial letters of the bacterial species was used (Tables 1 and 2), if not otherwise indicated. When the inoculation order was sequential, a dash sign (“–”) in the sample names indicates 48 h pre-colonization of the species corresponding to the first letter. For example, sample “ASL” indicates inoculation of the three species at the same time, while in sample “S–AL” S. thermosulfidooxidans was used as pre-colonizer and 48 h later A. caldus and L. ferriphilum were added.
Table 2.
Identification rate of MS/MS spectra for the different samples.
| Condition | Combination of strains* | MS/MS submitted | MS/MS Identified (%) |
|---|---|---|---|
| Cn | AXX | 185932 | 24.63 |
| M | LXX | 166215 | 14.14 |
| M | S–AL** | 263940 | 20.61 |
| M | S–LX** | 78849 | 30.16 |
| P | ASX | 530117 | 16.78 |
| P | AXX | 152847 | 14.91 |
| P | LSX | 347396 | 29.66 |
| P | LXX | 229079 | 20.01 |
| P | S–AL** | 168878 | 25.32 |
| P | SXX | 161726 | 24.63 |
| Average | 22.09 |
*The combination of strains were composed of A. caldus (A), L. ferriphilum (L) and S. thermosulfidooxidans (S) and used as pure or mixed cultures, resulting in the following categories: A, L, S, AS, LS, and ASL (X stands for no species). A description corresponding to the conditions is found in the FAIRDOMHub repository (“SysMetEx – Dataset collection”55, “Sample Identification Code”).
**The dash sign (“–”) in the combination of strain names indicates sequential inoculation for pre-colonization by the species corresponding to the letter before the dash sign.
Cell counts were obtained using a Neubauer improved counting chamber. Experiments using single, binary, and tertiary combinations of the microbial species along with a sterile control were carried out in triplicates. Cultures were incubated at 38 ± 2 °C with slow shaking at 120 rpm. Bioleaching experiments were monitored during a period of 14–20 days by measuring the redox potential with respect to an Ag/AgCl electrode. At the termination of the experiment, cell mass for protein and RNA extraction were obtained.
RNA and protein extraction
To ensure no degradation of biomolecules occurred, the bioleaching vessels were allowed to settle for 5 min, before the supernatant (75 mL) was removed and immediately mixed with 75 mL ice cold sterile MAC medium. The resulting mixture was then centrifuged (20 min, 12,500 g, and 4 °C). The cell pellets were washed twice by re-suspending in sterile, ice-cold MAC medium, flash frozen in liquid nitrogen, and used for biomolecular extractions according to Roume et al.24 with the alteration that metabolites were not extracted. RNA samples were sequenced at Science for Life Laboratory (Stockholm, Sweden) while the precipitated protein fraction was analyzed by mass spectrometry.
RNA sequencing and transcript analysis
The Illumina TruSeq Stranded mRNA kit was used for rRNA depletion and library preparation. RNA sequencing reads with an average length of 126 bases were obtained by Illumina HiSeq2500. Raw reads were filtered with Trimmomatic v0.3225 and aligned to a concatenation of the three reference genomes (A. caldus DSM8584: GCF_000175575.2; S. thermosulfidooxidans DSM 9293: GCF_900176145.1; L. ferriphilum DSM 14647: GCF_900198525.1) using Bowtie-2 v2.3.226. Sequencing reads that mapped to protein coding sequences were quantified with FeatureCounts of the subread package v1.5.127 and expressed as transcripts per million (TPM) for the A. caldus, S. thermosulfidooxidans, and L. ferriphilum genomes, respectively. Similarly, to compare samples of different compositions, read counts were normalized per reference genome using DESeq 2 v1.16.128 and compared accordingly to obtain log2-fold changes (Log2FC)29.
Proteomics and protein identification
A total of five protein extracts from the continuous culture and batch experiments were precipitated in acetone, dried, and dissolved in 20 μL of 6 M urea - 2 M thiourea by vortexing. Cysteines reduction was achieved by 30 min incubation at room temperature with 1 μL of 1 M dithiothreitol, followed by 20 min alkylation with 1 μL of 550 mM iodoacetamide in the dark. Proteins were digested at room temperature for 3 h with lysyl endopeptidase (Wako) at a protease/protein ratio of 1:100, thereupon urea was diluted to 2 M with 50 mM ammonium bicarbonate and digestion continued with sequencing grade trypsin (Promega) for 12 h at room temperature at a protease/protein ratio of 1:100. The resulting peptides were extracted from the gel with acetonitrile and stored on stop-and-go extraction (STAGE) tips prior to mass spectrometry30.
Mass spectra were recorded with Xcalibur software 3.1.66.10 (Thermo Scientific). The continuous culture samples were analyzed with an EASY-nLC 1000 liquid chromatography (LC) system (Thermo Scientific) and a Q-Exactive HF mass spectrometer (Thermo Scientific), as described previously31. Mineral culture samples were analyzed by using a nanoACQUITY gradient ultraperformance liquid chromatography (UPLC) pump system (Waters, Milford, MA, USA) coupled to an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). An UPLC HSS T3 M-class column (1.8 μm, 75 μm by 150 mm; Waters, Milford, MA, USA) and an UPLC Symmetry C 18 trapping column (5 μm, 180 μm by 20 mm; Waters, Milford, MA, USA) were used for LC in combination with a PicoTip emitter (SilicaTip, 10 μm internal diameter [i.d.]; New Objective, Woburn, MA, USA). To elute the peptides, a linear gradient was employed with increasing concentrations of buffer B (0.1% formic acid in acetonitrile [ULC/MS grade]; Biosolve, Netherlands) from 1% to 95% within 166.5 min, followed by a linear gradient from 1% acetonitrile within 13.5 min (1% buffer B from 0 to 10 min, 5% buffer B from 10 to 161 min, 40% buffer B from 161 to 161.5 min, 85% buffer B from 161.5 to 166.5 min, 95% buffer B from 166.5 to 167.1 min, and 1% buffer B from 167.1 to 180 min) at a flow rate of 400 nL min−1 and a spray voltage of 1.5 to 1.8 kV. Finally, 2% buffer B was used to re-equilibrate the column in 15 min using an oven set to 55 °C and a heated desolvation capillary set to 275 °C. Xcalibur (Rev.2.1.0) was used to operate the LTQ Orbitrap Elite mass spectrometer via its instrument method files in the positive-ion mode. Linear ion trap and Orbitrap instruments were operated in parallel in which a full Orbitrap MS scan detected tandem MS (MS/MS) spectra of the ten most intense precursors, from the most to least intense, in the range of 150 to 2,000 m/z at a resolution of 60,000. The relative collision energy for rapid collision-induced dissociation (rCID) was set to 35% using dynamic exclusion with a repeat count of 1 and a 45-s exclusion duration window. Singly and unknown charged ions were rejected for MS/MS and the corresponding mass spectra recorded with Xcalibur software 2.2 SP1.48 (Thermo Scientific).
The Andromeda software32 was used to identify proteins from mineral and continuous cultures, and MaxQuant 1.5.3.17531 label-free protein quantifications (LFQ) algorithm33 used for quantification. The genomes of the three bacteria (see above) were used for protein identification based on the corresponding FASTA files. After quantification, the Perseus software v1.5.8.534 was used to discard rows with <2 values of either condition (mineral or continuous), and to compare the intensities with two-sample Welch’s t test.
Microscopy imaging
Mineral sample preparation for microscopy imaging
Chalcopyrite flotation concentrate obtained from Boliden AB, Sweden was wet sieved (Retsch, Germany) to obtain the 50–100 μm particle size fraction. The resulting mineral grains were washed twice for 30 min in 10 volumes of a solution of 0.1 M EDTA and 0.4 M NaOH, stirred twice in approximately five volumes of acetone for 30 min to remove soluble sulfur compounds13, and finally sealed and heated in a nitrogen atmosphere for 10 h at 125 °C.
Microbial species cultivation for microscopy imaging
Epifluorescence microscopy (EFM) pictures were taken of biofilms formed by the three bacterial strains used for transcriptomics and proteomics. A. caldus, S. thermosulfidooxidans, and L. ferriphilum were cultured in sterile MAC medium with soluble electron donors for inoculation of chalcopyrite cultures as described below. For L. ferriphilum, 4 g/L iron(II)-ions were provided as FeSO4 · 7H2O and the pH was adjusted to pH 1.6–1.8 with H2SO4 to prevent precipitation of the electron donor. A. caldus and S. thermosulfidooxidans were pre-cultured using 0.9 g/L potassium tetrathionate (K2S4O6) with the further addition of 0.02% yeast extract (YE) and 0.1 g/L iron(II)-ions for S. thermosulfidooxidans11. Cultivation proceeded until reaching stationary growth phase after 4–5 days of incubation. Cells were harvested by centrifugation at 11,270 g for 10 min, washed in sterile medium, and used to inoculate chalcopyrite-containing Erlenmeyer flasks (150 mL MAC medium and 2% (wt/vol) chalcopyrite grains of size 50–100 μm) at an initial cell density of 107 cells/mL (in equal proportions for mixed cultures). Mineral cultures were grown for a different time span of 1, 3, 7, 14, and 21 days after inoculation.
Microscopy sample preparation
Fixation of mineral-attached cells was achieved by transferring about 25 mg of mineral material from the cultures to 1 mL sterile MAC medium at pH 1.8 with 4% formaldehyde and incubation at room temperature for 1 h. The samples were then washed twice with water followed by a single wash with 1 mL phosphate-buffered saline (PBS) and frozen at −20 °C in 50% ethanol in PBS until ready to be examined. For visualization, the mineral particles were incubated in 200 μL 0.01% 4′,6-diamidine-2′-phenylindole dihydrochloride in 2% formaldehyde. The mineral grains were then washed with 1 mL PBS before and after staining mineral-attached cells, and mounted on 10-well diagnostic glass slides (10-well, 6.7 mm; Thermo Scientific) using a glycerol-based mounting medium (CitiFluor AF2) and covered with glass coverslips14. Observations of biofilms before and after fixation did not show any disruptive effects of the fixation.
High-throughput epifluorescence microscopy
An EFM platform AxioImager M2m (Zeiss) with a motorized microscopy stage (IM SCAN 130 × 85 - DC 1 mm, Märzhäuser Wetzlar) and an AxioCam MRm camera was used to automate image acquisition in order to generate sets of images for different acidophile microbial cultures. The individual images of stained microbes on the mineral grains were recorded using a Zeiss Plan-Neofluar (20 × 0.50) objective and corresponded to an imaged area of 450 × 335 μm. The obtained images were taken with a 2-μm step size and stacked to cover the entire maximum grain depth of 100 μm (i.e., a total of 50 layers). The extended-focus module of the Zen 2 software (blue edition, 2011; Carl Zeiss GmbH) was used to calculate projection images using the Wavelet option and the resulting projections were exported as JPEG files. For each mineral sample and time point, quantification of the cells on the mineral surfaces used a minimum of 36 images. Images were taken at days 1, 3, 7, 14, and 21 (Fig. 2).
Fig. 2.

Example of EFM images of a mixed microbial community composed of L. ferriphilum, S. thermosulfidooxidans, and A. caldus taken at different time points.
Data Records
The raw reads of the RNAseq data for 12 continuous culture samples and 49 bioleaching samples were deposited at ENA35–40. Proteomics data for three continuous culture samples and 61 bioleaching samples was deposited at the PRIDE database41–53. A per-sample overview of the available omics data with the respective accessions can be found in table “omics samples” at the FAIRDOMHub54 repository “SysMetEx – Dataset collection”55, in which raw omics data are summarized in a structured format (Fig. 3). Sample identifiers provide information, such as laboratory of origin, inoculated strains, and inoculum size, cellular fraction, as well as run time of the cultures. Additional information to decode the contained data is also provided in the same repository. The number of samples for which RNAseq or proteomics data was generated is summarized in Table 1.
Fig. 3.
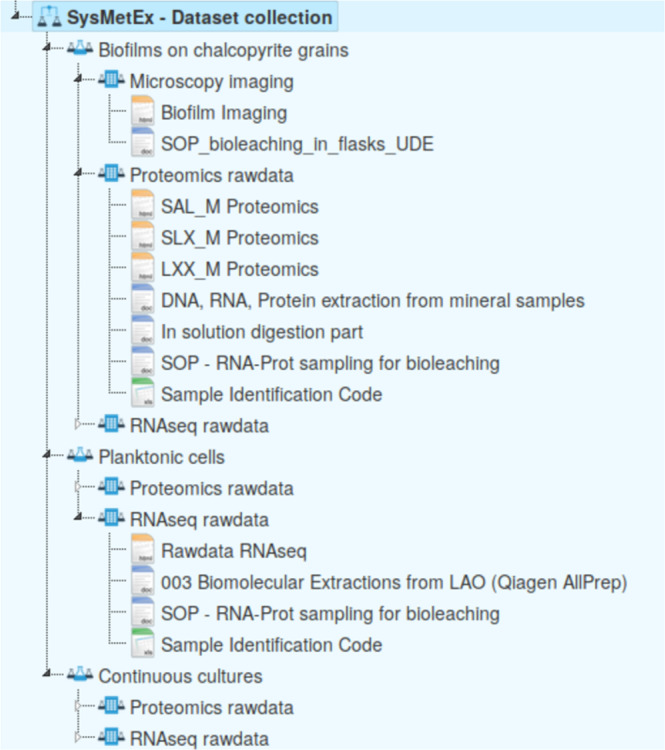
Overview of the FAIRDOMHub raw data repository. Raw data files are accessible for each cellular fraction, i.e., biofilm on mineral particles, planktonic cells from leaching experiments, and continuous culture samples alongside standard operating procedures applied when processing the tables. Links to the raw data repositories for the respective data types are provided.
Technical Validation
Cell pellets from the bioleaching experiments were sampled aseptically and changes in nucleic acid and protein immediately inhibited by rapid cooling followed by flash freezing in liquid nitrogen (as described in the methods). All samples were shipped on dry ice to ensure the samples remained frozen. Biomolecular extractions were performed according to standard procedures following best practices to avoid RNA degradation and contamination. Quality and concentration of extracted biomolecules were checked before proceeding to library preparation for RNAseq or proteomics analyses (see methods).
Quality of sequencing reads for the RNAseq data was assessed with FastQC and summarized across all samples with multiQC56. Some samples failed the FastQC check for quality scores. However, after preprocessing, all reads showed high quality scores (FAIRDOMHub repository “SysMetEx – Dataset collection”55, files “qc raw reads” and “qc processed reads”) and successful adapter removal. Overall read duplication levels might appear excessively elevated. However, this is likely due to incomplete rRNA removal. Especially for the continuous culture samples, for which no rRNA removal was performed and duplicate read counts constitute a large portion of the total raw reads. This is also reflected in reads mapping to reference genomes. These samples show an elevated number of multi-mapping reads in contrast to depleted bioleaching samples (Fig. 4). Overall, the amount of reads mapping to the reference genomes provides sufficient depth for detailed analyses with 7 to 35 million reads assigned to the reference protein coding genes in bioleaching samples (FAIRDOMHub repository “SysMetEx – Dataset collection”55, file “mapping statistics”).
Fig. 4.

Summary of mapping files. Preprocessed RNAseq reads were mapped to a concatenation of the three reference genomes. The resulting alignments were summarized and classified with samtools flagstat indicated by bar color. The plot is an excerpt of the multiqc report for the mapping files and featurecount results (FAIRDOMHub repository “SysMetEx – Dataset collection”55, file “mapping statistics”). A description corresponding to the sample names on the left is found in the FAIRDOMHub repository (“SysMetEx – Dataset collection”55, “Sample Identification Code”).
Quality of proteome data was assessed using MaxQuant output. First, the identification rate of MS/MS spectra was surveyed (Table 2). The identification rates varied between conditions, without clear bias towards any of them. The average identification rate was 22%, which is in the typical range for high-throughput proteomics using CID as fragmentation technique. As in shotgun proteomics, peptide sequences are used to infer protein presence and abundance and a common problem is shared peptide sequences – here, not only in one organism but up to three organisms per sample. To gauge the magnitude of shared peptides and their detrimental effect, an example proteome from a cultivation comprising all three organisms was analyzed. It can be seen (Fig. 5) that shared peptides did not severely impair protein inference. Altogether 96% of all peptides were unique. Interestingly, of the 4% shared peptides, 3% belonged to the same organism. Only about 1% and 0.2% were shared between 2 and 3 organisms, respectively. Therefore, in this study the individual proteomes of the used three organisms were dissimilar enough to avoid potential complications of inter species-shared peptides.
Fig. 5.
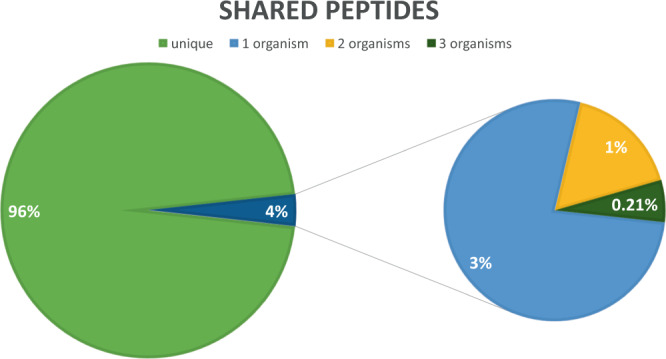
Overview of identified peptide sequences shared between two and more proteins for a cultivation condition comprising all three organisms. The 4% shared peptides were further broken down into peptides shared by proteins from 1, 2, or 3 organisms.
In this project we used a motorized EFM for automated image acquisition coupled to automated image analysis using algorithms that allowed quantification of mineral-attached cells. The evaluation of the method’s statistical accuracy depended on the number of images considered. After manual removal of extreme values, representing the top and bottom deciles of images with extremely low or high cell counts, metal sulfide colonization values (cells per mm2) of at least 36 images were used. Then, the values were randomly sorted using Microsoft Excel’s random function and grouped in four arbitrarily chosen classes with data from nine microscopy images in order to calculate the mean of each class. These four classes can be understood as four sets of equal mineral areas used for averaging of the naturally non-homogeneous mineral colonization over a larger area than that represented in a single microscopy image. The coefficient of variation was found to not exceed 16 ± 8% when at least 36 images per sample were analyzed. In order to take into account the fact that the mineral grains were viewed only from the top, the resulting values were doubled in order to account for the unobserved bottom side, while no correction factor was used for extrapolation from two-dimensional areas to the true three-dimensional mineral objects.
Acknowledgements
This project was supported by Bundesministerium für Bildung und Forschung (BMBF, 031A600A and B), Vetenskapsrådet (contract 2014-6545), the Luxembourg National Research Fund (FNR) (INTER/SYSAPP/14/05), and the Swiss Initiative in Systems Biology (SystemsX.ch, SysMetEx) under the frame of ERASysAPP. The calculations were performed on resources provided by the Swiss National Supercomputing Centre (CSCS). IVP acknowledges the support from Swiss National Science Foundation and Swiss Platform for Advanced Scientific Computing. ABD and IVP acknowledge support from the Swiss National Science Foundation grant 205321_173020. MV acknowledges support from Fondecyt 1161007 and 1190892 grants. Nucleic acids were sequenced at the National Genomics Infrastructure hosted by Science for Life Laboratory. Bioinformatic analysis employed the Uppsala Multidisciplinary Center for Advanced Computational Science resource at Uppsala University (project b2013127) and the HPC facilities of the University of Luxembourg. Open access funding provided by Linnaeus University.
Author contributions
M.D. and M.V. conceived the study. M.H. and A.B.D. performed the analysis of the data and drafted the initial manuscript. M.H., S.C., M.E.H. and S.B. generated the experimental data. M.H. carried out OMICs data preprocessing. All authors contributed to write the manuscript. All authors read and approved the final manuscript.
Code availability
The workflow utilized for quality filtering the RNAseq reads, alignment to references genomes, and counting of mapped reads has previously been described11, and can be found in the “SysMetEx – Data analysis” repository57. Reference genomes of the three strains used in the project and auxiliary files can be accessed at the “SysMetEx – Reference genomes” repository58. Proteomics data processing was carried out according to the MaxQuant parameter (FAIRDOMHub repository “SysMetEx – Dataset collection”55, file “maxquant parameters”).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Antoine Buetti-Dinh, Malte Herold, Stephan Christel, Mohamed El Hajjami, Sören Bellenberg.
Contributor Information
Antoine Buetti-Dinh, Email: antoine.buetti@lnu.se.
Malte Herold, Email: Malte.Herold@lns.etat.lu.
Mark Dopson, Email: mark.dopson@lnu.se.
References
- 1.Brierley CL, Brierley JA. Progress in bioleaching: part B: applications of microbial processes by the minerals industries. Appl. Microbiol. Biotechnol. 2013;97:7543–7552. doi: 10.1007/s00253-013-5095-3. [DOI] [PubMed] [Google Scholar]
- 2.Petersen J. Heap leaching as a key technology for recovery of values from low-grade ores – a brief overview. Hydrometallurgy. 2016;165:206–212. doi: 10.1016/j.hydromet.2015.09.001. [DOI] [Google Scholar]
- 3.Bonnefoy V, Holmes DS. Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments. Environ. Microbiol. 2012;14:1597–1611. doi: 10.1111/j.1462-2920.2011.02626.x. [DOI] [PubMed] [Google Scholar]
- 4.Vera M, Schippers A, Sand W. Progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation–part A. Appl. Microbiol. Biotechnol. 2013;97:7529–7541. doi: 10.1007/s00253-013-4954-2. [DOI] [PubMed] [Google Scholar]
- 5.Dopson M, Johnson DB. Biodiversity, metabolism and applications of acidophilic sulfur-metabolizing microorganisms. Environ. Microbiol. 2012;14:2620–2631. doi: 10.1111/j.1462-2920.2012.02749.x. [DOI] [PubMed] [Google Scholar]
- 6.Khoshkhoo M, Dopson M, Engström F, Sandström Å. New insights into the influence of redox potential on chalcopyrite leaching behaviour. Min. Engin. 2017;100:9–16. doi: 10.1016/j.mineng.2016.10.003. [DOI] [Google Scholar]
- 7.Li Y, Kawashima N, Li J, Chandra A, Gerson A. A review of the structure, and fundamental mechanisms and kinetics of the leaching of chalcopyrite. Adv. in Colloid Interface Sci. 2013;197–198:1–32. doi: 10.1016/j.cis.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Panda S, Akcil A, Pradhan N, Deveci H. Current scenario of chalcopyrite bioleaching: A review on the recent advances to its heap-leach technology. Biores. Technol. 2015;196:694–706. doi: 10.1016/j.biortech.2015.08.064. [DOI] [PubMed] [Google Scholar]
- 9.Gericke, M., Govender, Y. & Pinches, A. Tank bioleaching of low-grade chalcopyrite concentrates using redox control. Hydrometallurgy104, 414–419, 18th International Biohydrometallurgy Symposium, IBS2009, Bariloche-Argentina, 13–17 September 2009 (2010).
- 10.Masaki Y, Hirajima T, Sasaki K, Miki H, Okibe N. Microbiological redox potential control to improve the efficiency of chalcopyrite bioleaching. Geomicrobiol. 2018;35:648–656. doi: 10.1080/01490451.2018.1443170. [DOI] [Google Scholar]
- 11.Christel S, et al. Weak iron oxidation by Sulfobacillus thermosulfidooxidans maintains a favorable redox potential for chalcopyrite bioleaching. Front. Microbiol. 2018;9:3059. doi: 10.3389/fmicb.2018.03059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellenberg S, et al. Biofilm formation, communication and interactions of leaching bacteria during colonization of pyrite and sulfur surfaces. Res. Microbiol. 2014;165:773–781. doi: 10.1016/j.resmic.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Christel, S. et al. Multi-omics reveal the lifestyle of the acidophilic, mineral-oxidizing model species Leptospirillum ferriphilumT. Appl. Environ. Microbiol. 84, e02091-17 (2017). [DOI] [PMC free article] [PubMed]
- 14.Bellenberg, S. et al. Automated microscopic analysis of metal sulfide colonization by acidophilic microorganisms. Appl. and Environ. Microbiol.84, e01835-18 (2018). [DOI] [PMC free article] [PubMed]
- 15.Buetti-Dinh A, et al. Deep neural networks outperform human expert’s capacity in characterizing bioleaching bacterial biofilm composition. Biotechnol. Rep. 2019;22:e00321. doi: 10.1016/j.btre.2019.e00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buetti-Dinh, A. et al. Reverse engineering directed gene regulatory networks from transcriptomics and proteomics data of biomining bacterial communities with approximate bayesian computation and steady-state signalling simulations. BMC Bioinformatics21, 23 (2020). [DOI] [PMC free article] [PubMed]
- 17.Forssberg, K. S. E. Flotation of sulphide minerals/edited by Eric Forssberg, K. S. (Elsevier; Distributors for the U.S. and Canada, Elsevier Science Pub. Co Amsterdam; New York: New York, N.Y, 1985).
- 18.Clarke P, Fornasiero D, Ralston J, Smart R. A study of the removal of oxidation products from sulfide mineral surfaces. Min. Engin. 1995;8:1347–1357. doi: 10.1016/0892-6875(95)00101-U. [DOI] [Google Scholar]
- 19.Dopson M, Sundkvist J-E, Lindström EB. Toxicity of metal extraction and flotation chemicals to Sulfolobus metallicus and chalcopyrite bioleaching. Hydrometallurgy. 2006;81:205–213. doi: 10.1016/j.hydromet.2005.12.005. [DOI] [Google Scholar]
- 20.Coram NJ, Rawlings DE. Molecular relationship between two groups of the genus Leptospirillum and the finding that Leptospirillum ferriphilum sp. nov. dominates South African commercial biooxidation tanks that operate at 40 degrees C. Appl. Environ. Microbiol. 2002;68:838–845. doi: 10.1128/AEM.68.2.838-845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golovacheva RS, Karavaiko GI. Sulfobacillus, a new genus of thermophilic sporulating bacteria. Mikrobiologiia. 1978;47:815–822. [PubMed] [Google Scholar]
- 22.Hallberg KB, Lindstrom EB. Characterization of Thiobacillus caldus sp. nov., a moderately thermophilic acidophile. Microbiology (Reading, Engl.) 1994;140:3451–3456. doi: 10.1099/13500872-140-12-3451. [DOI] [PubMed] [Google Scholar]
- 23.Mackintosh E. M. Nitrogen fixation by Thiobacillus ferrooxidans. Microbiology. 1978;105:215–218. [Google Scholar]
- 24.Roume H, et al. A biomolecular isolation framework for eco-systems biology. ISME J. 2013;7:110–121. doi: 10.1038/ismej.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq 2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klingenberg H, Meinicke P. How to normalize metatranscriptomic count data for differential expression analysis. PeerJ. 2017;5:e3859. doi: 10.7717/peerj.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 31.Kaur H, et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 2016;118:1906–1917. doi: 10.1161/CIRCRESAHA.116.308643. [DOI] [PubMed] [Google Scholar]
- 32.Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 33.Cox J, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 35.Leinonen R, et al. The European Nucleotide Archive. Nucleic Acids Res. 2011;39:28–31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buetti-Dinh A, 2018. RNAseq SysMetEx, mixed and single species cultures European Nucleotide Archive. PRJEB30001
- 37.Buetti-Dinh A, 2018. RNAseq of binary cultures of S. thermosulfidooxidans and A. caldus with varied inoculum sizing. European Nucleotide Archive. PRJEB29980
- 38.Bellenberg S, 2018. RNAseq of chalcopyrite leaching cultures for L.ferriphilum and L.ferriphilum + S.thermosulfidooxidans cultures. European Nucleotide Archive. PRJEB27815
- 39.Christel S, 2018. RNAseq of binary and tertiary mixtures of microorganisms in chalcopyrite bioleaching. European Nucleotide Archive. PRJEB27534
- 40.Christel S, 2017. RNA sequencing for L. ferriphilum cultures. European Nucleotide Archive. PRJEB21842
- 41.Perez-Riverol Y, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buetti-Dinh A, 2020. Sysmetex proteome data comparison for ASX and LSX planktonic growth. PRIDE Archive. PXD011502
- 43.Buetti-Dinh A, 2020. Sysmetex proteome data for SAL mineral growth. PRIDE Archive. PXD011503
- 44.Buetti-Dinh A, 2020. Sysmetex proteome data for SLX mineral growth. PRIDE Archive. PXD011504
- 45.Buetti-Dinh A, 2020. Sysmetex proteome data for SAX planktonic growth. PRIDE Archive. PXD011506
- 46.Buetti-Dinh A, 2020. Sysmetex proteome data for Acidithiobacillus caldus in continuous culture. PRIDE Archive. PXD011507
- 47.Buetti-Dinh A, 2020. Sysmetex proteome data for AAS. PRIDE Archive. PXD011508
- 48.Buetti-Dinh A, 2020. Sysmetex proteome data for ASS. PRIDE Archive. PXD011509
- 49.Buetti-Dinh A, 2020. Sysmetex proteome data for SAL planktonic growth. PRIDE Archive. PXD011540
- 50.Buetti-Dinh A, 2020. Sysmetex proteome data for Acidithiobacillus caldus planktonic growth. PRIDE Archive. PXD011863
- 51.Buetti-Dinh A, 2020. Sysmetex proteome data for Sulfobacillus thermosulfidooxidans planktonic growth. PRIDE Archive. PXD011921
- 52.Buetti-Dinh A, 2020. Sysmetex proteome data for Leptospirillum ferriphilum mineral growth. PRIDE Archive. PXD012780
- 53.Buetti-Dinh A, 2020. Sysmetex proteome data for Leptospirillum ferriphilum planktonic growth. PRIDE Archive. PXD012783
- 54.Wolstencroft K, et al. FAIRDOMHub: a repository and collaboration environment for sharing systems biology research. Nucleic Acids Res. 2017;45:D404–D407. doi: 10.1093/nar/gkw1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herold, M. SysMetEx – Dataset collection. FAIRDOMHub, 10.15490/fairdomhub.1.investigation.292.2 (2020).
- 56.Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.SysMetEx – Data analysis, https://git-r3lab.uni.lu/malte.herold/sysmetex_data_analysis.
- 58.SysMetEx – Reference genomes, https://git-r3lab.uni.lu/malte.herold/SysMetEx_ReferenceGenomes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Buetti-Dinh A, 2018. RNAseq SysMetEx, mixed and single species cultures European Nucleotide Archive. PRJEB30001
- Buetti-Dinh A, 2018. RNAseq of binary cultures of S. thermosulfidooxidans and A. caldus with varied inoculum sizing. European Nucleotide Archive. PRJEB29980
- Bellenberg S, 2018. RNAseq of chalcopyrite leaching cultures for L.ferriphilum and L.ferriphilum + S.thermosulfidooxidans cultures. European Nucleotide Archive. PRJEB27815
- Christel S, 2018. RNAseq of binary and tertiary mixtures of microorganisms in chalcopyrite bioleaching. European Nucleotide Archive. PRJEB27534
- Christel S, 2017. RNA sequencing for L. ferriphilum cultures. European Nucleotide Archive. PRJEB21842
- Buetti-Dinh A, 2020. Sysmetex proteome data comparison for ASX and LSX planktonic growth. PRIDE Archive. PXD011502
- Buetti-Dinh A, 2020. Sysmetex proteome data for SAL mineral growth. PRIDE Archive. PXD011503
- Buetti-Dinh A, 2020. Sysmetex proteome data for SLX mineral growth. PRIDE Archive. PXD011504
- Buetti-Dinh A, 2020. Sysmetex proteome data for SAX planktonic growth. PRIDE Archive. PXD011506
- Buetti-Dinh A, 2020. Sysmetex proteome data for Acidithiobacillus caldus in continuous culture. PRIDE Archive. PXD011507
- Buetti-Dinh A, 2020. Sysmetex proteome data for AAS. PRIDE Archive. PXD011508
- Buetti-Dinh A, 2020. Sysmetex proteome data for ASS. PRIDE Archive. PXD011509
- Buetti-Dinh A, 2020. Sysmetex proteome data for SAL planktonic growth. PRIDE Archive. PXD011540
- Buetti-Dinh A, 2020. Sysmetex proteome data for Acidithiobacillus caldus planktonic growth. PRIDE Archive. PXD011863
- Buetti-Dinh A, 2020. Sysmetex proteome data for Sulfobacillus thermosulfidooxidans planktonic growth. PRIDE Archive. PXD011921
- Buetti-Dinh A, 2020. Sysmetex proteome data for Leptospirillum ferriphilum mineral growth. PRIDE Archive. PXD012780
- Buetti-Dinh A, 2020. Sysmetex proteome data for Leptospirillum ferriphilum planktonic growth. PRIDE Archive. PXD012783
Data Availability Statement
The workflow utilized for quality filtering the RNAseq reads, alignment to references genomes, and counting of mapped reads has previously been described11, and can be found in the “SysMetEx – Data analysis” repository57. Reference genomes of the three strains used in the project and auxiliary files can be accessed at the “SysMetEx – Reference genomes” repository58. Proteomics data processing was carried out according to the MaxQuant parameter (FAIRDOMHub repository “SysMetEx – Dataset collection”55, file “maxquant parameters”).