

Abstract
Aporphine alkaloids containing a C10 nitrogen motif were synthesized and evaluated for affinity at 5-HT1AR, 5-HT2AR, 5-HT6R and 5-HT7AR. Three series of racemic aporphines were investigated: 1,2,10-trisubstituted, C10 N-monosubstituted and compounds containing a C10 benzofused aminothiazole moiety. The 1,2,10-trisubstituted series of compounds as a group displayed modest selectivity for 5-HT7AR and also had moderate 5-HT7AR affinity. Compounds from the C10 N-monosubstituted series generally lacked affinity for 5-HT2AR and 5-HT6R and showed strong affinity for 5-HT1A or 5-HT7AR. Compounds in this series that contained an N6-methyl group were up to 27-fold selective for 5-HT7AR over 5-HT1AR, whereas compounds with an N6-propyl substituent showed a reversal in this selectivity. The C10 benzofused aminothiazole analogues showed a similar binding profile as the C10 N-monosubstituted series i.e. strong affinity for 5-HT1AR or 5-HT7AR, with selectivity between the two receptors being similarly influenced by N6-methyl or N6-propyl substituents. Compounds 29 and 34a exhibit high 5-HT7AR affinity, excellent selectivity versus dopamine receptors and function as antagonists in 5-HT7AR cAMP-based assays. Compounds 29 and 34a have been identified as new lead molecules for further tool and pharmaceutical optimization.
Keywords: 5-HT7, 5-HT1A, aporphine, serotonin, dopamine, CNS, SAR
Graphical Abstract
1. Introduction
The neurotransmitter serotonin (5-HT) is involved in numerous physiological functions. Seven 5-HT receptors types are known (5-HT1R – 5-HT7R) of which all except 5-HT3R (a ligand-gated ion channel) are metabotropic G-protein coupled receptors (GPCRs). In all, there are at least 14 5-HT receptors owing to the occurrence of sub-types for some of the receptors. The 5-HT7R is the most recently described serotonergic receptor [1–3] and it is involved in various pathophysiological functions such as regulation of circadian rhythm, body temperature and sleep. [4–6] There are four known 5-HT7R isoforms: 5-HT7AR, 5-HT7BR and 5-HT7CR isoforms are found in rats whereas 5-HT7AR, 5-HT7BR and 5-HT7DR are present in humans [7]. The 5-HT7AR is a full-length receptor comprising 445 amino acid residues, while the 5-HT7BR is a truncated variant with 432 amino acid residues. The 5-HT7DR is a distinct isoform containing 479 amino acid residues. The 5-HT7AR is the most abundant of the human isoforms. 5-HT7Rs signal via Gαs; agonist activation of 5-HT7Rs leads to adenylate cyclase activation and cAMP production. [8]
5-HT7R antagonists appear to be useful agents to remediate neuropsychiatric conditions including obsessive-compulsive disorder, schizophrenia, anxiety, depression and substance abuse. [9–12] Agonists of the 5-HT7R are able to restore deficient locomotory activity and may be useful as therapeutics for spinal cord injury, amyotrophic lateral sclerosis and sleep apnea. The role of 5-HT7R in the regulation of nociception, mitogenesis, gastrointestinal disorders, cardiopulmonary disorders and cognition has been less clear as both agonists and antagonists have shown positive outcomes in animal studies.[11]
A number of scaffolds have yielded potent 5-HT7R ligands.[9] Among these are the so-called “long chain” arylpiperazines (e.g. 1, Fig. 1), arylsulfonamides (e.g. 2), biarylalkylamines (e.g. 3), azepines (e.g. 4) and aminotriazines (e.g. 5). However, selectivity of 5-HT7R ligands especially versus the closely related 5-HT1A receptor has been challenging. The 5-HT7R antagonist JNJ-18038683 (4) is currently undergoing clinical trials for the treatment of cognitive impairment in bipolar disorder.[9] JNJ-1803863 is the only selective 5-HT7R ligand that has been evaluated in clinical trials. The discovery of new potent and selective 5-HT7R ligands promises to expand our armamentarium of useful pharmacological tools and drugs pertinent to various neuropsychiatric disorders. Thus, the identification of new 5-HT7R ligands remains an area of current interest.
Figure 1.
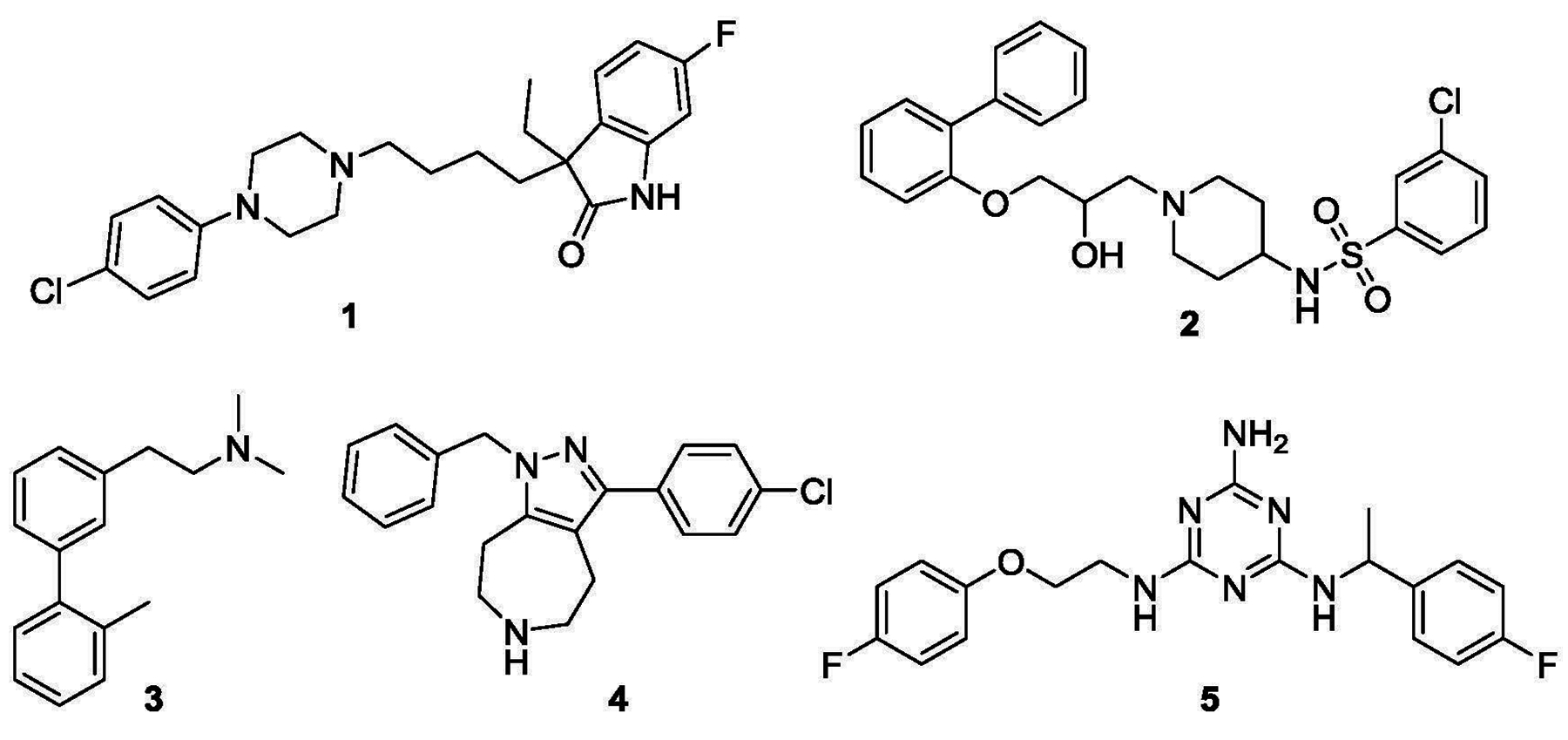
Structures of typical 5-HT7R ligands with “long chain” arylpiperazine (1), arylsulfonamide (2), biarylalkylamine (3), azepine (4, JNJ-18038683) and aminotriazine (5) scaffolds.
Aporphine alkaloids are known to have affinity across a range of serotonergic and dopaminergic receptors. In particular, structure-affinity relationship studies on aporphines have resulted in the identification of potent and selective ligands for serotonin 5-HT1AR [13–16] and dopamine D2R [17, 18] as well as compounds with interesting multi-receptor profiles [19–22]. In contrast to their evaluation as 5-HT1A receptor ligands, the evaluation of aporphines as 5-HT7R ligands has been little studied. In that regard, Johannson et al have described a number of atropisomeric 11-phenyl aporphines and 1,11-methylene aporphines (e.g. compounds 6 and 7, Fig. 2) with 5-HT7R receptor potency and selectivity versus the 5-HT1AR. [23, 24] Recently we conducted an SAR study on the aporphine alkaloid N- methyllaurotetanine and identified a number of compounds with moderate 5-HT7R affinity and selectivity. (e.g. compound 8, Figure 2). [25]
Figure 2.
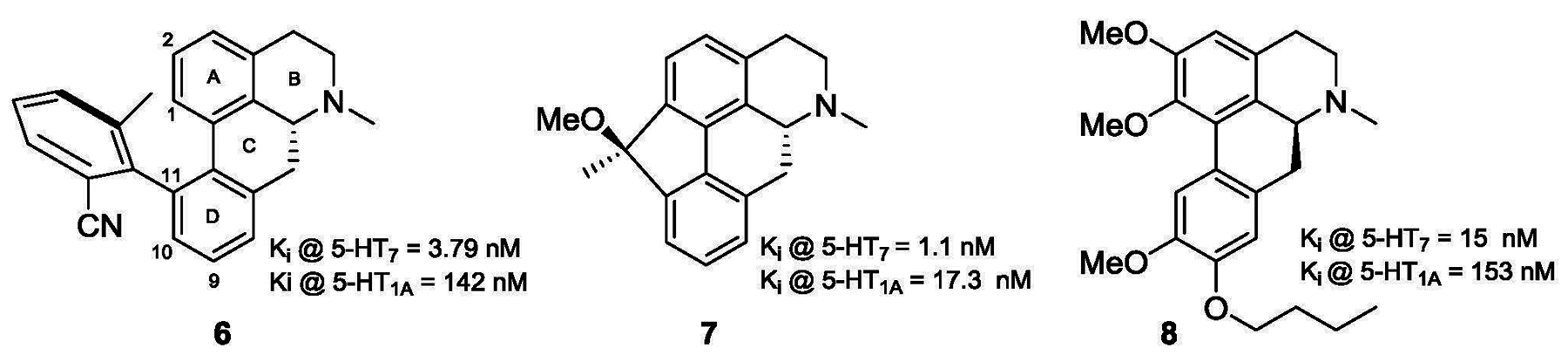
Structures of aporphine 5-HT7 receptor ligands
We regard the aporphine template as a privileged scaffold for the discovery of new CNS receptor targeting agents and envisage that structural modification of the aporphine core may lead to ligands that selectively target individual or multiple CNS receptors. Although numerous SAR studies have been performed on aporphines in relation to their affinity and activity at CNS receptors, there have been no previous SAR studies examining the effect of a C-10 nitrogen substituent in this context. Thus, in this exploratory SAR study, we were curious to determine the extent to which C10 nitrogen substituents would impact affinity and selectivity of the scaffold across a range of serotonin receptors, including 5-HT7R. In this manuscript we describe the synthesis and pharmacological evaluation of novel aporphines that bear a C10 nitrogen substituent group at a subset of 5-HT receptors which has led to the identification of novel selective 5-HT7R ligands. In addition, we describe computational docking experiments that shed light on the observed 5-HT7R affinity of the newly identified ligands.
2. Results and discussion
Synthesis
As mentioned earlier in this study we sought to examine compounds that contain 1,2,10-trisubstituted and 10-substituted patterns on the racemic aporphine core. Compounds with 9,10- and 10,11- benzo-fused aminothiazole moieties were also targeted for synthesis and evaluation as these represented cyclized C10 nitrogen motifs. Additionally, we were interested in examining the impacts of N-methyl and N-propyl variations on 5-HT7R affinity and selectivity of the aporphines as such N-substituent groups have featured prominently in the selectivity of aporphines at other CNS receptor targets. [17, 18, 26, 27]
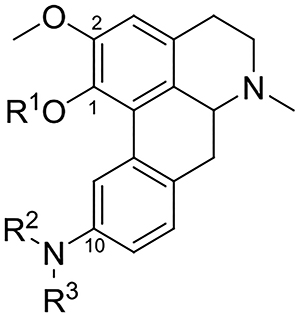
The synthesis of compounds bearing 1,2,10-trisubstituted and 10-monosubstituted patterns required the preparation of the corresponding C10 aniline substrates. These aniline precursors were prepared as we have previously described (in a study evaluating the activity of C10 nitro-, amide- and aniline-substituted aporphine analogues at dopamine receptors).[28] Synthesis of the 1,2,10-trisubstituted analogues was accomplished by treatment of the aniline precursors 9a-e with various carboxylic anhydrides, methanesulfonic anhydride and potassium cyanate to give the corresponding amides (11-14), sulfonamides (15-18) and urea analogues (19-21) respectively (Scheme 1). Reductive methylation of aniline precursors gave the N,N-dimethylated analogues (22-24). In a similar fashion to synthesis of the 1,2,10-trisubstituted analogues, aniline precursors 10a and 10b were transformed into the corresponding 10-monosubstituted amide, sulfonamide, urea and N,N-dimethylated analogues 25–26, 27–28, 29–30 and 31–32 respectively.
Scheme 1. Reagents and conditions:
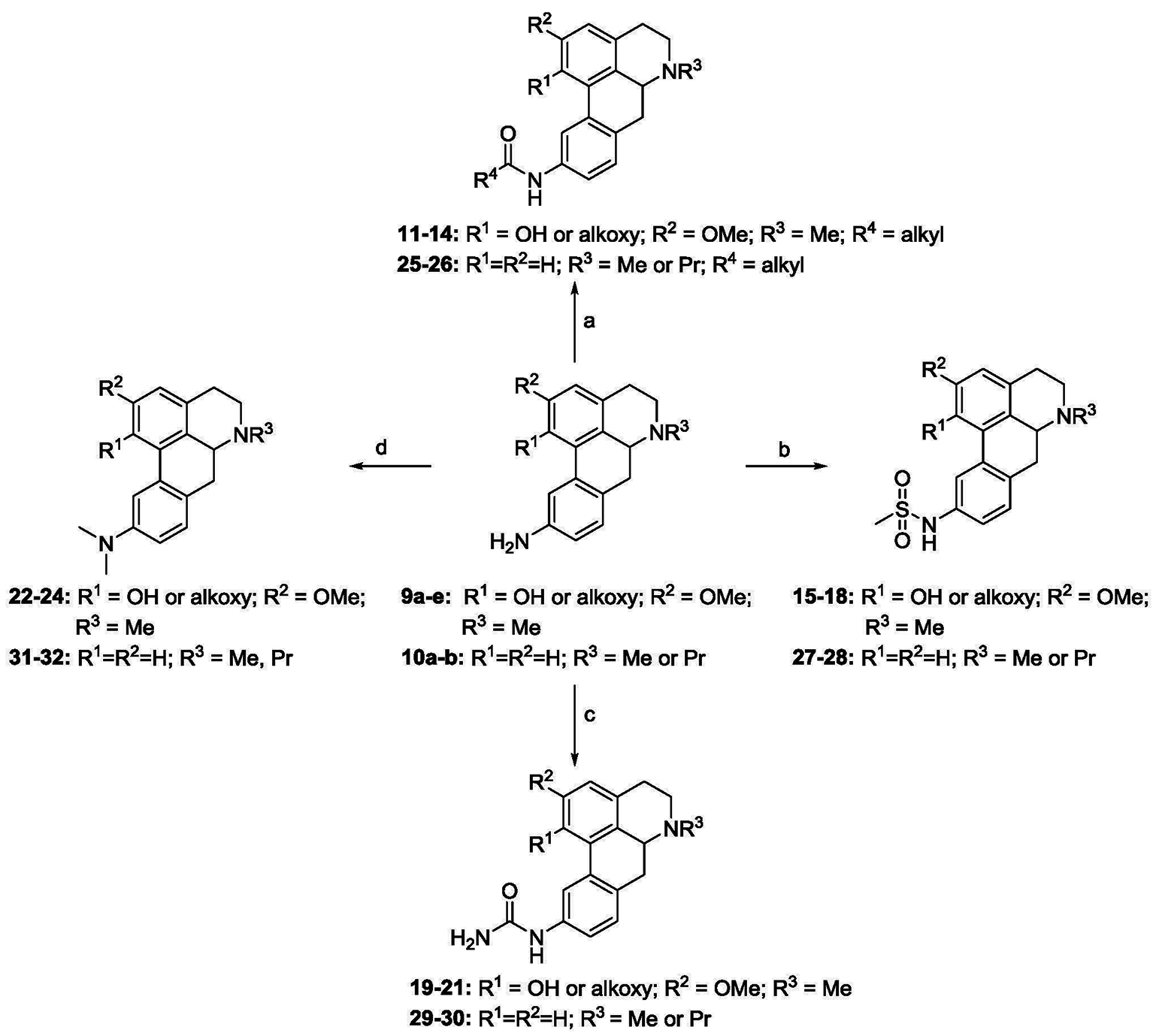
(a) corresponding anhydride, triethylamine, DCM, 2h-14h, rt, 37–73%; (b) methanesulfonic anhydride, triethylamine, DCM, rt, 14h, 33–48%; (c) potassium cyanate, acetic acid, water, 1h, rt, 30–86%; (d) formaldehyde, NaBH(OAc)3, DCM, rt, 18h, 30–63%
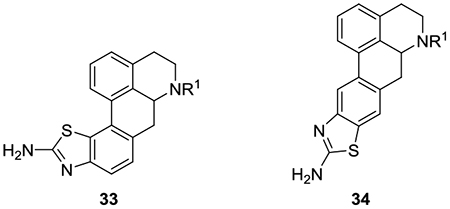
Compounds 10a and 10b were reacted with potassium thiocyanate and bromine to afford the C10/C11 benzofused aminothiazole derivatives 33a and 33b and their respective C9/C10 regioisomers 34a and 34b (Scheme 2). In each case, the pair of regioisomers was present in an approximately 1:1 ratio after the reaction (as determined by 1H NMR). The tedious purification of each pair of regioisomers was accomplished by prep TLC. The 9/10 regioisomers were slightly more polar than their corresponding 10/11 congeners.
Scheme 2. Reagents and conditions:
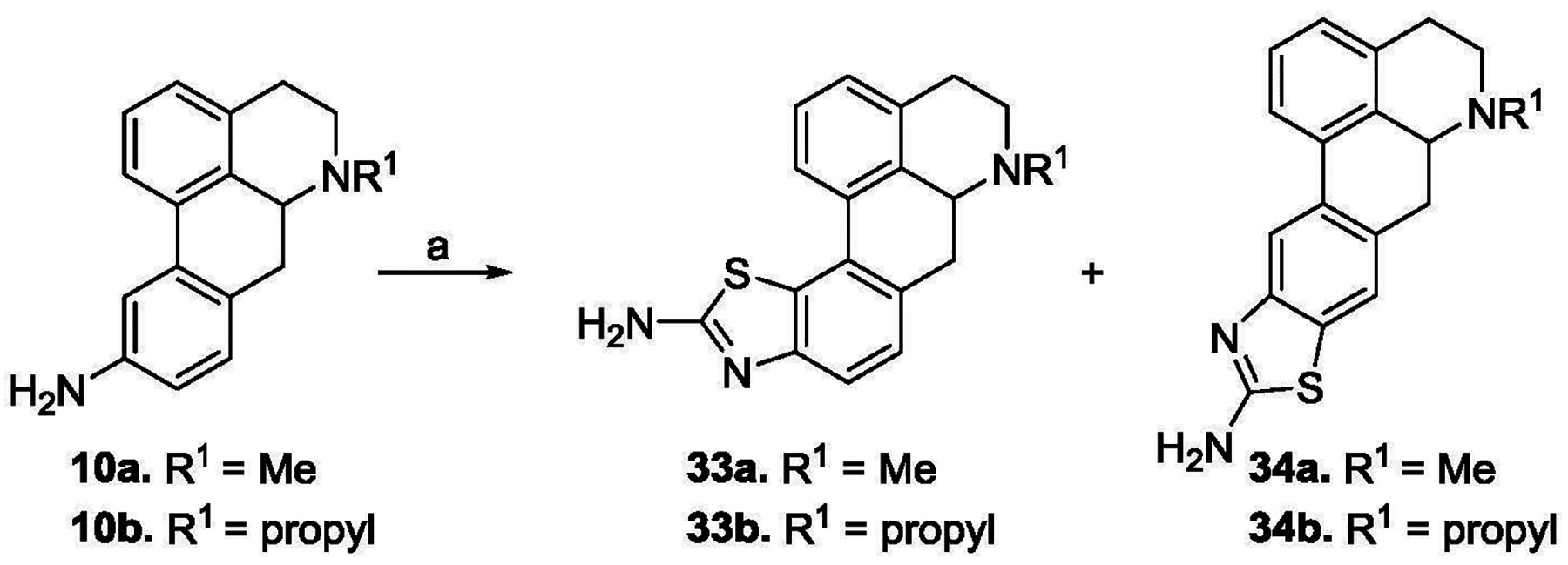
KSCN, Br2, acetic acid, 0 °C - rt, 12h, 30–35%
Biological Evaluation
Table 1 summarizes data from evaluation of compounds in the 1,2,10-trisubstituted series of compounds. As a group, the analogues in Table 1 showed low affinity for the 5-HT receptors evaluated. The highest affinity observed at the 5-HT1AR was for compound 16 (385 nM). Compounds with C1 methoxy groups in tandem with C10 methanesulfonamide or urea groups were poorly tolerated for affinity at the 5-HT1AR (i.e. compounds 15 and 19 – no affinity in a primary assay). Compounds with a C10 butanamide group (e.g. 11, 12b, 13, 14) or a C10 methanesulfonamide group (e.g. 16-18) show slightly better affinity for the 5-HT1AR than compounds with a C10 urea motif (e.g. 19-21). A similar situation ensued at the 5-HT2AR as was evident from the lack of affinity of compounds 15 and 19 for the 5-HT2AR. For compounds with C10 amide, sulfonamide or urea groups, C1 allyl or cyclopropylmethyl groups are better tolerated than C1 propargyloxy groups (e.g. compare 13 versus 14; 16 versus 18; 20 versus 21). Whereas a C1 methyl group was not beneficial for 5-HT1AR and 5-HT2AR affinity, affinity for the 5-HT6R was maintained (e.g. compound 15, 86 nM at 5-HT6R; no affinity at 5-HT1AR and 5-HT2AR). Although the affinities of the compounds for the 5-HT7AR was moderate, in most cases individual compounds displayed higher affinity for the 5-HT7AR than for the other receptors tested indicating selectivity of the group of compounds on a whole for the 5-HT7AR.
Table 1.
Affinity of 1,2,10-trisubstituted analogues at 5-HT receptors
 |
|||||||
|---|---|---|---|---|---|---|---|
| Ki (nM)a | |||||||
| Cmpd. # | R1 | R2 | R3 | 5-HT1ARb | 5-HT2ARc | 5-HT6Rd | 5-HT7ARe |
| 9c |  |
H | H | naf | 110±14 | 132±17 | 104±13 |
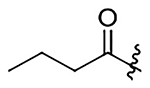
| 11 | Me |  |
H | 469±61 | 417±54 | 356±46 | 67±8.6 |
| 12a | 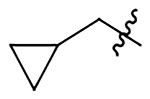 |
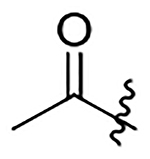 |
H | 1540±200 | 974±130 | 1500±190 | 636±82 |
| 12b | 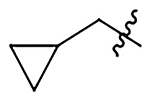 |
 |
H | 429±55 | 665±86 | 322±42 | 395±51 |
| 13 | 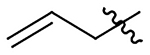 |
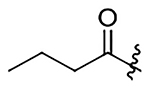 |
H | 463±60 | 443±57 | 198±26 | 188±24 |
| 14 | 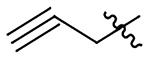 |
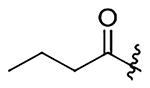 |
H | 661±85 | 1038±130 | 354±46 | 455±59 |
| 15 | Me | 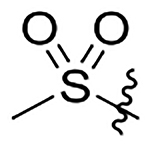 |
H | na | na | 86±11 | 89±11 |
| 16 | 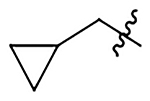 |
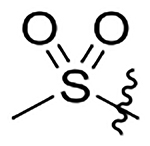 |
H | 385±50 | 433±56 | 505±65 | 114±15 |
| 17 | 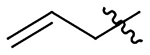 |
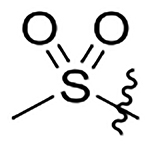 |
H | 403±52 | 668±86 | 590±76 | 42±5.4 |
| 18 | 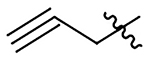 |
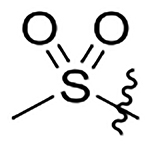 |
H | 511±66 | 1331±170 | 218±28 | 31±4.0 |
| 19 | Me | 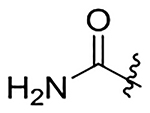 |
H | na | na | 415±54 | 289±37 |
| 20 | 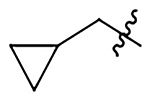 |
 |
H | 1280±170 | 857±110 | 527±68 | 353±46 |
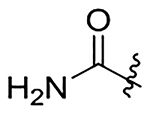
| 21 | 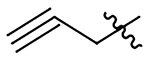 |
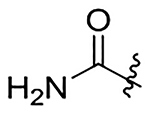 |
H | 911±120 | na | 410±53 | 123±16 |
| 22 | Me | Me | Me | 1570±200 | 295±38 | 127±16 | 73±9.4 |
| 23 | 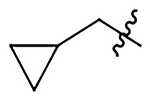 |
Me | Me | 1440±190 | 878±110 | 329±42 | 752±97 |
| 24 | 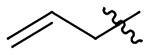 |
Me | Me | na | 318±41 | 102±13 | 309±40 |
| 8-OH DPAT | 0.8 ±0.01 | ||||||
| Clozapine | 3.0±0.4 | 4.0±0.5 | 11.4±1.5 | ||||
Experiments carried out in triplicate;
[3H]WAY100635 used as radioligand;
[3H]Ketanserin used as radioligand;
[3H]LSD used as radioligand;
[3H]LSD used as radioligand;
na – not active (compounds displayed < 50% inhibition in a primary assay)
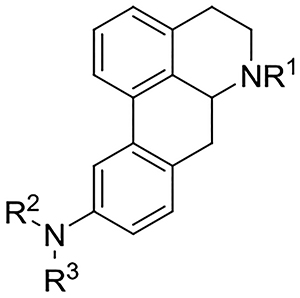
Data for evaluation of the C10 monosubstituted analogues are compiled in Table 2. As a group the compounds display stronger affinity for 5-HT1AR and 5-HT7AR than for 5-HT2AR and 5-HT6R. In fact, compound 31 was the only compound in this series to display affinity for 5-HT2AR and 5-HT6R (this being albeit fairly weak affinity). In this series, the compounds with a C10 amide motif seem to prefer binding to 5-HT1AR over 5-HT7AR (e.g. 25 and 26). The N-propyl-containing compound 25 showed good 5-HT1AR affinity (Ki = 21 nM) and was selective over all other 5-HT receptors tested (no affinity at other 5-HT receptors). The C10 sulfonamide-containing compound 28 which also bears an N-propyl group, also showed selectivity for 5-HT1AR over 5-HT7AR; the C10 urea-containing N-propyl analogue 30 similarly displayed 5-HT1AR versus 5-HT7AR selectivity. Overall, it appears that irrespective of the C10 functionality, an N-propyl group is more favored for binding to the 5-HT1AR than 5-HT7AR in this series (e.g. compare the N-propyl analogues 25, 26, 28, 30 and 32 with the N-methyl analogues 27, 29 and 31). The compound with the highest 5-HT7AR affinity in this series was compound 29 (Ki = 4.5 nM); 29 is 10- fold selective for 5-HT7AR versus 5-HT1AR and is among the most potent aporphinoid 5-HT7R ligands identified to date.
Table 2.
Affinity of 10-monosubstituted analogues at 5-HT receptors
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Ki (nM)a | ||||||||
| Cmpd # | R1 | R2 | R3 | 5-HT1ARb | 5-HT2ARc | 5-HT6Rd | 5-HT7ARd | clogPe |
| 10b | Pr | H | H | 30±3.8 | naf | na | 63±8.2 | 3.6 |
| 25 | Pr | 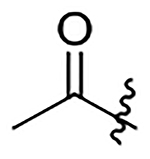 |
H | 21±2.7 | na | na | na | 3.8 |
| 26 | Pr | 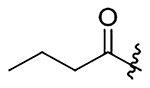 |
H | 15±2.0 | na | na | 311±40 | 4.9 |
| 27 | Me | 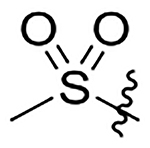 |
H | 133±17 | na | na | 27±3.5 | 2.6 |
| 28 | Pr | 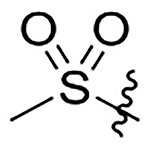 |
H | 23±3.0 | na | na | 199±26 | 3.6 |
| 29 | Me | 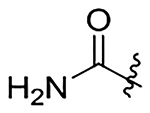 |
H | 49±6.3 | na | na | 4.5±0.6 | 2.8 |
| 30 | Pr |  |
H | 236±30 | na | na | na | 3.8 |
| 31 | Me | Me | Me | 458±59 | 566±73 | 1588±200 | 17±2.2 | 3.9 |
| 32 | Pr | Me | Me | 256±33 | na | na | 984±130 | 5.0 |
| 8-OH DPAT | 0.8 ±0.01 | |||||||
| Clozapine | 3.0±0.4 | 4.0±0.5 | 11.4±1.5 | |||||
Experiments carried out in triplicate;
[3H]WAY100635 used as radioligand;
[3H]Ketanserin used as radioligand;
[3H]LSD used as radioligand;
calculated with ChemDraw Professional v. 16.0.1.4;
na – not active (compounds displayed < 50% inhibition in a primary assay)
Data for evaluation of the benzofused aminothiazole analogues is shown in Table 3. As with the 10-monosubstituted derivatives, the aminothiazole analogues showed stronger affinity for 5-HT1AR and 5-HT7AR than for 5-HT2AR and 5-HT6R. The compounds all lacked affinity for the 5-HT6R. Another similarity in the SAR of the benzofused aminothiazole series with the C10-monosubstituted series is that an N6-methyl moiety confers 5-HT7AR selectivity versus 5-HT1AR, whereas an N6-propyl moiety is preferred for 5-HT1AR selectivity. Thus, the N-methyl analogues 33a and 34a are approximately 19- and 4-fold selective respectively for 5-HT7AR, whereas the N-propyl analogues 33b and 34b are roughly 2-and 17-fold more selective respectively for the 5-HT1AR.
Table 3.
Affinity of aminothiazole analogues at 5-HT receptors
 |
||||||
|---|---|---|---|---|---|---|
| Ki (nM)a | ||||||
| Cmpd # | R1 | 5-HT1ARb | 5-HT2ARc | 5-HT6Rd | 5-HT7ARd | clogPe |
| 33a | Me | 187±24 | 1040±130 | naf | 9.9±1.3 | 3.5 |
| 33b | Pr | 93±12 | na | na | 150±19 | 4.5 |
| 34a | Me | 23±3.0 | na | na | 6.5±0.8 | 3.5 |
| 34b | Pr | 7.5±0.97 | na | na | 132±17 | 4.5 |
| 8-OH DPAT | 0.8 ±0.01 | |||||
| Clozapine | 3.0±0.4 | 4.0±0.5 | 11.4±1.5 | |||
Experiments carried out in triplicate;
[3H]WAY100635 used as radioligand;
[3H]Ketanserin used as radioligand;
[3H]LSD used as radioligand;
calculated with ChemDraw Professional v. 16.0.1.4;
na – not active (compounds displayed < 50% inhibition in a primary assay)
The compounds with the highest 5-HT7AR and 5-HT1A affinity (29, 33a, 34a and 34b) were further profiled for affinity at dopamine receptors (D1R - D5R). Table 4 shows these results. Compound 29 lacked affinity for all receptors except D3R, where low affinity was seen (Ki = 2460 nM). Compound 33a showed low affinity for D1R, D2R, D3R and D4R (Ki = 1150, 1740, 381 and 481 nM respectively) and had no affinity for D5R. Compound 34a lacked affinity for all dopamine receptors. Similar dopamine receptor binding studies on the 5-HT1AR selective compound 34b revealed a lack of affinity for D1R, D2R, D4R and D5R; low affinity (Ki = 2110 nM) was observed for D3R
Table 4.
Affinity of 29, 33a, 34a and 34b at dopamine receptors
| Ki (nM)a | |||||
|---|---|---|---|---|---|
| Cmpd # | D1Rb | D2Rc | D3Rc | D4Rc | D5R |
| 29 | nad | na | 2460±320 | na | na |
| 33a | 115 0±150 | 1740±220 | 381±49 | 481±62 | na |
| 34a | na | na | na | na | na |
| 34b | na | na | 2110±270 | na | na |
| (+)-butaclamol | 4.30±0.55 | ||||
| Haloperidol | 5.58±0.72 | ||||
| Nemonapride | 1.18±0.15 | 0.86±0.11 | |||
Experiments carried out in triplicate;
[3H]SCH23390 used as radioligand;
[3H]N-Methylspiperone used as radioligand;
na – not active (compounds displayed < 50% inhibition in a primary assay)
Compounds 29 and 34a were evaluated for 5-HT7R functional activity using Hit Hunter® agonist and antagonist cAMP assays at Eurofins Pharma Discovery Services. Data from these assays is provided in Table 5. Both compounds were found to be 5-HT7R antagonists with EC50 values of 0.125 and 0.26 μM respectively for 29 and 34a, which was comparable to the positive control spiperone; the compounds lacked agonist activity in this assay.
Table 5.
Functional activity of compounds 29 and 34a in cAMP assays
| Compound | Assay mode | Agonista EC50(μM) | Antagonista IC50 (μM) |
|---|---|---|---|
| 29 | agonist | >10 | - |
| 29 | antagonist | - | 0.125 |
| 34a | agonist | >10 | - |
| 34a | antagonist | - | 0.26 |
| Serotonin | agonist | 0.07 | - |
| Spiperone.HCl | antagonist | - | 0.31 |
Experiments performed in duplicate
Docking studies
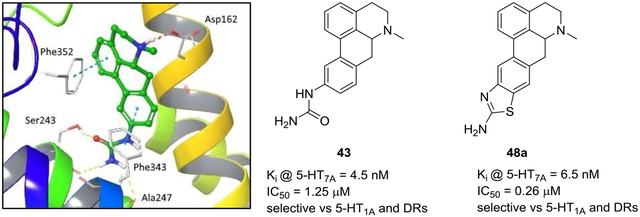
Computational docking simulations were performed in order to rationalize the high measured 5-HT7R binding affinities for compounds 29, 33a and 34a. In this context, we investigated the docked ligand poses and identified key receptor-ligand interactions that influence binding to the 5-HT7R and provide a deeper appreciation of the observed high affinity towards this receptor system. A homology model of the 5-HT7R was generated from the high-resolution crystal structure of the human serotonin 5-HT1B G protein-coupled receptor with pdb code 4IAQ [29]. This approach involved utilization of the Schrödinger Prime Structure Prediction and Glide software modules and manual intervention to support the generation of known key receptor-ligand interactions. The model for the 5-HT7R structure, therefore, comprised suitable backbone and side-chain orientations within the binding site. The docking simulations of compounds 29, 33a and 34a into the 5-HT7R binding site exploited the Schrödinger Induced Fit and Glide methodologies in Standard Precision (SP) mode. Using this docking protocol, the Glidescore scoring function was used to give an estimate of the ligand binding affinities for the highest ranked poses of compounds 29, 33a and 34a in the 5-HT7R target. The ligand binding poses are depicted in Figure 3.
Figure 3.
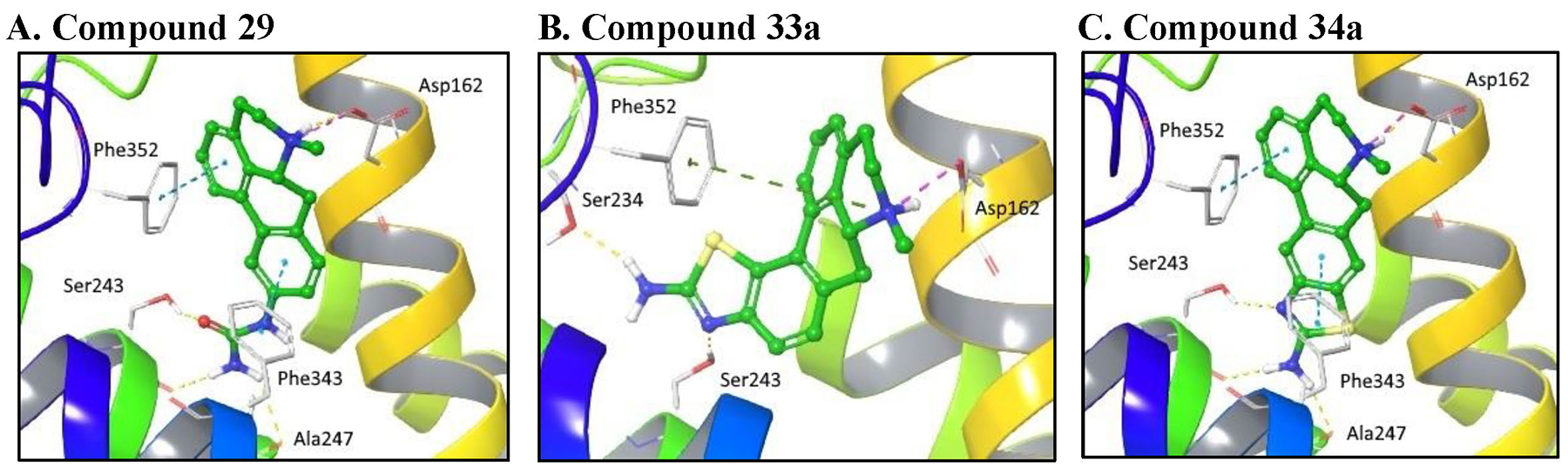
Docked poses of A. compound 29 (with binding energy of −8.9 kcal/mol), B. compound 33a (with binding energy of −8.7 kcal/mol) and C. compound 34a (with binding energy of −10.0 kcal/mol) shown by green carbon atoms in the serotonin 5-HT7 receptor target shown by secondary structure elements and grey carbon atoms for select residues. Key quaternary N – Asp salt bridges are depicted by the pink dashed lines, H-bonding interactions by the yellow dashed lines, π-π stacking by the blue dashed lines and corresponding π-cation interactions by the green dashed lines.
Compounds 29 and 34a give very similar binding poses in the serotonin 5-HT7R binding pocket as shown in Figures 3A and C. The bound structures with these ligands comprise the key salt bridge between the quaternary N atom and Asp162, hydrogen bonding interactions to the Ser243 side chain and Ala247 backbone and π-stacking interactions between the ligand aromatic rings and receptor Phe343 and Phe352 residues. Compound 34a gives a better predicted binding energy (−10.0 kcal/mol) compared to 29 (−8.9 kcal/mol), due to a slightly stronger protonated N-Asp salt bridge and better π-π interactions involving the fused thiazole ring in 34a.
For compound 33a, in order to accommodate the modified position of the fused thiazole ring, the best binding pose identified by induced fit docking involves a refined 5-HT7 receptor structure with a rotated Asp162 side-chain orientation. As depicted in Figure 3B, key receptor-ligand interactions include the quaternary N – Asp162 salt bridge, H-bonds to Ser234 and Ser243 and a π-cation interaction between the same quaternary N and Phe352. As a consequence of the modified binding pose, the binding energy for compound 33a (−8.7 kcal/mol) is worse than that for 34a (−10.0kcal/mol) primarily due to the lack of complementary π-π interactions involving the ligand aromatic rings in 33a.
3. Conclusions
In this study, we sought to evaluate the extent to which C10 nitrogen substituents on the aporphine core impact affinity and selectivity across a range of 5-HT receptors, including 5-HT7AR. Aporphines with 1,2,10-trisubstituted and 10-substituted substitution patterns as well as ring D aminothiazole moieties were investigated.
We found that affinities of the 1,2,10-trisubstituted analogues across the four receptors evaluated (5-HT1AR, 5-HT2AR, 5-HT6R and 5-HT7AR) was low to moderate; C10 N-substitution here did not result in pronounced high affinity and selectivity for any of the 5-HT receptors, although there was generally a slight preference for 5-HT7R binding.
In contrast, the C10 monosubstituted group of compounds as well as the benzofused aminothiazole group of compounds were found to selectively target 5-HT1AR and 5-HT7AR and largely lacked affinity for 5-HT2AR and 5-HT6R. Taken together with the results from the trisubstituted series, it would appear that C1 and C2 alkoxy substituents on the aporphine core are not required for affinity to 5-HT7AR and 5-HT1AR. Furthermore, the SAR results indicate that in general, an N6-methyl substituent engenders 5-HT7AR selectivity over 5-HT1AR whereas this selectivity is reversed with an N6-propyl substituent.
Overall, our study provides valuable SAR information by revealing that the inclusion of a C10 nitrogen substituent group on the ring A unsubstituted aporphine core, allows for selective binding of the scaffold to 5-HT7R and 5-HT1AR. Compounds 29 and 34a were found to function as 5-HT7R antagonists in assays that measured cAMP production. These compounds do not display any appreciable affinity for dopamine receptors, have moderate selectivity versus 5-HT1AR and represent good starting points from which to further optimize C10 nitrogenated aporphines as selective 5-HT7R antagonists. The impact of the configuration of the chiral center of the aporphines on affinity and selectivity will need to be investigated in future. Optimization of the ligands for 5-HT7R affinity is expected to be challenging given the 5-HT1AR affinity of the series, but is nevertheless a promising dimension for future studies.
4. Experimental
4.1. Synthetic experimental procedures
General Procedures:
All glass apparatus was oven-dried prior to use. HRESIMS spectra were obtained using an Agilent 6520 Q-TOF instrument. 1H NMR and 13C NMR spectra were recorded using a Bruker DPX-500 spectrometer (operating at 500 and 600 MHz for 1H; 125 and 150 MHz, respectively, for 13C) using CDCl3 as solvent. Tetramethylsilane (δ 0.00 ppm) served as an internal standard in 1H NMR and 13C NMR unless stated otherwise. Chemical shift (δ 0.00 ppm) values are reported in parts per million and coupling constants in Hertz (Hz). Splitting patterns are described as singlet (s), doublet (d), triplet (t), and multiplet (m). Reactions were monitored by TLC with Whatman Flexible TLC silica gel G/UV 254 precoated plates (0.25 mm). TLC plates were visualized in UV light (254 nm) and by staining with phosphomolybdate spray reagent, vanillin or iodine. Flash column chromatography was performed with silica gel 60 (EMD Chemicals, 230–400 mesh, 0.04–0.063 mm particle size). Preparative thin layer chromatography was performed with silica gel GF plates (Analtech, catalog # 02003). All chemicals and reagents were obtained from Sigma-Aldrich and Fischer Scientific (USA) in reagent grade and were used without further purification. Yields reported are after purification.
General procedure for synthesis of amide analogues:
To a stirred solution of the corresponding aniline (0.2 mmol) in DCM (10 mL) was added the respective anhydride (0.4 mmol) and the resulting reaction mixture was stirred at rt for 2 h under nitrogen atmosphere. After completion of the reaction (monitored by TLC) the reaction mixture was concentrated in-vacuo and a solution of NaOH (0.2 g) in methanol (10 mL) was added and stirred for another 30 min at the same temperature. The reaction mixture was concentrated in-vacuo and the obtained crude mass was dissolved in water and extracted with DCM (3×10 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in-vacuo to give crude product, which was purified by preparative TLC purification eluting in 5% MeOH/DCM.
N-(1,2-dimethoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)butyramide (11)
37% yield. Yellow oil. 1H NMR (600 MHz, CDCl3): δ 8.25 (d, J = 8.6 Hz, 1H), 7.66 (s, 1H), 7.28 (s, 1H), 7.18 – 7.13 (m, 1H), 6.54 (s, 1H), 3.81 (s, 3H), 3.56 (s, 3H), 3.12 – 3.05 (m, 1H), 3.02 (m, 1H), 2.99 – 2.92 (m, 2H), 2.61 (m, 1H), 2.54 (m, 1H), 2.48 – 2.41 (m, 4H), 2.29 (t, J = 7.4 Hz, 2H), 1.74 – 1.67 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3): δ 171.4, 152.0, 144.8, 137.6, 136.9, 128.9, 128.8, 128.1, 127.5, 126.6, 119.0, 117.9, 110.8, 62.2, 60.2, 55.8, 53.3, 44.0, 39.8, 35.2, 29.2, 19.2, 13.8 ppm. HRESIMS m/z 381.2173 [M+H]+ (calcd. for C23H28N2O3, 381.2177)
N-(1-(cyclopropylmethoxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)acetamide (12a).
48% yield. Dark yellow oil. 1H NMR (600 MHz, CDCl3): δ 8.29 (d, J = 8.6 Hz, 1H), 7.62 – 7.51 (m, 1H), 7.45 (s, 1H), 7.20 (d, J = 8.4 Hz, 1H), 6.45 (s, 1H), 3.72 (s, 3H), 3.55 (m, 1H), 3.25 (m, 1H), 3.18 – 3.07 (m, 2H), 3.07 – 2.99 (m, 1H), 2.98 – 2.91 (m, 1H), 2.64 – 2.52 (m, 3H), 2.48 (s, 3H), 2.05 (s, 3H), 0.98 (m, 1H), 0.34 – 0.27 (m, 2H), 0.03 – −0.07 (m, 2H) ppm.13C NMR (125 MHz, CDCl3): δ 168.3, 152.4, 143.9, 136.9, 136.9, 129.6, 128.4, 128.4, 128.0, 127.1, 118.9, 117.8, 110.8, 77.8, 62.2, 55.8, 53.2, 43.4, 34.8, 29.7, 24.8, 11.0, 3.4, 3.1 ppm. HRESIMS m/z 393.2179 [M+H]+ (calcd. for C24H28N2O3, 393.2170)
N-(1-(cyclopropylmethoxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)butyramide (12b)
40% yield. White solid. Melting point: 89.1 °C – 90.1 °C. 1H NMR (500 MHz, CDCl3): δ 8.31 (d, J = 8.6 Hz, 1H), 7.57 (s, 1H), 7.20 (s, 1H), 7.11 (dd, J = 8.5, 1.8 Hz, 1H), 6.46 (s, 1H), 3.72 (s, 3H), 3.56 (m, 1H), 3.25 (m, 1H), 3.08 – 2.98 (m, 1H), 2.98 – 2.87 (m, 3H), 2.55 (m, 1H), 2.48 (m, 1H), 2.43 – 2.36 (m, 4H), 2.23 (t, J = 7.4 Hz, 2H), 1.65 (m, 2H), 1.03 – 0.94 (m, 1H), 0.89 (t, J = 7.4 Hz, 3H), 0.30 (m, 2H), −0.02 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ 171.4, 152.1, 143.7, 137.4, 136.8, 129.6, 128.5, 128.4, 127.2, 127.1, 118.9, 117.6, 110.8, 77.8, 62.2, 55.8, 53.3, 43.9, 39.8, 35.2, 29.1, 19.1, 13.8, 11.0, 3.4, 3.0 ppm. HRESIMS m/z 421.2490. [M+H]+ (calcd. for C26H33N2O3, 421.2486)
N-(1-(allyloxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)butyramide (13)
55% yield. Brown solid. Melting point: 99.1 °C – 99.6 °C. 1H NMR (500 MHz, CDCl3): δ 8.27 (d, J = 8.5 Hz, 1H), 7.65 (s, 1H), 7.18–7.10 (m, 2H), 6.53 (s, 1H), 5.88 (m, 1H), 5.16 (m, 1H), 5.05 (m, 1H), 4.28 (m, 1H), 4.09 (m, 1H), 3.79 (s, 3H), 3.10 (s, 1H), 3.05 – 2.93 (m, 3H), 2.61 (m, 1H), 2.54 (m, 1H), 2.45 (m, 4H), 2.29 (t, J = 7.4 Hz, 2H), 1.71 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 171.3, 152.1, 143.3, 137.5, 136.9, 134.2, 129.3, 128.7, 128.7, 128.2, 127.0, 118.9, 117.6, 117.5, 110.8, 73.7, 62.2, 55.9, 53.3, 43.9, 39.8, 35.1, 29.1, 19.2, 13.8 ppm. HRESIMS m/z 407.2329 [M+H]+ (calcd. for C25H30N2O3, 407.2177)
N-(2-methoxy-6-methyl-1-(prop-2-yn-1-yloxy)-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)butyramide (14)
52% yield. Yellow oil. 1H NMR (500 MHz, CDCl3): δ 8.27 (d, J = 8.6 Hz, 1H), 7.67 (s, 1H), 7.19 (s, 1H), 7.14 (d, J = 8.5 Hz, 1H), 6.54 (s, 1H), 4.46 (dd, J = 15.0, 2.3 Hz, 1H), 4.33 (dd, J = 15.0, 2.4 Hz, 1H), 3.80 (s, 3H), 3.16 – 3.06 (m, 1H), 3.05 – 2.95 (m, 3H), 2.62 (m, 1H), 2.55 (m, 1H), 2.51 – 2.43 (m, 4H), 2.31 – 2.25 (m, 3H), 1.72 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ 171.3, 152.0, 142.3, 137.5, 137.1, 129.4, 128.0, 127.3, 127.2, 118.9, 117.8, 110.9, 79.2, 74.9, 62.1, 59.6, 55.9, 53.2, 43.8, 39.8, 35.1, 29.0, 19.1, 13.8 ppm. HRESIMS m/z 405.2173 [M+H]+ (calcd. for C25H28N2O3, 405.2177)
N-(6-propyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)acetamide (25)
73% yield. Off-white solid. Melting point: 99.8 °C – 100.3 °C. 1H NMR (600 MHz, CDCl3): δ 7.56 (d, J = 8.4 Hz, 1H), 7.51 (s, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.30 (m, 1H), 7.22 (t, J = 11.5 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 3.44 (m, 1H), 3.18 – 3.09 (m, 2H), 3.07 (m, 1H), 2.87 (m, 1H), 2.72 – 2.67 (m, 1H), 2.63 (m, 1H), 2.52 – 2.40 (m, 2H), 2.11 (s, 3H), 1.63 – 1.48 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H) ppm.13C NMR (125 MHz, CDCl3): δ 168.4, 137.3, 136.1, 133.6, 133.3, 130.4, 127.7, 127.0, 124.3, 121.6, 119.5, 119.4, 118.7, 59.3, 56.2, 49.4, 33.9, 28.9, 24.7, 18.9, 12.0 ppm. HRESIMS m/z 329.1964 [M+H]+ (calcd. for C21H25N2O, 321.1961)
N-(6-propyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)butyramide (26)
50% yield. Pale yellow solid. Melting point: 104.9 °C – 106.5 °C. 1H NMR (600 MHz, CDCl3): δ 7.66 (d, J = 7.4 Hz, 2H), 7.51 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.0 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 7.4 Hz, 1H), 3.61 – 3.50 (m, 1H), 3.31 – 3.15 (m, 3H), 3.01 – 2.92 (m, 1H), 2.84 – 2.71 (m, 2H), 2.66 – 2.52 (m, 2H), 2.37 (t, J = 7.4 Hz, 2H), 1.80 (m, 2H), 1.74 – 1.59 (m, 2H), 1.05 (t, J = 7.3 Hz, 3H), 1.00 (t, J = 7.3 Hz, 3H) ppm.13C NMR (150 MHz, CDCl3): δ 171.5, 137.3, 136.2, 133.5, 133.2, 133.1, 130.4, 127.7, 126.9, 124.4, 121.5, 119.6, 118.6, 61.9, 53.5, 53.4, 43.9, 39.7, 34.2, 29.7, 28.9, 19.1, 13.8 ppm. HRESIMS m/z 349.2279 [M+H]+ (calcd. for C23H29N20, 349.2274)
General procedure for preparation of methanesulfonamide analogues:
To a stirred solution of corresponding aniline (0.1 mmol) in DCM (10 mL), methanesulfonic anhydride (0.25 mmol) and triethylamine (0.3 mmol) were added. The resulting mixture was stirred at rt for 12h under nitrogen atmosphere. After completion of the reaction, the reaction mixture was concentrated in-vacuo and a solution of NaOH (0.2 g) in methanol (10 mL) was added to the flask and stirring was continued for another 30 min. The reaction mixture was concentrated in-vacuo, dissolved in water and extracted with DCM (3×10 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in-vacuo. The crude product thus obtained was purified using preparative TLC eluting in 5% MeOH/DCM to give respective sulfonamide analogues.
N-(1,2-dimethoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (15)
45% yield. Off-white solid. Melting point: 153.0 °C – 154.6 °C. 1H NMR (600 MHz, CDCl3): δ 8.26 (d, J = 8.5 Hz, 1H), 7.09 (s, 1H), 7.05 – 7.02 (m, 1H), 6.55 (s, 1H), 3.80 (s, 3H), 3.59 (s, 3H), 3.13 – 3.04 (m, 1H), 3.03 – 2.93 (m, 6H), 2.64 – 2.59 (m, 1H), 2.52 (m, 1H), 2.48 – 2.41 (m, 4H) ppm. 13C NMR (150 MHz, CDCl3): δ 152.0, 144.9, 138.3, 135.6, 129.7, 129.3, 128.8, 127.3, 126.1, 119.6, 118.7, 111.3, 62.0, 60.2, 55.8, 53.2, 43.9, 39.6, 35.1, 29.1 ppm. HRESIMS m/z 389.1529 [M+H]+ (calcd. for C20H25N2O4S, 389.4895)
N-(1-(cyclopropylmethoxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (16)
46% yield. Brown solid. Melting point: 140.3 °C – 141.5 °C. 1H NMR (500 MHz, CDCl3): δ 8.41 (d, J = 8.5 Hz, 1H), 7.18 (s, 1H), 7.08 (s, 1H), 7.03 (d, J = 7.0 Hz, 1H), 6.53 (s, 1H), 3.78 (s, 3H), 3.63 (m, 1H), 3.34 (m, 1H), 3.16 – 3.06 (m, 2H), 3.06 – 2.97 (m, 5H), 2.63 (m, 2H), 2.58 – 2.44 (m, 4H), 1.08 – 0.99 (m, 1H), 0.37 (d, J = 8.1 Hz, 2H), 0.10 – −0.03 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ 152.2, 143.8, 137.9, 135.4, 130.4, 129.7, 128.7, 128.1, 126.6, 119.4, 118.4, 111.1, 77.9, 62.0, 55.8, 53.5, 53.2, 39.6, 35.0, 11.0, 3.4, 3.1 ppm.
N-(1-(allyloxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (17)
33% yield. Light-yellow oil. 1H NMR (500 MHz, CDCl3): δ 8.30 (d, J = 8.5 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.01 (dd, J = 8.5, 2.3 Hz, 1H), 6.55 (s, 1H), 5.88 (ddt, J = 16.4, 10.4, 5.9 Hz, 1H), 5.17 (dd, J = 17.2, 1.5 Hz, 1H), 5.06 (dd, J = 10.4, 1.2 Hz, 1H), 4.31 (dd, J = 12.0, 5.9 Hz, 1H), 4.12 (dd, J = 12.0, 5.9 Hz, 1H), 3.80 (s, 3H), 3.14 – 3.07 (m, 1H), 3.07 – 2.95 (m, 6H), 2.63 (dd, J = 16.3, 3.1 Hz, 1H), 2.55 – 2.44 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 152.2, 143.5, 138.2, 135.5, 134.2, 130.1, 129.4, 128.8, 127.1, 126.5, 119.4, 118.4, 117.6, 111.3, 73.8, 62.0, 55.9, 53.2, 43.8, 39.6, 35.0, 29.0 ppm. HRESIMS m/z 415.1690 [M+H]+ (calcd. for C22H26N2O4S, 415.1686)
N-(2-methoxy-6-methyl-1-(prop-2-yn-1-yloxy)-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (18)
33% yield. Yellow oil. 1H NMR (500 MHz, CDCl3): δ 8.29 (d, J = 8.5 Hz, 1H), 7.10 (d, J = 1.9 Hz, 1H), 7.05 (dd, J = 8.5, 2.2 Hz, 1H), 6.55 (s, 1H), 4.50 (m, 1H), 4.37 (m, 1H), 3.80 (s, 3H), 3.12 – 3.04 (m, 1H), 3.02 – 2.92 (m, 6H), 2.62 (m, 1H), 2.49 (m, 5H), 2.26 (t, J = 2.4 Hz, 1H) ppm. 13C NMR (125 MHz, CDCl3): δ 152.0, 142.3, 138.3, 135.7, 130.1, 129.6, 129.3, 127.4, 126.9, 119.5, 118.7, 111.2, 79.1, 75.01, 62.0, 59.8, 55.9, 53.2, 44.0, 39.5, 35.1, 29.1 ppm. HRESIMS m/z 413.1532 [M+H]+ (calcd. for C22H24N2O4S, 413.1530)
N-(6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (27)
48% yield. Light brown oil. 1H NMR (500 MHz, CDCl3): δ 7.61 (d, J = 8.1 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.21 – 7.17 (m, 1H), 7.12 – 7.07 (m, J = 8.5 Hz, 2H), 7.02 (d, J = 7.6 Hz, 1H), 3.18 (m, 2H), 3.14 – 3.02 (m, 2H), 2.99 (s, 3H), 2.74 – 2.67 (m, 1H), 2.63 (m, 1H), 2.56 – 2.47 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 135.9, 135.0, 132.6, 132.1, 131.7, 130.6, 127.2, 126.2, 124.1, 120.7, 119.5, 118.6, 60.8, 52.4, 42.9, 38.5, 33.0, 27.8 ppm. HRESIMS m/z 329.1322 [M+H]+ (calcd. for C18H21N2O2S, 329.1318)
N-(6-propyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)methanesulfonamide (28)
38% yield. Dark brown oil. 1H NMR (500 MHz, CDCl3): δ 7.61 (d, J = 8.3 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 7.10 (s, 1H), 7.07 (dd, J = 8.3, 2.1 Hz, 1H), 7.01 (d, J = 7.6 Hz, 1H), 3.39 (dd, J = 14.0, 3.8 Hz, 1H), 3.17 – 3.05 (m, 3H), 3.00 (s, 3H), 2.86 (m, 1H), 2.69 (m, 1H), 2.59 (m, 1H), 2.47 – 2.35 (m, 2H), 1.63 – 1.48 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ 136.2, 134.9, 133.0, 131.9, 130.9, 127.7, 127.2, 126.0, 124.1, 120.7, 119.4, 118.6, 58.2, 55.4, 48.4, 38.5, 33.2, 28.1, 18.3, 11.1 ppm. HRESIMS m/z 357.1636 [M+H]+ (calcd. for C20H25N2O2S, 357.1631)
General procedure for synthesis of urea analogues:
To a magnetically stirred solution of amine (100 mg, 1 equiv.) in 10% acetic acid solution (10 mL) (v/v), a solution of potassium cyanate (2 equiv.) in water (5 mL) was added. The reaction was stirred at rt for 2h. After completion of the reaction (monitored by TLC), the reaction was quenched with saturated sodium bicarbonate and extracted with EtOAc (3×15 mL). The combined organic layer was dried over Na2SO4, filtered, concentrated under reduced pressure and the crude product thus obtained was purified using preparative TLC, eluting in 5% MeOH/DCM to give the respective urea analogues.
1-(1,2-dimethoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)urea(19):
40 % yield. Brown solid. Melting point: 208 °C – 209 °C. 1H NMR (600 MHz, CDCl3): δ 8.15 (d, J = 8.5 Hz, 1H), 7.53 (s, 1H), 7.22 (s, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.50 (s, 1H), 5.12 (bs, 2H), 3.77 (s, 3H), 3.53 (s, 3H), 3.15 – 3.06 (m, 1H), 3.00 (m, 2H), 2.93 (m, 1H), 2.59 (m, 1H), 2.53 (m, 1H), 2.45 (m, 4H). 13C NMR (150 MHz, CDCl3): δ 156.8, 152.2, 144.8, 137.9, 137.1, 129.1, 128.3, 127.2, 126.6, 126.3, 119.5, 118.8, 110.8, 62.0, 60.2, 55.8, 53.0, 43.3, 34.7, 28.5 ppm. HRESIMS m/z 354.1811 [M+H]+ (calcd. for C20H24N3O3, 354.4295)
1-(1-(cyclopropylmethoxy)-2-methoxy-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)urea (20):
30 % yield. Brown oil. 1H NMR (600 MHz, CDCl3): δ 8.26 (d, J = 8.5 Hz, 1H), 8.22 (s, 1H), 7.28 – 7.22 (m, 1H), 6.46 (s, 1H), 5.48 (bs, 2H), 3.75 (s, 3H), 3.57 – 3.52 (m, 1H), 3.34 – 3.28 (m, 1H), 3.22 – 3.08 (m, 3H), 2.94 (m, 1H), 2.62 (m, 3H), 2.54 (s, 3H), 1.01 (m, 1H), 0.34 (d, J = 6.5 Hz, 2H), 0.07 – −0.04 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3): δ 157.1, 152.6, 143.7, 138.49, 136.1, 129.5, 128.6, 127.2, 126.6, 124.6, 118.7, 118.0, 110.4, 77.8, 61.9, 55.8, 52.8, 42.4, 34.1, 27.7, 11.0, 3.4, 3.2 ppm. HRESIMS m/z 394.2123 [M+H]+ (calcd. for C23H28N3O3, 394.4945).
1-(2-methoxy-6-methyl-1-(prop-2-yn-1-yloxy)-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)urea (21):
30 % yield. Light brown solid. Melting point: 172.4 – 172.8 °C. 1H NMR (600 MHz, Acetone): δ 8.25 (d, J = 8.6 Hz, 1H), 8.09 (s, 1H), 7.55 (s, 1H), 7.32 (d, J = 8.3 Hz, 1H), 6.70 (s, 1H), 5.44 (bs, 2H), 4.60 (m, 1H), 4.44 (m, 1H), 3.86 (s, 3H), 3.08 – 3.04 (m, 2H), 2.98 (m, 1H), 2.87 – 2.83 (m, 2H), 2.64 (m, 1H), 2.47 (s, 3H), 2.41 (m, 2H) ppm. 13C NMR (150 MHz, Acetone): δ 155.7, 152.1, 142.1, 139.9, 137.5, 129.6, 128.9, 127.7, 127.4, 125.6, 117.3, 116.1, 110.8, 79.5, 75.3, 62.6, 59.6, 59.1, 55.3, 53.1, 43.4, 35.3 ppm. HRESIMS m/z 378.1819 [M+H]+ (calcd. for C22H24N3O3, 378.4515).
1-(6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)urea (29)
83% yield. White solid. Melting point: 125.6 °C – 126.9 °C. 1H NMR (500 MHz, CDCl3): δ 7.56 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 7.34 (bs, 1H), 7.26 (s, 1H), 7.20 (m, 2H), 7.04 (d, J = 7.5 Hz, 1H), 5.12 (bs, 2H), 3.25 – 3.16 (m, 2H), 3.08 (m, 2H), 2.74 (m, 1H), 2.65 (m, 1H), 2.54 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 156.7, 137.9, 136.0, 133.1, 133.0, 132.5, 130.0, 127.7, 127.2, 124.6, 121.5, 120.6, 119.9, 61.7, 53.3, 43.5, 33.8, 28.6 ppm. HRESIMS m/z 294.1604 [M+H]+ (calcd. for C18H19N3O, 294.1601)
1-(6-propyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-yl)urea (30)
86% yield. Brown Solid. Melting point: 114.5 °C – 114.9 °C. 1H NMR (600 MHz, Acetone-d6) δ 8.26 (dd, J = 2.2, 1.0 Hz, 1H), 8.20 (ddd, J = 8.6, 2.5, 0.9 Hz, 1H), 8.05 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.34 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 3.60 – 3.55 (m, 1H), 3.44 (m, 1H), 3.24 (m, 1H), 3.08 (m, 1H), 3.02 (m, 1H), 2.90 (bs, 2H), 2.79 (m, 1H), 2.62 (m, 1H), 2.50 – 2.42 (m, 2H), 1.69 – 1.56 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (125 MHz, Acetone-d6): δ 147.7, 141.8, 138.4, 136.5, 135.6, 132.5, 130.8, 128.0, 125.6, 124.3, 123.9, 123.3, 59.9, 56.8, 50.0, 34.6, 30.1, 20.5, 12.2 ppm. HRESIMS m/z 322.1922 [M+H]+ (calcd. for C20H24N3O, 322.4315)
General procedure for synthesis of N,N-dimethylated analogues:
To a stirred solution of the corresponding aniline (0.1 mmol) in DCM (10 mL) was added formaldehyde (37% aqueous solution, 5 μL, 0.1 mmol) and the reaction mixture was stirred at room temperature for 1h. NaBH(OAc)3 (200 mg, 1 mmol) was added and the resulting reaction mixture was stirred at rt for 12h. After completion of the reaction (monitored by TLC) the reaction mixture was quenched with saturated sodium bicarbonate solution and extracted with DCM (3×15 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated in-vacuo. The residue thus obtained was purified using preparative TLC eluting in 5% MeOH/DCM to give the N-methylated analogues.
1,2-dimethoxy-N,N,6-trimethyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-amine (22)
30% yield. Light purple solid. Melting point: 150.1 °C – 150.9 °C. 1H NMR (600 MHz, CDCl3): δ 8.20 (d, J = 8.8 Hz, 1H), 6.62 (dd, J = 8.8, 2.7 Hz, 1H), 6.54 (d, J = 2.5 Hz, 1H), 6.46 (s, 1H), 3.80 (s, 3H), 3.58 (s, 3H), 3.15 – 3.07 (m, 1H), 3.05 – 2.95 (m, d H), 2.94 (s, 6H), 2.65 – 2.57 (m, 2H), 2.50–2.52 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 152.0, 149.6, 144.2, 137.7, 129.4, 128.5, 127.5, 127.0, 120.6, 111.3, 110.9, 109.5, 62.6, 60.0, 55.8, 53.4, 44.1, 40.4, 35.9, 29.3 ppm. HRESIMS m/z 339.2065 [M+H]+ (calcd. for C21H27N2O2, 339.2067)
1-(cyclopropylmethoxy)-2-methoxy-N,N,6-trimethyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-amine (23)
40% yield. Orange oil. 1H NMR (500 MHz, CDCl3): δ 8.24 (d, J = 8.8 Hz, 1H), 6.53 (dd, J = 8.8, 2.7 Hz, 1H), 6.44 (d, J = 2.6 Hz, 1H), 6.35 (s, 1H), 3.69 (s, 3H), 3.48 (m, 1H), 3.27 (m, 1H), 3.03 – 2.96 (m, 1H), 2.90 – 2.86 (m, 3H), 2.84 (s, 6H), 2.53 – 2.45 (m, 2H), 2.39 (s, 3H), 2.39 – 2.33 (m, 1H), 1.07 – 0.98 (m, 1H), 0.34 – 0.27 (m, 2H), 0.03 – −0.04 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3): δ 152.1, 149.5, 143.1, 137.5, 129.9, 128.3, 127.9, 126.9, 121.0, 111.2, 110.7, 109.5, 77.4, 62.6, 55.8, 53.4, 44.0, 40.4, 35.9, 29.2, 11.1, 3.3, 3.1 ppm. HRESIMS m/z 379.2378 [M+H]+ (calcd. for C24H31N2O2, 379.2380)
1-(allyloxy)-2-methoxy-N,N,6-trimethyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-amine (24)
48% yield. Brown oil. 1H NMR (600 MHz, CDCl3): δ 8.22 (d, J = 8.8 Hz, 1H), 6.59 (dd, J = 8.8, 2.6 Hz, 1H), 6.53 (d, J = 2.2 Hz, 1H), 6.45 (s, 1H), 5.94 (ddt, J = 10.7, 5.8 Hz, 1H), 5.19 (dd, J = 17.2, 1.4 Hz, 1H), 5.06 (d, J = 10.4 Hz, 1H), 4.25 (dd, J = 12.0, 5.9 Hz, 1H), 4.14 (dd, J = 12.0, 5.7 Hz, 1H), 3.78 (s, 3H), 3.14 – 3.06 (m, 1H), 3.03 – 2.95 (m, 3H), 2.92 (s, 6H), 2.58 – 2.62 (m, 2H), 2.48 (m, 4H) ppm. 13C NMR (150 MHz, CDCl3): δ 152.2, 149.6, 142.9, 137.4, 134.6, 129.7, 128.3, 127.9, 126.6, 120.7, 117.2, 111.3, 110.8, 109.6, 73.4, 62.5, 55.8, 53.3, 43.8, 40.4, 35.7, 29.0 ppm. HRESIMS m/z 365.2222 [M+H]+ (calcd. for C23H29N2O2, 365.2224)
N,N,6-trimethyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-amine (31)
63% yield. Brown oil. 1H NMR (500 MHz, CDCl3): δ 7.52 (d, J = 8.6 Hz, 1H), 7.38 (d, J = 7.7 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.61 (dd, J = 8.6, 2.6 Hz, 1H), 6.54 (d, J = 2.2 Hz, 1H), 3.17 – 3.08 (m, 2H), 3.07 – 3.02 (m, 1H), 3.02 – 2.96 (m, 1H), 2.91 (s, 6H), 2.69 – 2.65 (m, 1H), 2.63 (m, 1H), 2.50 (s, 3H), 2.46 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3): δ 150.1, 136.4, 134.1, 133.3, 132.6, 126.8, 126.3, 124.8, 123.1, 120.6, 111.9, 111.5, 62.4, 53.6, 44.2, 40.6, 35.0, 29.2 ppm. HRESIMS m/z 279.1861 [M+H]+ (calcd. for C19H23N2, 279.1856)
N,N-dimethyl-6-propyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-10-amine (32)
62%. Light brown solid. Melting point: 164.5 °C – 165.8 °C. 1H NMR (500 MHz, CDCl3): δ 7.63 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.73 (dd, J = 8.6, 2.6 Hz, 1H), 6.66 (d, J = 2.4 Hz, 1H), 3.59 – 3.51 (m, 1H), 3.29 – 3.22 (m, 1H), 3.20 – 3.12 (m, 2H), 3.02 (s, 6H), 3.01 – 2.95 (m, 1H), 2.80 (s, 1H), 2.76 (s, 1H), 2.58 (m, 2H), 1.77 – 1.58 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ 150.1, 136.6, 134.3, 133.6, 133.1, 126.7, 126.3, 124.8, 123.2, 120.6, 111.9, 111.5, 59.6, 56.4, 49.4, 40.6, 34.9, 29.2, 19.2, 12.2 ppm. HRESIMS m/z 307.2168 [M+H]+ (calcd. for C21H27N2, 307.2169)
General Procedure for synthesis of benzofused aminothiazole analogues - compounds 33–34 (a-b)
To a magnetically stirred solution of respective aniline (100 mg, 1 equiv.) in acetic acid was added potassium thiocyanate (2 equiv.) at 0 °C. This was followed by addition of a solution of bromine (1.2 equiv, vol?.) in acetic acid (1 mL) in a dropwise manner. The resulting mixture was allowed to warm to rt and stirred at the same temperature for 12h. After completion of the reaction, the reaction mixture was concentrated in-vacuo, treated with saturated sodium bicarbonate solution and extracted with EtOAc (3×20 mL). The combined organic layer was dried using anhydrous Na2SO4 and concentrated in-vacuo. The crude compounds thus obtained were purified by preparative TLC using 5% MeOH/DCM.
7-methyl-6a,7,8,9-tetrahydro-6H-benzo[de]thiazolo[4’,5’:5,6]benzo[1,2-g]quinolin-2-amine (33a):
30% yield. Orange solid. Melting point: 215 °C – 216 °C. 1H NMR (500 MHz, CDCl3): δ 7.65 (d, J = 8.5 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.22 – 7.16 (m, 1H), 6.99 (d, J = 7.3 Hz, 1H), 5.30 (bs, 2H), 3.29 (d, J = 10.4 Hz, 1H), 3.20 – 3.12 (m, 1H), 3.08 – 2.97 (m, 2H), 2.78 (m, 1H), 2.70 (m, 1H), 2.61 – 2.47 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 165.6, 151.8, 133.6, 133.5, 132.4, 131.8, 128.5, 128.1, 127.6, 127.1, 122.3, 121.8, 118.2, 61.6, 53.4, 44.1, 34.2, 29.0 ppm. HRESIMS m/z 308.1219 [M+H]+ (calcd. for C18H18N3S, 308.1216)
6-methyl-5,6,6a,7-tetrahydro-4H-benzo[de]thiazolo[5’,4’:4,5]benzo[1,2-g]quinolin-10-amine (33b):
32% yield. Light yellow oil. 1H NMR (600 MHz, CDCl3): δ 7.80 (s, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.32 (s, 1H), 7.14 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 5.86 (s, 2H), 3.41 (d, J = 11.8 Hz, 1H), 3.13 (dd, J = 13.8, 4.1 Hz, 2H), 3.11 – 3.03 (m, 1H), 2.88 (m, 1H), 2.67 (m, 2H), 2.49 – 2.38 (m, 2H), 1.55 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3): δ 166.8, 151.5, 134.1, 134.0, 133.9, 133.7, 130.6, 129.5, 127.6, 126.8, 121.6, 118.4, 116.2, 59.5, 56.3, 49.3, 34.5, 29.1, 19.2, 12.2 ppm. HRESIMS m/z 336.1582 [M+H]+ (calcd. for C20H22N3S, 336.1529)
7-propyl-6a,7,8,9-tetrahydro-6H-benzo[de]thiazolo[4’,5’:5,6]benzo[1,2-g]quinolin-2-amine (34a):
35% yield. Brownish-white solid. Melting point: 198.1 °C – 199.5 °C. 1H NMR (500 MHz, CDCl3): δ 7.84 (s, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.34 (s, 1H), 7.17 (t, J = 7.6 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 5.50 (bs, 2H), 3.17 (m, 3H), 3.03 (m, 1H), 2.69 (m, 2H), 2.52 – 2.49 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ 166.4, 151.6, 133.9, 133.6, 133.5, 133.5, 130.9, 129.4, 127.6, 127.0, 121.6, 118.6, 116.3, 62.2, 53.4, 44.0, 34.6, 29.1 ppm. HRESIMS m/z 308.1219 [M+H]+ (calcd. for C18H18N3S, 308.1216)
6-propyl-5,6,6a,7-tetrahydro-4H-benzo[de]thiazolo[5’,4’:4,5]benzo[1,2-g]quinolin-10-amine (34b):
35% yield. Off-yellow solid. Melting point: 156.6 °C – 157.5 °C. 1H NMR (600 MHz, CDCl3): δ 7.80 (s, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.32 (s, 1H), 7.14 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 5.86 (s, 2H), 3.41 (d, J = 11.8 Hz, 1H), 3.17 – 3.11 (m, 2H), 3.07 (m, 1H), 2.88 (m, 1H), 2.66 (m, 2H), 2.49 – 2.38 (m, 2H), 1.55 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3): δ 166.8, 151.5, 134.1, 134.0, 133.9, 133.7, 130.6, 129.5, 127.6, 126.8, 121.6, 118.4, 116.2, 59.5, 56.3, 49.3, 34.5, 29.1, 19.2, 12.2 ppm. HRESIMS m/z 336.1534 [M+H]+ (calcd. for C20H22N3S, 336.1529)
4.2. Biological assay procedures
4.2.1. Receptor binding assays
All receptor binding assays were performed by the Psychoactive Drug Screening Program (PDSP). Complete details of the assays performed may be found online in the PDSP assay protocol book. (http://pdsp.med.unc.edu/PDSP%20Protocols%20II%202013-03-28.pdf).
4.2.2. 5-HT7R Hit Hunter® cAMP assays
Cell Handling
cAMP Hunter cell lines were expanded from freezer stocks according to standard procedures. Cells were seeded in a total volume of 20 μL in white-walled 384-well microplates and incubated at 37 °C for the appropriate time prior to testing. cAMP modulation was determined using the DiscoverX Hit Hunter cAMP XS+ assay
Gs agonist format
For agonist determination, cells were incubated with sample to induce response. Media was aspirated from cells and replaced with 15 μL 2:1 HBSS/10 mM Hepes: cAMP XS + Ab reagent. Intermediate dilution of sample stocks was performed to generate 4X sample in assay buffer. 5 μL of 4X sample was added to cells and incubated at 37 °C or room temperature for 30 or 60 minutes. Vehicle concentration was 1%.
Gs antagonist format
For antagonist determination, cells were pre-incubated with sample followed by agonist challenge at the EC80 concentration. Media was aspirated from cells and replaced with 10 μL 1:1 HBSS/HEPES:cAMP XS + Ab reagent. 5 μL of 4X compound was added to the cells and incubated at 37 °C or room temperature for 30 minutes. 5 μL of 4X EC80 agonist was added to the cells and incubated at 37 °C or room temperature for 30 or 60 minutes
Signal detection
After appropriate compound incubation, assay signal was generated through incubation with 20 μL cAMP XS + ED/CL lysis cocktail for one hour followed by incubation with 20 μL cAMP XS + EA reagent for three hours at room temperature. Microplates were read following signal generation with a Perkin-Elmer Envision TM instrument for chemiluminescent signal detection.
Data analysis
Compound activity was analyzed using CBIS data analysis suite (ChemInnovation, CA). For Gs agonist mode assays, percentage activity is calculated using the following formula: %Activity = 100% × (mean RLU of test sample – mean RLU of vehicle control)/(mean RLU of MAX control – mean RLU of vehicle control). For Gs antagonist mode, percentage inhibition is calculated using the following formula: %Inhibition =100%×(1-(mean RLU of test sample – mean RLU of vehicle control)/(mean RLU of EC80 control – mean RLU of vehicle control)).
Supplementary Material
Acknowledgements
This publication was made possible by Grant Number 1SC1DA049961-01 from the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions. Ki determinations, and receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP website http://pdsp.med.unc.edu/ and click on “Binding Assay” or “Functional Assay” on the menu bar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bard JA, Zgombick J, Adham N, Vaysse P, Branchek TA, Weinshank RL, Cloning of a novel human serotonin receptor (5-HT7) positively linked to adenylate cyclase, J Biol Chem, 268 (1993) 23422–23426. [PubMed] [Google Scholar]
- [2].Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, Foye PE, Racke M, Slone AL, Siegel BW, et al. , A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms, Neuron, 11 (1993) 449–458. [DOI] [PubMed] [Google Scholar]
- [3].Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC, Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation, Proc Natl Acad Sci U S A, 90 (1993) 8547–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sprouse J, Li X, Stock J, McNeish J, Reynolds L, Circadian rhythm phenotype of 5-HT7 receptor knockout mice: 5-HT and 8-OH-DPAT-induced phase advances of SCN neuronal firing, J Biol Rhythms, 20 (2005) 122–131. [DOI] [PubMed] [Google Scholar]
- [5].Faure C, Mnie-Filali O, Scarna H, Debonnel G, Haddjeri N, Effects of the 5-HT7 receptor antagonist SB-269970 on rat hormonal and temperature responses to the 5-HT1A/7 receptor agonist 8-OH-DPAT, Neurosci Lett, 404 (2006) 122–126. [DOI] [PubMed] [Google Scholar]
- [6].Monti JM, Jantos H, The role of serotonin 5-HT7 receptor in regulating sleep and wakefulness, Rev Neurosci, 25 (2014) 429–437. [DOI] [PubMed] [Google Scholar]
- [7].Heidmann DE, Metcalf MA, Kohen R, Hamblin MW, Four 5-hydroxytryptamine7 (5-HT7) receptor isoforms in human and rat produced by alternative splicing: species differences due to altered intron-exon organization, J Neurochem, 68 (1997) 1372–1381. [DOI] [PubMed] [Google Scholar]
- [8].Giulietti M, Vivenzio V, Piva F, Principato G, Bellantuono C, Nardi B, How much do we know about the coupling of G-proteins to serotonin receptors?, Mol Brain, 7 (2014) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thirumaran SL, Lepailleur A, Rochais C, Structure-activity relationships of serotonin 5-HT7 receptors ligands: A review, Eur J Med Chem, 183 (2019) 111705. [DOI] [PubMed] [Google Scholar]
- [10].Balcer OM, Seager MA, Gleason SD, Li X, Rasmussen K, Maxwell JK, Nomikos G, Degroot A, Witkin JM, Evaluation of 5-HT7 receptor antagonism for the treatment of anxiety, depression, and schizophrenia through the use of receptor-deficient mice, Behav Brain Res, 360 (2019) 270–278. [DOI] [PubMed] [Google Scholar]
- [11].Nikiforuk A, Targeting the Serotonin 5-HT7 Receptor in the Search for Treatments for CNS Disorders: Rationale and Progress to Date, CNS Drugs, 29 (2015) 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mnie-Filali O, Lambas-Senas L, Scarna H, Haddjeri N, Therapeutic potential of 5-HT7 receptors in mood disorders, Curr Drug Targets, 10 (2009) 1109–1117. [DOI] [PubMed] [Google Scholar]
- [13].Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL, R-(−)-N-alkyl-11-hydroxy-10-hydroxymethyl- and 10-methyl-aporphines as 5-HT1A receptor ligands, Bioorg Med Chem Lett, 17 (2007) 4128–4130. [DOI] [PubMed] [Google Scholar]
- [14].Gafner S, Dietz BM, McPhail KL, Scott IM, Glinski JA, Russell FE, McCollom MM, Budzinski JW, Foster BC, Bergeron C, Rhyu MR, Bolton JL, Alkaloids from Eschscholzia californica and their capacity to inhibit binding of [3H]8-Hydroxy-2-(di-N-propylamino)tetralin to 5-HT1A receptors in Vitro, J Nat Prod, 69 (2006) 432–435. [DOI] [PubMed] [Google Scholar]
- [15].Hedberg MH, Jansen JM, Nordvall G, Hjorth S, Unelius L, Johansson AM, 10-substituted 11-oxygenated (R)-aporphines: synthesis, pharmacology, and modeling of 5-HT1A receptor interactions, J Med Chem, 39 (1996) 3491–3502. [DOI] [PubMed] [Google Scholar]
- [16].Cannon JG, Moe ST, Long JP, Enantiomers of 11-hydroxy-10-methylaporphine having opposing pharmacological effects at 5-HT1A receptors, Chirality, 3 (1991) 19–23. [DOI] [PubMed] [Google Scholar]
- [17].Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL, Synthesis and binding studies of 2-O- and 11-O-substituted N-alkylnoraporphines, Bioorg Med Chem Lett, 18 (2008) 3971–3973. [DOI] [PubMed] [Google Scholar]
- [18].Hedberg MH, Linnanen T, Jansen JM, Nordvall G, Hjorth S, Unelius L, Johansson AM, 11-substituted (R)-aporphines: synthesis, pharmacology, and modeling of D2A and 5-HT1A receptor interactions, J Med Chem, 39 (1996) 3503–3513. [DOI] [PubMed] [Google Scholar]
- [19].Zhang H, Ye N, Zhou S, Guo L, Zheng L, Liu Z, Gao B, Zhen X, Zhang A, Identification of N-propylnoraporphin-11-yl 5-(1,2-dithiolan-3-yl)pentanoate as a new anti-Parkinson’s agent possessing a dopamine D2 and serotonin 5-HT1A dual-agonist profile, J Med Chem, 54 (2011) 4324–4338. [DOI] [PubMed] [Google Scholar]
- [20].Zhao R, Lu W, Fang X, Guo L, Yang Z, Ye N, Zhao J, Liu Z, Jia J, Zheng L, Zhao B, Zhang A, Zhen X, (6aR)-11-amino-N-propyl-noraporphine, a new dopamine D2 and serotonin 5-HT1A dual agonist, elicits potent antiparkinsonian action and attenuates levodopa-induced dyskinesia in a 6-OHDA-lesioned rat model of Parkinson’s disease, Pharmacol Biochem Behav, 124 (2014) 204–210. [DOI] [PubMed] [Google Scholar]
- [21].Liu Z, Chen X, Yu L, Zhen X, Zhang A, Synthesis and pharmacological investigation of novel 2-aminothiazole-privileged aporphines, Bioorg Med Chem, 16 (2008) 6675–6681. [DOI] [PubMed] [Google Scholar]
- [22].Liu Z, Chen X, Sun P, Yu L, Zhen X, Zhang A, N-Propylnoraporphin-11-O-yl carboxylic esters as potent dopamine D(2) and serotonin 5-HT(1A) receptor dual ligands, Bioorg Med Chem, 16 (2008) 8335–8338. [DOI] [PubMed] [Google Scholar]
- [23].Linnanen T, Brisander M, Unelius L, Rosqvist S, Nordvall G, Hacksell U, Johansson AM, Atropisomeric derivatives of 2’,6’-disubstituted (R)-11-phenylaporphine: selective serotonin 5-HT(7) receptor antagonists, J Med Chem, 44 (2001) 1337–1340. [DOI] [PubMed] [Google Scholar]
- [24].Linnanen T, Brisander M, Unelius L, Sundholm G, Hacksell U, Johansson AM, Derivatives of (R)-1,11-methyleneaporphine: synthesis, structure, and interactions with G-protein coupled receptors, J Med Chem, 43 (2000) 1339–1349. [DOI] [PubMed] [Google Scholar]
- [25].Madapa S, Harding WW, Semisynthetic Studies on and Biological Evaluation of NMethyllaurotetanine Analogues as Ligands for 5-HT Receptors, J Nat Prod, 78 (2015) 722–729. [DOI] [PubMed] [Google Scholar]
- [26].Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL, Synthesis and dopamine receptor affinities of N-alkyl-11-hydroxy-2-methoxynoraporphines: N-alkyl substituents determine D1 versus D2 receptor selectivity, J Med Chem, 51 (2008) 983–987. [DOI] [PubMed] [Google Scholar]
- [27].Gao YG, Ram VJ, Campbell A, Kula NS, Baldessarini RJ, Neumeyer JL, Synthesis and structural requirements of N-substituted norapomorphines for affinity and activity at dopamine D-1, D-2, and agonist receptor sites in rat brain, J Med Chem, 33 (1990) 39–44. [DOI] [PubMed] [Google Scholar]
- [28].Karki A, Juarez R, Namballa HK, Alberts I, Harding WW, Identification of C10 nitrogen-containing aporphines with dopamine D1 versus D5 receptor selectivity, Bioorg Med Chem Lett, 30 (2020) 127053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE, Structural basis for molecular recognition at serotonin receptors, Science, 340 (2013) 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.